")
Back to Archived Journals » Metalloproteinases In Medicine » Volume 2
Metalloproteinases as modulators of allergic asthma: therapeutic perspectives
Received 15 July 2015
Accepted for publication 2 September 2015
Published 23 October 2015 Volume 2015:2 Pages 61—74
DOI https://doi.org/10.2147/MNM.S63614
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yoshifumi Itoh
Jennifer L Ingram,1 Monica Kraft2
1Department of Medicine, Duke University, Durham, NC, 2Department of Medicine, University of Arizona, Tucson, AZ, USA
Abstract: Matrix metalloproteinases (MMPs) are proteolytic enzymes that facilitate extracellular matrix turnover; thus, these enzymes are important in both normal wound repair and pathologic processes, such as inflammation and fibrosis. In addition, MMPs regulate cell signaling through proteolytic shedding of bioactive growth factors, cytokines, and other molecules. Therefore, the expression and activation of MMPs and their endogenous inhibitors, tissue inhibitors of metalloproteinases, play roles in the development of a variety of diseases. Allergic asthma is a chronic inflammatory disease of the airways that affects millions of patients, both children and adults, worldwide. Allergen-induced airway injury and immune responses stimulate airway inflammation, hyperresponsiveness, and remodeling in these patients. The action of MMPs links these pathologic processes through a complex interaction of airway inflammatory cells and structural cells. This review explores the important roles of MMPs and tissue inhibitors of metalloproteinases in mediating the pathobiology of allergic asthma and highlights the potential for these enzymes to serve as biomarkers or therapeutic targets in allergic asthma.
Keywords: matrix metalloproteinase, MMP, asthma, allergy, airway
Introduction
Asthma is a chronic inflammatory disease that is estimated to affect 334 million adults and children worldwide, with nearly 14% of the world’s children experiencing symptoms associated with asthma.1 These symptoms include wheezing, chest tightness, and coughing, with some patients experiencing diminished lung function over time, resulting in substantially reduced quality of life.1 The primary long-term therapy for asthma is inhaled corticosteroids, sometimes combined with long-acting beta-agonists and oral corticosteroids in the most severe asthma patients.2 However, not all therapies are beneficial for all asthma patients. Asthma is a heterogeneous disease with different causes, manifestations, and severities. Much recent research has allowed for classification of the disease into various subphenotypes based upon clinical criteria, biomarkers, and genetic and molecular profiles.3 Therefore, more personalized therapies tailored for specific subphenotypes of asthma are currently being developed that may provide benefits where other, more standard therapies are not efficacious.
The most common and best-understood subphenotype of asthma, allergic or type 2 asthma, is characterized by a pathologic airway immune response in atopic individuals resulting from exposure to inhaled allergens. This response involves many different cell types and a complex cascade of cell signaling events, contributing to chronic airway inflammation, airways hyperresponsiveness (AHR), and remodeling. The allergic cascade in asthma has been reviewed elsewhere.4,5 To briefly summarize here, allergic asthma is characterized by an early asthmatic response (EAR), which occurs immediately upon inhaled allergen exposure and lasts 30–60 minutes, and a late asthmatic response (LAR), which manifests several hours after exposure.6 In the EAR, repeated exposure to a sensitizing allergen triggers recognition by specific type 2 cells such as T-helper 2 lymphocytes and innate lymphoid cells, which initiate secretion of type 2 cytokines (eg, interleukins [ILs]-4, -5, and -13) and stimulates immunoglobulin (Ig) E isotype switching in B-cells.7 Binding of allergen to specific IgE activates basophils and mast cells to release histamine, prostaglandins, leukotrienes, and other inflammatory mediators.8,9 Allergen exposure also promotes airway inflammation in the LAR by disrupting the protective airway epithelium layer and increasing permeability between epithelial cells.10–12 Allergen-induced damage and triggering of epithelial cell pattern recognition receptors lead to mucous secretion13 and production of IL-33,14 IL-25,15 and thymic stromal lymphopoietin,16 which activate dendritic cells and promote release of type 2 cytokines from innate lymphoid cells.17 Type 2 cytokines direct inflammation in the airway by promoting the survival and migration of eosinophils,18 activation of macrophages,19 and production of inducible nitric oxide synthase by airway epithelial cells.20,21
Ongoing inflammation contributes to AHR in asthma. Variable or episodic AHR results from acute allergen exposure and the effects of airway inflammation.22 IgE-mediated release of type 2 cytokines,23–25 leukotrienes,26,27 serine proteases,28 histamine,26 and complement cleavage products29 by mast cells and other inflammatory cells in the airway has been linked to acute onset of AHR following allergen challenge in both human and animal models. In addition to variable AHR, which is related directly to allergen exposure, a second component of AHR in asthma, persistent or baseline AHR, is present in most patients with asthma and is associated with structural alterations of the airways, termed airway remodeling.22,30
Airway remodeling is thought to result from chronic inflammation in the airways of atopic asthmatics, which involves mucous cell metaplasia31 and mucous hypersecretion,23,32 thickening of the airway smooth muscle layer,33,34 and an impaired wound healing response that results in increased airway fibroblast proliferation,35 invasion,36 and transformation of airway fibroblasts to myofibroblasts,37,38 leading to subepithelial collagen deposition.39,40 These permanent structural changes in the airway result in decreased airway luminal area41 and irreversible airway obstruction42 and is associated with persistent AHR.43,44
Matrix metalloproteinases (MMPs) are a family of zinc-dependent endopeptidases that play key roles in both normal processes, such as wound healing,45,46 and pathogenic processes, such as inflammation47 or fibrosis.48 Although MMPs are well known for their ability to degrade extracellular matrix (ECM), these enzymes also serve to regulate cell signaling through the cleavage and processing of bioactive molecules. MMPs are secreted by a number of cell types in the allergic asthmatic airway, including both inflammatory cells, such as macrophages49 and leukocytes,49–51 and airway structural cells, such as airway fibroblasts,36,39 epithelial cells,52,53 and smooth muscle cells.54 Expression and activity of MMPs are tightly regulated at the transcriptional and posttranslational levels and by the action of endogenous inhibitors of MMPs, the tissue inhibitors of metalloproteinase (TIMPs). Thus, the relative ratios of MMPs to TIMPs play a role in the pathobiology of various diseases.55,56
This review will focus on the functions of specific MMPs and TIMPs in each of the pathologic processes of allergic asthma and explore the potential of MMPs to serve as therapeutic targets or biomarkers in allergic asthma.
MMPs in airway inflammation in allergic asthma
Both acute and chronic airway inflammation play major roles in the pathobiology of allergic asthma. Upon stimulation with inhaled allergen, inflammatory cells are activated to migrate from the circulation to sites of injury in the airway wall and lumen. Much research has focused on the role of MMPs in facilitating this extravasation of inflammatory cells across the vascular and airway membranes. Cells that are key to the allergic inflammatory response secrete a variety of MMPs with diverse functions (Table 1), and these enzymes act to proteolytically degrade ECM to promote cellular migration to sites of inflammation in the airway. Ohno et al first demonstrated that MMP9 mRNA expression was significantly increased in the submucosal regions of airway biopsy tissue of patients with asthma as compared to normal subjects. They further localized the major source of expression of MMP9 in asthma patients’ tissue to airway eosinophils,57 although other studies have since shown that neutrophils also secrete MMP-9 following allergen challenge.50,51 In the follow-up studies, the same group used a murine model of allergic airways disease to determine that inhaled allergen stimulated significantly increased airway secretion of gelatinases, including MMP-9, and this effect was associated with an accumulation of eosinophils in bronchoalveolar lavage (BAL) fluid from ovalbumin-challenged animals. Administration of recombinant TIMP-2 prior to allergen challenge significantly reduced the numbers of inflammatory cells in BAL fluid, the numbers of eosinophils in the airway lumen and wall, and the degradation of type IV collagen and other ultrastructural effects of basement membrane injury, suggesting that MMP-9 and MMP-2 were important mediators of eosinophil infiltration of the airway wall in asthma.58,59 Schwingshackl et al60 extended these studies by demonstrating that addition of tumor necrosis factor-alpha (TNF-α), a proinflammatory cytokine, to human peripheral blood eosinophils, cultured from atopic asthmatic patients, stimulated a 95% increase in MMP-9 activity above baseline. Similarly, studies by Kelly et al51 and Mattos et al61 showed that airway secretion of MMP-9 significantly increased 24–48 hours following segmental allergen challenge of allergic patients with asthma, as compared to saline treatments; however, the immunoreactivity of MMP-9 in airway cells was predominantly associated with neutrophils, not eosinophils in the Kelly et al’s study.51 Furthermore, the concentration of MMP-9 and the MMP-9/TIMP-1 ratio in induced sputum at baseline was significantly increased in severe asthma patients as compared to healthy controls and patients with mild asthma, but the levels of MMP-9 did not correlate with numbers of sputum eosinophils.61 Similarly, a study of atopic preschool children showed that MMP-9 protein levels in BAL fluid were significantly increased in wheezers as compared to healthy controls, and this MMP-9 concentration was significantly and positively correlated with numbers of neutrophils in the BAL fluid. Follow-up studies 23 months after the initial bronchoscopy identified which of the children were persistent wheezers and determined that TIMP-1 protein levels in the initial BAL fluid were significantly elevated in the persistent wheezing children as compared to children who had outgrown wheezing.62 In contrast to these reports, one study found that MMP-9 protein and activity levels as well as MMP-9/TIMP-1 ratios were significantly reduced in BAL fluid collected from 34 atopic asthmatic children as compared to 35 nonatopic healthy children and that the MMP-9 levels were significantly associated with numbers of neutrophils in the BAL fluid, which were reduced in the atopic asthma patients.63 However, the majority of studies have shown MMP-9 is released by inflammatory cells in response to allergen provocation in human asthma.
