Back to Archived Journals » Metalloproteinases In Medicine » Volume 2
Metalloproteinases and neurodegenerative diseases: pathophysiological and therapeutic perspectives
Authors Rosenberg G
Received 26 May 2015
Accepted for publication 23 July 2015
Published 3 September 2015 Volume 2015:2 Pages 39—50
DOI https://doi.org/10.2147/MNM.S68849
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor William Parks
Gary A Rosenberg,1–4
1Department of Neurology, 2Department of Neurosciences, 3Department of Cell Biology and Physiology, 4Department of Mathematics and Statistics, University of New Mexico Health Sciences Center, Albuquerque, NM, USA
Abstract: Matrix metalloproteinases (MMPs) are important in the central nervous system from growth and development to brain injury and repair. Cerebral blood vessels are central to the role of the MMPs in the brain. MMPs are a final common pathway for disruption of the blood–brain barrier through proteolysis of the extracellular matrix proteins in the basal lamina surrounding cerebral capillaries and in the unraveling of the essential structure formed by tight-junction proteins that maintain the protected microenvironment necessary for neuronal function. MMPs are major factors in the pathological processes that occur in the brain with hypoxia/ischemic injury, multiple sclerosis, infection, and vascular causes of dementia. In each of these neurological disorders, the MMPs disrupt the blood–brain barrier and damage the myelinated nerve fibers. Inhibitors of MMPs block injury in each of these illnesses in animal models. It is important to develop MMP inhibitors that can be translated from animal studies to human treatments.
Keywords: blood–brain barrier, matrix metalloproteinases, multiple sclerosis, stroke, vascular cognitive impairment, Alzheimer's disease, hypoxia inducible factor-1α
Introduction
Matrix metalloproteinases (MMPs) are present in many cells of the central nervous system. Several of the MMP family members play important roles in development, injury, and repair in neurological diseases. Following the isolation of the human type IV collagenase from a metastatic melanoma cell and the realization of its importance in the spread of cancer cells from the blood to the brain, there was a major effort by many pharmaceutical companies to develop inhibitors with the intent of blocking invasion of tissues by metastatic cells.1,2 MMPs were first shown to play a role in brain cell cultures where astrocytes were found to secrete MMPs.3 In an earlier study, we showed that direct injection of MMP-2 into the brain resulted in opening of the blood–brain barrier (BBB) and that tissue inhibitor to metalloproteinases 2 (TIMP-2) blocked the barrier opening.4 Subsequently, we showed that MMPs are produced during an ischemic injury, providing the basis for the proteolytic disruption of the BBB, which is a final common response in many neuroinflammatory conditions, and is the target of therapeutic attempts to inhibit the action of the MMPs.5 Because of the MMPs’ central role in neuroinflammation and the large number of neurological disease processes related to damage to the blood vessels, they have been a subject of great research interest.
Extracellular matrix (ECM) is found in the extracellular space where it surrounds the cerebral vessels. Blood vessels have a basal lamina with several molecules that are targets for the action of the MMPs. Cerebral endothelial cells are unique highly metabolic structures that maintain the cerebral microenvironment. The first layer of protection that prevents serum proteins from entering the brain is formed by several unique tight-junction proteins, which are susceptible to proteolysis by the MMPs. Other layers beyond the endothelial cells include the basal lamina, the pericytes, and astrocyte endfeet; these components form the neurovascular unit (NVU). The major brain diseases involving the MMPs are stroke, multiple sclerosis (MS), and dementia; in each of these disorders, the MMPs are a key factor in the neuroinflammatory response. The focus of this review will be selectively on the role of the MMPs in the pathological changes in the blood vessels during various injuries. Several earlier reviews describe additional roles of the MMPs.6–10
Common features of the MMPs in nervous tissues and cerebral blood vessels
The basic structure of the MMP molecule has a propeptide that maintains latency of the enzyme by blocking the active zinc site. In the full molecule, there are multiple other regions, as shown in Figure 1A. The gelatinases, MMP-2 and MMP-9, have fibronectin binding sites that attach to the basement membrane, while stromelysin-1 (MMP-3) is a truncated version of the full molecule. The membrane type-1 metalloproteinase (MMP-14) has a transmembrane domain that is attached to the membrane (Figure 1B). In the brain, the major MMPs involved are MMP-2, MMP-3, MMP-9, and MMP-14. While other MMPs, such as MMP-8 and MMP-13, are found in the brain, they appear to play limited roles.
 | Figure 1 MMP molecular structure and major classes of MMPs. |
Secreted as latent enzymes, the MMPs require activation by either cleavage of a portion of the molecule or rearrangement without cleavage, with inactivation of the active molecule carried out mainly by TIMPs.11,12 Complicated activation mechanisms and rapid deactivation provide a protective mechanism to control proteolysis and to prevent unwanted tissue damage.11 To maintain latency, a complex series of interactions are required for activation, including the intervention of other enzymes and free radicals. Several MMPs are constitutively expressed and found normally in brain and cerebrospinal fluid (CSF). The two main constitutive enzymes are MMP-2 and MMP-14, which act close to the cell membranes, and are most likely involved in continual remodeling of the perivascular ECM proteins. Latent MMP-2 is found in astrocytes that are an essential component of the NVU. Astrocytes have processes that wrap around cerebral blood vessels; the close proximity of the foot processes to the basal lamina is an important feature of the control of permeability of the BBB by the MMPs. Activation of proMMP-2 occurs through the action of a trimolecular complex formed by proMMP-2, TIMP-2, and MMP-14. This unique arrangement causes the complex to be tethered to the extracellular space between the endfeet processes and the basal lamina, limiting the action of the active MMP-2 to the space next to the blood vessels. The proprotein convertase, furin, is an activator of MMP-14, which in turn activates the proMMP-2 to a 62 kDa active form.13
MMPs can be toxic if released untethered into the brain, and a mechanism exists that constrains the activity of the active MMP-2 to localized regions of the brain. TIMP-2 is one of four TIMPs, and is the type more closely associated with MMP-2, since it joins with MMP-14 to bind with MMP-2, thereby providing the mechanism of activation. In this manner, the active form of MMP-2 is contained in the contiguous space next to the basal lamina of the cerebral blood vessel.13 Plasmin is another enzyme involved in activation of MMP-2, and plasmin is released from plasminogen by the action of urokinase plasminogen activator (uPA) and tissue PA (tPA).14 Latent MMP-9 is activated by MMP-3 and free radicals.
