Back to Archived Journals » Metalloproteinases In Medicine » Volume 4
Metalloproteinases in liver fibrosis: current insights
Authors Zbodakova O, Chalupsky K ![]() , Tureckova J, Sedlacek R
, Tureckova J, Sedlacek R
Received 11 October 2016
Accepted for publication 15 December 2016
Published 3 February 2017 Volume 2017:4 Pages 25—35
DOI https://doi.org/10.2147/MNM.S124363
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yoshifumi Itoh
Olga Zbodakova, Karel Chalupsky, Jolana Tureckova, Radislav Sedlacek
Laboratory of Transgenic Models of Diseases, Division BIOCEV, Institute of Molecular Genetics of the ASCR, v. v. i., Prague, Czech Republic
Abstract: Liver fibrosis is defined by excessive deposition of extracellular matrix. The fibrotic conditions result from the imbalance between synthesis and deposition of fibrous tissues and decomposition of these matrix proteins. This process can be reversed as a regular part of the healing process after hepatic damage or can become chronic. When the protein matrix synthesis predominates and the decomposition is suppressed, fibrosis will progress into irreversible cirrhosis or steatosis. Among the molecular players involved in fibrotic liver diseases, metalloproteinases of matrix metalloproteinase (MMP) and a disintegrin and metalloproteinase (ADAM) families are critical in the development of liver fibrosis and its resolution. Previously, MMPs were recognized as extracellular matrix degrading enzymes. Currently, they are also known as mediators in a variety of processes related to immunity and tissue repair. In this article, we have reviewed the models of liver fibrosis and findings on MMPs and ADAMs in hepatic fibrosis conditions.
Keywords: matrix metalloproteinase, ADAM, liver, fibrosis
Introduction
Studies on metalloproteinases have mainly focused on their role in the extracellular matrix decomposition and cell migration. More recently, it has come to light that the function of metalloproteinases is not limited to the degradation of extracellular matrix.1,2 This is exemplified by the family of a disintegrin and metalloproteinase (ADAM), which executes very specific proteolytic activities known as shedding.1,3
Metalloproteinases are involved in many distinct processes in the liver, the central organ for metabolism of carbohydrates, proteins, and lipids; removal of different pharmaceutical compounds and toxins from the blood; and for regulation of immune responses. The liver consists of different types of cells, mainly hepatocytes, endothelial cells, hepatic stellate cells (HSCs), bile duct cells (cholangiocytes), and Kupffer cells (liver specific resident macrophages). Hepatocytes are responsible for the majority of metabolic functions in the liver and are the most numerous cells of the liver, building a front line of detoxification and regeneration.4
The liver can develop a range of pathological conditions such as viral hepatitis, classic fibrosis, alcoholic and nonalcoholic fatty liver disease (NAFLD), steatosis, cirrhosis, biliary obstruction, and others. However, the liver also has a vast regenerative capacity as seen following a hepatectomy, toxic or ischemic injury, or acute infection.5 All the mentioned diseases involve multiple cellular and molecular players,6 including metalloproteinases, especially matrix metalloproteinase (MMP) and ADAM families. MMPs and ADAMs are facilitating extracellular matrix remodeling, cell migration and adhesion, as well as immune responses and regeneration.4,7,8
In all fibrotic models as well as human diseases, the induction of metalloproteinases expression and/or activity is documented. This is accompanied by elevated expression of their inhibitors, the tissue inhibitor of metalloproteinases (TIMPs), especially TIMP-1 and TIMP-2.9,10 Expression of MMPs in the liver is not restricted to a certain cell type; they are produced by Kupffer cells, all immune cells,11,12 HSCs, cholangiocytes,13 as well as hepatocytes.14 MMPs act as a part of a proteolytic network, in which proteases activate each other by processing inactive proforms. For instance, it has been shown that proMMP-2 is activated by MT-MMP-1 (MMP-14) in complex with TIMP-2,15 and further MMP-2 and MT-MMP-1 are activated by proMMP-13.16 In general, MMPs are important for initiation and establishment of the liver fibrosis and they are involved in the activation and invasiveness of HSCs, the producers of fibrotic matrix.17,18 Overall, the MMP and ADAM families of metalloproteinases orchestrate not only the processes of fibrosis development and its resolution but also the inflammatory and regeneration of the liver.
Liver models
Animal models of liver fibrosis
The liver has a complex architecture with many specialized cell types and niches. Moreover, the liver is extremely well interconnected with the whole body, maintaining body’s homeostasis and controlling central immune responses. The complex of many functionally different cell types and their different reactions to body conditions are the main reasons why it is impossible to accurately mimic liver function and cell-cell interaction in vitro and why animal models are needed to study liver pathophysiology.
Fibrosis in animal models can be induced by administration of hepatotoxins, bile duct ligation (BDL), and using genetic models. To date, several reviews describing animal models for fibrosis induction have been published.4,8,19–21 In this review, we recapitulate their findings regarding metalloproteinases of MMP and ADAM families on the background of function of various liver cell types.
Models for centrizonal hepatocyte damage
These models primarily target hepatocytes in the area around the central vein where the first hepatocytes with the highest concentration of hydroxylating cytochrome P450 involved in detoxification are located.22 The models include chemically induced fibrosis with toxic compounds, mostly carbon tetrachloride (CCL4), thioacetamide (TAA), dialkylated nitrosamines, and others (Table 1). These chemicals are usually applied intraperitoneally and their major advantage is that after the acute phase caused by the single-dose application, a full recovery of liver tissue can be achieved. Repeated applications of these substances result in pericentral fibrosis, later bridging pericentral zones, which further develop into irreversible cirrhosis and carcinoma.4,19
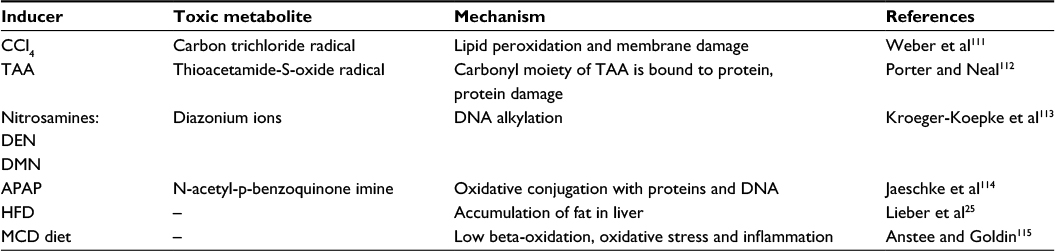 | Table 1 Hepatic toxins used in animal models Abbreviations: CCl4, carbon tetrachloride; TAA, thioacetamide; DEN, diethylnitrosamine; DMN, dimethylnitrosamine; APAP, acetaminophen; HFD, high-fat diet; MCD diet, a diet deprived of methionine and choline. |
The next group of models is based on special dietary conditions, under which the damaging compounds target the liver via blood stream. These models are mainly mimicking the chronic liver damage, which is more reminiscent of liver injury in humans. Modified mouse diet is mainly used to induce nonalcoholic steatohepatitis (NASH) and NAFLD.23,24 In general, NASH and NAFLD are called silent diseases with few or no symptoms during their progression but resulting in irreversible liver damage after years in humans. Since NASH and NAFLD manifest mostly in adults and the only cure is liver transplantation, extensive efforts have been made to reveal the mechanisms involved in the development of these disorders and their treatment.