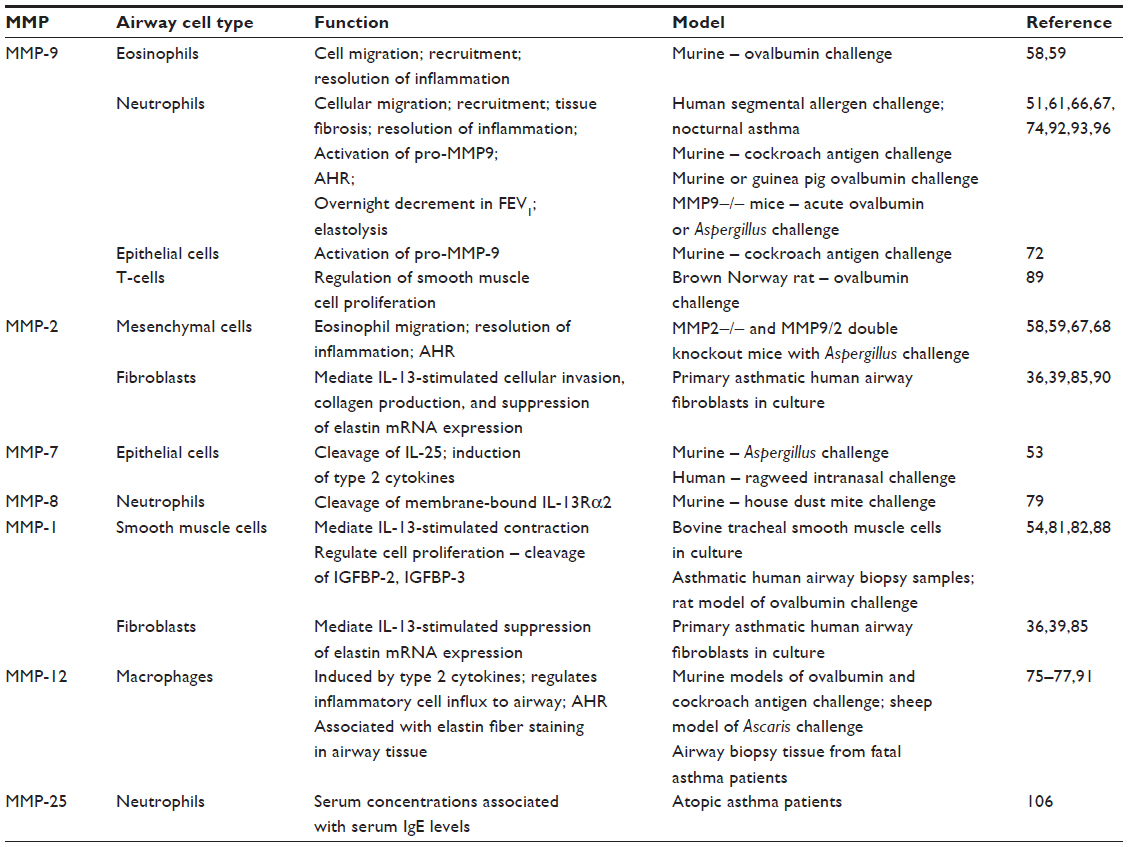 | Table 1 Airway secretion of MMPs and their functions in asthma pathobiology |
Studies of allergen-induced airway disease in MMP9- and MMP2-deficient mice have revealed a role of these gelatinases in airway inflammation in asthma. Cataldo et al64 reported significantly decreased peribronchiolar inflammation, reduced numbers of lymphocytes, and reduced secretion of IL-13 in BAL fluid of MMP9-deficient mice as compared to wild type following acute ovalbumin challenge. Furthermore, allergen-challenged mice that are deficient for TIMP1, an endogenous inhibitor of MMP-9, MMP-2, and MMP-12, exhibited significantly enhanced peribronchiolar inflammation and increased lung type 2 cytokine expression associated with significantly increased lung MMP-9 activity, suggesting that TIMP-1 regulates allergen-induced inflammation in asthma by inhibiting MMP-9 activity.65 In contrast, a subsequent study of MMP9-deficient mice using a similar model of acute ovalbumin challenge demonstrated significantly increased airway eosinophils, lymphocytes, and macrophages, as well as secretion of type 2 cytokines and eotaxin in both BAL fluid and lung tissue, and increased peribronchiolar inflammation in the MMP9-deficient mice compared to wild type. Further investigation showed that resolution of inflammation was impaired in the MMP9-deficient mice as compared to wild type, as concentrations of eotaxin in BAL fluid and numbers of T-helper 2 lymphocytes in the lungs of MMP9-deficient mice were still significantly elevated as compared to wild type 6 days after the last ovalbumin challenge.66 In addition, Corry et al demonstrated that MMP-2 and MMP-9 expression and secretion in murine BAL fluid and lung tissue were significantly increased following allergen challenge and that IL-13 alone was sufficient to induce this effect. MMP2 was expressed primarily by mesenchymal cells, and MMP9 was expressed primarily by inflammatory cells. Challenge with Aspergillus fumigatus allergen revealed significantly decreased eosinophils in BAL fluid but significantly increased inflammatory cells and type 2 cytokines in the lung tissue of MMP2- or MMP9-deficient mice as compared to wild type. Measurement of chemokines in BAL fluid showed significantly reduced eotaxin, CCL7 (MARC), and CCL17 (TARC [thymus and activation related chemokine]) chemokines suggesting an abnormal chemotactic gradient that impaired egression of inflammatory cells to the airway lumen.67–69 Dendritic cell trafficking to the airway lumen was also impaired in MMP9-deficient mice.69 The considerable accumulation of inflammatory cells in the lung tissue contributed to increased mortality of the MMP2-deficient mice due to asphyxiation68 and increased numbers of dead and apoptotic cells in MMP9-deficient mice.67 Likewise, ovalbumin challenge of mice with targeted overexpression of MMP9 in tissue macrophages resulted in reduced levels of IL-13 in serum, decreased numbers of lymphocytes in BAL fluid, but increased numbers of lymphocytes in draining lymph nodes, suggesting that MMP-9 was involved in lymphocyte trafficking.70 Study of double MMP2/MMP9 knockout mice provided no additive effect on the MMP9-deficiency-alone phenotype, suggesting that MMP-9 has the primary role in regulating inflammatory cell egression from lung tissue to the airway lumen. MMP-2 has an overlapping role that is specific for eosinophilia.67 Finally, Greenlee et al71 identified three substrates of MMP-2 and MMP-9 in the BAL fluid of allergic mice, Ym1, S100A8, and S100A9. These proteins serve as chemokines for inflammatory cells, and cleavage of these proteins provides a potential mechanism to explain the impaired chemotactic responses observed in MMP2- and MMP9-deficient mice. Taken together, these studies, although somewhat contradictory, show that MMP-9 and MMP-2 are key mediators of inflammatory cell egression in allergic airways disease and may have an influence on type 2 cytokine expression.
Allergens with protease activity, such as tree pollen, fungi (A. fumigatus), cat, house dust mite, and cockroach allergens, are clinically important as allergic asthma triggers and have specific effects on activation of MMPs. Page et al72 found that serine protease activity in German cockroach frass augmented TNF-α-induced MMP9 expression by bronchial epithelial cells and promoted cleavage of the pro-MMP-9 into the active form of MMP-9.73 This group further showed that German cockroach frass induced MMP9 expression in a murine model of allergic airways disease and that release of MMP-9 from neutrophils was mediated by a toll-like receptor 2 (TLR2) ligand in frass. Also, when challenged with German cockroach frass, MMP9-deficient mice exhibited significantly increased airway eosinophils but reduced airway neutrophils, increased type 2 cytokines in the lung, and increased serum IgE levels as compared to saline controls and wild-type animals. These data suggested that TLR2 agonists in complex allergens may serve to protect against type 2 airway responses in asthma by mediating release of MMP-9 on neutrophils.74 Considering the overlapping functions of MMP-9 and MMP-2 as reported by Corry et al,67 it is plausible to consider that MMP-2 is compensating for the lack of MMP-9 in this model to stimulate eosinophilic inflammation in response to the cockroach allergen.