Normally, CSF contains latent MMP-2, which can be measured by gelatin zymography or enzyme-linked immunosorbent assay (ELISA). Since the interstitial fluid is contiguous with the CSF, the concentration of MMPs in the CSF reflects production in the brain. Normally, when the BBB is intact, there is no leakage from the blood into the CSF compartment. However, when the BBB is disrupted by inflammation, ischemia, or trauma, some of the MMP-2 detected in the brain may come from the blood.15 To determine the endogenously produced MMP-2, it is necessary to use the MMP-2 index, which is formed by dividing MMP-2 in the CSF and blood by albumin in each compartment. In this way, leakage across a damaged cerebral blood vessel can be taken into account, and the endogenous production of the MMPs can be determined.15,16
Inducible MMPs are mainly present during inflammation, where they can act over a wider area than the tethered MMP-2, which is constrained to act close to the membrane by MMP-14. During inflammation, there is release of cytokines and other factors that induce the transcription of the main inducible MMPs, MMP-3 and MMP-9, by stimulating the activator protein-1 (AP-1) and the nuclear factor-κB (NF-κB) sites in the genes. Tumor necrosis factor-α (TNF-α) is a potent inducer of MMP-9. Secreted in the latent 92 kDa form, activation reduces MMP-9 to a 84 kDa form; MMP-3 and free radicals activate MMP-9.14 Active MMP-9 moves untethered between cells, and requires an efficient mechanism of rapid deactivation; TIMP-1 is the main inhibitor of MMP-9.12
MMPs and the NVU
The BBB is now referred to as the NVU to indicate that in addition to the endothelial cells, there is a basal lamina around the endothelial cells, pericytes, and astrocytic endfeet (Figure 2A). The brain microenvironment is protected by a series of interfaces between the blood and brain. Tight-junction proteins in the clefts between endothelial cells form the first defense. For many years, the only component discussed as part of the BBB was the endothelial cell. Tight junctions in brain endothelial cells create a high electrical resistance, similar to that found in epithelial sheets, such as the toad bladder.17 The major molecules that create the tight junctions are claudin, occludin, and zonula occludens, which join the endothelial cells together.18 During an injury, the MMPs attack both the molecules in the basal lamina and the tight-junction proteins, resulting in disruption of the BBB.
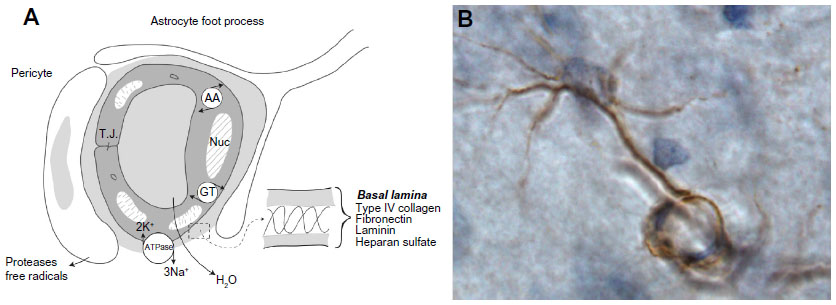 | Figure 2 Components in the neurovascular unit and an astrocyte immunostained with MMP-2. |
Astrocytes mainly produce MMP-2, which is found in the endfeet that surround the blood vessels (Figure 2B). Astrocyte endfeet also have MMP-14. MMP-9 is found in the endothelial cells. Pericytes produce MMP-3; they are macrophage-like cells embedded in the basal lamina. Microglia contain MMP-9 and MMP-3. During hypoxic injury, hypoxia-inducible factor-1α (HIF-1α) is induced, leading to the transcription of a large number of genes that aid the removal of damaged tissues and initiate the repair process.19 One of the genes activated is fur, which leads to the production of furin, an activator of MMP-14 and MMP-3.20,21 Thus, furin links hypoxia with the activation of MMP-9 through MMP-3 (Figure 3). More damage is done to the cells of the brain by the inducible enzymes, MMP-3 and MMP-9, than by MMP-2, since MMP-3 and MMP-9 are released into the extracellular space without the constraint of being attached to the membranes.
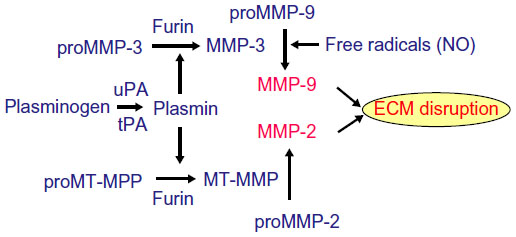 | Figure 3 Multiple mechanisms of MMP activation. |
In the acute damaging process, the released and activated MMPs are either destroyed or inactivated by TIMPs, bringing the damaging phase to a halt. However, in some still poorly understood situations, the inflammation is ongoing, with resultant chronic release of the MMPs. In lung diseases, neutrophils release MMP-9 when stimulated with N-acetyl Pro-Gly-Pro, a fragment of collagen generated by MMP-9 proteolysis; this feed-forward cycle could have implications for chronic brain inflammation.22 Much needs to be learned both about the mechanism of deactivation of MMPs and the processes that convert an acute inflammation into a chronic one.
