")
Back to Archived Journals » Metalloproteinases In Medicine » Volume 3
Metalloproteinases and their inhibitors in the pathophysiology of heritable connective tissue disorders: current evidence
Authors Delhon L, Cormier-Daire V, Le Goff C
Received 22 September 2015
Accepted for publication 3 December 2015
Published 4 February 2016 Volume 2016:3 Pages 1—9
DOI https://doi.org/10.2147/MNM.S63634
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yoshifumi Itoh
Laure Delhon, Valerie Cormier-Daire, Carine Le Goff
INSERM UMR 1163, Laboratory of Molecular and Physiopathological Bases of Osteochondrodysplasias, Imagine Institute, Paris Descartes University, Paris, France
Abstract: Extracellular matrix is composed of macromolecules constituting a complex network that allows the regulation of major cellular functions and maintenance of normal tissue homeostasis. The turnover of extracellular matrix is mediated by matrix-degrading enzymes such as metalloproteinases during normal and pathological conditions. Matrix metalloproteinases and a disintegrin and metalloproteinase with thrombospondin repeats (ADAMTS) are metalloproteinases that have been identified in distinct genetic disorders. The aim of our review was to emphasize the role of the metalloproteinases based on heritable connective tissue disorder phenotypes identified in relation with ADAMTS, ADAMTS-like, or metalloproteinases mutations.
Keywords: MMP, ADAMTS, connective tissue, collagen, disorder
Introduction
Extracellular matrix (ECM) is composed of macromolecules such as collagens, proteoglycans, elastin, fibronectin, laminins, and several other glycoproteins, which form a complex network around cells in all tissues and organs. This network plays a role in signal transduction allowing the regulation of major cellular functions and maintenance of normal tissue homeostasis. The remodeling of ECM is mediated by matrix-degrading enzymes such as metalloproteinases during normal and pathological conditions.
Matrix metalloproteinase (MMP), a disintegrin and metalloproteinase (ADAM) and a disintegrin and metalloproteinase with thrombospondin repeats (ADAMTS) are proteases that belong to the metzincin family, named after the presence of zinc ion and the conserved Met residue at the active site.
Mammalian MMPs share a conserved domain structure that consists of a catalytic domain and an autoinhibitory prodomain.1 The removal of the prodomain is mandatory to activate the MMPs. Their C-termini also contain a hemopexin domain that mediates protein–protein interactions.2 The C-terminal domain has a role in substrate recognition, enzyme activation, and protease localization, internalization, and degradation. MMP-2 and MMP-9 contain three fibronectin type-II repeats that mediate binding of collagens.1 All MMPs are not secreted, six MMPs are transmembrane. MMPs are divided into six subgroups according to their similarities in tridimensional structure and substrate affinity: collagenases, gelatinases, stromelysins, matrilysins, membrane-type metalloproteinases, and others.3 The primary function of MMPs is to degrade the structural components of ECM. MMPs have also a major role in the regulation and turnover of ECM. MMPs can impact on cellular function by regulating the ECM proteins. For example, MMP-2 and MMP-9 are known to cleave transforming growth factor (TGF)β, and through this activation module they promote tumor invasion.4 Studies on mmp-mutant mice have pointed out the requirement of MMPs in postnatal development and tissue remodeling but not in embryogenesis.
The superfamily of ADAMTS contains 19 ADAMTS and 7 ADAMTS-like (ADAMTSL) proteins, which are secreted enzymes. ADAMTS are organized into two domains: catalytic domain and ancillary domain.5 The catalytic domain, at the N-terminus, includes a metalloproteinase domain and a disintegrin-like domain. The ancillary domain, at the C-terminus, is organized with a modular structure comprising, at least, one or more thrombospondin type-I repeats. ADAMTS precursors are activated intracellularly in the Golgi or extracellularly by the cleavage of furin, a pro-protein convertase.6 They are distinct from the membrane-anchored ADAMTS mainly involved in protein ectodomain shedding from the cell surface. ADAMTS proteases are implicated in different biological processes: procollagen propeptidase (ADAMTS2, ADAMTS3, and ADAMTS14), aggrecanase in arthritis (ADAMTS4, ADAMTS5), other aggrecanases (ADAMTS1, ADAMTS8, ADAMTS9, ADAMTS15, and ADAMTS20), von Willebrand factor proteolysis (ADAMTS13), and cartilage oligomeric matrix protein proteolysis (ADAMTS7, ADAMTS12).5,7–12
ADAMTSL proteins have an ancillary domain similar to ADAMTS proteins but do not possess enzymatic activity because of the lack of a catalytic domain. Similar to ADAMTS, they are ECM proteins, but their functions are unknown.13–16 However, it was observed that papilin, another ECM glycoprotein that shares similar structures with ADAMTSL, has an inhibitory action on ADAMTS proteinase similar to ADAMTS2.17,18
ADAMTSs and MMPs are regulated by tissue inhibitors of metalloproteinases (TIMPs). The family comprises four protease inhibitors (TIMP1–4).5
Deregulation of ECM composition and structure is associated with the development and progression of several pathologic conditions such as connective tissue disorders. Connective tissue disease refers to a group of disorders involving fibrous tissues that maintain the form of organs and other parts of the body such as bones, tendons, and cartilages. Through the past decades, molecular bases of numerous connective tissue disorders have been identified. This review focuses on a group of heritable connective tissue disorders related to metalloproteinases and their inhibitors.