Murine models of ovalbumin, Ascaris, or cockroach antigen-induced allergic airways disease were used to assess the role of macrophage-derived metalloelastase (MMP-12) in allergic inflammation in asthma. MMP12 mRNA, protein, and activity were upregulated in airway tissues following allergen challenge.52,75 Pouladi et al demonstrated reduced numbers of eosinophils in BAL fluid, but not in tissue, in MMP12-deficient mice as compared to wild-type mice following ovalbumin challenge. Also, MMP12 mRNA expression and eosinophilia were significantly reduced in IL-13-deficient mice, suggesting that IL-13 stimulated MMP12 expression in response to allergen challenge.75 Warner et al reported that mice deficient for MMP12 exhibited significantly reduced airway and peribronchiolar inflammatory cell infiltration and reduced BAL fluid levels of proinflammatory cytokines and chemokines, TNF-α, IL-5, and TARC, as compared to MMP12 replete animals following repeated challenges with cockroach antigen. Levels of MMP-9 and MMP-2 in BAL fluid were significantly induced by cockroach antigen in the MMP12-expressing animals, but this effect was not observed in the MMP12-deficient mice.76 Additionally, administration of an MMP-12-specific inhibitor (MMP408) to Ascaris-challenged sheep resulted in significantly fewer inflammatory cells in BAL fluid, suggesting that the inhibitor blocked the influx of inflammatory cells to the airway.77 Thus, MMP-12 may be regulated by type 2 cytokines and act to direct inflammatory cell responses, which effects release of MMP-9 and MMP-2 in the airway.
Proteolytic allergens directly damage airway epithelium, and Goswami et al demonstrated that Aspergillus allergen strongly upregulated MMP7 mRNA expression in airway epithelial cells and that MMP-7 activity cleaved IL-25, leading to enhanced type 2 responses. Mice deficient for MMP7 exhibited significantly reduced airway eosinophilia and reduced concentrations of IL-4, IL-5, IL-13, and eotaxin in BAL fluid and reduced IL25 mRNA in the lung tissue. Moreover, MMP-7 expression was significantly increased in airway biopsy tissue from patients with asthma as compared to healthy patients, and intranasal challenge with another proteolytic allergen, ragweed, resulted in increased MMP-7 secretion as compared to saline controls.53 Therefore, allergen-activated MMP-7 may act to stimulate type 2 cytokine responses in asthma.
MMP-8 also plays a role in the regulation of type 2 cytokine activity in allergic asthma. Chen et al78 demonstrated that levels of soluble IL-13 receptor alpha 2 (IL-13Rα2), a decoy receptor and a negative regulator of IL-13,79 were increased in both serum and BAL fluid of house dust mite allergen-challenged mice. MMP-8 was found to be the enzyme that specifically cleaves IL-13Rα2 from mouse and human cells, and levels of soluble IL-13Rα2 in BAL fluid were attenuated in house dust mite allergen-challenged MMP8-deficient mice as compared to wild-type mice.78 Gueders et al80 also found increased numbers of eosinophils in airway walls and increased IL-4 concentration in BAL fluid of ovalbumin-challenged MMP8-deficient mice. These data suggest that MMP-8 acts to regulate type 2 cytokine signaling by shedding membrane-bound IL-13Rα2 to increase the soluble IL-13Rα2 concentration in the airway that binds to IL-13 with high affinity and sequester IL-13 from its signaling receptor subunits. Thus, allergen induction of MMPs in airway epithelium (MMP-7), neutrophils (MMP-9/MMP-8), macrophages (MMP-12), or even mesenchymal cells (MMP-2) contributes to airway inflammation in asthma, either through modulation of type 2 cytokine production and signaling or through regulation of leukocyte egress and infiltration of airway tissues.
Contribution of MMPs to AHR in allergic asthma
AHR is a hallmark feature of allergic asthma, but the development of AHR is linked to airway inflammation and remodeling, so the effects on either of the latter processes in asthma may also affect AHR. In addition, much of the current knowledge of the role that MMPs play in the development and persistence of AHR in asthma is derived from animal models of allergic airways disease, which makes overall interpretation of the role of these enzymes in human AHR difficult since these reports use models that utilize several different allergens and measure airways reactivity using different bronchoconstrictors and systems. One of the first studies to provide data on MMPs and AHR reported that administration of TIMP-2 to mice prior to ovalbumin challenge significantly attenuated methacholine-induced airways reactivity as compared to vehicle control-treated mice.59 These data suggested that MMP-2 plays a role in AHR in asthma. Similarly, Sands et al65 showed that methacholine-induced airways reactivity was enhanced in ovalbumin-challenged TIMP1-deficient mice as compared to saline control-treated mice, and these responses were accompanied by significantly increased MMP9 expression in lung homogenates. However, in ovalbumin-challenged mice with targeted overexpression of MMP9 in tissue macrophages, methacholine-induced airways resistance was attenuated as compared to wild-type mice.70 And, in ovalbumin-challenged MMP9-deficient mice, methacholine-induced airways reactivity was enhanced.66 These effects of MMP-9 on AHR are likely associated with the role that MMP-9 plays in airway inflammatory cell egression and resolution of inflammation as described earlier.
The development of AHR in both MMP8- and MMP7-deficient mice is likely related to the reported effects of each of these enzymes on IL-13 production and signaling. In A. fumigatus allergen-challenged MMP7-deficient mice, IL-13 levels in BAL fluid were reduced, and acetylcholine-induced airway resistance was significantly lower than wild-type mice.53 In contrast, house dust mite allergen-challenged MMP8-deficient mice exhibited increased acetylcholine-induced airway resistance as compared to wild-type mice,79 suggesting that these enzymes modulate IL-13 signaling to regulate AHR in allergic asthma.
An Ascaris-challenged sheep model was used by Mukhopadhyay et al to evaluate the effectiveness of MMP408, an MMP-12 inhibitor, on airway resistance and carbachol-induced reactivity. Pretreatment of either intravenous or orally administered inhibitor prior to allergen challenge significantly abrogated both early-phase and late-phase allergic airway resistance and AHR.77
More evidence of the contribution of MMPs to AHR in allergic asthma may be derived from the effects of MMPs on airway smooth muscle contraction in cell culture models. Several studies have demonstrated the importance of MMP-1, a collagenase, in airway smooth muscle cell contraction. Ohta et al demonstrated that IL-4 and IL-13 stimulate MMP1 expression in bovine tracheal smooth muscle cells but have opposing effects on contraction of collagen gels containing smooth muscle cells, with IL-4 inhibiting contraction and IL-13 stimulating contraction. Both of these effects were mediated by MMP-1, and the authors provided evidence of changes in the collagen fiber structure surrounding the smooth muscle cells in response to either IL-4 or IL-13, suggesting that MMP-1 acts to either degrade or reorganize collagen/smooth muscle cell interactions to modulate contraction responses to each cytokine.54 Similarly, Rogers et al found that ECM proteins, collagen type I and tenascin-C, stimulated significantly increased MMP1 expression by airway smooth muscle cells in asthma as compared to cells from healthy patients. MMP-1 and tenascin-C co-localized to airway smooth muscle cells in biopsy tissue from asthma patients, but not in tissue from healthy control patients. Furthermore, pretreatment of airway smooth muscle cells with a broad-spectrum MMP inhibitor, GM6001, or small interfering RNA specific for MMP1 significantly reduced agonist-stimulated gel contraction, suggesting that matrix-induced MMP-1 mediates airway smooth muscle contraction in asthma.81 In addition, Margulis et al demonstrated that human airway smooth muscle cells co-cultured with human mast cells in collagen gels undergo increased contraction in response to IgE cross-linking or IL-13 treatment and that this effect was dependent on MMPs. Activation of MMP-1 and MMP-2 by serine proteases was implicated in the mast cell-directed smooth muscle cell contraction.82 Taken together, these studies highlight the actions of MMP-1 to stimulate airway smooth muscle contraction and implicate a role for this collagenase in AHR in the setting of type 2 cytokine-driven inflammation and allergen-induced asthma.
To this end, Bruce and Thomas published results of a small pilot clinical trial to investigate the effects of marimastat (British Biotech, Oxford, UK), a broad-spectrum MMP inhibitor, in nine steroid-naïve patients with mild atopic asthma. In this randomized, double-blind crossover trial, patients received a placebo twice daily for 3 weeks separated by a 6-week washout period, with allergen challenges at the beginning and end of each treatment phase. The authors found that house dust mite allergen-provoked airways hyperreactivity was significantly reduced in patients receiving marimastat as compared to placebo control, but no significant changes were observed with regard to effects of marimastat on airway inflammation, forced expiratory volume in 1 second (FEV1), or asthma symptoms as compared to placebo control.83 Although this trial was very small, the effect of MMP inhibition on AHR is somewhat promising and merits further investigation to determine if inhibition of specific MMPs, particularly MMP-1, may serve as therapies for airway inflammation or AHR in allergic asthma.