Cerebral vessels differ from systemic vessels in multiple ways; they are joined by specialized proteins that form tight junctions between the endothelial cells. A higher number of mitochondria provide the energy for sodium-potassium adenosine triphosphatase pumps that remove sodium and bring in potassium to maintain ionic equilibrium, while forming water by the osmotic action of removing three sodium atoms for every two potassium atoms. Pericytes are embedded in the basal lamina layer in a strategic location to influence the endothelial cells, tight junctions, and astrocytes; they secrete MMP-3, which both disrupts proteins directly and acts indirectly by activating MMP-9. Microglia cells are the brain’s innate macrophage-like cells that are the first responders in an injury, and once activated, they change shape and secrete primarily MMP-9. Thus, in an injury, multiple MMPs are released into the interstitial space, where they act on many structural proteins and alter cell surface receptors.10
There is some evidence that MMP-3 plays a role in the hypothalamo-neurohypophysial system (HNS), which synthesizes arginine vasopressin (AVP) and oxytocin and is well known to show structural plasticity during chronic physiological stimulation, such as salt loading and lactation.23 The HNS is an important site of regulation of water balance through the control of AVP. Chronic stimulation increases protein levels of uPA in the supraoptic nucleus, but the stimulation does not change protein levels of MMP-3 in the supraoptic nucleus or the neurohypophysis. Depolarizing agent potassium chloride releases tPA from isolated neurosecretosomes, and this depolarization-dependent release is abolished by verapamil, a calcium (2+) channel blocker. These results demonstrate that tPA and MMP-3 are localized mainly at dendrites and terminals of AVP-expressing magnocellular neurons and that tPA is released in an activity-dependent manner, suggesting that matrix-degrading proteases are candidate molecules to be concerned with in investigating the structural plasticity in the HNS.23
MMPs in neurological diseases
MMPs in ischemia/hypoxia
Loss of blood flow and oxygen initiates molecular cascades leading to cell death.24 Focal loss of blood flow occurs when an individual artery is occluded by a thrombus or emboli. This causes damage to the tissue supplied by that vessel because the brain has a very poor system of collateral flow vessels, and the tissue undergoes infarction. Cardiac arrest leads to global hypoxia/ischemia, and multiple areas of the brain undergo cell death, depending on the length of time without oxygen. In chronic conditions, there can be a reduction in blood flow without complete loss; this partial loss of oxygen injures selective areas that are more vulnerable than others. The most common cause of restricted blood flow is narrowing of the vessel lumen due to hypertension.24
HIF-1α, which is increased during low oxygen states, triggers transcription of a large number of genes.19 The fur gene is induced by HIF-1α, leading to the production of the convertase, furin, which is important because of the large number and variety of bioactive proteins and peptides that can be generated through its activity, including key elements involved in normal and pathophysiological conditions, such as MMP-14 and the transforming growth factor-β; both mediators are well-characterized furin substrates that have been shown to profoundly affect many aspects of injury progression.25
The sequence of MMP activation during an episode of hypoxia is well regulated. While the mechanisms acting on the MMPs in ischemia are not well understood in humans, animal studies in rats using the suture model of reperfusion provide insight into the molecular cascades involved in the expression and activation of the MMPs in ischemia. Initially, the constitutive enzymes, MMP-2 and MMP-14, are activated, beginning the process of proteolysis in a controlled fashion, which can be reversed when the insult is lifted. As time passes and the extent of hypoxia worsens, the inducible enzymes, MMP-3 and MMP-9, are induced and activated, leading to more extensive tissue damage that is now irreversible (Figure 4).26 As the molecular injury cascade progresses, cytokines are formed, triggering the transcription of the inducible MMPs that cause the irreversible protein damage. TNF-α and interleukin-1β (IL-1β) act at the AP-1 and NF-κB sites to form latent MMP-9 and MMP-3. The initial damage can begin within hours of the injury, while the secondary release of MMPs usually occurs 24–48 hours later as the genes involved are activated.27
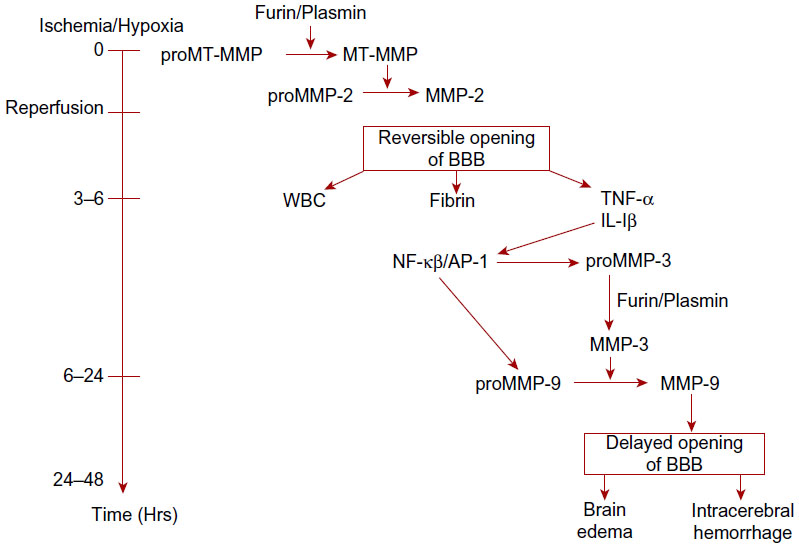 | Figure 4 The role of MMPs in the molecular cascades occurring with reperfusion of brain tissue after acute stroke in rat models. |
MMPs participate in the repair processes that begin from 3–7 days after the insult. After the cells are damaged by ischemia/hypoxia, the membranes are destroyed in the neurons, which are removed by macrophages, leaving a glia scar surrounding a cyst. Astrocytes move into the site of injury to form a glial scar that surrounds the cystic cavity. Blood vessels are more resistant to hypoxia and form scaffolding for the injured tissue to rebuild the tissue. Angiogenesis and neurogenesis require MMPs for their action, which is similar to the role of blood vessels in other tissues undergoing repair.28 Pericytes are important in angiogenesis, breaking down the ECM, permitting the regrowing vessels to expand into the surrounding tissues.29 MMP-9 is also important in the process.
MMPs in intracerebral hemorrhage
A major role of MMPs in intracerebral hemorrhage is related to hemorrhagic transformation of an infarct and to the use of tPA, which improves outcome in stroke if given within 4.5 hours after onset, but also has the undesirable side effect of hemorrhage, which depending on many circumstances, can range from 3% to 6% of patients affected.30 The mechanism of bleeding with tPA involves the activation of the MMPs by plasmin. If the tPA remains in the blood after intravenous injection, the rate of bleeding is low. However, the damage done by the infarct leads to opening of the BBB early in the process, allowing the tPA to enter the brain. Once in the brain, the plasmin activates the MMPs, which cause the bleeding. MMPs play an important role in the hemorrhage by attacking the basal lamina and tight junctions.31 When an MMP inhibitor is given with tPA, the rate of bleeding is reduced.32 This reduction has been demonstrated in animals and in preliminary studies in humans where minocycline was used as the MMP inhibitor.33 Many potential agents are now undergoing testing in animals to block the MMP damage to the BBB in order to prolong the therapeutic window for tPA treatment.
MMPs in MS
MS is a disease of the central nervous system that affects young adults in their most productive years.34 The illness generally begins with a relapsing-remitting course and eventually enters a secondary progressive stage. In spite of studies over many decades related to toxic and infectious etiologies, the cause of the progression remains unclear. Early investigators observed inflammation around the blood vessels, particularly the veins. Pathological studies by the Scottish physician, James Dawson, provided evidence for the vascular theory of MS by showing inflammation centered around veins, particularly those lining the cerebral ventricles, forming a pattern of demyelination referred to as “Dawson’s fingers”.35 Prior to modern neuroimaging, less emphasis was placed on the role of inflammation, because the pathological studies failed to show inflammatory lesions, but instead revealed gliotic scars called plaques. These were regions of inactive tissue, which are prominent in the late stages of the illness.