NAFLD is a multiorgan disease, which also affects the cardiovascular system, kidney, and pancreas. Accumulation of excess fat in the liver cells, which initiates NAFLD, is the first clinical sign. Thus, high-fat diet is used to induce fatty liver progression in rodents,25 which then exhibits obesity, high insulin levels, and insulin resistance.23 Upon this diet, steatosis develops not only in pericentral zones but also in periportal zones, which are specialized in gluconeogenesis.26
In the NASH studies, a diet deprived of methionine and choline (MCD diet), which are needed for beta-oxidation, is used. Rodents fed with MCD develop pericentral fibrosis with necrosis. As NASH is characterized by liver damage involving inflammation paralleled with oxidative stress, this model also displays metabolic syndrome-like features.24,27 MCD-induced NASH is considered to be a more severe NAFLD model, which develops irreversible damage of the liver in a short time.
Liver injury induced by Schistosoma sp. is another model that could be used to study periportal fibrosis. Parasite eggs are deposited in an area with high blood flow – small portal venules.28 Deposition of parasite eggs triggers accumulation of collagen that initially blocks the release of antigen from parasites and later causes periportal fibrosis, liver cirrhosis, and organ failure.29 Schistosoma mansoni infection of mice mimics the pathological observations in humans.30
Models to target cholangiocytes
Cholangiocytes are epithelial cells of bile ducts that bring the bile, a toxic product of liver, into the gall bladder. BDL is a well-known model of biliary fibrosis where the bile outflow from the liver is interrupted and the bile accumulates in the bile ducts.13 The backpressure and toxicity of bile acids are the major causes of proliferation of the duct cells and their spreading into liver parenchyma. Subsequently, Kupffer cells migrate to the bile ducts causing portal inflammation and necrosis, thus leading to fibrosis. This model is irreversible in mice; however, it is possible to reconnect the bile duct in rat models.31 Due to wound healing after surgery, this model is usually performed in studies focused on chronic liver injury. Probably, the most important modification of the BDL model is partial BDL (pBDL).32 Decreased necrosis and proliferation rates have been reported in pBDL model, which makes pBDL more relevant to human bile duct obstruction than classical BDL. In addition, pBDL is used to simulate acute cholestasis, whereas BDL is considered to be the model for chronic stage. Another nonsurgical model where obstruction of bile ducts and proliferation of cholangiocytes occur is based on dietary model using 3,5-diethoxycarbonyl-1,4-dihydrocollidine-enriched diet.33 In this model, porphyrin crystals block small intrahepatic bile ducts and large bile ducts remain usually unaffected.
Targeting Kupffer cells
Kupffer cells are liver-specific macrophages responsible for the release of many cytokines during liver injury, especially tumor necrosis factor alpha (TNFa). Experimental treatment of animals with antibodies specific to TNFa reduced induced liver injury.34 Similarly, dexamethasone, coupled to mannosylated albumin (Dexa(5)-Man(10)-HSA), specifically delivered to Kupffer cells, reduced inflammation and liver damage.35 On the contrary, nonspecific activation of all immune cells including Kupffer cells is a hallmark of autoimmune human diseases. To stimulate immune cells in the liver, concanavalin A can be applied intraperitoneally.36 High levels of cytokines generated during the immune response cause acute hepatocyte damage. Such an acute immune cell activation may serve as a model of autoimmune hepatitis37 and for studying the role of immune cells in liver injury and regeneration.38
Unspecific overall liver damage
During viral hepatitis, no primary cell target can be determined in the liver. Human and chimpanzee are the only two species susceptible to human hepatitis B virus (HBV) infection; murine hepatocytes cannot be infected by human hepatitis as mice lack receptors for human viral particles.39 The first model of HBV is transgenic mouse, in which the surface antigen (HBsAg) and HBV e antigen were found in the serum.40 Another model is based on injection of high amounts of plasmid bearing the viral genome into the blood stream.41 Similar to toxic models, where the liver is the first affected organ, high replication rate is preserved up to 80 days in immunodeficient mice. Infection with adenovirus containing HBV genome is a model consisting of both adaptive and innate systems.42 An alternative to this model is human liver chimeric mouse model,43 in which the mouse hepatocytes are replaced by transplanted human hepatocytes; however, preparation of this chimeric mouse is challenging. Finally, humanized mice with human immune system and hepatocytes is the only model, which upon infection triggers responses similar to humans, i.e., hepatitis and fibrosis.44–47
Genetically modified mice
Genetically modified animals are becoming a more frequent tool to study liver pathophysiology as well as the roles of metalloproteinases. The liver consists of several cell types and it is not easy to target them specifically. To study specific functions in hepatocytes, the albumin promoter is often used in combination with Cre recombinase, although it partially affects cholangiocytes.48 The transgenic mouse with the Cre-driven lecithin-retinol acyltransferase promoter targets 99% of HSCs. This recent model of cell-specific transgene has been used to demonstrate that HSCs give rise to 82%–96% of myofibroblasts in models of toxic, cholestatic, and fatty liver disease.49 The Mx1-Cre model50 expresses Cre recombinase under the control of an inducible Mx1 promoter, which is silent in healthy mice and active in the liver and in lymphocytes after stimulation with interferon alpha or polyinosinic:polycytidylic acid. Thus, mice with gene deficiencies in both hepatocytes and immune cells can be studied. To target myeloid cell lineage (monocytes, mature macrophages, and granulocytes) including the Kupffer cells, a model with Cre recombinase activity under the control of Lyz2 promoter/enhancer elements is often used.51 However, since Lyz2 promoter-based recombinase is not specific only for Kupffer cells but also affects floxed alleles in all myeloid cells, targeting the immune compartment cannot be completely restricted just to the liver or to Kupffer cells only.
Regarding the ablation of gene functions in the liver, only a few genetically modified mice develop spontaneous liver injury. Interestingly, metalloproteinase-deficient mice do not display any obvious liver damage; ubiquitous deletion is either lethal or mostly with minor or no effects.6 The reason for that is likely the redundancy of metalloproteinases and relatively large substrate promiscuity. Genes whose deletion leads to spontaneous liver phenotype are listed in Table 2.