MMPs in airway remodeling in allergic asthma
Key cellular features of airway remodeling in allergic asthma include mucous cell metaplasia, airway smooth muscle cell hyperplasia and hypertrophy, airway fibroblast proliferation, invasion, and differentiation to a myofibroblast phenotype, and deposition of collagen and other ECM components in the submucosal region of the airway. IL-13 is a type 2 cytokine that directs most of these processes in allergic asthma. Elias et al, who developed mice with inducible lung-targeted overexpression of IL-13, elegantly demonstrated the effects of IL-13 on airway remodeling and inflammation. They found that IL-13 overexpression alone was sufficient to induce most of the features of allergic asthma seen in human patients and other murine models of allergen challenge, but they also observed an emphysema-like phenotype. A characteristic of these IL-13 transgenic mice significantly increased MMP9, MMP12, MMP2, MMP13, MMP14, and TIMP1 mRNA expression and MMP-9, MMP-12, and MMP-2 activity in the lung tissue as compared to nontransgenic animals. Furthermore, treatment of the IL-13 transgenic mice with GM6001 significantly reduced lung and alveolar size, reduced total numbers of cells in BAL fluid, and reduced numbers of eosinophils and lymphocytes in BAL fluid by >90%; however, GM6001 had no effect on IL-13-induced mucous cell metaplasia.84 Taken together, this study demonstrated that MMP expression and activity mediated the IL-13-induced emphysema and effects on airway inflammation and remodeling.
Reports focusing on airway remodeling in human allergic asthma have shown that IL-13 stimulates significantly increased MMP-1 and MMP-2 protein and activity in airway fibroblasts derived from patients with mild allergic asthma as compared to healthy controls.39,85 Treatment of airway fibroblasts derived from patients with asthma with GM6001 significantly reduced IL-13-induced airway fibroblast invasion, a process implicated in proteolytic penetration of the airway basement membrane and submucosa.36 In addition to this direct role in degrading ECM, MMP-1 and MMP-2 were found to mediate IL-13 effects on ECM gene expression by airway fibroblasts in allergic asthma. Firszt et al showed that the action of MMP-2 was required for IL-13-induced COL1A2 gene expression and collagen type I protein expression by airway fibroblasts in asthma. Furthermore, inhibition of MMP-2 blocked IL-13-induced activation of the profibrotic cytokine, transforming growth factor-β1 (TGFβ1), in airway fibroblasts in allergic asthma,39 suggesting that MMP-2 is required for IL-13-induced processing of TGFβ1 from a latent form, bound to latency-associated protein, to the dissociated and active form of TGFβ1. Thus, MMP-2 serves to regulate COL1A2 gene expression and collagen production by activating an upstream mediator. Similarly, Ingram et al found that both MMP-1 and MMP-2 mediate IL-13-induced suppression of elastin (ELN) gene expression by airway fibroblasts in allergic asthma, which is associated with loss of elastic fiber staining in patients with increased AHR. Treatment of airway fibroblasts with GM6001 or inhibitors specific for MMP-1 or MMP-2 in combination with IL-13 significantly increased ELN gene expression as compared to IL-13 treatment alone.85 Regulation of ELN gene expression may be related to proteolytic shedding of heparin-binding epidermal growth factor (HB-EGF), which has been shown to downregulate ELN mRNA expression in pulmonary fibroblasts.86,87 Therefore, MMP-1 and MMP-2 play roles in mediating IL-13 effects on ECM degradation as well as modulation of gene expression to direct airway remodeling in allergic asthma.
A similar role for MMP-1 was reported by Rajah et al, in which they demonstrated that airway smooth muscle cell hyperplasia in allergic asthma was regulated by MMP-1-mediated proteolysis of insulin-like growth factor binding proteins (IGFBPs). These proteins suppress airway smooth muscle cell growth in healthy subjects, but in the setting of allergic asthma, increased secretion of MMP-1 by airway smooth muscle cells leads to increased cleavage of IGFBP-2 and IGFBP-3, rendering them inactive in the regulation of cell proliferation.88 In addition, Al Heialy et al demonstrated that allergen-stimulated CD4+ T-cells adhere to airway smooth muscle cells and stimulate cell proliferation. The mechanism for this CD4+ T-cell-mediated mitogenic effect involves allergen-stimulated release of HB-EGF from the cell surface of the T-cells by MMP-9. The shed HB-EGF then activates epidermal growth factor receptor on the surface of airway smooth muscle cells to stimulate proliferation.89 Thus, MMPs regulate both airway fibrosis and airway wall thickening in allergic asthma by directly modulating the action of growth factors and cell signaling.
Direct effects of both structural cell-derived and inflammatory cell-derived MMPs on airway remodeling through ECM proteolysis are well documented. Activity of MMP-2 and MMP-3, but not MMP-1, MMP-9, or TIMP-1, was significantly increased in bronchial fibroblasts from patients with asthma as compared to cells derived from healthy patients. These MMP-2 and MMP-3 activity levels were also found to be significantly and positively correlated with procollagen I and proteoglycan levels secreted from fibroblasts as well as negatively correlated with FEV1 in asthma patients.90 Araujo et al found that the fractional area of elastic fibers present in airway smooth muscle in large and small airway biopsy tissue from patients with fatal asthma was significantly increased as compared to cases of nonfatal asthma and nonasthma controls. The fractional areas of MMP-9-positive immunostaining within the airway smooth muscle were also increased in fatal asthma as compared to nonasthma controls, and MMP-12 staining significantly and positively correlated with elastic fiber staining and age of asthma onset in the fatal asthma patients.91 These data suggest that MMP-2, -3, and -12 participate in abnormal ECM turnover and degradation in asthma.
MMP-9 is also implicated in airway remodeling in allergic asthma. Tang et al reported significantly increased positive staining for MMP-9 in the lung tissue of ovalbumin-challenged guinea pigs as compared to saline-challenged controls. The positive staining was observed in both inflammatory cells and structural cells in the lung tissue.92 Allergen-challenged MMP9-deficient mice exhibited significantly reduced peribronchiolar fibrosis and total lung collagen as compared to allergen-challenged wild-type mice.93 Airway vascular wall remodeling in a model of allergen-induced guinea pig airways disease was also mediated by MMP-9. Percent collagen and elastic fibers were significantly increased in ovalbumin-challenged guinea pigs as compared to saline controls, but treatment with 1400W, a specific inhibitor of inducible nitric oxide synthase, significantly reduced this response as well as reduced immunostaining of MMP-9, TIMP-1, and TGFβ1 in airway vascular wall.94 In human allergic asthma, MMP-9 activity and secretion in BAL fluid were significantly increased in asthma patients with mucus hypersecretion and in subjects with severe asthma as compared to mild/moderate asthma subjects and healthy controls.95 Also, Pham et al demonstrated that MMP-9 protein and MMP-9/TIMP-1 ratios were significantly elevated in the BAL of subjects with nocturnal asthma as compared to asthmatic subjects without nocturnal symptoms. These effects were significantly correlated with percentage overnight fall in FEV1 and with reduced airway elastin density.96 Thus, MMP-9 secretion by inflammatory cells links airway inflammation and remodeling in allergic asthma.
