Back to Journals » Neuropsychiatric Disease and Treatment » Volume 13
Metabolic screening and its impact in children with non-syndromic intellectual disability
Authors Ali YF ![]() , EL-Morshedy S, Elsayed RM
, EL-Morshedy S, Elsayed RM ![]() , EL-sherbini AM, El-Sayed SAM, Abdrahman NIA, Imam AA
, EL-sherbini AM, El-Sayed SAM, Abdrahman NIA, Imam AA ![]()
Received 14 December 2016
Accepted for publication 17 February 2017
Published 19 April 2017 Volume 2017:13 Pages 1065—1070
DOI https://doi.org/10.2147/NDT.S130196
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Yasser F Ali,1 Salah EL-Morshedy,1 Riad M Elsayed,2 Amr M EL-sherbini,3 Saber AM El-Sayed,4 Nasser Ismail A Abdelrahman,1 Abdulbasit Abdulhalim Imam5
1Department of Pediatrics, Faculty of Medicine, Zagazig University, Zagazig, 2Pediatric Neurology Unit, Department of Pediatrics, Mansoura University, Mansoura, 3Department of Psychiatry, Faculty of Medicine, El-Minia University, El-Minia, 4Department of Pediatrics, National Research Center, 5Department of Pediatrics, Al-Azhar Faculty of Medicine for Girls, Cairo, Egypt
Objective: The objective of this study was to analyze the value of routine metabolic screening tests in children with an intellectual disability (ID) and its impact on improving their outcome and quality of life through appropriate intervention and treatment.
Patients and methods: This cross-sectional study was conducted in the Pediatric Neurology Clinic, Al Khafji Joint Operations Hospital, Kingdom of Saudi Arabia. A total of 150 children with nonsyndromic ID (66% males) in the age range of 5–17 years were compared with 50 apparently healthy age- and sex-matched controls. All studied groups were subjected to detailed history taking, family pedigree, thorough clinical examination, anthropometric measurements, routine laboratory investigations and urine metabolic screening tests (ferric chloride test and toluidine blue spot test and gas chromatography–mass spectrometry). Electroencephalography, IQ, psychiatric assessment and chromosomal study were done for the patient group only.
Results: Positive consanguineous marriage, older maternal or paternal age and family history of mental disabilities in other siblings were considered as risk factors for the development of mental disabilities. History of admission to neonatal intensive care unit was significantly higher among the patient group than among the controls (P<0.05). Metabolic screening tests showed that up to 35% of patients were positive for ferric chloride test, 9% of patients were positive for gas chromatography–mass spectrometry, and only 7 out of 150 (4.7%) patients were toluidine blue test positive.
Conclusion: Metabolic testing should be considered in the workup of individuals with nonsyndromic ID, which will need further specific investigations to confirm the diagnosis and determine the possible treatable cases.
Keywords: inborn error of metabolism, mentally retarded children, quality of life
Introduction
Intellectual disability (ID) is characterized by a significant impairment in cognitive ability. Within the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-V; American Psychiatric Association, 2000), for a diagnosis of ID to be made, symptoms should be present before the age of 18 years and IQ should be <70 (ie, more than two standard deviations [SDs] below the population mean in the typical scoring system for IQ that uses a mean of 100 and an SD of 15). ID can be further divided into syndromic and nonsyndromic forms. In syndromic ID, cognitive deficits are caused by identified medical problems such as phenylketonuria and fetal alcohol exposure. Nonsyndromic ID is characterized by a lack of known pathology.1 ID is a common problem in children with a reported frequency of ~2%–3%.2 The severity of ID has been based on the IQ score (ID defined as an IQ score of <70, at the age of ≥5 years). The various groups as per the World Health Organization classification are mild (IQ 55–69), moderate (IQ 40–54), severe (IQ 25–39) and profound (IQ <25).3
ID is a condition with varying etiology. It may have genetic or nongenetic (environmental) origin.3 Genetic causes of ID are broadly classified into single gene disorders, including all the inborn errors of metabolism (IEM), and chromosomal disorders, which are multifactorial in origin.4 ID can be due to static or progressive encephalopathy. Progressive encephalopathy is where the child loses already acquired milestones and regresses from the higher level of central nervous system (CNS) functioning to a lower level. It is due to neurodegenerative disorders that are very often metabolic in origin.4