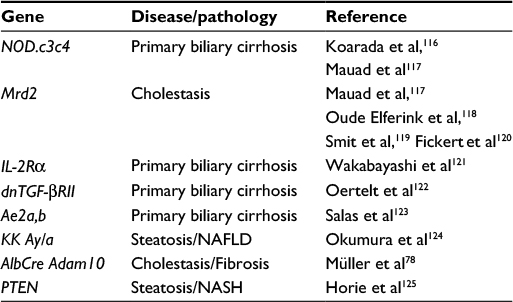 | Table 2 List of genes involved in spontaneous liver damage Abbreviations: NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis. |
Metalloproteinases in the liver
Metalloproteinases in the development of liver fibrosis
All fibrotic models are accompanied by elevated expression of MMP-2 and MMP-9 (Table 3). These two MMPs are called gelatinases as they cleave collagen-IV besides other basal membrane proteins. MMP-2 has been reported to be important not only in establishment of CCL4-induced fibrosis52 but also in the resolution phase when fibrotic matrix is degraded and then replaced by the natural matrix components.53,54 Thus, MMP-2-deficient mice show enhanced fibrosis after CCL4 treatment and MMP-2 could be therefore critical for inhibiting type I collagen synthesis by activated HSCs, which strongly express this MMP.55,56 MMP-9 and MMP-2 are elevated in serum and livers of patients with chronic hepatitis type B and C.57–59 Elevated expression of MMPs in the mentioned fibrotic conditions was shown both in liver parenchyma60 and portal macrophages.57,61 Especially, MMP-9 seems to be involved in ongoing liver inflammation,62 and its level drops after antiviral treatment.57,63 Induction of MMP-2 is significantly stimulated in the liver of HCV-infected patients.64
 | Table 3 MMPs and TIMPs involved in mouse fibrosis Abbreviations: MMP, matrix metalloproteinase; TIMP, tissue inhibitor of metalloproteinase; ADAM, a disintegrin and metalloproteinase; BDL, bile duct ligation; DDC, 5-diethoxycarbonyl-1,4-dihydrocollidine; MCD diet, a diet deprived of methionine and choline. |
Although it appears that MMP-9 and MMP-2 induction in fibrosis is the most prominent among MMPs, there are also other MMPs that are able to cleave collagen-IV and other basement membrane components. MMP-19 deficiency causes fibrosis exacerbation in chronic fibrosis models induced by CCL4 application.65 MMP-12 is also involved in the regulation of fibrotic matrix processing as the oral administration of TAA leads not only to accumulation of elastin and increased fibrosis in MMP-12-deficient mice, but also to higher overall fibrosis.66 Interestingly, these mice did not exhibit difference in overall fibrosis compared to control animals if treated by CCL4, although increased perisinusoidal elastin was still detected. This points to a distinct mode of action of toxins used to induce fibrosis. The role of MMP-8 in fibrosis development is still not clearly understood, but it could facilitate HSC migration and invasion, suggesting a potential profibrotic role.67
Apart from being matrix cleavage proteases, MMPs also participate directly in certain signaling cascades. Both MMP-9 and MMP-2, with their ability to activate transforming growth factor beta (TGFβ), participate in the induction of this main profibrogenic pathway2 as TGFβ is crucial for the differentiation of quiescent HSC into collagen-I producing myofibroblast. It also appears that the activation of HSC in three-dimensional culture is dependent on gelatinolytic activity.68 MMP-19 seems to be involved in TGFβ signaling as well, as MMP-19 knockout hepatocytes exhibit lower response to the cytokine.65 Also, MT1-MMP participates in the regulation of fibrosis progression as it cleaves intramolecular RGD motifs of collagen produced by activated HSCs and thus controls αVβ1 activation, providing a survival signal via PI3K/AKT/IκB.69
During the entire progress of liver fibrosis, the expression of main inhibitor of MMPs, TIMP-1, is increased to regulate the elevated MMP activity. TIMP-1 has distinct functions in establishment and resolution of fibrosis. This inhibitor may be beneficial at the beginning of the fibrotic tissue formation; yet by inhibiting MMPs during the recovery phase, it may slow down decomposition of collagen-I. Results from animal models are intriguing as both transgenic mice, the overexpressing TIMP-1 mice70 and TIMP-1 knockout mice, developed more prominent fibrosis after CCL4 intoxication.9 Those contradictory observations might be explained by specific engagement of TIMP-1 in different signaling pathways. TIMP-1 is able to inhibit HSC apoptosis,71 suggesting profibrotic function of TIMP-1. Yet, silencing of TIMP-1 reduces HSC proliferation72 and, moreover, TIMP-1 inhibits apoptosis of hepatocytes.9 Probably, loss of this hepatoprotective function in TIMP-1 knockout results in exacerbated fibrosis. Still, expression of TIMP-1 and TIMP-2 is elevated in HCV patients,57 and some studies examined blockage of TIMP-1 as potential therapeutic treatment for patients with cirrhosis. In one case, TIMP-1 expression was reduced by adenoviral delivery of siRNA.73 Another approach was inhibiting TIMP-1 activity by MMP-9 proteolytic inactive mutants.74 In both studies, the treated animals developed less severe fibrosis, which indicates that inhibition of TIMP-1 can be beneficial in liver fibrosis. By contrast, complete ablation leads to opposite result. TIMP-1 appears to be a key player in liver fibrosis, and an important potential therapeutic target as TIMP-1 might promote liver fibrosis by yet another means than its previously described antiapoptotic effect on HSC.72
ADAM10 plays an important role in development and organization of epithelial tissues and is the main alpha-secretase cleaving Notch75,76 The whole-body ADAM10-deficient mice die prenatally due to limited Notch signaling that results in vasculature failure and numerous other developmental problems.77 However, specific ablation of ADAM10 in hepatocytes did not exhibit alteration in Notch signal,78 although some percentage of those animals have problems with liver homeostasis manifested by necrosis of hepatocytes and subsequent fibrosis.78
MMPs in recovery of liver fibrosis
During recovery from fibrosis, removal of the scar tissue formed by collagen-I is needed. In rodents, collagen-I is cleaved predominantly by MMP-1379 and MMP-8.80 The expression of MMP-13 peaks during resolution phase.81 Other MMPs such as MMP-2 and MMP-14 are also upregulated in this phase,82 by likely participating in restoration of the healthy tissue. MMP-13 is mainly expressed by macrophages that infiltrate the fibrotic lesions.81 MMP-13-deficient mice suffer from hampered recovery of CCl4-induced fibrosis.81 This finding is in line with adenovirus-mediated MMP-13 expression, which help to resolve the established fibrosis.83 Apart from direct collagenolytic activity, overexpression of MMP-13 includes healing promoting signals as hepatocyte growth factor (HGF) expression is elevated and activity of MMP-2 and MMP-9 is increased. Similarly, application of adenoviruses carrying MMP-884 or MMP-1, the main human collagenase,85 had beneficial effect in resolution of induced fibrosis.