MMPs as biomarkers of allergic asthma pathobiology
Investigators of the pathobiology of allergic asthma can utilize several relatively noninvasive techniques to search for potential measurable markers of disease diagnosis, classification, and severity that may serve to guide medical treatment. These noninvasive techniques include collection and analyses of cell types and protein content of induced sputum, blood serum and plasma, and exhaled breath condensate (EBC) in both children and adults with allergic asthma.97 In order for a biomarker to be useful, it must be 1) reliably measured in biological fluid or tissue secretion; 2) consistently correlated with a biological process or disease state; and 3) measurable using noninvasive techniques that can be easily and inexpensively translated to routine clinic use. In studies of allergic asthma, several measurements have emerged as accepted markers of airway inflammation and atopy that have allowed for classification of asthma into subphenotypes based on molecular, inflammatory, or cellular profiles. These markers include the measurement of fraction of exhaled nitric oxide in exhaled breath, inflammatory cell profiles of induced sputum, numbers of peripheral blood eosinophils, serum IgE levels, and molecular profiling of type 2 or “T2 high” gene signatures in blood.3
Recent studies have begun to investigate the association of MMPs in induced sputum, EBC, and serum with markers of airway inflammation and atopy in allergic asthma. The majority of these reports have measured MMP-9 protein or activity or MMP-9/TIMP-1 ratios and found associations with airway inflammation in allergic asthma. First, MMP-9 protein and activity levels in plasma were shown to be significantly associated with severity and inflammatory profile of disease, as patients hospitalized with acute asthma exacerbations had significantly increased MMP-9 in plasma as compared to atopic nonasthmatic patients and healthy controls. MMP-9 protein levels were positively and significantly associated with numbers of neutrophils in the plasma.98
Analyses of MMP-9 protein levels in induced sputum at baseline and following allergen challenge have provided evidence that MMP-9 has the potential to serve as a biomarker for the classification of allergic asthma patients into subphenotypes based on airway inflammation and remodeling indices. Zhang et al found that baseline MMP-9 protein levels in induced sputum collected from asthma patients with irreversible airway obstruction (as defined by prebronchodilator FEV1 of <80% predicted and was improved by <15% predicted after albuterol administration) were significantly increased as compared to patients with reversible airway obstruction (as defined by prebronchodilator FEV1 of <80% predicted and was improved by >15% predicted after albuterol administration), nonobstructed airways (defined by prebronchodilator FEV1 of >80% predicted), and healthy controls.99 In addition, baseline MMP-9 and TIMP-1 protein levels in induced sputum of atopic children with intermittent or moderate asthma were significantly increased compared to healthy controls, but no significant difference was reported between the two asthma groups. The MMP-9 measurements in the induced sputum of moderate asthmatic children were significantly correlated with the numbers of eosinophils.100 Furthermore, Cataldo et al showed that MMP-9 protein and activity in induced sputum were significantly increased in allergic asthma patients following house dust mite allergen challenge relative to patients receiving the sham challenge. TIMP-1 levels were not significantly different between groups. Also, the change in FEV1 during the EAR was significantly and positively associated with sputum MMP-9 activity, while the percentage of sputum neutrophils was significantly and positively associated with MMP-9 protein levels.101 In contrast, this group also reported that MMP9 mRNA expression in sputum cell pellets was not changed in mild asthma as compared to normal controls; however, no information was given regarding atopy in the subjects and the patients included in the study exhibited a predominantly eosinophilic inflammatory profile in the sputum.102 Similarly, Boulay et al collected induced sputum and plasma from mild allergic asthma patients before and after allergen provocation. MMP-9 protein and MMP9/TIMP1 ratio levels in sputum were significantly increased 6 hours and 24 hours after allergen provocation as compared to baseline and were significantly associated with numbers of neutrophils, but not eosinophils in sputum. This group also found that MMP-9 protein concentrations in induced sputum were significantly and positively correlated with the change in FEV1 during the LAR.103
More information on the relationship of MMP-9 secretion and pathobiology of allergic asthma has been gathered through the analyses of EBC. MMP-9 protein concentrations in EBC from children with persistent allergic asthma were significantly greater than children with intermittent allergic asthma or healthy controls, and both groups with asthma were significantly higher than healthy controls. These measurements of MMP-9 protein were significantly and inversely correlated with both FEV1 and peak expiratory flow measurements in the children, but positively correlated with levels of IL-4 and IL-10 cytokines in EBC.104 In addition, Barbaro et al first classified subjects in their study based on eosinophil and neutrophil counts in induced sputum of mild and severe allergic asthma patients. Although no information is given regarding the levels of MMP-9 in the induced sputum, the group measured MMP-9 protein levels in EBCs from the patients. MMP-9 was significantly increased in severe allergic asthma subjects as compared to mild/moderate asthma subjects, and the greatest concentrations of MMP-9 were found in severe asthma patients that had predominantly neutrophils in their sputum as compared to eosinophilic severe or mild/moderate asthma patients. Significant and positive associations between MMP-9 in EBC and percentage of neutrophils in sputum and exhaled nitric oxide suggest the MMP-9 is reflective of airway inflammation.105 Taken together, these studies suggest that MMP-9 protein concentrations and activity levels in induced sputum and EBC may be a useful tool to indicate airway inflammation and airway remodeling in allergic asthmatic patients and to facilitate classification of patients based on inflammatory profiles, severity, or type 2 cytokine-driven inflammation.
In addition to MMP-9, two other MMPs have been evaluated in either induced sputum or serum from allergic asthma patients. MMP1 and TIMP1 mRNA were significantly increased in sputum pellets from patients with mild asthma compared to controls, and the fold change in MMP1 mRNA expression was significantly and positively correlated with monocyte chemoattractant protein-1 (MCP1) mRNA in the sputum pellets of asthma patients, but negatively correlated with FEV1 in asthma,102 suggesting that MMP1 gene expression in induced sputum of allergic asthmatics may serve as a marker of airway remodeling. Finally, a report by Blumenthal et al106 found a significant association between concentrations of serum leukolysin (MMP-25), which is a membrane-type MMP expressed primarily on neutrophils and serum IgE levels in atopic asthma patients, suggesting that neutrophilic release of MMP-25 may be associated with atopy and allergen sensitization. Therefore, MMPs important for airway remodeling may also be measured noninvasively and could serve as biomarkers for the process in allergic asthma.
Polymorphisms of MMPs associated with allergic asthma
Several recent studies have identified polymorphisms in MMP9, MMP1, MMP2, and MMP12 genes as potential risk factors for the development of allergic asthma in various populations. First, Nakashima et al identified 17 polymorphisms in MMP9 in a Japanese population of 290 pediatric atopic asthma patients and 638 healthy controls. Two of the polymorphisms, 2127G>T (rs2274755) and 5546G>A (rs2274756), were found to be significantly associated with the risk for childhood atopic asthma. Also, two common haplotypes of these polymorphisms were found in significantly higher frequencies in atopic asthmatic children as compared to nonasthma children.107 Likewise, the rs2274755 polymorphism of MMP9 was identified, along with another polymorphism, rs17577, to be associated with risk for atopic asthma in a study of 403 Mexican pediatric patients as compared to healthy, nonatopic controls.108
In addition, two studies have investigated associations of MMP1 and MMP2 with asthma in Asian populations. A study of 131 Taiwanese adults with asthma as well as healthy controls identified significantly increased risk for persistent airflow obstruction (defined as FEV1/forced vital capacity <75%) in asthma patients with at least one 1G allele at position −1607 relative to the transcription start site in the promoter of the MMP1 gene as compared to patients homozygous for the 2G allele.109 Also, Birbian et al reported a C/T single nucleotide polymorphism in the promoter of MMP2 at position −1306 relative to the transcription start site that was associated with increased risk for allergic asthma in a Northern Indian population. The allele frequency of the C allele was significantly increased in allergic asthmatic patients as compared to the T allele, and overall, the homozygous CC genotype was significantly more frequent in patients with asthma than healthy control patients. The heterozygous CT and the homozygous TT genotypes were more prevalent in healthy controls, suggesting that the T allele offers some protective effect in allergic asthma.110
Finally, a polymorphism in MMP12 was also implicated with increased risk for asthma. In a study of Scottish children and young adult asthma patients, presence of the serine variant of rs652548 in the MMP12 gene conferred increased risk of asthma severity and exacerbations.77 Taken together, all of these studies point to the potential for polymorphisms of genes encoding MMPs to be associated with allergic asthma risk, but investigations of more diverse populations are required in order to draw definitive conclusion.
Conclusion
Allergic asthma is a complex, chronic disease that involves airway inflammation, hyperresponsiveness, and remodeling. The activity of MMPs and their endogenous inhibitors plays roles in all of these processes through degradation of ECM and/or modulation of cell signaling (Figure 1). It is clear that MMPs are secreted/released by inflammatory cells following allergen provocation and in response to type 2 cytokine signaling, and MMP-9, in particular, serves to facilitate inflammatory cell egress from the tissues to the airway lumen. Inflammatory cell- and structural cell-derived MMPs also contribute to AHR and remodeling by altering ECM turnover, which affects smooth muscle contraction, airway fibroblast invasion, and submucosal accumulation of collagen. Furthermore, MMP-induced regulation of cell signaling through proteolytic shedding and activation of key growth factors, such as TGFβ1, stimulates airway cell proliferation and modulates matrix production, contributing to airway fibrosis. Because MMPs link these three processes, inhibition of their activity, either with pharmacologic inhibitors or overexpression of TIMPs, may have potential for providing benefit in certain patients with allergic asthma, and a limited number of studies suggest that MMP inhibition attenuates inflammation and AHR. But, it is important to understand which patients may be most responsive to those therapies, and to that end, the usefulness of MMPs as biomarkers is just beginning to be investigated, particularly with regard to MMP-9 as a marker of neutrophilic airway inflammation in allergic asthma. Similarly, studies of genetic polymorphisms in MMP genes suggest that, in certain populations, these polymorphisms confer increased risk for allergic asthma, but more information is needed to determine the impact of these polymorphisms in more broad populations. Therefore, MMPs are critical mediators of all three pathologic processes in allergic asthma, and more studies in the future will help us to gain a better understanding of how measurement and inhibition of these proteolytic enzymes in the airway may benefit patients with allergic asthma.