The presence of inflammation at autopsy in the earlier studies may possibly be due to the fact that the lack of antibiotics resulted in patients dying at a younger age when inflammation was still active, and the sclerotic plaque was not yet formed. As patients lived longer with improved medical care, the chronic changes of gliosis were more commonly seen, and the role of inflammation was neglected. The emphasis on gliosis rather than inflammation changed dramatically with the introduction of computed tomography (CT) and magnetic resonance imaging (MRI), which revealed a large number of enhancing lesions that fluctuated over time, as did recurrent inflammatory lesions.36
However, disease progression is not necessarily related to the times of vessel leakage, suggesting multiple factors are involved in the MS lesions. It is now recognized that there are multiple forms of MS with different pathological findings.37 Recently, MRIs with field strengths of up to 7 Tesla have become available; they show clearly that the sites of inflammation are centered around the blood vessels, which is not surprising based on the pathological studies of Dawson and others who followed his ideas.38
MMPs contribute to the inflammatory opening of the BBB seen in acute episodes of relapsing/remitting MS.39 Veins appear to be the main site of inflammation, which explains the pattern of periventricular lesions that form Dawson’s fingers. While the pathological process has been intensively studied, the factors that trigger the inflammation are less well understood. T lymphocytes release the proteases that disrupt the tight junctions in the endothelial cells and break down the ECM. MMPs are detected in the brains of patients with MS.40 In the animal model of experimental allergic encephalomyelitis (EAE), there is evidence of MMPs in the vascular abnormalities seen in the brains of animals, and evidence that MMP inhibitors can reduce damage to the brain.41
During acute attacks of MS, MMP-9 levels are increased in the CSF, reflecting the changes that are occurring in the brain. Leakage of blood-derived proteins into the extracellular space leads to brain edema, hypoxia, and loss of myelin. While many factors contribute to the initiation of injury in MS, the MMPs play an important role in the final common pathway of tissue damage. Contrast-enhanced MRI shows opening of the BBB at various stages in the relapsing-remitting form of MS. Steroids are used to restore BBB integrity; the injection of high doses of steroids closes the BBB rapidly, but the effect is not long lasting.42
During an acute exacerbation with inflammatory opening of the BBB, there is a marked increase in MMP-9 in the CSF.42 When these patients are treated with high-dose intravenous methylprednisolone, levels of MMP-9 are dramatically reduced in the CSF. It is well established that high-dose steroids rapidly restore BBB integrity, suggesting a direct effect of the drug on the production of MMP-9. uPA is increased in the CSF, which could possibly release plasmin to activate the MMPs. Steroids block the AP-1 site in the MMP-9 gene, which is responsive to c-fos and cytokines, such as TNF-α. The steroids bind with a cytosolic factor to block the AP-1 site, reducing the production of MMP-9.43 Treatment with steroids increases levels of TIMP-3 in the CSF. Thus, either blocking of the AP-1 site by the steroid complex or inhibition by TIMP-3 could contribute to the closing of the BBB.
MMPs have another important effect that could influence the MS attack, namely, they attack the myelin and break it down into basic myelin protein fragments.41 While MMPs appear to play a major role in the pathology of MS, the complexity of the molecular events argues for a number of mechanisms acting in tandem. MS has a number of possible mechanisms involved in myelin injury and oligodendrocyte regeneration. While a direct genetic link has not been found, there are several candidate genes in families with multiple members with the disease. The potential environmental factors also remain speculative, with the leading candidates including infections and metabolic factors. Injury to the BBB allows sensitized T cells from the systemic circulation to enter the central nervous system; this process is facilitated by MMPs, which degrade the ECM and facilitate migration of immune cells. Once the T cells have entered the brain, they can release cytokines, which can attack the oligodendrocyte and can lead to its death. Injury to the myelin membrane results in axons that are no longer able to transmit action potentials efficiently within the central nervous system. Repair processes also require natural inhibitors of MMPs, such as the TIMPs, for resolution of the inflammatory response with restoration of function by remyelination.
Several proteases in the brains of MS patients can attack myelin. Lysosomal enzymes have been identified in inflammatory cells.34 Phospholipase A attacks membrane lipid components, such as lysolecithin, which has cytolytic action on myelin membranes. Acid and neutral proteases have been detected in the CSF of patients with MS. Neutral proteases have been shown to attack the structure of the myelin molecule. Stimulated macrophages secrete proteases that attack myelin.44 The active substance in the macrophage supernatant is plasmin, which has been shown to disrupt myelin basic protein in frozen specimens of tissue. Similarly, myelin breakdown occurs in fresh tissue when both complement and plasmin are added.44
Treatment of animals with EAE with MMP inhibitors reduces the severity of the illness.45 Direct inhibitors of MMPs have not been tested in the treatment of MS patients. However, a tetracycline derivative, minocycline, which has anti-inflammatory actions including inhibition of MMPs, benefited patients with MS in an uncontrolled clinical trial involving a small number of subjects.46 Minocycline suppresses production of MMP-9 but also has other effects on the inflammatory response, including inhibiting the microglia response, making it difficult to determine its mechanism of action.47
MMP-7 knockout mice display reduced inflammation and white blood cell entry into the brain as well as across an in vitro cell culture system, suggesting that MMP-7 may contribute to the inflammatory response.24,48 Immunomodulation with β-interferon reduces MMPs, as does treatment with the anti-inflammatory agent minocycline; however, minocycline causes only a minor reduction in disease severity.49 Free radicals contribute to the disruption of the ECM and the opening of the BBB. Nitrogen-free radicals are produced in the site of inflammation. Co-cultures of neurons and microglia that have been stimulated with the inflammatory-inducing agents, interferon gamma and lipopolysaccharide, lead to the death of the neurons. Dexamethasone reduces cell death by blocking the production of nitric oxide through inhibition of the gene that produces inflammatory nitric oxide synthetase. The mechanism of steroid action involves interfering with the steroid receptor and blocking the action of the c-fos/c-jun dimer at the AP-1 site.43 These cell studies have been paralleled in humans by showing that the contrast enhancement in both CT and MRI can be drastically attenuated with high-dose steroids.
The presence of MMPs in the CSF can either reflect the endogenous production or transport into the central nervous system across a disrupted BBB. This is true for all proteins detected in the CSF, and normalizing proteins with albumin in the CSF and blood has solved it. Albumin is a large protein molecule that is made in the liver and is prevented from entering the central nervous system by the BBB; while 4 g of albumin are normally found in the blood, only 40 mg are present in the CSF. Formation of a ratio between albumin in the CSF and that in the blood, the albumin ratio, is a biomarker of the intactness of the BBB. IgG is a protein normally found in the blood, but in MS it can be produced in the CSF. An IgG index has been used to separate endogenous IgG production from transport into the brain.14,15 An MMP-2 index can be formed in a similar fashion to the IgG index, by the following formula:
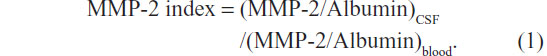
Similarly, the MMP index can be formed with albumin in both compartments. When that is done, it is found that the MMPs in the CSF are endogenously produced. Detection of changes in MMP-2 in the CSF is difficult, because it is normally found at a high level in the CSF, and small changes in the levels are obscured. MMP-3 and MMP-9, on the other hand, are present in the CSF normally at very low levels, making it possible to detect small changes in either protein.