Another part of recovery from fibrosis is elimination of activated HSCs by apoptosis, and some MMPs are suggested to participate in this process according to several in vitro studies.86,87 In agreement with the mentioned role of TIMP-1 in HSC apoptosis,71 MMP-2 can promote HSC apoptosis through cleavage of N-cadherin,86 typical marker of activated myofibroblasts. Recently, the role of degradation products produced by MMPs in regulation of fibrosis has been proposed; cleaved fibronectin peptides induced apoptosis in HSC line and stimulated MMP-9 secretion from human monocytes.87
Inflammation and regulation of TNFα signaling in liver injury
ADAM17 was originally discovered as a protease effectively cleaving TNFα and is therefore also known as TNFα-converting enzymes. TNFα is pivotal in many pathological conditions of the liver and has a distinct effect on hepatocytes depending on the strength and duration of a signal. Experimental approaches against TNFα in models of liver fibrosis have shown reduction of liver injury.34,35,88 TNFα in liver, mainly produced by Kupffer cells,89 binds to TNFα receptors (TNFR) on hepatocytes and Kupffer cells after its release. ADAM17 is a key player in this signaling, and mice lacking ADAM17 specifically in myeloid lineage release almost no TNFα into serum after lipopolysaccharide application.90 Interestingly, ADAM17 ablation in hepatocytes also resulted in reduced shedding of TNFα into serum, suggesting that both cell types cooperate during release of TNFα in the liver. ADAM17 also mediates shedding of TNFR and thus affects activation of the TNFα downstream targets. Subsequently, only coordinated shedding of both, TNFα and TNFR, dictates outcome of the signaling.91
In liver diseases, TNFα plays both protective and damaging roles. High levels of TNFα produced in liver after CCl4 injection contribute to apoptosis of hepatocytes,92 and neutralization with TNFα antibody showed to be beneficial in this type of injury. Interestingly, with too high doses of neutralizing antibody, beneficial effect disappeared resulting in even higher damage than without antibody.93 This suggests that a small amount of TNFα has protective effect during liver intoxication. ADAM17 mice deficient in myeloid lineage shed lower amount of TNFα after CCl4 application compared to WT, leading to lower release of hepatocyte damage markers into serum.91 However, since this disruption of TNFα signaling could not prevent liver damage as apoptosis and necrosis of hepatocytes were not altered in ADAM17 null mice, CCL4 causes more complex reaction, which is not controlled by TNFα signaling.
Regulator of growth factor signaling in the liver
Epidermal growth factor receptor (EGFR) pathway represents an important signaling pathway in the liver as it protects hepatocytes from apoptosis,94 and it is necessary for hepatocyte division.95 Epidermal growth factor (EGF), amphiregulin, heparin-binding EGF (HB-EGF), TGFα, epiregulin, and betacellulin are all ligands of EGFR and their bioavailability is provided by ADAM proteases.96 Individual ligands have different sources; EGF is mainly produced in duodenum, HB-EGF is produced by Kupffer cells, and amphiregulin and TGFα are made by hepatocytes and HSCs, but generally all have similar effects in liver pathology. They promote proliferation of hepatocytes after liver challenge as reported in many rodent models.97–102
EGFR is expressed in both the parenchymal and non-parenchymal liver cells, and several studies have reported that amphiregulin enhances liver fibrosis through its mitogenic effect on HSCs.102,103 These effects, protecting hepatocytes as well as profibrotic HSCs, could account for the fact that ADAM17 hepatocyte-deficient mice do not develop exaggerated fibrosis after intoxication with CCl4 (our observation, unpublished). Similarly, despite certain dysregulations, no major liver regeneration phenotypes were observed in mice with ADAM17 deficiency in either hepatocytes or Kupffer cells.91
Regulation of the EGFR signaling by ADAMs in liver is complex and is not fully understood so far. One issue is certain redundancy between EGFR ligands, some of which can be cleaved not only by ADAM17 but also by other ADAMs, e.g., ADAM10104 and ADAM12.105 Moreover, EGFR ligands are produced and shed by different liver cell types, not just hepatocytes. Thus, deficient models that would distinguish among hepatocytes, cholangiocytes, and HSCs would be useful.
Even though the EGFR signaling plays a significant role in liver homeostasis, it can be quite efficiently compensated by the pathway of HGF and its receptor MET, which is regulated via shedding by both ADAM10 and ADAM17.106 This regulation does not occur through shedding of ligands as in EGFR but occurs via shedding of the receptor itself. By this action, HGF/MET signaling is disrupted as MET is no longer available on the targeted cells, although the soluble MET can serve as a decoy receptor.106
Recent studies have shown that combined disruption of both EGFR and MET signaling but not individual pathways, leads to dramatic effects compromising liver recovery from partial hepatectomy and aggravating necrosis after intoxication.105,107–109 Even though MET and EGFR pathways both provide mitogenic signals to hepatocytes, they have distinct roles in differentiation of hepatic progenitor cells (HPCs), also called oval cells. Kitade et al110 showed that MET induced HPC differentiation into hepatocytes, while EGFR signaling led to Notch1-dependent differentiation of cholangiocytes. Roles of ADAMs in this process remain to be established. First hints can be seen in study of ADAM10 knockout targeted to hepatocytes, cholangiocytes, and HPC.78 These animals developed spontaneous fibrosis, which results from hepatocyte necrosis caused by high bile acid content in the blood.
Conclusion
Metalloproteinases are involved in many different processes, playing the central role in tissue damage and remodeling. However, they can function as a double-edged sword. Inhibiting their activity could prevent inflammation and accumulation of the extracellular matrix, but it could also hamper the healing process. Therefore, comprehensive knowledge of the individual functions of metalloproteinases is needed. Nevertheless, as the MMP and ADAM families of metalloproteinases work in the proteolytic network and exhibit number of functional redundancies and substrate promiscuity, it could be beneficial to study several metalloproteinases in parallel to decipher their functional complexity. Moreover, specific attention should be devoted to functional differences in various cell types. All this knowledge could be translated into more feasible therapeutic interventions.
Acknowledgment
The authors are grateful to Nicole Chambers for proofreading the article. Financial support was given to RS by Ministry of Education, Youth and Sports of the Czech Republic, (NPU II project LQ1604).
Disclosure
The authors report no conflicts of interest in this work.