MMPs
MMP-9 and MMP-13 mutations and metaphyseal anadysplasia
Metaphyseal anadysplasia (MAD) is characterized by an early-onset regressive form of metaphyseal dysplasia.19 It is reported in the first months when distal metaphyses of long bones are found to be very irregular. The main clinical features are hypoplastic femoral necks and vertical edges of the metaphyses. These anomalies disappear after 2 years of age. Stature is usually not affected. The first patient with this disorder was reported by Wiedemann and Spranger and four other boys were diagnosed by Maroteaux et al.19,20
Two types of MAD have been described: type 1 (Spranger type), a clinically more severe type with reduced stature and autosomal dominant inheritance, and type 2 (Maroteaux type), a milder type with normal stature and autosomal recessive inheritance. The radiographic features of both types are indistinguishable.21
The autosomal recessive form of MAD (MIM 613073) is caused by homozygous loss of function of gene MMP-9.21 The autosomal dominant form of MAD (MANDP1 [MIM 602111]) is caused by heterozygous mutations in gene MMP-13.22 MMP-9 and MMP-13 are both involved in the degradation of ECM in the context of bone development.
mmp-9-null mice exhibited an abnormal endochondral bone development with a decreased hypertrophic chondrocyte apoptosis and trabecular bone formation.23 The growth plate of mmp-9-null mice presented an accumulation of galectin 3, the substrate of MMP-9. Galectin 3 is a regulator of MMP-9 which acts on the recruitment of osteoclast at the chondro-osseous junction. The growth retardation was compensated after 3 weeks and the mmp-9-null mice had a normal axial skeleton at adult age.24 MMP-2 and MMP-9 deregulation leads to severe elastic fiber fragmentation and degradation and may explain the thoracic aortic aneurysm observed in mutant fbn1C1039G/+, a model of Marfan syndrome.25 MMP-9 is also involved in immune system development and potentially in autoimmune disease development.26
Considering the multiple function of MMP-9, the restricted phenotype of MAD patients is due to the major role of MMP-9 in bone homeostasis.
MMP-13 is expressed by both terminal hypertrophic chondrocytes and osteoblasts, supporting its crucial role in bone formation and remodeling.27 MMP-13 is considered as a collagenase. Its substrates are collagen I and II, two major collagens in chondrogenesis.28 MMP-13 is also involved in human osteoarthritis cartilage pathophysiology.29 mmp-13-null mice that appeared healthy at birth showed defects in the growth plate. These mice showed normal differentiation of chondrocytes but the hypertrophic zone was larger as compared to the wild-type. Like mmp-9-null mice, the growth plate defect increased after 5 weeks and was resolved by 12 weeks of age. This model showed that collagen II and aggrecan are degraded by MMP-13,30 those two substrates being shared by MMP-9 and MMP-13. The double mmp-13-and mmp-9-null mice also exhibited endochondral ossification impairment, ECM remodeling defect, increased chondrocyte survival, delayed vascular recruitment, and defective trabecular bone formation leading to shortened long bones.30 These findings showed the requirement of balance between the assembly and degradation of collagen in endochondral ossification.
MMP-13 mutations and Missouri-type spondyloepimetaphyseal dysplasia
Spondyloepimetaphyseal dysplasia is characterized by vertebral, epiphyseal, and metaphyseal anomalies.31 The Missouri type (MIM 602111), an autosomal dominant disorder, displays moderate to severe metaphyseal changes, mild epiphyseal involvement, pear-shaped vertebrae in childhood, and rhizomelic shortening of the lower limbs.32 The modeling defects improved spontaneously by early adolescence, but the affected individuals remained shorter than their age-matched unaffected sibs. The bowing deformities predispose to osteoarthritis of the knees.33 MMP-13 mutations are responsible for spondyloepimetaphyseal dysplasia – Missouri type. The missense mutation in MMP-13, F56S leads to the degradation of misfolded proenzyme.32 MAD and Missouri type appear clinically distinct but are allelic conditions.
MMP-14 mutations and Winchester syndrome
Winchester syndrome (WNCHRS; MIM 277950) belongs to a group of osteolytic syndromes, which are rare skeletal disorders characterized by osteolysis and joint destruction. Generalized osteolysis of the carpals and tarsals, arthropathy, generalized osteoporosis, corneal opacities, and myocardial damages are the main features of this syndrome.34
This autosomal recessive syndrome is caused by mutations in MMP-14. Mutations are localized in the hydrophobic region of the signal peptide resulting in a defect of MMP-14 transport to the membrane. MMP-14 is an activator of MMP-2. Due to the mislocalization of MMP-14, MMP-2 is not activated.35
The mmp-14-null mice displayed skeletal dysplasia with craniofacial dysmorphism and dwarfism, arthritis, severe osteopenia, and fibrosis of the dermis.36 The mice were normal at birth but developed multiple defects and died by 3–12 weeks of age. The mmp-14-null mouse model is the only lethal mmp-mutant mice.37 This model confirmed the importance of collagenolytic activity for the development and growth.