IEM are a heterogeneous group of disorders caused by single gene defect, which may manifest immediately after birth or within few days or weeks after birth.5
Most of the IEM result from defects in any of the key enzymes of various metabolic pathways that lead to accumulation of compounds that follow an alternative pathway of metabolism, resulting in the production of toxic metabolites and deficiency of biologically important compounds.6
Among children with ID, it was reported that 5.75% of them were due to inherited metabolic disorders.7,8
Most IEM are inherited as autosomal recessive traits. Hence, a history of parental consanguinity and/or sibling deaths should increase the suspicion of IEM. IEM causing clinical manifestations in the neonatal period are usually severe and often lethal.9 Early diagnosis of the condition by various laboratory tests and initiation of prompt therapy are very much essential in order to prevent lethal complications that are irreversible. Various simple preliminary laboratory tests can aid in the diagnosis of IEM, which can be further confirmed by advanced diagnostic techniques.8
The aim of this study was to analyze the value of routine metabolic screening tests in children with ID and its impact on improving their outcome and quality of life through appropriate intervention and treatment.
Patients and methods
This case–control study was carried out at Al Khafji Joint Operations Hospital, Kingdom of Saudi Arabia (KSA), during the period from May 2014 to January 2016. Initially, the study enrolled 180 children of ages ranging from 5 to 17 years with suspected unexplained nonsyndromic ID. They were screened for psychiatric assessment at the Pediatric Neurology Clinic. A total of 30 children were excluded from the study based on the normal IQ level and clinical assessment. So, a total of only 150 (100 males and 50 females) children were actually included and diagnosed to have ID.
Exclusion criteria were children with syndromic mental disability, hypothyroidism, CNS malformations or history of CNS injury or infection and history of prenatal, perinatal and postnatal insults.
A total of 50 children (30 males and 20 females) of age, sex and socioeconomic status matched with normal mentality and normal IQ were studied as the control group. They were selected from children of general pediatric clinic attending for nongenetic causes.
Ethical approval was obtained from the local research ethics committee of Al Khafji Joint Operations Hospital, KSA, and parents of all children gave an informed written consent prior to the study.
All studied children were subjected to the following:
- Studied children were subjected to history taking, including socioeconomic status, residence, order of birth, perinatal history, obstetric history (parity, abortions, gestational age, stillbirths, live births and unexplained neonatal deaths), family history (consanguinity, similar cases of mental retardation [MR] and age of the parents at the time of conception) and developmental history (history of teething, motor milestones, social milestones and toilet control).
- Studied children were subjected to family pedigree. It is a graphical record of the family health history. Relationships of family members are represented schematically and demonstrate patterns of transmission of familial disorders.
- Studied children were subjected to detailed clinical examination. Anthropometric measurements, including height, weight and head circumference, were taken based on the national growth reference charts. They were also subjected to general and local examination, with specific attention to dysmorphic features that may suggest genetic or syndromic etiologies. Chromosomal study was done for all cases to exclude syndromic cases. They also underwent complete neurologic and neurodevelopmental assessment and psychological and psychiatric assessment, including attention, activity, disordered social communication and any associated psychiatric comorbidity.
- Special tests included the following:
- IQ assessment using the Wechsler Revised Intelligence Scale for preschool- and school-aged children.
- Aberrant Behavior Checklist (ABC; 40). All patients and controls were evaluated for behavioral disturbances using ABC. It is a 58-item scale that resolves into five subscales: 1) irritability and agitation; 2) lethargy and social withdrawal; 3) stereotypic behavior; 4) hyperactivity and noncompliance and 5) inappropriate speech.