References
Weber S, Saftig P. Ectodomain shedding and ADAMs in development. Development. 2012;139(20):3693–3709. | ||
Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14(2):163–176. | ||
van Goor H, Melenhorst WB, Turner AJ, Holgate ST. Adamalysins in biology and disease. J Pathol. 2009;219(3):277–286. | ||
Duarte S, Baber J, Fujii T, Coito AJ. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015;44–46:147–156. | ||
Suzuki Y, Katagiri H, Wang T, et al. Ductular reactions in the liver regeneration process with local inflammation after physical partial hepatectomy. Lab Invest. 2016;96(11):1211–1222. | ||
Giannandrea M, Parks WC. Diverse functions of matrix metalloproteinases during fibrosis. Dis Model Mech. 2014;7(2):193–203. | ||
Arthur MJ. Fibrogenesis II. Metalloproteinases and their inhibitors in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2000;279(2):G245–G249. | ||
Tuñón MJ, Alvarez M, Culebras JM, González-Gallego J. An overview of animal models for investigating the pathogenesis and therapeutic strategies in acute hepatic failure. World J Gastroenterol. 2009;15(25):3086–3098. | ||
Wang H, Lafdil F, Wang L, Yin S, Feng D, Gao B. Tissue inhibitor of metalloproteinase 1 (TIMP-1) deficiency exacerbates carbon tetrachloride-induced liver injury and fibrosis in mice: involvement of hepatocyte STAT3 in TIMP-1 production. Cell Biosci. 2011;1(1):14. | ||
Hemmann S, Graf J, Roderfeld M, Roeb E. Expression of MMPs and TIMPs in liver fibrosis – a systematic review with special emphasis on anti-fibrotic strategies. J Hepatol. 2007;46(5):955–975. | ||
Nielsen BS, Timshel S, Kjeldsen L, et al. 92 kDa type IV collagenase (MMP-9) is expressed in neutrophils and macrophages but not in malignant epithelial cells in human colon cancer. Int J Cancer. 1996;65(1):57–62. | ||
Yu Z, Xie M, Fan X, Jia J. Interferon α2b increases MMP-13 and IL-10 expression in Kupffer cells through MAPK signaling pathways. Hepatogastroenterology. 2015;62(138):350–354. | ||
Tag CG, Sauer-Lehnen S, Weiskirchen S, et al. Bile duct ligation in mice: induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J Vis Exp. 2015;(96):52438. | ||
Calabro SR, Maczurek AE, Morgan AJ, et al. Hepatocyte produced matrix metalloproteinases are regulated by CD147 in liver fibrogenesis. PLoS One. 2014;9(7):e90571. | ||
Wang Z, Juttermann R, Soloway PD. TIMP-2 is required for efficient activation of proMMP-2 in vivo. J Biol Chem. 2000;275(34):26411–26415. | ||
Knäuper V, Will H, López-Otin C, et al. Cellular mechanisms for human procollagenase-3 (MMP-13) activation. Evidence that MT1-MMP (MMP-14) and gelatinase a (MMP-2) are able to generate active enzyme. J Biol Chem. 1996;271(29):17124–17131. | ||
Galli A, Svegliati-Baroni G, Ceni E, et al. Oxidative stress stimulates proliferation and invasiveness of hepatic stellate cells via a MMP2-mediated mechanism. Hepatology. 2005;41(5):1074–1084. | ||
Han S, Han L, Yao Y, Sun H, Zan X, Liu Q. Activated hepatic stellate cells promote hepatocellular carcinoma cell migration and invasion via the activation of FAK-MMP9 signaling. Oncol Rep. 2014;31(2):641–648. | ||
Liedtke C, Luedde T, Sauerbruch T, et al. Experimental liver fibrosis research: update on animal models, legal issues and translational aspects. Fibrogenesis Tissue Repair. 2013;6(1):19. | ||
Liu Y, Meyer C, Xu C, et al. Animal models of chronic liver diseases. Am J Physiol Gastrointest Liver Physiol. 2013;304(5):G449–G468. | ||
Rahman TM, Hodgson HJ. Animal models of acute hepatic failure. Int J Exp Pathol. 2000;81(2):145–157. | ||
Jungermann K, Kietzmann T. Zonation of parenchymal and nonparenchymal metabolism in liver. Annu Rev Nutr. 1996;16:179–203. | ||
Nakamura A, Terauchi Y. Lessons from mouse models of high-fat diet-induced NAFLD. Int J Mol Sci. 2013;14(11):21240–21257. | ||
Larter CZ, Yeh MM. Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol. 2008;23(11):1635–1648. | ||
Lieber CS, Leo MA, Mak KM, et al. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. 2004;79(3):502–509. | ||
Willebrords J, Pereira IV, Maes M, et al. Strategies, models and biomarkers in experimental non-alcoholic fatty liver disease research. Prog Lipid Res. 2015;59:106–125. | ||
Gaemers IC, Stallen JM, Kunne C, et al. Lipotoxicity and steatohepatitis in an overfed mouse model for non-alcoholic fatty liver disease. Biochim Biophys Acta. 2011;1812(4):447–458. | ||
Boros DL. Immunopathology of Schistosoma mansoni infection. Clin Microbiol Rev. 1989;2(3):250–269. | ||
Wynn TA, Thompson RW, Cheever AW, Mentink-Kane MM. Immunopathogenesis of schistosomiasis. Immunol Rev. 2004;201:156–167. | ||
Andrade ZA. Schistosomiasis and liver fibrosis. Parasite Immunol. 2009;31(11):656–663. | ||
Popov Y, Sverdlov DY, Bhaskar KR, et al. Macrophage-mediated phagocytosis of apoptotic cholangiocytes contributes to reversal of experimental biliary fibrosis. Am J Physiol Gastrointest Liver Physiol. 2010;298(3):G323–G334. | ||
Heinrich S, Georgiev P, Weber A, Vergopoulos A, Graf R, Clavien PA. Partial bile duct ligation in mice: a novel model of acute cholestasis. Surgery. 2011;149(3):445–451. | ||
Fickert P, Stöger U, Fuchsbichler A, et al. A new xenobiotic-induced mouse model of sclerosing cholangitis and biliary fibrosis. Am J Pathol. 2007;171(2):525–536. | ||
Ponnappa BC, Israel Y. Targeting Kupffer cells with antisense oligonucleotides. Front Biosci. 2002;7:e223–e233. | ||
Melgert BN, Olinga P, Van Der Laan JM, et al. Targeting dexamethasone to Kupffer cells: effects on liver inflammation and fibrosis in rats. Hepatology. 2001;34(4 Pt 1):719–728. | ||
Gantner F, Leist M, Lohse AW, Germann PG, Tiegs G. Concanavalin A-induced T-cell-mediated hepatic injury in mice: the role of tumor necrosis factor. Hepatology. 1995;21(1):190–198. | ||
Wang HX, Liu M, Weng SY, et al. Immune mechanisms of concanavalin A model of autoimmune hepatitis. World J Gastroenterol. 2012;18(2):119–125. | ||
Heymann F, Hamesch K, Weiskirchen R, Tacke F. The concanavalin A model of acute hepatitis in mice. Lab Anim. 2015;49(1 Suppl):12–20. | ||
Yang J, Xi Q, Deng R, Wang J, Hou J, Wang X. Identification of interspecies recombination among hepadnaviruses infecting cross-species hosts. J Med Virol. 2007;79(11):1741–1750. | ||
Akbar SK, Onji M. Hepatitis B virus (HBV)-transgenic mice as an investigative tool to study immunopathology during HBV infection. Int J Exp Pathol. 1998;79(5):279–291. | ||
Yang PL, Althage A, Chung J, Chisari FV. Hydrodynamic injection of viral DNA: a mouse model of acute hepatitis B virus infection. Proc Natl Acad Sci U S A. 2002;99(21):13825–13830. | ||