MMP-2 mutations and Torg syndrome
Torg syndrome (MIM 259600), Winchester syndrome, and nodulus, arthropathy, osteolysis constitute a continuous clinical spectrum.38 Torg syndrome is characterized by the presence of multiple, painless, subcutaneous nodules, mild to moderate osteoporosis, and osteolysis of the carpal and tarsals. Radiographically, the osteolysis is associated with a widening of the metacarpal and metatarsal bones.39,40
This syndrome is transmitted with an autosomal recessive mode of inheritance and due to MMP-2 mutations.38
Mutant mmp-2-null mice showed attenuated features of human skeletal abnormalities.41 In contrast, mmp-2-and mmp-14-null mice died at birth. These double mutants resemble the much older mmp-14 single mutants, indicating that MMP-2 and MMP-14 function redundantly.42
MMP-20 mutations and pigmented hypomaturation-type amelogenesis imperfecta (type IIA)
Amelogenesis imperfecta (AI) is a group of hereditary defects of the enamel which are not associated with any other generalized defect.43 Pigmented hypomaturation-type amelogenesis imperfecta (MIM 612529) is characterized by anomalies of the maturation stage during the formation of enamel, resulting in an opaque and porous enamel. The enamel has a normal thickness but is hypomineralized. Radiographs show a lack of contrast between enamel and dentin.44,45 The enamel layer of the teeth was pigmented, showing an agar brown discoloration. Pigmented hypomaturation-type amelogenesis imperfecta is an autosomal recessive disorder associated with MMP-20 mutations.44,45
The exact function of MMP-20 is unknown. Recently, it has been shown that mmp-20-null mice presented with thin and soft enamel.46 Unexpectedly, introduction of transgene expressing mmp-20 in null mice, in order to compensate the thin enamel, resulted in decreased hardness. This suggests that an essential balance takes place between active and non-active MMP-20 in the formation of a properly hardened enamel layer.47
ADAMTS and ADAMTSL
ADAMTS2 mutations and Ehlers–Danlos syndrome – type VIIC
The Ehlers–Danlos syndrome (EDS) is a group of soft connective tissue disorders which are characterized by fragility in skin, ligaments and joints, blood vessels, and internal organs.48 The dermatosparaxis type of EDS (EDS type VIIC [MIM 225410]) is a recessively inherited connective tissue disorder, mainly characterized by extreme skin fragility. Joint laxity is moderate compared to other EDS.49 Other characteristics are reported such as orofacial features, umbilical hernia, and blue sclera after minor trauma.48,49
Mostly EDS are linked to mutations in one of the genes encoding fibrillar collagen proteins or enzymes involved in their maturation. EDS VIIA (MIM120150) and B (120160) autosomal dominant disorders caused by mutations in the COL1A1 and COL1A2 genes, respectively, are characterized by joint hyperlaxity and only moderate skin fragility.50 EDS VIIC is due to mutations in ADAMTS2 which encodes a procollagen N-proteinase (PNP), involved in the processing of type-I procollagen.51 In the absence of ADAMTS2 activity, the unprocessed procollagens interfere in their assembly and lead to the formation of “hieroglyphic” collagen, which is not functional and responsible for skin fragility in EDS patients.7 Interestingly, the difference in clinical manifestations between these three types of EDS VII seems related to variation in processing the type I procollagen in tissues or a potential PNP compensation.
Considering the common biosynthetic pathway, it was expected that defects in ADAMTS2 would have an impact on a wide number of tissues. The limited number of tissues involved in EDS type VIIC might be explained by redundant functions of other PNPs, with the ability to process procollagen I in tissues other than skin.
Two other enzymes, ADAMTS3 and ADAMTS14, belong to the same group of procollagen N-endopeptidase involved in the maturation of collagen fibrils.52 In the process of collagen maturation, the amino- and carboxyl-propeptide of procollagen are cleaved leading to proper assembly in collagen fibrils. The PNP processes the aminopeptide of procollagens I, II, and III in vitro.7 Their gene expression pattern during mouse embryogenesis has shown how closely related proteases with similar substrate may have distinct biological roles owing to tissue-specific gene expression.52 For example, during the mouse embryogenesis, ADAMTS3 seems able to process collagen I– and collagen II–rich tissues during the development. ADAMTS2 also plays a major role in the cleavage of aminopropeptide of type-III collagen.
While the first description of PNP activity was reported in a study of dermatosparaxis in Belgian cattle, studies on adamts2-deficient mice showed that these mice displayed skin fragility at 2 weeks of age.53
ADAMTS10 mutations and Weill-Marchesani syndrome 1
Weill–Marchesani syndrome (WMS) belongs to the acromelic dysplasia group which also includes geleophysic dysplasia (GD) and acromicric dysplasia (AD).54 Weill–Marchesani syndrome 1 (MIM 277600) is a rare connective tissue disorder characterized by short stature, brachydactyly, joint stiffness, thick skin, and lens abnormalities. WMS is distinct from GD and AD by the presence of dislocation of microspherophakic lens that causes severe myopia, glaucoma, or cataract.55,56 Two modes of inheritance have been reported: autosomal dominant and autosomal recessive. Weill–Marchesani syndrome 1 is an autosomal recessive disorder caused by mutations in ADAMTS10 which disrupt the metalloprotease domain.57 Our group also showed that FBN1 is responsible for the dominant form of WMS2.58 Another autosomal recessive form of WMS (WMS3) is caused by mutation in LTBP2 (latent TGFβ binding protein 2).59
The expression of adamts10 is ubiquitous in the mouse embryonic tissues until 12.5 days of gestation and then its expression is more specific with especially strong expression in developing lung, bone, and craniofacial region.60 Our genetic findings suggested a direct functional link between ADAMTS10 and fibrillins, extracellular microfibrils assembled from large glycoproteins. Indeed it was shown that ADAMTS10 binds to fibrillin 1 (FBN1) and fibrillin 2 (FBN2) and promotes microfibril deposition in the ECM of cultured fibroblasts.61 Despite this interaction, ADAMTS10 does not cleave FBN1 efficiently. This may be due to the absence of correct consensus sequence of the furin site in ADAMTS10 zymogen. These findings support a role of ADAMTS10 in microfibril biogenesis as a structural protein rather than as an enzyme.