- Child and Adolescent Psychiatric Assessment Schedule (CHA-PAS; 41) test was used to evaluate patients for the presence of any comorbid psychiatric illness. It is a 4-point scale constructed around the International Statistical Classification of Diseases and Related Health Problems-10th revision and the DSM-IV criteria. It is formed of a 97-item questionnaire covering eight domains, namely, anxiety disorder, depressive episode, manic episode, obsessive compulsive disorder, psychosis, attention-deficit hyperkinetic disorder, conduct disorder and autism spectrum disorder.
- Electroencephalography was done in suspected cases of comorbid epilepsy.
- Laboratory investigations included the following:
- Routine investigations as routine urine examination, stool analysis, complete blood count, renal function and hepatic function tests.
- Specific investigations included the following:
- Gas chromatography–mass spectrometry (GC–MS): one of the procedure involves the use of urine or eluates from urine on filter paper, stable isotope dilution and GC–MS. This procedure not only offers reliable and quantitative evidence for diagnosing, understanding and monitoring the diseases but also provides evidence for the diagnosis of new kinds of IEM.10 Not only organic acids or polar ones but also amino acids, sugars, polyols, purines, pyrimidines and other compounds are simultaneously analyzed and quantified.11
- Ferric chloride test: this test is used to determine the presence or absence of phenols in a given sample (for instance, natural phenols in a plant extract). Enols, hydroxamic acids, oximes, and sulfinic acids give positive results as well. The ferric chloride test can be used to detect metabolites in urine in case of IEM such as phenylketonuria.12
- Toluidine blue spot test: a screening test for urinary glycosaminoglycans as qualitative indicators for mucopolysaccharidoses.
This study was approved by the local ethical committee, and all parents of children give informed consent.
Statistical analysis
Results were collected, tabulated and statistically analyzed by an IBM-compatible personal computer with SPSS statistical package version 20. Two types of statistical analysis were done: 1) descriptive statistics, eg, was expressed in number (no), percentage (%), mean 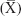 and SD, and 2) analytical statistics, eg, qualitative data were analyzed by chi-square test and whenever one cell of the expected was ≤5, Fisher’s exact test was done. Normally distributed quantitative data were analyzed by independent t-test. Not normally distributed data were analyzed by Mann–Whitney U-test. P-value of <0.05 was considered as statistically significant.
and SD, and 2) analytical statistics, eg, qualitative data were analyzed by chi-square test and whenever one cell of the expected was ≤5, Fisher’s exact test was done. Normally distributed quantitative data were analyzed by independent t-test. Not normally distributed data were analyzed by Mann–Whitney U-test. P-value of <0.05 was considered as statistically significant.
Results
We studied 150 children with ID with a mean age of 10.32±2.94 years (66.66 were males) compared to 50 apparently healthy age- and sex-matched children. Positive consanguinity occurred in a statistically significant frequency (30.66%, P=0.001) in children with ID compared to the control group. ID occurred in a significantly higher frequency among children who had a positive family history of ID (15.33%, P=0.001). ID occurred in a higher frequency among children who had older mother’s ages at birth of child with ID (37.47 years, P=0.001). Developmental history shows that among ID patient group, delayed motor milestones and delayed speech were statistically significant (90%, P=0.001; Table 1).
 | Table 1 Demographic data of studied groups |
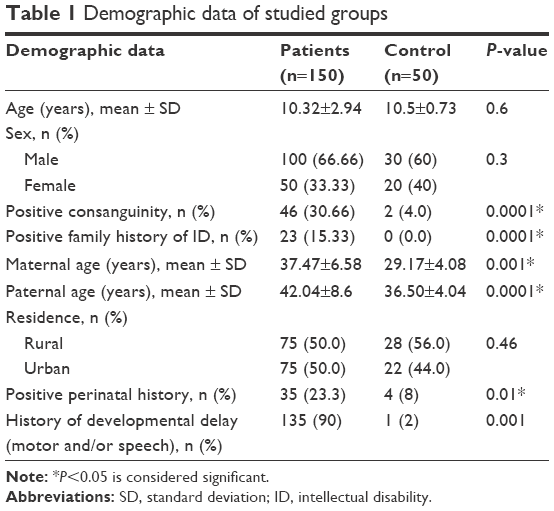
Patients with MR showed significantly more disturbed behavior in all the subscales of the ABC, which differentiated patients from controls (Table 2).