Sprinzl MF, Oberwinkler H, Schaller H, Protzer U. Transfer of hepatitis B virus genome by adenovirus vectors into cultured cells and mice: crossing the species barrier. J Virol. 2001;75(11):5108–5118. | ||
Ilan E, Burakova T, Dagan S, et al. The hepatitis B virus-trimera mouse: a model for human HBV infection and evaluation of anti-HBV therapeutic agents. Hepatology. 1999;29(2):553–562. | ||
Bility MT, Zhang L, Washburn ML, Curtis TA, Kovalev GI, Su L. Generation of a humanized mouse model with both human immune system and liver cells to model hepatitis C virus infection and liver immunopathogenesis. Nat Protoc. 2012;7(9):1608–1617. | ||
Keng CT, Sze CW, Zheng D, et al. Characterisation of liver pathogenesis, human immune responses and drug testing in a humanised mouse model of HCV infection. Gut. 2016;65(10):1744–1753. | ||
Billerbeck E, Mommersteeg MC, Shlomai A, et al. Humanized mice efficiently engrafted with fetal hepatoblasts and syngeneic immune cells develop human monocytes and NK cells. J Hepatol. 2016;65(2):334–343. | ||
Bility MT, Cheng L, Zhang Z, et al. Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: induction of human-specific liver fibrosis and M2-like macrophages. PLoS Pathog. 2014;10(3):e1004032. | ||
Postic C, Shiota M, Niswender KD, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using cre recombinase. J Biol Chem. 1999;274(1):305–315. | ||
Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. | ||
Kühn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427–1429. | ||
Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8(4):265–277. | ||
Takahara T, Furui K, Funaki J, et al. Increased expression of matrix metalloproteinase-II in experimental liver fibrosis in rats. Hepatology. 1995;21(3):787–795. | ||
Issa R, Zhou X, Constandinou CM, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology. 2004;126(7):1795–1808. | ||
Watanabe T, Niioka M, Ishikawa A, et al. Dynamic change of cells expressing MMP-2 mRNA and MT1-MMP mRNA in the recovery from liver fibrosis in the rat. J Hepatol. 2001;35(4):465–473. | ||
Onozuka I, Kakinuma S, Kamiya A, et al. Cholestatic liver fibrosis and toxin-induced fibrosis are exacerbated in matrix metalloproteinase-2 deficient mice. Biochem Biophys Res Commun. 2011;406(1):134–140. | ||
Radbill BD, Gupta R, Ramirez MC, et al. Loss of matrix metalloproteinase-2 amplifies murine toxin-induced liver fibrosis by upregulating collagen I expression. Dig Dis Sci. 2011;56(2):406–416. | ||
Latronico T, Mascia C, Pati I, et al. Liver fibrosis in HCV monoinfected and HIV/HCV coinfected patients: dysregulation of matrix metalloproteinases (MMPs) and their tissue inhibitors TIMPs and effect of HCV protease inhibitors. Int J Mol Sci. 2016;17(4):455. | ||
Akca G, Tunçbilek S, Sepici-Dinçel A. Association between matrix metalloproteinase (MMP)-2, MMP-9 and total antioxidant status of patients with asymptomatic hepatitis C virus infection. Lett Appl Microbiol. 2013;57(5):436–442. | ||
Sumer S, Aktug Demir N, Kölgelier S, et al. The clinical significance of serum apoptotic cytokeratin 18 neoepitope M30 (CK-18 M30) and matrix metalloproteinase 2 (MMP-2) levels in chronic hepatitis B patients with cirrhosis. Hepat Mon. 2013;13(6):e10106. | ||
D’Amico F, Consolo M, Amoroso A, et al. Liver immunolocalization and plasma levels of MMP-9 in non-alcoholic steatohepatitis (NASH) and hepatitis C infection. Acta Histochem. 2010;112(5):474–481. | ||
Gadd VL, Melino M, Roy S, et al. Portal, but not lobular, macrophages express matrix metalloproteinase-9: association with the ductular reaction and fibrosis in chronic hepatitis C. Liver Int. 2013;33(4):569–579. | ||
Reif S, Somech R, Brazovski E, et al. Matrix metalloproteinases 2 and 9 are markers of inflammation but not of the degree of fibrosis in chronic hepatitis C. Digestion. 2005;71(2):124–130. | ||
Kennedy A, Hennessy M, Bergin C, Mulcahy F, Hopkins S, Spiers JP. Ribavirin and interferon alter MMP-9 abundance in vitro and in HIV-HCV-coinfected patients. Antivir Ther. 2011;16(8):1237–1247. | ||
Li Y, Zhang Q, Liu Y, et al. Hepatitis C virus activates Bcl-2 and MMP-2 expression through multiple cellular signaling pathways. J Virol. 2012;86(23):12531–12543. | ||
Jirouskova M, Zbodakova O, Gregor M, et al. Hepatoprotective effect of MMP-19 deficiency in a mouse model of chronic liver fibrosis. PLoS One. 2012;7(10):e46271. | ||
Pellicoro A, Aucott RL, Ramachandran P, et al. Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology. 2012;55(6):1965–1975. | ||
Baig MS, Yaqoob U, Cao S, Saqib U, Shah VH. Non-canonical role of matrix metalloprotease (MMP) in activation and migration of hepatic stellate cells (HSCs). Life Sci. 2016;155:155–160. | ||
Han YP, Zhou L, Wang J, et al. Essential role of matrix metalloproteinases in interleukin-1-induced myofibroblastic activation of hepatic stellate cell in collagen. J Biol Chem. 2004;279(6):4820–4828. | ||
Birukawa NK, Murase K, Sato Y, et al. Activated hepatic stellate cells are dependent on self-collagen, cleaved by membrane type 1 matrix metalloproteinase for their growth. J Biol Chem. 2014;289(29):20209–20221. | ||
Yoshiji H, Kuriyama S, Miyamoto Y, et al. Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model. Hepatology. 2000;32(6):1248–1254. | ||
Murphy FR, Issa R, Zhou X, et al. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: implications for reversibility of liver fibrosis. J Biol Chem. 2002;277(13):11069–11076. | ||
Fowell AJ, Collins JE, Duncombe DR, Pickering JA, Rosenberg WM, Benyon RC. Silencing tissue inhibitors of metalloproteinases (TIMPs) with short interfering RNA reveals a role for TIMP-1 in hepatic stellate cell proliferation. Biochem Biophys Res Commun. 2011;407(2):277–282. | ||
Cong M, Liu T, Wang P, et al. Antifibrotic effects of a recombinant adeno-associated virus carrying small interfering RNA targeting TIMP-1 in rat liver fibrosis. Am J Pathol. 2013;182(5):1607–1616. | ||
Roderfeld M, Weiskirchen R, Wagner S, et al. Inhibition of hepatic fibrogenesis by matrix metalloproteinase-9 mutants in mice. FASEB J. 2006;20(3):444–454. | ||
Pan D, Rubin GM. Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell. 1997;90(2):271–280. | ||
Solanas G, Cortina C, Sevillano M, Batlle E. Cleavage of E-cadherin by ADAM10 mediates epithelial cell sorting downstream of EphB signalling. Nat Cell Biol. 2011;13(9):1100–1107. | ||
Hartmann D, de Strooper B, Serneels L, et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet. 2002;11(21):2615–2624. | ||