ADAMTS17 mutations and Weill–Marchesani-like syndrome
Patients with Weill–Marchesani-like syndrome (WMS-like; MIM 613195) have many characteristics of WMS such as short stature and lens abnormalities but they do not show brachydactyly, joint stiffness, or stiff skin. It is an autosomal recessive disorder due to mutations in ADAMTS17.62
The function of ADAMTS17 is unknown. Studies on ADAMTS17 tissue distribution showed a high expression in fetal brain, heart, kidney, and eye. Two mutations in ADAMTS17 associated with WMS-like lead to proteins without the C-terminal Thrombospondin (TS) repeats, which may be involved in the ECM structure.62 The absence of some WMS characteristics could be explained by the lack of ADAMTS17 expression in those tissues or the activity of another ADAMTS. ADAMTS10 and ADAMTS17 have structural similarities and both are expressed in the eyes supporting that ADAMTS10 and ADAMTS17 play a critical role in crystalline lens zonule.
ADAMTSL2 mutations and geleophysic dysplasia
GD (MIM231050) is the more severe form of acromelic dysplasia and is transmitted with an autosomal recessive mode of inheritance. The main distinctive features are short stature, short hands and feet, facial features, joint contractures, a progressive cardiac valvular thickening often leading to an early death, toe walking, tracheal stenosis, and respiratory insufficiency. Radiological manifestations include delayed bone age, cone-shaped epiphyses, shortened long tubular bones, and ovoid vertebral bodies.63,64
Mutations in ADAMTSL2 are responsible for GD autosomal recessive form. This gene encodes a secreted glycoprotein of unknown function.65
Functional studies in HEK293 cells showed that ADAMTSL2 mutations lead to a decreased secretion of the mutated proteins, possibly due to the misfolding of ADAMTSL2. A first partner – LTBP1– of ADAMTSL2 has been identified. Considering the role of LTBP1 in TGFβ signaling, we have studied the TGFβ signaling pathway in the culture medium of GD patient fibroblasts. Our findings suggested that a dysregulation of TGFβ signaling is the underlying mechanism of GD and that ADAMTSL2 is directly involved in TGFβ bioavailability.65
Interestingly, the identification of heterozygous FBN1 mutations was reported in a dominant form of GD, strictly fulfilling the diagnostic criteria for GD. We identified a total of 16 heterozygous FBN1 mutations (15 missense and 1 insertion), all mutations were located in exons 41–42, encoding TGFβ binding protein-like domain 5 (TB5) of FBN1 in 19 GD.66 Moreover, a direct interaction between ADAMTSL2 and FBN1 emphasizes the potential role of ADAMTS and ADAMTSL protein in microfibril formation and function.66 FBN1 encodes the FBN1, the principal component of the microfibrillar network.67 Microfibrillar network disorganization was also consistently observed in GD fibroblasts. Recently, it has been shown that FBN1 mutations in GD and WMS2 disrupt fibrillin binding to heparin sulfate proteoglycans.68
Hubmacher et al generated an adamtsl2-null mouse model, which showed a high level of mortality at birth with lung anomalies associated with an increased expression of FBN2. They showed that ADAMTSL2 binds FBN2, another extracellular component of fibrillin family.69
ADAMTSL4 mutations and isolated ectopia lentis
Isolated ectopia lentis 2 (ECTOL2; MIM 225100) is a lens dislocation due to the disruption of zonular fibers, which are microfibrillar structures maintaining the lens. The displacement of the lens leads to an acute or chronic visual impairment. It could be associated with complications including myopia, retinal detachment, cataract, and glaucoma.70
Two transmission modes were discovered for ECTOL. The autosomal dominant form of ECTOL, named ECTOL1, is caused by mutations in FBN1.71 ECTOL2 is an autosomal recessive form of ECTOL associated with nonsense mutations in ADAMTSL4.70 Ectopia lentis can also be found in other connective tissue disorders, such as Marfan syndrome, caused by FBN1 mutations.72 More recently, a homozygous deletion in ADAMTSL4 was identified causing craniosynostosis in association with ectopia lentis.73 The impact of these mutations on the protein and the role of ADAMTSL4 in zonular fibers are unknown. The pattern of ADAMTSL4 expression shows its presence not only in ciliary body but also in cornea, iris, lens, and retina.74 But, it was shown that the presence of ADAMTSL4 increases the biogenesis of microfibrils by addition of ADAMTSL4 in fetal bovine nuchal ligament cells culture.74
ADAMTS10, ADAMTS17, ADAMTSL4 are associated with eye pathology, but only ADAMTSL4 mutations lead to a restricted phenotype suggesting a specific role of ADAMTSL4 in the lens zonule which cannot be compensated by the two other ADAMTS.
ADAMTS18 mutations and microcornea
Microcornea with myopic chorioretinal atrophy and telecanthus (MIM 615458) is an ocular syndrome, recently described in five children from four consanguineous families.75 The children had also a slight posterior rotation of the ears. It was discovered that this disorder was caused by homozygous mutations in ADAMTS18.76 ADAMTS18 is expressed during the development of not only murine embryo eye but also adult mouse eye, mainly in the retina and the lens.77 The potential role of ADAMTS18 in the formation or maintenance of microfibrils remains to be established.
TIMP (inhibitor of MMP): TIMP3 and Sorsby’s fundus dystrophy
Sorsby’s fundus dystrophy (MIM 136900) is characterized by the loss of central color vision during the fourth to fifth decade and peripheral visual loss in late life due to untreatable submacular neovascularization. Some patients suffer from night blindness.78
Sorsby’s fundus dystrophy is an autosomal dominant retinal dystrophy caused by TIMP3 mutations.79 TIMP3 has been involved in different biological processes such as liver injury, cancer, and lung development.