Studies in pathological tissues from MS patients confirm the presence of the MMPs in the brain. There is extensive information from animal studies that implicate the MMPs in the pathological processes. An animal model of EAE is produced by the injection of myelin fragments into the footpad, setting up an allergic reaction that results in inflammation around the blood vessels, fibrin deposition, and myelin damage. Primarily a model of blood vessel damage with secondary injury to the myelin, it provides a convenient model in which to test treatments.40
Early enthusiasm for agents that block the MMPs and reduce the activation of TNF-α have failed to be translated into a treatment for similar reasons that MMP inhibitors have failed in cancer treatments.41 Recently, the tetracycline derivative that blocks MMPs and microglia activation, minocycline, has been used with limited success to treat patients with MS, indicating that as newer MMP inhibitors are developed, they may be useful in MS treatment.
MMPs in vascular cognitive impairment
Vascular risk factors predispose individuals to chronic vascular disease with a gradual worsening of cognitive function.50 Hypertension is the major risk factor, beginning to produce changes in the vasculature and brain microstructure in people aged in their mid-40s with blood pressure greater than 140/90.51 Long-term hypertension reduces the inner lumen of the blood vessels and thickens the outer wall, limiting cerebral blood flow and leading to hypoxic hypoperfusion.52 The action of MMPs contributes to the remodeling of the vessel wall.28 Fibrosis of the outer vessel wall stiffens the blood vessels, making them less responsive to the changes in caliber necessary to rapidly respond to changes in cardiac output, blood pressure, and metabolic tasks in the brain. The ultimate effect of hypertension is to both narrow the lumen, reducing flow, and to stiffen the vessel wall, impairing responsiveness; both effects restrict access of blood to the brain, producing damage to the poorly perfused deeper portions of the brain and a chronic rather than an acute form of hypoxia/ischemia referred to as “incomplete infarction”.53
Pathological studies show the presence of MMPs in the brain tissue of patients with vascular cognitive impairment (VCI).40,54 MMP-2 is found in astrocytes in the white matter, and pericytes surrounding the endothelial cells immunostain for MMP-3. Other MMPs are seen in the brain tissue, including MMP-13.55 The presence of MMPs in the inflammatory cells found around blood vessels provides strong evidence for a major role of MMPs in the pathophysiology of VCI. Additional support comes from studies of CSF in patients with VCI.16,56–58 Patients with extensive damage to the white matter, referred to as Binswanger’s disease, have elevated levels of CSF MMP-9, which is not seen in Alzheimer’s disease (AD).56 MMP indexes show an increase in MMP-9 in Binswanger’s disease patients and a decrease in MMP-2 index.15 This result is likely due to decrease in the latent form of MMP-2, which is measured by zymography, but the exact reason needs further study. Active forms of MMPs can be detected in CSF by either using molecular weights to separate latent and active forms in zymography, or by using the immunocapture technique with immunofluorecent cleavage of specially designed peptides that are sensitive to active enzyme.59
The pathophysiology of the chronic ischemic insult is difficult to understand from pathological studies in humans because of the accumulation of multiple pathological findings in the elderly brain at the time of autopsy.60 This situation has been remedied by the use of animal models, which are very helpful in unraveling the pathophysiology of the chronic changes.60 Two animal models have been most helpful: 1) the bilateral occlusion of the common carotid arteries (BCAO) in the normotensive rat; and 2) the administration of the modified diet in the spontaneously hypertensive stroke-prone rat (SHR/SP). With BCAO, there is an increase in MMP-2 that leads to BBB disruption, which can be seen both on MRI and histologically.61,62 The BCAO animals show disruption of the BBB 3 days after the occlusion associated with increased expression of MMP-2. Knockout mice lacking the MMP-2 gene have reduced disruption of the BBB, which is also seen with inhibitors to MMPs.61
Another model that is closer to the human clinical condition is the SHR/SP.63 Modifying the diet by reducing protein and adding salt, along with occluding one carotid artery, can accelerate damage to the white matter. A study using the SHR/SP model shows that starting the diet with carotid occlusion after 12 weeks of life results in death in 4 weeks after diet initiation, markedly shortening the normal lifespan in these rats.64 Analysis of the brain tissue shows an increase in HIF-1α, the induction of MMP-9, opening of the BBB, and breakdown of myelin. In the SHR/SP with dietary manipulation, there is hypoxia in the deep white matter as shown by electron paramagnetic resonance, which begins around the 12th week of life when hypertension reaches a maximum.65 Treatment with the anti-inflammatory MMP inhibitor, minocycline, reduces damage to the white matter, prolongs lifespan, and improves behavior in the Morris water maze.66 Thus, the SHR/SP with diet manipulation and carotid occlusion is a model that can be used to test drugs for eventual translation into human studies.
Hypoxia signals a series of molecular events that will cause both injury and initiate recovery (Figure 5).56 HIF-1 is induced by hypoxia, and in turn, triggers a cassette of hypoxia-responsive genes, ranging from fur an inducer of furin, to vascular endothelia growth factor, which is involved in angiogenesis. The effect of the induction of the MMPs in the chronically hypoxic brain is to disrupt the endothelial tight junctions and ECM,27 for acute ischemia. The difference is that the time frame is much slower for induction of MMPs in the chronically hypoxic brain, and the resulting opening of the BBB results in release of serum proteins into the white matter, with damage to the myelinated fibers that is seen in the MRI as white-matter hyperintensities.
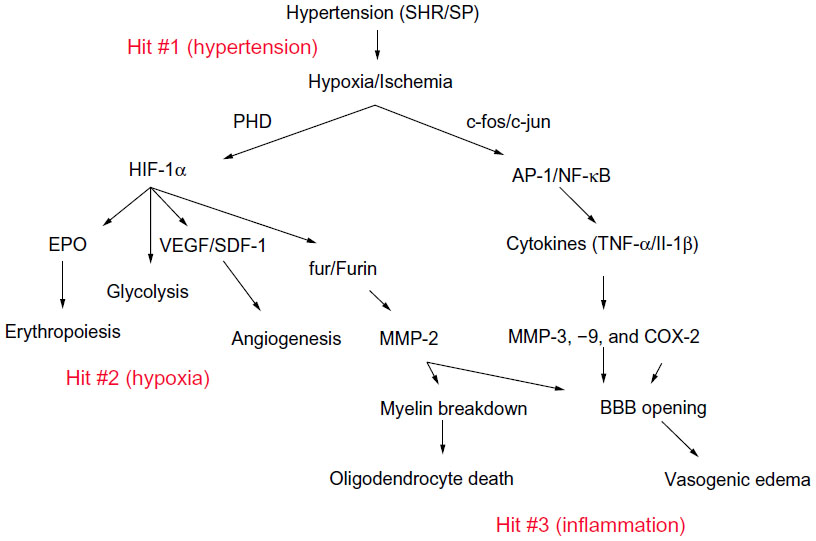 | Figure 5 Mechanism of white matter damage from chronic hypoxia secondary to hypertension. Hit #1 represents reduced cerebral blood flow, which leads to hypoxia with release of hypoxia-inducible factor-1α (HIF-1α). Hit #2 represents a cascade of molecular events, which follows activation of MMP-2 by furin. More severe hypoxia leads to induction of MMP-9 by cytokines. Hit #3 shows the breakdown of the BBB and demyelination secondary to the inflammatory response. |
An inflammatory response involving macrophages and microglia is seen around fibrous blood vessels in the white matter. Part of the macrophage response is a normal attempt at tissue repair triggered by the presence of increased collagen fibers, but the macrophages may act independently to release proteases and free radicals.66 In addition to attacking the tight junctions and opening the BBB, the proteases, if activated, attack the myelin, producing another mechanism of white matter damage.