 | Table 2 Mean scores on the ABC subscales in both groups |
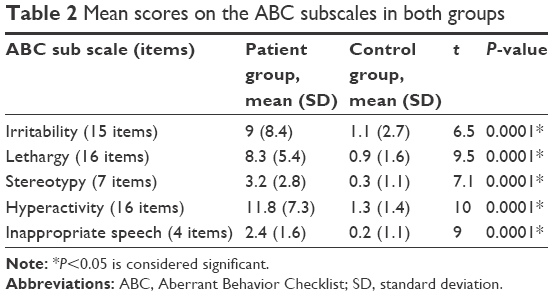
Comorbid psychiatric disorders associated with ID was found in 67 patients (44.6%) who had an additional psychiatric disorder. The most common comorbidity was attention-deficit hyperactivity disorder occurring in 16 patients (11.3%), followed by autistic disorder in 12 patients (8%; Table 3).
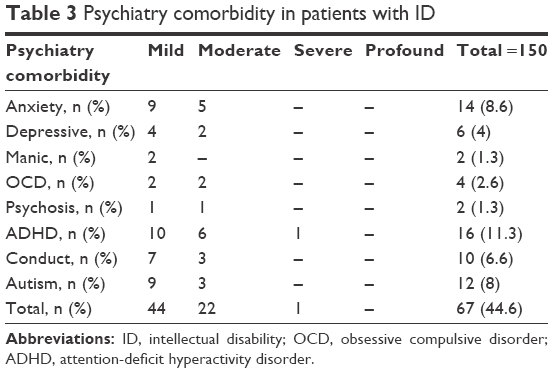 | Table 3 Psychiatry comorbidity in patients with ID |
Metabolic screening showed that positive ferric chloride test was significantly higher among the cases (53 cases [34.4%]) compared to the control group, while toluidine blue test did not show significant difference when comparing patient and control groups. Positive GC–MS was significantly higher among the cases (17 cases [11.03%]) compared to the control group. The metabolic urine screening tests and paper chromatography results in all control cases were negative (Table 4).
 | Table 4 The metabolic urine screening test and paper chromatography results among the studied groups |
Discussion
ID affects ~2%–3% of the general population. Nonsyndromic or idiopathic cases account for 30%–50% of cases.13 Metabolic disorders are another possible cause of MR. In some cases (eg, phenylketonuria [PKU] and hypothyroidism), retardation is preventable with early treatment. Other disorders (eg, mucopolysaccharidosis and sphingolipidoses) are less responsive to early intervention. Molecular medicine has made it possible to diagnose a number of conditions referred to as mitochondrial cell diseases.13,14 IEM are single-gene disorders resulting from the defects in the biochemical pathways of the body. Although these disorders are individually rare, collectively, they account for a significant portion of childhood disability and deaths.15
In the conducted study, we collected urine sample from children with ID for screening metabolic causes by using GC–MS, which can be used to detect amino acids in urine and needs no expensive outfits and tedious sample pretreatments.16 Two other tests were added: ferric chloride test for PKU and toluidine blue spot test for mucopolysaccharidoses.
Regarding demographic data of our patients, there was male predominance (69.5%) and parental consanguinity (27.8%). Sheffali et al17 reported in another study that the features suggestive of metabolic disorders included a male predominance (76.6%) with consanguinity in 10% of patients only. Parental consanguinity, which is highly significant in this study, is due to the traditional high incidence of consanguineous marriages in KSA.18
The male predominance can be explained by the fact that some IEMs are transmitted as X-linked recessive inheritance. The high incidence of IEMs among cases with a positive family history of parental consanguinity was also observed by Temtamy et al19 and Vasant et al,20 and this can be explained by the fact that consanguineous marriages are more likely to produce offsprings affected by autosomal recessive disorders because relatives more often share abnormal genes inherited from a common ancestor and most IEMs are autosomal recessive disorders.