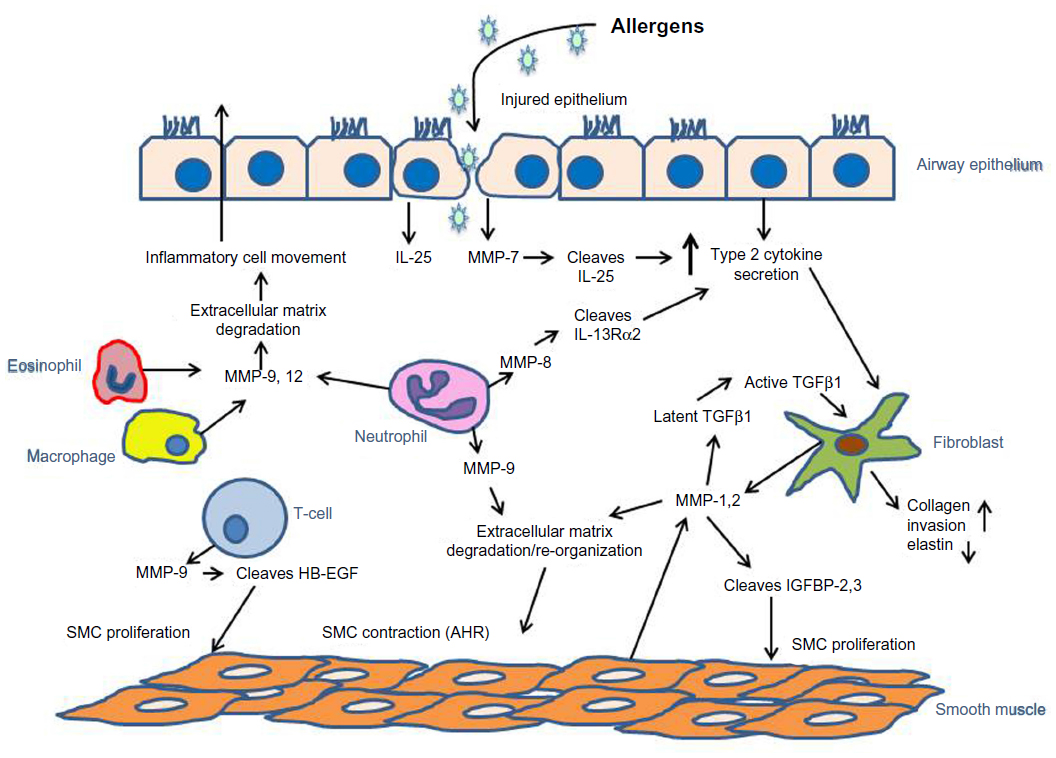 | Figure 1 MMPs mediate allergen-induced airway inflammation, hyperresponsiveness, and remodeling. |
Disclosure
The authors report no conflicts of interest in this work.
References
Asher I, Billo N, Bissell K, et al. The Global Asthma Report 2014. Auckland: New Zealand Global Asthma Network; 2014. | |
NHLBI. How is Asthma Treated and Controlled? 2014. [updated August 4, 2014; cited 2015]. Available from: nhlbi.nih.gov. | |
Ingram JL, Kraft M. IL-13 in asthma and allergic disease: asthma phenotypes and targeted therapies. J Allergy Clin Immunol. 2012;130(4):829–842. quiz 843–824. | |
Bloemen K, Verstraelen S, Heuvel R, Witters H, Nelissen I, Schoeters G. The allergic cascade: review of the most important molecules in the asthmatic lung. Immunol Lett. 2007;113:6–18. | |
Salazar F, Ghaemmaghami AM. Allergen recognition by innate immune cells: critical role of dendritic and epithelial cells. Front Immunol. 2013;4:356. | |
Picado C. Early and late-phase asthmatic reactions: a hypothesis. Allergy. 1992;47(4 pt 1):331–333. | |
Van der Pouw Kraan TC, Van der Zee JS, Boeije LC, De Groot ER, Stapel SO, Aarden LA. The role of IL-13 in IgE synthesis by allergic asthma patients. Clin Exp Immunol. 1998;111:129–135. | |
Amin K. The role of mast cells in allergic inflammation. Respir Med. 2012;106(1):9–14. | |
Murray JJ, Tonnel AB, Brash AR, et al. Release of prostaglandin D2 into human airways during acute antigen challenge. N Engl J Med. 1986;315(13):800–804. | |
Wan H, Winton HL, Soeller C, et al. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J Clin Invest. 1999; 104(1):123–133. | |
Vinhas R, Cortes L, Cardoso I, et al. Pollen proteases compromise the airway epithelial barrier through degradation of transmembrane adhesion proteins and lung bioactive peptides. Allergy. 2011;66(8):1088–1098. | |
Saatian B, Rezaee F, Desando S, et al. Interleukin-4 and interleukin-13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers. 2013;1(2):e24333. | |
Chen JC, Chuang JG, Su YY, Chiang BL, Lin YS, Chow LP. The protease allergen Pen c 13 induces allergic airway inflammation and changes in epithelial barrier integrity and function in a murine model. J Biol Chem. 2011;286(30):26667–26679. | |
Iijima K, Kobayashi T, Hara K, et al. IL-33 and thymic stromal lymphopoietin mediate immune pathology in response to chronic airborne allergen exposure. J Immunol. 2014;193(4):1549–1559. | |
Tamachi T, Maezawa Y, Ikeda K, et al. IL-25 enhances allergic airway inflammation by amplifying a TH2 cell-dependent pathway in mice. J Allergy Clin Immunol. 2006;118(3):606–614. | |
Allakhverdi Z, Comeau MR, Jessup HK, et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med. 2007;204:253–258. | |
Hallstrand TS, Hackett TL, Altemeier WA, Matute-Bello G, Hansbro PM, Knight DA. Airway epithelial regulation of pulmonary immune homeostasis and inflammation. Clin Immunol. 2014;151(1):1–15. | |
Pope SM, Brandt EB, Mishra A, et al. IL-13 induces eosinophil recruitment into the lung by an IL-5- and eotaxin-dependent mechanism. J Allergy Clin Immunol. 2001;108:594–601. | |
Robbe P, Draijer C, Borg TR, et al. Distinct macrophage phenotypes in allergic and nonallergic lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2015;308(4):L358–L367. | |
Roos AB, Mori M, Grönneberg R, et al. Elevated exhaled nitric oxide in allergen-provoked asthma is associated with airway epithelial iNOS. PLoS One. 2014;9(2):e90018. | |
Chibana K, Trudeau JB, Mustovich AT, et al. IL-13 induced increases in nitrite levels are primarily driven by increases in inducible nitric oxide synthase as compared with effects on arginases in human primary bronchial epithelial cells. Clin Exp Allergy. 2008;38:936–946. | |
Busse WW. The relationship of airway hyperresponsiveness and airway inflammation: airway hyperresponsiveness in asthma: its measurement and clinical significance. Chest. 2010;138(2 Suppl):4S–10S. | |
Kuperman DA, Huang X, Koth LL, et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med. 2002;8:885–889. | |
Tournoy KG, Kips JC, Pauwels RA. The allergen-induced airway hyperresponsiveness in a human-mouse chimera model of asthma is T cell and IL-4 and IL-5-dependent. J Immunol. 2001;166(11):6982–6991. | |
Kaur D, Gomez E, Doe C, et al. IL-33 drives airway hyper-responsiveness through IL-13-mediated mast cell: airway smooth muscle crosstalk. Allergy. 2015;70(5):556–567. | |
Riley JP, Fuchs B, Sjöberg L, et al. Mast cell mediators cause early allergic bronchoconstriction in guinea pigs in vivo: a model of relevance to asthma. Clin Sci (Lond). 2013;125(11):533–542. | |
Hamilton A, Faiferman I, Stober P, Watson RM, O’Byrne PM. Pranlukast, a cysteinyl leukotriene receptor antagonist, attenuates allergen-induced early- and late-phase bronchoconstriction and airway hyperresponsiveness in asthmatic subjects. J Allergy Clin Immunol. 1998;102(2):177–183. | |
Cui Y, Dahlin JS, Feinstein R, et al. Mouse mast cell protease-6 and MHC are involved in the development of experimental asthma. J Immunol. 2014;193(10):4783–4789. | |
Peng T, Hao L, Madri JA, et al. Role of C5 in the development of airway inflammation, airway hyperresponsiveness, and ongoing airway response. J Clin Invest. 2005;115(6):1590–1600. | |
Meurs H, Gosens R, Zaagsma J. Airway hyperresponsiveness in asthma: lessons from in vitro model systems and animal models. Eur Respir J. 2008;32:487–502. | |
Johnson JR, Wiley RE, Fattouh R, et al. Continuous exposure to house dust mite elicits chronic airway inflammation and structural remodeling. Am J Respir Crit Care Med. 2004;169(3):378–385. | |
Schroeder BW, Verhaeghe C, Park SW, et al. AGR2 is induced in asthma and promotes allergen-induced mucin overproduction. Am J Respir Cell Mol Biol. 2012;47(2):178–185. | |
Trian T, Allard B, Dupin I, et al. House dust mites induce proliferation of severe asthmatic smooth muscle cells via an epithelium-dependent pathway. Am J Respir Crit Care Med. 2015;191(5):538–546. | |
West AR, Syyong HT, Siddiqui S, et al. Airway contractility and remodeling: links to asthma symptoms. Pulm Pharmacol Ther. 2013; 26(1):3–12. | |
Kraft M, Lewis C, Pham D, Chu HW. IL-4, IL-13, and dexamethasone augment fibroblast proliferation in asthma. J Allergy Clin Immunol. 2001;107(4):602–606. | |
Ingram JL, Huggins MJ, Church TD, et al. Airway fibroblasts in asthma manifest an invasive phenotype. Am J Respir Crit Care Med. 2011; 183(12):1625–1632. | |
Kelly MM, Chakir J, Vethanayagam D, et al. Montelukast treatment attenuates the increase in myofibroblasts following low-dose allergen challenge. Chest. 2006;130(3):741–753. | |
Stumm CL, Halcsik E, Landgraf RG, Camara NO, Soqayar MC, Jancar S. Lung remodeling in a mouse model of asthma involves a balance between TGF-B1 and BMP-7. PLoS One. 2014;9(4):e95959. | |