Conclusion
Although there are 26 different MMPs and 26 different ADAMTS(L), only a few genetic disorders have been associated with metalloproteinases (Table 1). The metalloproteinases involved in connective tissues disorders constitute two main groups: one linked to collagen fibrils and another to fibrillin microfibrils. Unlike collagen, fibrillins are not known to be important components of the ECM. The first group includes proteins that degrade not only different collagens such as MMP-2, MMP-9, MMP-13, and MMP-14 but also ADAMTS2, which is involved in the process of collagen maturation. Studies on mutant mice have shown that metalloproteinases such as MMP-9, MMP-13, and MMP-14 play crucial roles in bone development and remodeling. Their association with human bone disorders confirms this finding and also reveals their function as active regulators of postnatal tissue formation.
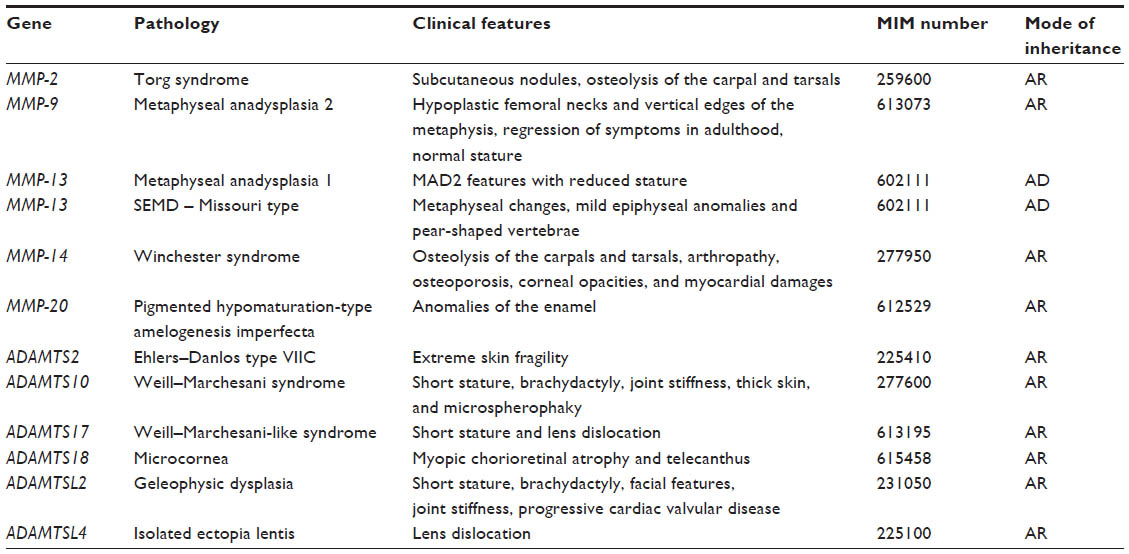 | Table 1 Main clinical features of human heritable disorders related to metalloproteinases and associated proteins |
The metalloproteinases, the ADAMTS or (-like) which are implicated or may implicate in the microfibril network as ADAMTSL2, ADAMTSL4, ADAMTS10, ADAMTS17, and ADAMTS18 form the second group (Figure 1). It is now established that ADAMTS10 directly binds FBN1 and FBN2, two principal components of microfibrils, and is involved in microfibril biogenesis. ADAMTSL2 interacts with FBN1, FBN2, and LTBP1 and accelerates the microfibril formation. ADAMTSL2 mutations are linked to the most severe human phenotype (patients present a cardiac disease leading to an early death) which can be explained by the unique relation between ADAMTSL2 and TGFβ. The short stature associated with ADAMTSL2, ADAMTS10, and ADAMTS17 mutations suggests a role of microfibril biogenesis in endochondral ossification even if no link has been demonstrated between ADAMTS17 and formation or maintenance of microfibrils.
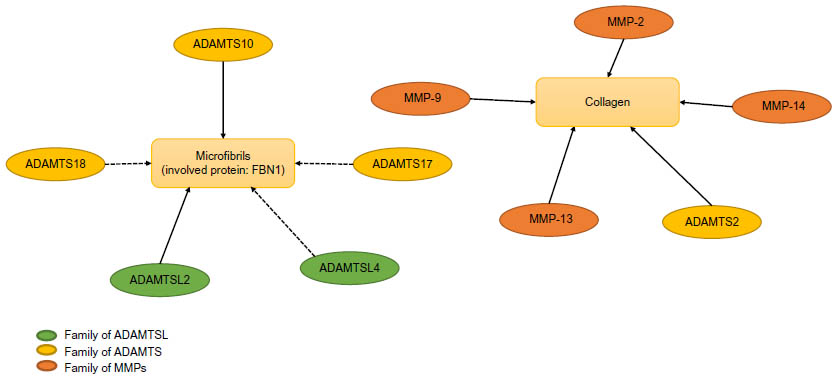 | Figure 1 Network between collagen and metalloproteinases/fibrillins and metalloproteinases. |
Moreover, the identification of ADAMTS10, ADAMTS17, and ADAMTSL4 mutations related to ectopia lentis supports an association between these proteins in zonule formation. As the eye is the only affected tissue in isolated ectopia lentis, it is considered that ADAMTSL4 seems to have a specific function in eyes even if ADAMTSL4 is widely expressed in other tissues.