MMPs in AD
AD involves the abnormal accumulation of amyloid proteins in cortical regions, which begins in midlife and continues with increasing age.67 Most patients have a sporadic form of AD. There are multiple forms of amyloid of differing molecular weights, which are formed from amyloid precursor protein, a transmembrane molecule with an extracellular component. Secretases degrade amyloid precursor protein, and the sites of cleavage determine the fate of these protein fragments. The physiological pathway results in the cleavage of the amyloid precursor by α-secretase, which produces a soluble component that can be broken down for clearance. ADAM10 and ADAM17 are possible candidates for α-secretase.68 Two other secretases (β-secretase and γ-secretase) act together to produce Aβ peptides, Aβ1–40 and Aβ1–42; cleavage of amyloid precursor protein by β-secretase produces a fragment of amyloid precursor protein that can be further processed to form Aβ1–42 by γ-secretase. Any type of Aβ can aggregate to form dimers, oligomers, and subsequently fibrils that are deposited in neuropils and in cells.69 When Aβ1–42 builds up in the ISF from failure of enzymatic breakdown of the molecule, the insoluble fibrils turn into neuritic plaques.
MMPs participate in the formation and clearance of Aβ in AD.70,71 Blood vessels, astrocytes, and microglia can be induced to produce MMPs by the amyloid molecules.72 Exposure of astrocytes to Aβ1–40 induces the secretion of MMP-2, MMP-3, and MMP-9.73 Activated microglia and astrocytes are seen around Aβ-containing plaques, indicating inflammation.74 MMP-2 release and activation differentially degrades Aβ species, delaying their toxic effects on endothelial cells. However, taking into consideration MMP ability to degrade basement membrane components, these protective effects might also undesirably compromise BBB integrity and precipitate a hemorrhagic phenotype.75 A genome-wide association study of CSF levels of 59 AD-related analytes shows significant single-nucleotide polymorphisms in angiotensin-converting enzyme, and MMP-3 shows an association with AD risk.74 Immunohistochemistry has also detected MMP-3 expression in hippocampal neurons, around amyloid plaques in the cortex.76
Inhibitors to the MMPs as therapeutic agents in neurological disorders
A number of MMP inhibitors have been studied as therapeutic agents for neurological disorders. Early studies were done with the hydroxymate-based zinc inhibitors that are non-specific MMP inhibitors active against a wide variety of MMPs.77 MMP inhibitors have been tested in models of MS, Guillain–Barré polyneuropathy, meningitis, and stroke; in each of these diseases, they show efficacy. Several clinical studies have also been done with non-hydroxymate drugs. The tetracycline derivative, minocycline, has multiple beneficial effects, including reduction in inflammation and inhibition of MMP-9.47,49 Short-term use in cerebral ischemia has been shown to block the opening of the BBB, thereby protecting the brain against tissue plasminogen activator-induced hemorrhage.33 A clinical trial has also been carried out in patients with MS.78 One unsuccessful trial of minocycline in amyotrophic lateral sclerosis made patients worse, probably because the dose used was twice the normal human dose.79 Long-term use of MMP inhibitors has been limited by stiffening of the joints due to lack of collagen breakdown. As newer, more specific MMP inhibitors are developed, they will need to be tested in patients.
Conclusion
We have reviewed the role of the MMPs in stroke, MS, VCI, and AD, which are the major neurological illnesses. In the current review, we have emphasized pathology of the cerebral vasculature. In the diseases discussed, the central role of the inflammatory response mediated by MMP production has been discussed. Stroke shows an early induction of MMPs, which play a prominent role in ischemic damage, the effects of which are reversed as recovery requires MMPs for angiogenesis and neurogenesis. During an acute episode of MS, the MMPs are important in the process of inflammatory damage done to the blood vessels, which eventually results in myelin loss. Finally, in chronic vascular disease due to hypertension, changes in the blood vessel walls lead to reduced cerebral blood flow, hypoxia, and BBB disruption over a much longer time frame than seen with acute ischemia. In each of these diseases, animal studies show that MMP inhibitors can reduce damage to the vessels and injury to the tissue. In spite of considerable experimental data having been gathered about animals receiving MMP inhibitors for treatment, these data have not reached the clinic. Now that the pathophysiology of these major neurological disorders has been shown to involve MMPs at multiple stages, the challenge will be to develop inhibitors of MMPs that can be safely used in human patients. Use of MMP inhibitors in the treatment of acute diseases may be a more reachable goal, assuming that long-term side effects can be avoided. Use of MMP inhibitors in chronic conditions, such as MS and VCI, will be more difficult because of the dual function of the MMPs both as acute perpetrators of injury and as initiators of repair processes during recovery.
Acknowledgments
The present study was supported by funding from the US National Institutes of Health (NIH) (grant number RO1 NS052305-07), the US–Israeli Binational Foundation, Bayer Pharmaceutical Corp, and the University of New Mexico NIH National Center for Advancing Translational Sciences (grant number UL1 TR000041).
Disclosure
The author reports no conflicts of interest in this work.