Firszt R, Francisco D, Church TD, Thomas JM, Ingram JL, Kraft M. Interleukin-13 induces collagen type-1 expression through matrix metalloproteinase-2 and transforming growth factor-beta1 in airway fibroblasts in asthma. Eur Respir J. 2014;43(2):464–473. | |
Roche WR, Beasley R, Williams JH, Holgate ST. Subepithelial fibrosis in the bronchi of asthmatics. Lancet. 1989;1(8637):520–524. | |
Brillet PY, Debray MP, Golmard JL, et al. Computed tomography assessment of airways throughout bronchial tree demonstrates airway narrowing in severe asthma. Acad Radiol. 2015;22(6):734–742. | |
Fish JE, Peters SP. Airway remodeling and persistent airway obstruction in asthma. J Allergy Clin Immunol. 1999;104(3 pt 1):509–516. | |
Shifren A, Witt C, Christie C, Castro M. Mechanisms of remodeling in asthmatic airways. J Allergy (Cairo). 2012;2012:316049. | |
Kariyawasam HH, Aizen M, Barkans J, Robinson DS, Kay AB. Remodeling and airway hyperresponsiveness but not cellular inflammation persist after allergen challenge in asthma. Am J Respir Crit Care Med. 2007;175(9):896–904. | |
Gill SE, Parks WC. Metalloproteinases and their inhibitors: regulators of wound healing. Int J Biochem Cell Biol. 2008;40(6–7):1334–1347. | |
Rohani MG, Parks WC. Matrix remodeling by MMPs during wound repair. Matrix Biol. 2015;44–46:113–121. | |
Nissinen L, Kahari VM. Matrix metalloproteinases in inflammation. Biochim Biophys Acta. 2014;1840(8):2571–2580. | |
Giannandrea M, Parks WC. Diverse functions of matrix metalloproteinases during fibrosis. Dis Model Mech. 2014;7(2):193–203. | |
Sergejeva S, Ivanov S, Lotvall J, Linden A. Interleukin-17 as a recruitment and survival factor for airway macrophages in allergic airway inflammation. Am J Respir Cell Mol Biol. 2005;33(3):248–253. | |
Ventura I, Vega A, Chacón P, et al. Neutrophils from allergic asthmatic patients produce and release metalloproteinase-9 upon direct exposure to allergens. Allergy. 2014;69(7):898–905. | |
Kelly EA, Busse WW, Jarjour NN. Increased matrix metalloproteinase-9 in the airway after allergen challenge. Am J Respir Crit Care Med. 2000; 162(3 pt 1):1157–1161. | |
Chiba Y, Yu Y, Sakai H, Misawa M. Increase in the expression of matrix metalloproteinase-12 in the airways of rats with allergic bronchial asthma. Biol Pharm Bull. 2007;30(2):318–323. | |
Goswami S, Angkasekwinai P, Shan M, et al. Divergent functions for airway epithelial matrix metalloproteinase 7 and retinoic acid in experimental asthma. Nat Immunol. 2009;10(5):496–503. | |
Ohta Y, Hayashi M, Kanemaru T, Abe K, Ito Y, Oike M. Dual modulation of airway smooth muscle contraction by Th2 cytokines via matrix metalloproteinase-1 production. J Immunol. 2008;180(6):4191–4199. | |
Diaz A, García F, Mozos A, et al. Lymphoid tissue collagen deposition in HIV-infected patients correlates with the imbalance between matrix metalloproteinases and their inhibitors. J Infect Dis. 2011;203(6):810–813. | |
Polyakova V, Loeffler I, Hein S, et al. Fibrosis in endstage human heart failure: severe changes in collagen metabolism and MMP/TIMP profiles. Int J Cardiol. 2011;151(1):18–33. | |
Ohno I, Ohtani H, Nitta Y, et al. Eosinophils as a source of matrix metalloproteinase-9 in asthmatic airway inflammation. Am J Respir Cell Mol Biol. 1997;16:212–219. | |
Kumagai K, Ohno I, Okada S, et al. Inhibition of matrix metalloproteinases prevents allergen-induced airway inflammation in a murine model of asthma. J Immunol. 1999;162:4212–4219. | |
Kumagai K, Ohno I, Imai K, et al. The involvement of matrix metalloproteinases in basement membrane injury in a murine model of acute allergic airway inflammation. Clin Exp Allergy. 2002;32:1527–1534. | |
Schwingshackl A, Duszyk M, Brown N, Moqbel R. Human eosinophils release matrix metalloproteinase-9 on stimulation with TNF-α. J Allergy Clin Immunol. 1999;104:983–990. | |
Mattos W, Lim S, Russell R, Jatakanon A, Chung KF, Barnes PJ. Matrix metalloproteinase-9 expression in asthma: effect of asthma severity, allergen challenge, and inhaled corticosteroids. Chest. 2002;122:1543–1552. | |
Erlewyn-Lajeunesse MD, Hunt LP, Pohunek P, et al. Bronchoalveolar lavage MMP-9 and TIMP-1 in preschool wheezers and their relationship to persistent wheeze. Pediatr Res. 2008;64(2):194–199. | |
Doherty GM, Kamath SV, de Courcey F, et al. Children with stable asthma have reduced airway matrix metalloproteinase-9 and matrix metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio. Clin Exp Allergy. 2005;35(9):1168–1174. | |
Cataldo DD, Tournoy KG, Vermaelen K, et al. Matrix metalloproteinase-9 deficiency impairs cellular infiltration and bronchial hyperresponsiveness during allergen-induced airway inflammation. Am J Pathol. 2002;161(2):491–498. | |
Sands MF, Ohtake PJ, Mahajan SD, et al. Tissue inhibitor of metalloproteinase-1 modulates allergic lung inflammation in murine asthma. Clin Immunol. 2009;130(2):186–198. | |
McMillan SJ, Kearley J, Campbell JD, et al. Matrix metalloproteinase-9 deficiency results in enhanced allergen-induced airway inflammation. J Immunol. 2004;172(4):2586–2594. | |
Corry DB, Kiss A, Song LZ, et al. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB J. 2004;18(9):995–997. | |
Corry DB, Rishi K, Kanellis J, et al. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nat Immunol. 2002;3(4):347–353. | |
Vermaelen KY, Cataldo D, Tournoy K, et al. Matrix metalloproteinase-9-mediated dendritic cell recruitment into the airways is a critical step in a mouse model of asthma. J Immunol. 2014;171(2):1016–1022. | |
Mehra D, Sternberg DI, Jia Y, et al. Altered lymphocyte trafficking and diminished airway reactivity in transgenic mice expressing human MMP-9 in a mouse model of asthma. Am J Physiol Lung Cell Mol Physiol. 2010;298(2):L189–L196. | |
Greenlee KJ, Corry DB, Engler DA, et al. Proteomic identification of in vivo substrates for matrix metalloproteinases 2 and 9 reveals a mechanism for resolution of inflammation. J Immunol. 2006;177(10):7312–7321. | |
Page K, Hughes VS, Bennett GW, Wong HR. German cockroach proteases regulate matrix metalloproteinase-9 in human bronchial epithelial cells. Allergy. 2006;61(8):988–995. | |
Hughes VS, Page K. German cockroach frass proteases cleave pro-matrix metalloproteinase-9. Exp Lung Res. 2007;33(3–4):135–150. | |
Page K, Ledford JR, Zhou P, Wills-Karp MA. TLR2 agonist in German cockroach frass activates MMP-9 release and is protective against allergic inflammation in mice. J Immunol. 2009;183(5):3400–3408. | |
Pouladi MA, Robbins CS, Swirski FK, et al. Interleukin-13-dependent expression of matrix metalloproteinase-12 is required for the development of airway eosinophilia in mice. Am J Respir Cell Mol Biol. 2004;30(1):84–90. | |
Warner RL, Lukacs NW, Shapiro SD, et al. Role of metalloelastase in a model of allergic lung responses induced by cockroach allergen. Am J Pathol. 2004;165(6):1921–1930. | |