Like ADAMTS1, ADAMTS15, or ADAMTS12, which display anti-angiogenic activities apart from their enzymatic activities, ADAMTS10 and ADAMTS17 may act as ADAMTSL proteins in the context of microfibrils. The function of ADAMTSL is not clearly defined yet; however, it appears that ADAMTSL2, ADAMTSL4, and ADAMTSL6 have a role in microfibril network at a structural level.
Different ADAMTS or ADAMTSL interacts with the proteins belonging to the fibrillin family such as LTBP1 and LTBP2 or FBN1 and FBN2. Ongoing studies will permit understanding of the eventual formation of complex constituted of ADAMTSL and one or more fibrillins in the manner of specific tissue.
Disclosure
LD has a PhD financing from FRM (Fondation de la RechercheMedicale-Prix Pomaret Delalande). CLG acknowledges the Association Française contre les Myopathies (AFM). The authors declare no other conflicts of interest in this work.
References
Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8(3):221–233. | |
Maskos K. Crystal structures of MMPs in complex with physiological and pharmacological inhibitors. Biochimie. 2005;87(3–4):249–263. | |
Birkedal-Hansen H, Moore WG, Bodden MK, et al. Matrix metalloproteinases: a review. Crit Rev Oral Biol Med. 1993;4(2):197–250. | |
Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14(2):163–176. | |
Apte SS. A disintegrin–like and metalloprotease (reprolysin type) with thrombospondin type 1 motifs: the ADAMTS family. Intern J Biochem Cell Biol. 2004;36:981–985. | |
Apte SS. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem. 2009;284:31493–31497. | |
Bekhouche M, Colige A. The procollagen N-proteinases ADAMTS2, 3 and 14 in pathophysiology. Matrix Biol. 2015;44–46:46–53. | |
Stanton H, Rogersoon FM, East CJ, et al. ADAMTS5 is the major aggreganase in mouse cartilage in vivo and in vitro. Nature. 2005;434:648–652. | |
Sandy JD, Verscharen C. Analysis of aggregan in human knee cartilage and synovial indicates that aggreganase (ADAMTS) activity is responsible for the catabolic turnover and loss of whole aggregan whereas other protease activity is required for C-terminal processing in vivo. Biochem J. 2001;358:615–626. | |
Asada Y, Sumiyoshi A, Hayashi T, Suzumiya J, Kaketami K. Immunohistochemistry of vascular lesion in thrombotic thromcytopenic purpura with special reference to factor VIII related antigen. Thromb Res. 1985;38:469–479. | |
Luan Y, Kong L, Howell DR, et al. Inhibition of ADAMTS-7 and ADAMTS-12 degradation of cartilage oligomeric matrix protein by alpha-2-macroglobulin. Osteoarthritis Cartilage. 2008;16(11):1413–1420. | |
Liu CJ, Kong W, Ilalov K, et al ADAMTS-7: a metalloproteinase that directly binds to and degrades cartilage oligomeric matrix protein. FASEB J. 2006;20(7):988–990. | |
Hirohata S, Wang LW, Miyagi M, et al. Punctin, a novel ADAMTS-like molecule, ADAMTSL-1, in extracellular matrix. J Biol Chem. 2002;277:12182–12189. | |
Hall NG, Klenotic P, Anand-Apte B, Apte SS. ADAMTSL-3/punctin-2, a novel glycoprotein in extracellular matrix related to the ADAMTS family of metalloproteases. Matrix Biol. 2003;22:501–510. | |
Koo BH, Le Goff C, Jungers KA, et al. ADAMTS-like 2 (ADAMTSL2) is a secreted glycoprotein that is widely expressed during mouse embryogenesis and is regulated during skeletal myogenesis. Matrix Biol. 2007;26:431–441. | |
Tsutsui K, Manabe R, Yamada T, et al. ADAMTSL-6 is a novel extracellular matrix protein that binds to fibrillin-1 and promotes fibrillin-1 fibril formation. J Biol Chem. 2010;285:4870–4882. | |
Kramerova IA, Kawaguchi N, Fessler LI, et al. Papilin in development; a pericellular protein with a homology to the ADAMTS metalloproteinases. Development. 2000;127:5475–5485. | |
Fessler JH, Kramerova I, Kramerov A, Chen Y, Fessler LI. Papilin, a novel component of basement membranes, in relation to ADAMTS metalloproteases and ECM development. Int J Biochem Cell Biol. 2004;36(6):1079–1084. | |
Maroteaux P, Verloes A, Stanescu V, Stanescu R. Metaphysealanadysplasia: a metaphyseal dysplasia of early onset with radiological regression and benign course. Am J Med Genet. 1991;39:4–10. | |
Wiedemann H-R, Spranger J. Chondrodysplasia metaphysaria (Dysostosismetaphysaria) – ein neuer Typ? Z Kinderheilk. 1970;108:171–186. | |
Lausch E, Keppler R, Hilbert K. Mutations in MMP9 and MMP13 determine the mode of inheritance and the clinical spectrum of metaphysealanadysplasia. Am J Hum Genet. 2009;85:168–178. | |
Bonafe L, Liang J, Gorna MW, et al. MMP13 mutations are the cause of recessive metaphyseal dysplasia, Spahr type. Am J Med Genet. 2014;164A:1175–1179. | |
Ortega N, Behonick DJ, Colnot C, Cooper DN, Werb Z. Galectin-3 is a downstream regulator of matrix metalloproteinase-9 function during endochondral bone formation. Mol Biol Cell. 2005;16(6):3028–3039. | |