References
Liotta LA, Tryggvason K, Garbisa S, Hart I, Foltz CM, Shafie S. Metastatic potential correlates with enzymatic degradation of basement membrane collagen. Nature. 1980;284:67–68. | |
Brinckerhoff CE, Matrisian LM. Matrix metalloproteinases: a tail of a frog that became a prince. Nat Rev Mol Cell Biol. 2002;3(3):207–214. | |
Apodaca G, Rutka JT, Bouhana K, et al. Expression of metalloproteinases and metalloproteinase inhibitors by fetal astrocytes and glioma cells. Cancer Res. 1990;50:2322–2329. | |
Rosenberg GA, Kornfeld M, Estrada E, Kelley RO, Liotta LA, Stetler-Stevenson WG. TIMP-2 reduces proteolytic opening of blood-brain barrier by type IV collagenase. Brain Res. 1992;576:203–207. | |
Rosenberg GA, Navratil M, Barone F, Feuerstein G. Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb Blood Flow Metab. 1996;16(3):360–366. | |
Lukes A, Mun-Bryce S, Lukes M, Rosenberg GA. Extracellular matrix degradation by metalloproteinases and central nervous system diseases. Mol Neurobiol. 1999;19(3):267–284. | |
Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. 2005;50(4):329–339. | |
Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6(12):931–944. | |
Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009;8(2):205–216. | |
Candelario-Jalil E, Yang Y, Rosenberg GA. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience. 2009;158(3):983–994. | |
Nagase H. Activation mechanisms of matrix metalloproteinases. Biol Chem. 1997;378(3–4):151–160. | |
Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 2010;1803(1):55–71. | |
Yana I, Weiss SJ. Regulation of membrane type-1 matrix metalloproteinase activation by proprotein convertases. Mol Biol Cell. 2000;11(7):2387–2401. | |
Ramos-DeSimone N, Hahn-Dantona E, Sipley J, et al. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J Biol Chem. 1999;274(19):13066–13076. | |
Liuzzi GM, Trojano M, Fanelli M, et al. Intrathecal synthesis of matrix metalloproteinase-9 in patients with multiple sclerosis: implication for pathogenesis. Mult Scler. 2002;8(3):222–228. | |
Candelario-Jalil E, Thompson J, Taheri S, et al. Matrix metalloproteinases are associated with increased blood-brain barrier opening in vascular cognitive impairment. Stroke. 2011;42(5):1345–1350. | |
Engelhardt B, Sorokin L. The blood-brain and the blood-cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol. 2009;31(4):497–511. | |
Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57(2):173–185. | |
Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol. 2014;9:47–71. | |
Cao J, Rehemtulla A, Pavlaki M, Kozarekar P, Chiarelli C. Furin directly cleaves proMMP-2 in the trans-Golgi network resulting in a nonfunctioning proteinase. J Biol Chem. 2005;280(12):10974–10980. | |
Remacle AG, Shiryaev SA, Oh ES, et al. Substrate cleavage analysis of furin and related proprotein convertases. A comparative study. J Biol Chem. 2008;283(30):20897–20906. | |
Xu X, Jackson PL, Tanner S, et al. A self-propagating matrix metalloprotease-9 (MMP-9) dependent cycle of chronic neutrophilic inflammation. PLoS One. 2011;6(1):e15781. | |
Miyata S, Nakatani Y, Hayashi N, Nakashima T. Matrix-degrading enzymes tissue plasminogen activator and matrix metalloprotease-3 in the hypothalamo-neurohypophysial system. Brain Res. 2005; 1058(1–2):1–9. | |
Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4(5):399–415. | |
McMahon S, Grondin F, McDonald PP, Richard DE, Dubois CM. Hypoxia-enhanced expression of the proprotein convertase furin is mediated by hypoxia-inducible factor-1: impact on the bioactivation of proproteins. J Biol Chem. 2005;280(8):6561–6569. | |
Rosenberg GA, Cunningham LA, Wallace J, et al. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures. Brain Res. 2001;893(1–2):104–112. | |
Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27(4):697–709. | |
Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008;75(2):346–359. | |
Bell RD, Winkler EA, Sagare AP, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68(3):409–427. | |
Hacke W, Donnan G, Fieschi C, et al; ATLANTIS Trials Investigators; ECASS Trials Investigators; NINDS rt-PA Study Group Investigators. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet. 2004; 363(9411):768–774. | |
Fan X, Lo EH, Wang X. Effects of minocycline plus tissue plasminogen activator combination therapy after focal embolic stroke in type 1 diabetic rats. Stroke. 2013;44(3):745–752. | |
Pfefferkorn T, Rosenberg GA. Closure of the blood-brain barrier by matrix metalloproteinase inhibition reduces rtPA-mediated mortality in cerebral ischemia with delayed reperfusion. Stroke. 2003;34(8):2025–2030. | |
Fagan SC, Waller JL, Nichols FT, et al. Minocycline to improve neurologic outcome in stroke (MINOS): a dose-finding study. Stroke; A Journal of Cerebral Circulation. 2010;41(10):2283–2287. | |
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343(13):938–952. | |
Dawson JW. The histology of disseminated sclerosis. Trans Roy Soc Edin. 1916;50:517–525. | |
Filippi M, Rocca MA, Barkhof F, et al; Attendees of the Correlation between Pathological MRI findings in MS workshop. Association between pathological and MRI findings in multiple sclerosis. Lancet Neurol. 2012;11(4):349–360. | |
Lucchinetti C, Brϋck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47(6):707–717. | |
Tallantyre EC, Dixon JE, Donaldson I, et al. Ultra-high-field imaging distinguishes MS lesions from asymptomatic white matter lesions. Neurology. 2011;76(6):534–539. | |
Leppert D, Ford J, Stabler G, et al. Matrix metalloproteinase-9 (gelatinase B) is selectively elevated in CSF during relapses and stable phases of multiple sclerosis. Brain. 1998;121 (Pt 12):2327–2334. | |
Anthony DC, Ferguson B, Matyzak MK, Miller KM, Esiri MM, Perry VH. Differential matrix metalloproteinase expression in cases of multiple sclerosis and stroke. Neuropathol Appl Neurobiol. 1997;23(5):406–415. | |
Clements JM, Cossins JA, Wells GM, et al. Matrix metalloproteinase expression during experimental autoimmune encephalomyelitis and effects of a combined matrix metalloproteinase and tumour necrosis factor-alpha inhibitor. J Neuroimmunol. 1997;74(1–2):85–94. | |
Rosenberg GA, Dencoff JE, Correa N Jr, Reiners M, Ford CC. Effect of steroids on CSF matrix metalloproteinases in multiple sclerosis: Relation to blood-brain barrier injury. Neurology. 1996;46:1626–1632. | |
Jonat C, Rahmsdorf HJ, Park KK, et al. Antitumor promotion and antiinflammation: down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell. 1990;62:1189–1204. | |
Cammer W, Bloom BR, Norton WT, Gordon S. Degradation of basic protein in myelin by neutral proteases secreted by stimulated macrophages: a possible mechanism of inflammatory demyelination. Proc Natl Acad Sci U S A. 1978;75:1554–1558. | |