Mukhopadhyay S, Sypek J, Tavendale R, et al. Matrix metalloproteinase-12 is a therapeutic target for asthma in children and young adults. J Allergy Clin Immunol. 2010;126(1):70. e–76. e. | |
Chen W, Tabata Y, Gibson AM, et al. Matrix metalloproteinase 8 contributes to solubilization of IL-13 receptor alpha2 in vivo. J Allergy Clin Immunol. 2008;122(3):625–632. | |
Zheng T, Liu W, Oh SY, et al. IL-13 receptor alpha2 selectively inhibits IL-13-induced responses in the murine lung. J Immunol. 2008;180(1):522–529. | |
Gueders MM, Balbin M, Rocks N, et al. Matrix metalloproteinase-8 deficiency promotes granulocytic allergen-induced airway inflammation. J Immunol. 2005;175(4):2589–2597. | |
Rogers NK, Clements D, Dongre A, Harrison TW, Shaw D, Johnson SR. Extra-cellular matrix proteins induce matrix metalloproteinase-1 (MMP-1) activity and increase airway smooth muscle contraction in asthma. PLoS One. 2014;9(2):e90565. | |
Margulis A, Nocka KH, Brennan AM, et al. Mast cell-dependent contraction of human airway smooth muscle cell-containing collagen gels: influence of cytokines, matrix metalloproteases, and serine proteases. J Immunol. 2009;183(3):1739–1750. | |
Bruce C, Thomas PS. The effect of marimastat, a metalloprotease inhibitor, on allergen-induced asthmatic hyper-reactivity. Toxicol Appl Pharmacol. 2005;205(2):126–132. | |
Zheng T, Zhu Z, Wang Z, et al. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest. 2000;106(9):1081–1093. | |
Ingram JL, Slade D, Church TD, et al. Matrix metalloproteinases-1 and -2 mediate IL-13-induced suppression of elastin in airway fibroblasts in asthma. Am J Respir Cell Mol Biol. Epub 2015 Jun 12. | |
Liu J, Rich CB, Buczek-Thomas JA, Nugent MA, Panchenko MP, Foster JA. Heparin-binding EGF-like growth factor regulates elastin and FGF-2 expression in pulmonary fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2003;285(5):L1106–L1115. | |
DiCamillo SJ, Carreras I, Panchenko MV, et al. Elastase-released epidermal growth factor recruits epidermal growth factor receptor and extracellular signal-regulated kinases to down-regulate tropoelastin mRNA in lung fibroblasts. J Biol Chem. 2002;277(21):18938–18946. | |
Rajah R, Nachajon RV, Collins MH, Hakonarson H, Grunstein MM, Cohen P. Elevated levels of the IGF-binding protein protease MMP-1 in asthmatic airway smooth muscle. Am J Respir Cell Mol Biol. 1999;20(2):199–208. | |
Al Heialy S, Risse PA, Zeroual MA, et al. T cell-induced airway smooth muscle cell proliferation via the epidermal growth factor receptor. Am J Respir Cell Mol Biol. 2013;49(4):563–570. | |
Todorova L, Bjermer L, Miller-Larsson A, Westergren-Thorsson G. Relationship between matrix production by bronchial fibroblasts and lung function and AHR in asthma. Respir Med. 2010;104(12):1799–1808. | |
Araujo BB, Dolhnikoff M, Silva LF, et al. Extracellular matrix components and regulators in the airway smooth muscle in asthma. Eur Respir J. 2008;32(1):61–69. | |
Tang LF, Du LZ, Zou CC, Gu WZ. Levels of matrix metalloproteinase-9 and its inhibitor in guinea pig asthma model following ovalbumin challenge. Fetal Pediatr Pathol. 2005;24(2):81–87. | |
Lim DH, Cho JY, Miller M, McElwain K, McElwain S, Broide DH. Reduced peribronchial fibrosis in allergen-challenged MMP-9-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2006;291(2):L265–L271. | |
Prado CM, Yano L, Rocha G, et al. Effects of inducible nitric oxide synthase inhibition in bronchial vascular remodeling-induced by chronic allergic pulmonary inflammation. Exp Lung Res. 2011;37(5):259–268. | |
Ko FW, Diba C, Roth M, et al. A comparison of airway and serum matrix metalloproteinase-9 activity among normal subjects, asthmatic patients, and patients with asthmatic mucus hypersecretion. Chest. 2005;127(6):1919–1927. | |
Pham DN, Chu HW, Martin RJ, Kraft M. Increased matrix metalloproteinase-9 with elastolysis in nocturnal asthma. Ann Allergy Asthma Immunol. 2003;90(1):72–78. | |
Munoz X, Bustamante V, Lopez-Campos JL, Cruz MJ, Barreiro E. Usefulness of noninvasive methods for the study of bronchial inflammation in the control of patients with asthma. Int Arch Allergy Immunol. 2015;166(1):1–12. | |
Belleguic C, Corbel M, Germain N, et al. Increased release of matrix metalloproteinase-9 in the plasma of acute severe asthmatic patients. Clin Exp Allergy. 2002;32(2):217–223. | |
Zhang L, Gang J, Zhigang C, et al. Irreversible airway obstruction assessed by high-resolution computed tomography (HRCT), exhaled nitric oxide (FENO), and biological markers in induced sputum in patients with asthma. Wien Klin Wochenschr. 2014;126(17–18):515–523. | |
Gagliardo R, La Grutta S, Chanez P, et al. Non-invasive markers of airway inflammation and remodeling in childhood asthma. Pediatr Allergy Immunol. 2009;20(8):780–790. | |
Cataldo DD, Bettiol J, Noel A, Bartsch P, Foidart JM, Louis R. Matrix metalloproteinase-9, but not tissue inhibitor of matrix metalloproteinase-1, increases in the sputum from allergic asthmatic patients after allergen challenge. Chest. 2002;122(5):1553–1559. | |
Cataldo DD, Gueders M, Munaut C, et al. Matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases mRNA transcripts in the bronchial secretions of asthmatics. Lab Invest. 2004;84(4):418–424. | |
Boulay ME, Prince P, Deschesnes F, Chakir J, Boulet LP. Metalloproteinase-9 in induced sputum correlates with the severity of the late allergen-induced asthmatic response. Respiration. 2004;71(3):216–224. | |
Karakoc GB, Yukselen A, Yilmaz M, Altintas DU, Kendirli SG. Exhaled breath condensate MMP-9 level and its relationship with asthma severity and interleukin-4/10 levels in children. Ann Allergy Asthma Immunol. 2012;108(5):300–304. | |
Barbaro MP, Spanevello A, Palladino GP, Salerno FG, Lacedonia D, Carpagnano GE. Exhaled matrix metalloproteinase-9 (MMP-9) in different biological phenotypes of asthma. Eur J Intern Med. 2014;25(1):92–96. | |
Blumenthal MN, Zhong W, Miller M, Wendt C, Connett JE, Pei D. Serum metalloproteinase leukolysin (MMP-25/MT-6):a potential metabolic marker for atopy-associated inflammation. Clin Exp Allergy. 2010;40(6):859–866. | |
Nakashima K, Hirota T, Obara K, et al. A functional polymorphism in MMP-9 is associated with childhood atopic asthma. Biochem Biophys Res Commun. 2006;344(1):300–307. | |
Jiménez-Morales S, Martínez-Aguilar N, Gamboa-Becerra R, et al. Polymorphisms in metalloproteinase-9 are associated with the risk for asthma in Mexican pediatric patients. Hum Immunol. 2013;74(8):998–1002. | |
Huang CD, Lin SM, Chang PJ, et al. Matrix metalloproteinase-1 polymorphism is associated with persistent airway obstruction in asthma in the Taiwanese population. J Asthma. 2009;46(1):41–46. | |
Birbian N, Singh J, Jindal SK. Highly protective association of MMP-2-1306C/T promoter polymorphism with asthma in a North Indian population: a pilot study. Allergy Asthma Immunol Res. 2014;6(3):234–241. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.