Vu TH, Shipley JM, Bergers G, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. | |
Chung AWY, Yeung KA, Sandor GG, et al. Loss of elastic fiber integrity and reduction of vascular smooth muscle contraction resulting from the upregulated activities of matrix metalloproteinase-2 and -9 in the thoracic aortic aneurysm in Marfan syndrome. Circ Res. 2007;101:512–522. | |
Dubois B, Masure S, Hurtenbach U, et al. Resistance of young gelatinase B-deficient mice to experimental autoimmune encephalomyelitis and necrotizing tail lesions. J Clin Invest. 1999;104:1507–1515. | |
Engsig MT, Chen QJ, Vu TH, et al. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J Cell Biol. 2000;151(4):879–889. | |
Mitchell PG, Magna HA, Reeves LM, et al. Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J Clin Invest. 1996;97:761–768. | |
Reboul P, Pelletier J-P, Tardif G, Cloutier J-M, Martel-Pelletier J. The new collagenase, collagenase-3, is expressed and synthesized by human chondrocytes but not by synoviocytes: a role in osteoarthritis. J Clin Invest. 1996;97:2011–2019. | |
Stickens D, Behonick DJ, Ortega N, et al. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development. 2004;131(23):5883–5895. | |
Borochowitz ZU, Scheffer D, Adir V, Dagoneau N, Munnich A, Cormier-Daire V. Spondylo-epi-metaphyseal dysplasia (SEMD) matrilin 3 type: homozygote matrilin 3 mutation in a novel form of SEMD. J Med Genet. 2004;41:366–372. | |
Kennedy AM, Inada M, Krane SM, et al. MMP13 mutation causes spondyloepimetaphyseal dysplasia, Missouri type (SEMD(MO)). J Clin Invest. 2005;115:2832–2842. | |
Patel AC, McAlister WH, Whyte MP. Spondyloepimetaphyseal dysplasia: clinical and radiologic investigation of a large kindred manifesting autosomal dominant inheritance, and a review of the literature. Medicine. 1993;72:326–342. | |
Winchester P, Grossman H, Lim WN, Danes BS. A new acid mucopolysaccharidosis with skeletal deformities simulating rheumatoid arthritis. Am J Roentgen. 1969;106:121–128. | |
Evans BR, Mosig RA, Lobl M, et al. Mutation of membrane type-1 metalloproteinase, MT1-MMP, causes the multicentric osteolysis and arthritis disease Winchester syndrome. Am J Hum Genet. 2012;91:572–576. | |
Holmbeck K, Bianco P, Caterina J, et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99:81–92. | |
Zhou Z, Apte SS, Soininen R, et al. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc Natl Acad Sci U S A. 2000;97(8):4052–4057. | |
Zankl A, Pachman L, Poznanski A, et al. Torg syndrome is caused by inactivating mutations in MMP2 and is allelic to NAO and Winchester syndrome. J Bone Miner Res. 2007;22:329–333. | |
Torg JS, DiGeorge AM, Kirkpatrick JA Jr, Trujillo MM. Hereditary multicentric osteolysis with recessive transmission: a new syndrome. J Pediat. 1969;75:243–252. | |
Eisenstein DM, Poznanski AK, Pachman LM. Torgosteolysis syndrome. Am J Med Genet. 1998;80:207–212. | |
Inoue K, Mikuni-Takagaki Y, Oikawa K, et al. A crucial role for matrix metalloproteinase 2 in osteocyticcanalicular formation and bone metabolism. J Biol Chem. 2006;281(44):33814–33824. | |
Oh J, Takahashi R, Adachi E, et al. Mutations in two matrix metalloproteinase genes, MMP-2 and MT1-MMP, are synthetic lethal in mice. Oncogene. 2004;23(29):5041–5048. | |
Witkop CJ Jr. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J Oral Path. 1989;17:547–553. | |
Kim J-W, Simmer JP, Hart TC, et al. MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet. 2005;42:271–275. | |
Ozdemir D, Hart PS, Ryu OH, et al. MMP20 active-site mutation in hypomaturation amelogenesis imperfecta. J Dent Res. 2005;84:1031–1035. | |
Caterina JJ, Skobe Z, Shi J, et al. Enamelysin (matrix metalloproteinase 20)-deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem. 2002;277:49598–49604. | |
Shin M, Hu Y, Tye CE. Matrix metalloproteinase-20 over-expression is detrimental to enamel development: a Mus musculus model. PLoS One. 2014;9(1):e86774. | |
Lapiere CM, Nusgens BV. Ehlers DAnlos type VIIC or human dermatosparaxis. The offspring of a union between basic and clinical research. Arch Dermatol. 1993;129:1316–1319. | |
Nusgens BV, Verellen-Dumoulin C, Hermanns-Le T, et al. Evidence for a relationship between Ehlers-Danlos type VII C in humans and bovine dermatosparaxis. Nat Genet. 1992;1:214–217. | |
Malfait F, De Paepe A. The Ehlers-Danlos syndrome. Adv Exp Med Biol. 2014;802:129–143. | |
Colige A, Sieron AL, Li SW, et al. Human Ehlers-Danlos syndrome type VIIc and bovine dermatosparaxis are caused by mutations in the procollagen I N proteinase gene. Am J Hum Genet, 1999;65:308–317. | |
Le Goff C, Somerville RP, Kesteloot F. Regulation of procollagen amino-propeptide processing during mouse embryogenesis by specialization of homologous ADAMTS proteases: insights on collagen biosynthesis and dermatosparaxis. Development. 2006;133:1587–1596. | |