Hewson AK, Smith T, Leonard JP, Cuzner ML. Suppression of experimental allergic encephalomyelitis in the Lewis rat by the matrix metalloproteinase inhibitor Ro31-9790. Inflamm Res. 1995;44(8):345–349. | |
Metz LM, Zhang Y, Yeung M, et al. Minocycline reduces gadolinium-enhancing magnetic resonance imaging lesions in multiple sclerosis. Ann Neurol. 2004;55(5):756. | |
Yong VW, Wells J, Giuliani F, Casha S, Power C, Metz LM. The promise of minocycline in neurology. Lancet Neurol. 2004;3(12):744–751. | |
Buhler LA, Samara R, Guzman E, et al. Matrix metalloproteinase-7 facilitates immune access to the CNS in experimental autoimmune encephalomyelitis. BMC Neurosci. 2009;10:17. | |
Yong VW, Zabad RK, Agrawal S, Goncalves Dasilva A, Metz LM. Elevation of matrix metalloproteinases (MMPs) in multiple sclerosis and impact of immunomodulators. J Neurol Sci. 2007;259(1–2):79–84. | |
Gorelick PB, Scuteri A, Black SE, et al; American Heart Association Stroke Council, Council on Epidemiology and Prevention, Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, and Council on Cardiovascular Surgery and Anesthesia. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(9):2672–2713. | |
Maillard P, Seshadri S, Beiser A, et al. Effects of systolic blood pressure on white-matter integrity in young adults in the Framingham Heart Study: a cross-sectional study. Lancet Neurol. 2012;11(12):1039–1047. | |
Pires PW, Rogers CT, McClain JL, Garver HS, Fink GD, Dorrance AM. Doxycycline, a matrix metalloprotease inhibitor, reduces vascular remodeling and damage after cerebral ischemia in stroke-prone spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2011;301(1):H87–H97. | |
Brun A. Pathology and pathophysiology of cerebrovascular dementia: pure subgroups of obstructive and hypoperfusive etiology. Dementia. 1994;5(3–4):145–147. | |
Rosenberg GA, Sullivan N, Esiri MM. White matter damage is assoiciated with matrix metalloproteinases in vascular dementia. Stroke. 2001;32:1162–1168. | |
Cuadrado E, Rosell A, Borrell-Pagès M, et al. Matrix metalloproteinase-13 is activated and is found in the nucleus of neural cells after cerebral ischemia. J Cereb Blood Flow Metab. 2009;29(2):398–410. | |
Adair JC, Charlie J, Dencoff JE, et al. Measurement of gelatinase B (MMP-9) in the cerebrospinal fluid of patients with vascular dementia and Alzheimer disease. Stroke. 2004;35(6):e159–e162. | |
Bjerke M, Zetterberg H, Edman å, Blennow K, Wallin A, Andreasson U. Cerebrospinal fluid matrix metalloproteinases and tissue inhibitor of metalloproteinases in combination with subcortical and cortical biomarkers in vascular dementia and Alzheimer’s disease. J Alzheimers Dis. 2011;27(3):665–676. | |
Bjerke M, Jonsson M, Nordlund A, et al. Cerebrovascular biomarker profile Is related to white matter disease and ventricular dilation in a LADIS substudy. Dement Geriatr Cogn Dis Extra. 2014;4(3):385–394. | |
Hawkins KE, DeMars KM, Yang C, Rosenberg GA, Candelario-Jalil E. Fluorometric immunocapture assay for the specific measurement of matrix metalloproteinase-9 activity in biological samples: application to brain and plasma from rats with ischemic stroke. Mol Brain. 2013;6:14. | |
Sonnen JA, Santa Cruz K, Hemmy LS, et al. Ecology of the aging human brain. Arch Neurol. 2011;68(8):1049–1056. | |
Nakaji K, Ihara M, Takahashi C, et al. Matrix metalloproteinase-2 plays a critical role in the pathogenesis of white matter lesions after chronic cerebral hypoperfusion in rodents. Stroke. 2006;37(11):2816–2823. | |
Sood R, Yang Y, Taheri S, et al. Increased apparent diffusion coefficients on MRI linked with matrix metalloproteinases and edema in white matter after bilateral carotid artery occlusion in rats. J Cereb Blood Flow Metab. 2009;29(2):308–316. | |
Guerrini U, Sironi L, Tremoli E, et al. New insights into brain damage in stroke-prone rats: a nuclear magnetic imaging study. Stroke. 2002;33(3):825–830. | |
Jalal FY, Yang Y, Thompson J, Lopez AC, Rosenberg GA. Myelin loss associated with neuroinflammation in hypertensive rats. Stroke. 2012; 43(4):1115–1122. | |
Weaver J, Jalal FY, Yang Y, Thompson J, Rosenberg GA, Liu KJ. Tissue oxygen is reduced in white matter of spontaneously hypertensive-stroke prone rats: a longitudinal study with electron paramagnetic resonance. J Cereb Blood Flow Metab. 2014;34(5):890–896. | |
Jalal FY, Yang Y, Thompson JF, Roitbak T, Rosenberg GA. Hypoxia-induced neuroinflammatory white-matter injury reduced by minocycline in SHR/SP. J Cereb Blood Flow Metab. 2015;35(7):1145–1153. | |
Jack CR Jr, Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron. 2013;80(6):1347–1358. | |
Buxbaum JD, Liu KN, Luo Y, et al. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273(43):27765–27767. | |
LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci. 2007;8(7):499–509. | |
Yin KJ, Cirrito JR, Yan P, et al. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J Neurosci. 2006;26(43):10939–10948. | |
Walsh DM, Minogue AM, Sala Frigerio C, Fadeeva JV, Wasco W, Selkoe DJ. The APP family of proteins: similarities and differences. Biochem Soc Trans. 2007;35(Pt 2):416–420. | |
Yan P, Hu X, Song H, et al. Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ. J Biol Chem . 2006;281(34):24566–24574. | |
Deb S, Gottschall PE. Increased production of matrix metalloproteinases in enriched astrocyte and mixed hippocampal cultures treated with beta- amyloid peptides. J Neurochem. 1996;66(4):1641–1647. | |
Kauwe JS, Bailey MH, Ridge PG, et al. Genome-wide association study of CSF levels of 59 alzheimer’s disease candidate proteins: significant associations with proteins involved in amyloid processing and inflammation. PLoS Genet. 2014;10(10):e1004758. | |
Hernandez-Guillamon M, Mawhirt S, Fossati S, et al. Matrix metalloproteinase 2 (MMP-2) degrades soluble vasculotropic amyloid-beta E22Q and L34V mutants, delaying their toxicity for human brain microvascular endothelial cells. J Biol Chem. 2010;285(35):27144–27158. | |
Yoshiyama Y, Asahina M, Hattori T. Selective distribution of matrix metalloproteinase-3 (MMP-3) in Alzheimer’s disease brain. Acta Neuropathol. 2000;99(2):91–95. | |
Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295(5564):2387–2392. | |
Zhang Y, Metz LM, Yong VW, et al. Pilot study of minocycline in relapsing-remitting multiple sclerosis. Can J Neurol Sci. 2008;35(2):185–191. | |
Gordon PH, Moore DH, Miller RG, et al; Western ALS Study Group. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6(12):1045–1053. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.