Müller M, Wetzel S, Köhn-Gaone J, et al. A disintegrin and metalloprotease 10 (ADAM10) is a central regulator of murine liver tissue homeostasis. Oncotarget. 2016;7(14):17431–17441. | ||
Yata Y, Takahara T, Furui K, Zhang LP, Watanabe A. Expression of matrix metalloproteinase-13 and tissue inhibitor of metalloproteinase-1 in acute liver injury. J Hepatol. 1999;30(3):419–424. | ||
Harty MW, Huddleston HM, Papa EF, et al. Repair after cholestatic liver injury correlates with neutrophil infiltration and matrix metalloproteinase 8 activity. Surgery. 2005;138(2):313–320. | ||
Fallowfield JA, Mizuno M, Kendall TJ, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178(8):5288–5295. | ||
Zhou X, Hovell CJ, Pawley S, et al. Expression of matrix metalloproteinase-2 and -14 persists during early resolution of experimental liver fibrosis and might contribute to fibrolysis. Liver Int. 2004;24(5):492–501. | ||
Endo H, Niioka M, Sugioka Y, et al. Matrix metalloproteinase-13 promotes recovery from experimental liver cirrhosis in rats. Pathobiology. 2011;78(5):239–252. | ||
Siller-López F, Sandoval A, Salgado S, et al. Treatment with human metalloproteinase-8 gene delivery ameliorates experimental rat liver cirrhosis. Gastroenterology. 2004;126(4):1122–1133. | ||
Iimuro Y, Brenner DA. Matrix metalloproteinase gene delivery for liver fibrosis. Pharm Res. 2008;25(2):249–258. | ||
Hartland SN, Murphy F, Aucott RL, et al. Active matrix metalloproteinase-2 promotes apoptosis of hepatic stellate cells via the cleavage of cellular N-cadherin. Liver Int. 2009;29(7):966–978. | ||
Mòdol T, Brice N, Ruiz de Galarreta M, et al. Fibronectin peptides as potential regulators of hepatic fibrosis through apoptosis of hepatic stellate cells. J Cell Physiol. 2015;230(3):546–553. | ||
Simeonova PP, Gallucci RM, Hulderman T, et al. The role of tumor necrosis factor-alpha in liver toxicity, inflammation, and fibrosis induced by carbon tetrachloride. Toxicol Appl Pharmacol. 2001;177(2):112–120. | ||
Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol. 2013;3(2):785–797. | ||
Horiuchi K, Kimura T, Miyamoto T, et al. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol. 2007;179(5):2686–2689. | ||
McMahan RS, Riehle KJ, Fausto N, Campbell JS. A disintegrin and metalloproteinase 17 regulates TNF and TNFR1 levels in inflammation and liver regeneration in mice. Am J Physiol Gastrointest Liver Physiol. 2013;305(1):G25–G34. | ||
Sato A, Nakashima H, Nakashima M, et al. Involvement of the TNF and FasL produced by CD11b Kupffer cells/macrophages in CCl4-induced acute hepatic injury. PLoS One. 2014;9(3):e92515. | ||
Dong Y, Kase S, Dong Z, et al. Regulation of vascular endothelial growth factor-C by tumor necrosis factor-α in the conjunctiva and pterygium. Int J Mol Med. 2016;38(2):545–550. | ||
Svinka J, Pflügler S, Mair M, et al. Epidermal growth factor signaling protects from cholestatic liver injury and fibrosis. J Mol Med (Berl). Epub 2016 Aug 27. | ||
Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci U S A. 2007;104(43):17081–17086. | ||
Sahin U, Weskamp G, Kelly K, et al. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164(5):769–779. | ||
Takemura T, Yoshida Y, Kiso S, et al. Conditional loss of heparin-binding EGF-like growth factor results in enhanced liver fibrosis after bile duct ligation in mice. Biochem Biophys Res Commun. 2013;437(2):185–191. | ||
Takemura T, Yoshida Y, Kiso S, et al. Conditional knockout of heparin-binding epidermal growth factor-like growth factor in the liver accelerates carbon tetrachloride-induced liver injury in mice. Hepatol Res. 2013;43(4):384–393. | ||
Tomita K, Haga H, Mizuno K, et al. Epiregulin promotes the emergence and proliferation of adult liver progenitor cells. Am J Physiol Gastrointest Liver Physiol. 2014;307(1):G50–G57. | ||
Webber EM, Wu JC, Wang L, Merlino G, Fausto N. Overexpression of transforming growth factor-alpha causes liver enlargement and increased hepatocyte proliferation in transgenic mice. Am J Pathol. 1994;145(2):398–408. | ||
Huang G, Besner GE, Brigstock DR. Heparin-binding epidermal growth factor-like growth factor suppresses experimental liver fibrosis in mice. Lab Invest. 2012;92(5):703–712. | ||
Perugorria MJ, Latasa MU, Nicou A, et al. The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology. 2008;48(4):1251–1261. | ||
McKee C, Sigala B, Soeda J, et al. Amphiregulin activates human hepatic stellate cells and is upregulated in non alcoholic steatohepatitis. Sci Rep. 2015;5:8812. | ||
Miller MA, Meyer AS, Beste MT, et al. ADAM-10 and -17 regulate endometriotic cell migration via concerted ligand and receptor shedding feedback on kinase signaling. Proc Natl Acad Sci U S A. 2013;110(22):E2074–E2083. | ||
Higashiyama S, Nanba D. ADAM-mediated ectodomain shedding of HB-EGF in receptor cross-talk. Biochim Biophys Acta. 2005;1751(1):110–117. | ||
Chalupský K, Kanchev I, Žbodáková O, et al. ADAM10/17-dependent release of soluble c-Met correlates with hepatocellular damage. Folia Biol (Praha). 2013;59(2):76–86. | ||
Paranjpe S, Bowen WC, Mars WM, et al. Combined systemic elimination of MET and epidermal growth factor receptor signaling completely abolishes liver regeneration and leads to liver decompensation. Hepatology. 2016;64(5):1711–1724. | ||
Scheving LA, Zhang X, Stevenson MC, Threadgill DW, Russell WE. Loss of hepatocyte EGFR has no effect alone but exacerbates carbon tetrachloride-induced liver injury and impairs regeneration in hepatocyte Met-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2015;308(5):G364–G377. | ||
Seki E, Kondo Y, Iimuro Y, et al. Demonstration of cooperative contribution of MET- and EGFR-mediated STAT3 phosphorylation to liver regeneration by exogenous suppressor of cytokine signalings. J Hepatol. 2008;48(2):237–245. | ||
Kitade M, Factor VM, Andersen JB, et al. Specific fate decisions in adult hepatic progenitor cells driven by MET and EGFR signaling. Genes Dev. 2013;27(15):1706–1717. | ||
Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol. 2003;33(2):105–136. | ||
Porter WR, Neal RA. Metabolism of thioacetamide and thioacetamide S-oxide by rat liver microsomes. Drug Metab Dispos. 1978;6(4):379–388. | ||
Kroeger-Koepke MB, Koepke SR, McClusky GA, Magee PN, Michejda CJ. Alpha-hydroxylation pathway in the in vitro metabolism of carcinogenic nitrosamines: N-nitrosodimethylamine and N-nitroso-N-methylaniline. Proc Natl Acad Sci U S A. 1981;78(10):6489–6493. | ||
Jaeschke H, Williams CD, McGill MR, Xie Y, Ramachandran A. Models of drug-induced liver injury for evaluation of phytotherapeutics and other natural products. Food Chem Toxicol. May 2013;55:279–289. | ||
Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol. 2006;87(1):1–16. | ||
Koarada S, Wu Y, Fertig N, et al. Genetic control of autoimmunity: protection from diabetes, but spontaneous autoimmune biliary disease in a nonobese diabetic congenic strain. J Immunol. 2004;173(4):2315–2323. | ||
Mauad TH, van Nieuwkerk CM, Dingemans KP, et al. Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am J Pathol. 1994;145(5):1237–1245. | ||
Oude Elferink RP, Ottenhoff R, van Wijland M, Smit JJ, Schinkel AH, Groen AK. Regulation of biliary lipid secretion by mdr2 P-glycoprotein in the mouse. J Clin Invest. 1995;95(1):31–38. | ||
Smit JJ, Schinkel AH, Oude Elferink RP, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75(3):451–462. | ||
Fickert P, Fuchsbichler A, Wagner M, et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2004;127(1):261–274. | ||
Wakabayashi K, Lian ZX, Moritoki Y, et al. IL-2 receptor alpha(-/-) mice and the development of primary biliary cirrhosis. Hepatology. 2006;44(5):1240–1249. | ||
Oertelt S, Lian ZX, Cheng CM, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol. 2006;177(3):1655–1660. | ||
Salas JT, Banales JM, Sarvide S, et al. Ae2a,b-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology. 2008;134(5):1482–1493. | ||
Okumura K, Ikejima K, Kon K, et al. Exacerbation of dietary steatohepatitis and fibrosis in obese, diabetic KK-A(y) mice. Hepatol Res. 2006;36(3):217–228. | ||
Horie Y, Suzuki A, Kataoka E, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113(12):1774–1783. | ||
Chung H, Hong DP, Kim HJ, et al. Differential gene expression profiles in the steatosis/fibrosis model of rat liver by chronic administration of carbon tetrachloride. Toxicol Appl Pharmacol. 2005;208(3):242–254. | ||
Jiang Y, Liu J, Waalkes M, Kang YJ. Changes in the gene expression associated with carbon tetrachloride-induced liver fibrosis persist after cessation of dosing in mice. Toxicol Sci. 2004;79(2):404–410. | ||
Marsillach J, Ferré N, Camps J, Rull A, Beltran R, Joven J. Changes in the expression of genes related to apoptosis and fibrosis pathways in CCl4-treated rats. Mol Cell Biochem. 2008;308(1–2):101–109. | ||
Park SY, Shin HW, Lee KB, Lee MJ, Jang JJ. Differential expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in thioacetamide-induced chronic liver injury. J Korean Med Sci. 2010;25(4):570–576. | ||
Chen IS, Chen YC, Chou CH, Chuang RF, Sheen LY, Chiu CH. Hepatoprotection of silymarin against thioacetamide-induced chronic liver fibrosis. J Sci Food Agric. 2012;92(7):1441–1447. | ||
Subramanian P, Arul D. Attenuation of NDEA-induced hepatocarcinogenesis by naringenin in rats. Cell Biochem Funct. 2013;31(6):511–517. | ||
Prakobwong S, Yongvanit P, Hiraku Y, et al. Involvement of MMP-9 in peribiliary fibrosis and cholangiocarcinogenesis via Rac1-dependent DNA damage in a hamster model. Int J Cancer. 2010;127(11):2576–2587. | ||
Mandal AK, Ghosh D, Sarkar S, Ghosh A, Swarnakar S, Das N. Nanocapsulated quercetin downregulates rat hepatic MMP-13 and controls diethylnitrosamine-induced carcinoma. Nanomedicine (Lond). 2014;9(15):2323–2337. | ||
Ito Y, Abril ER, Bethea NW, McCuskey RS. Inhibition of matrix metalloproteinases minimizes hepatic microvascular injury in response to acetaminophen in mice. Toxicol Sci. 2005;83(1):190–196. | ||
Kato H, Kuriyama N, Duarte S, Clavien PA, Busuttil RW, Coito AJ. MMP-9 deficiency shelters endothelial PECAM-1 expression and enhances regeneration of steatotic livers after ischemia and reperfusion injury. J Hepatol. 2014;60(5):1032–1039. | ||
Stanton MC, Chen SC, Jackson JV, et al. Inflammatory signals shift from adipose to liver during high fat feeding and influence the development of steatohepatitis in mice. J Inflamm (Lond). 2011;8:8. | ||
de Meijer VE, Sverdlov DY, Le HD, Popov Y, Puder M. Tissue-specific differences in inflammatory infiltrate and matrix metalloproteinase expression in adipose tissue and liver of mice with diet-induced obesity. Hepatol Res. 2012;42(6):601–610. | ||
Wang C, Tao Q, Wang X, Wang X, Zhang X. Impact of high-fat diet on liver genes expression profiles in mice model of nonalcoholic fatty liver disease. Environ Toxicol Pharmacol. 2016;45:52–62. | ||
Lee HS, Son WC, Ryu JE, Koo BA, Kim YS. Standardized salvia miltiorrhiza extract suppresses hepatic stellate cell activation and attenuates steatohepatitis induced by a methionine-choline deficient diet in mice. Molecules. 2014;19(6):8189–8211. | ||
Wang RQ, Nan YM, Wu WJ, et al. Induction of heme oxygenase-1 protects against nutritional fibrosing steatohepatitis in mice. Lipids Health Dis. 2011;10:31. | ||
Velayudham A, Dolganiuc A, Ellis M, et al. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology. 2009;49(3):989–997. | ||
Uchinami H, Seki E, Brenner DA, D’Armiento J. Loss of MMP 13 attenuates murine hepatic injury and fibrosis during cholestasis. Hepatology. 2006;44(2):420–429. | ||
Buryova H, Chalupsky K, Zbodakova O, et al. Liver protective effect of ursodeoxycholic acid includes regulation of ADAM17 activity. BMC Gastroenterol. 2013;13:155. | ||
Popov Y, Patsenker E, Stickel F, et al. Integrin alphavbeta6 is a marker of the progression of biliary and portal liver fibrosis and a novel target for antifibrotic therapies. J Hepatol. 2008;48(3):453–464. | ||
Schrading S, Schild H, Kühr M, Kuhl C. Effects of tamoxifen and aromatase inhibitors on breast tissue enhancement in dynamic contrast-enhanced breast MR imaging: a longitudinal intraindividual cohort study. Radiology. 2014;271(1):45–55. | ||
Díaz-Gil JJ, García-Monzón C, Rúa C, et al. The anti-fibrotic effect of liver growth factor is associated with decreased intrahepatic levels of matrix metalloproteinases 2 and 9 and transforming growth factor beta 1 in bile duct-ligated rats. Histol Histopathol. 2008;23(5):583–591. | ||
Kossakowska AE, Edwards DR, Lee SS, et al. Altered balance between matrix metalloproteinases and their inhibitors in experimental biliary fibrosis. Am J Pathol. 1998;153(6):1895–1902. | ||
Ishikawa T, Factor VM, Marquardt JU, et al. Hepatocyte growth factor/c-met signaling is required for stem-cell-mediated liver regeneration in mice. Hepatology. 2012;55(4):1215–1226. | ||
Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19(5):725–737. | ||
Okazaki I, Watanabe T, Hozawa S, Arai M, Maruyama K. Molecular mechanism of the reversibility of hepatic fibrosis: with special reference to the role of matrix metalloproteinases. J Gastroenterol Hepatol. 2000;15(Suppl):D26–D32. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.