Li SW, Arita M, Fertala A, et al. Transgenic mice with inactive alleles for procollagen N-proteinase (ADAMTS-2) develop fragile skin and male sterility. Biochem J. 2001;355(Pt 2):271–278. | |
Le Goff C, Cormier-Daire V. Genetic and molecular aspects of acromelic dysplasia. Pediatr Endocrinol Rev. 2009;6(3):418–423. | |
Weill G. Ectopie du cristallin et malformations générales. Ann Occul. 1932;169:21–44. | |
Marchesani O. Brachydaktylie und angerobene kugellines als systemerkrnakung. Klin Mibl Augenheilk. 1939;103:392–406. | |
Dagoneau N, Benoist-Lasselin C, Huber C, et al. ADAMTS 10 mutations in autosomal recessive Weill-Marchesani syndrome. Am J Hum Genet. 2004;75:801–806. | |
Faivre L, Gorlin RJ, Wirtz MW, et al. In-frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndrome. J Med Genet. 2003;240:34–35. | |
Haji-Seyed-Javadi R, Jelodari-Mamaghani S, Paylakhi SH. LTBP2 mutations cause Weill-Marchesani and Weill-Marchesani-like syndrome and affect disruptions in the extracellular matrix. Hum Mutat. 2012;33(8):1182–1187. | |
Somerville RP, Jungers KA, Apte SS. Discovery and characterization of a novel, widely expressed metalloprotease, ADAMTS10, and its proteolytic activation J Biol Chem. 2004;279(49):51208–51217. | |
Kutz WE, Wang LW, Bader HL, et al. ADAMTS10 protein interacts with fibrillin-1 and promotes its deposition in extracellular matrix of cultured fibroblasts. J Biol Chem. 2011;286:17156–17167. | |
Morales J, Al-Sharif L, Khalil DS, et al. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopialentis, glaucoma, spherophakia, and short stature. Am J Hum Genet. 2009;85:558–568. | |
Spranger JW, Gilbert EF, Tuffli GA, Rossiter FP, Opitz JM. Geleophysic dwarfism – a “focal” mucopolysaccharidosis? Lancet. 1971; 10:97–98. | |
Allali S, Le Goff C, Pressac-Diebold I, et al. Molecular screening of ADAMTSL2 gene in 33 patients reveals the genetic heterogeneity of geleophysic dysplasia. J Med Genet. 2011;48:417–421. | |
Le Goff C, Morice-Picard F, Dagoneau N, et al. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat Genet. 2008;40:1119–1123. | |
Le Goff C, Mahaut C, Wang LW, et al. Mutations in the TGFβ binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am J Hum Genet. 2011;89:7–14. | |
Ramirez F, Sakai LY. Biogenesis and function of fibrillin assemblies. Cell Tissue Res. 2010;339(1):71–82. | |
Cain SA, McGovern A, Baldwin AK, Baldock C, Kielty CM. Fibrillin-1 mutations causing Weill-Marchesani syndrome and acromicric and geleophysicdysplasias disrupt heparan sulfate interactions. PLoS One. 2012;7(11):e48634. | |
Hubmacher D, Wang LW, Mecham RP, Reinhardt DP, Apte SS. Adamtsl2 deletion results in bronchial fibrillin microfibril accumulation and bronchial epithelial dysplasia – a novel mouse model providing insights into geleophysic dysplasia. Dis Model Mech. 2015;8(5):487–499. | |
Ahram D, Sato TS, Kohilan A, et al. A homozygous mutation in ADAMTSL4 causes autosomal-recessive isolated ectopialentis. Am J Hum Genet. 2009;84:274–278. | |
Lonqvist L, Child A, Kainulainen K, Davidson R, Puhakka L, Peltonen L. A novel mutation of the fibrillin gene causing ectopialentis. Genomics. 1994;19:573–576. | |
Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. | |
Chandra A, Aragon-Martin JA, Sharif S, Parulekar M, Child A, Arno G. Craniosynostosis with ectopialentis and a homozygous 20-base deletion in ADAMTSL4. Ophthalmic Genet. 2013;34(1–2):78–82. | |
Gabriel LA, Wang LW, Bader H. ADAMTSL4, a secreted glycoprotein widely distributed in the eye, binds fibrillin-1 microfibrils and accelerates microfibril biogenesis. Invest Ophthalmol Vis Sci. 2012;53(1):461–469. | |
Khan AO. Microcornea with myopic chorioretinal atrophy, telecanthus and posteriorly-rotated ears: a distinct clinical syndrome. Ophthalmic Genet. 2012;33:196–199. | |
Aldahmesh MA, Alshammari MJ, Khan AO, Mohamed JY, Alhabib FA, Alkuraya FS. The syndrome of microcornea, myopic chorioretinal atrophy, and telecanthus (MMCAT) is caused by mutations in ADAMTS18. Hum Mutat. 2013;34:1195–1199. | |
Peluso I, Conte I, Testa F. The ADAMTS18 gene is responsible for autosomal recessive early onset severe retinal dystrophy. Orphanet J Rare Dis. 2013;8:16. | |
Sorsby A, Mason MEJ, Gardner N. A fundus dystrophy with unusual features (late onset and dominant inheritance of a central retinal lesion showing oedema, haemorrhage and exudates developing into generalized choroidal atrophy with massive pigment proliferation). Brit J Ophthal. 1949;33:67–97. | |
Weber, BHF, Vogt G, Pruett RC, Stohr H, Felbor U. Mutations in the tissue inhibitor metalloproteinases-3 (TIMP3) in patients with Sorsby’s fundus dystrophy. Nature Genet. 1994;8:352–356. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.