In this study, 21 patients (~13.6%) had a positive family history of MR, in the form of first-degree relative, which is concomitant with the study of Temtamy et al19 who reported that a positive family history of MR is a risk factor.
Developmental history showed that there were 139 cases (90.0%) of patients who had a positive history of delayed motor milestone This comes in agreement with Sheffali et al17 who reported that signs suggestive of metabolic disease are developmental delay. Speech disorders in this study occurred in 89.6% of cases in the patient group with a significant difference between patients and control. Comorbid psychiatric disorders were reported in 44.6% of cases, which is comparable to the results of other different studies.21,22
In the conducted study, results of the metabolic screening tests revealed that the ferric chloride test was positive in 53 (34.4%) of cases, which points to PKU or tyrosinemia type 1. The test is also highly sensitive and less specific. The ferric chloride test may give false-positive results due to bilirubin, acidity and ketones, as reported by Blau et al.23 GC–MS gave positive result in 14 cases out of 150 (9.3%) of the studied patients. This is similar to a similar study that reported 8% cases of MR due to metabolic disorders.22 However, this is higher than a similar study done by Indian Council of Medical Research24 that revealed that 4.9% of genetic causes of MR were due to metabolic disorders. This is also higher than a similar study done by Kumta9 who reported that ~6.1% of MR children were diagnosed with a metabolic disorder. This may be due to lack of early detection and intervention or due to the traditional high incidence of consanguineous marriages in KSA.18
Although symptoms emerge at discrete points in childhood, these disorders result from abnormal brain maturation that likely precedes clinical impairment. As a result, research has focused on the identification of predictive biological and behavioral markers, with the ultimate goal of initiating treatments that may either alter developmental trajectories or lessen clinical severity.25 Yap et al26 suggest that early treatment with good biochemical control (lifetime plasma-free homocystine median <11 μmol/L) may prevent MR. In some cases (eg, PKU and hypothyroidism), retardation is preventable with early treatment. Other disorders (eg, mucopolysaccharidosis and sphingolipidoses) are less responsive to early intervention. Molecular medicine has made it possible to diagnose a number of conditions referred to as mitochondrial cell diseases.13,14 Early diagnosis of IEM is very important since the condition is rapidly progressive and results into irreversible damage in early childhood. It also helps in initiating the treatment in the earlier stages of development as it will be more effective and will improve the outcome at a later stage. The most important aspect of early diagnosis is genetic counseling.9
Conclusion
This study focused on increasing awareness of general practitioners, pediatricians and neonatologists about the importance of suspecting the possibility of IEM in children with ID and promoting for their early detection by using a set of rapid, easy and inexpensive simple urine tests for screening, especially in a high-risk group. The screening tests should be adopted by most of governments as routine neonatal screening, as this will give early diagnosis, intervention, minimal complications and better lifestyle for children with ID.
Disclosure
The authors report no conflicts of interest in this work.
References
American Association on Mental Retardation (AAMR). Mental Retardation: Definition, Classification, and Systems of Supports. 10th ed. Washington, DC: American Association on Mental Retardation; 2000. | ||
Puri RD, Tuteja M, Verma IC. Genetic approach to diagnosis of intellectual disability. Indian J Pediatr. 2016;83(10):1141–1149. | ||
Petrovic BB, Peric TO, Markovic DL, et al. Unmet oral health needs among persons with intellectual disability. Res Dev Disabil. 2016;59:370–377. | ||
Kumta NB. Genetic etiology of mental retardation. In: Shastri PC, Parikh AP, editors. Role of Genetics in Mental Retardation. 1998:1–13. | ||
Choudhuri T, Sengupta S. Inborn error of metabolism-an Indian perspective. Int J Hum Genet. 2006;6:89–91. | ||
Childs B, Valle D, Jimenez-Sanchez G. The inborn error and biochemical variability. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: Mc-Graw Hill; 2001:155–166. | ||
Chiaratti de Oliveira A, dos Santos AM, Martins AM, D’Almeida V. Screening for inborn errors of metabolism among newborns with metabolic disturbance and/or neurological manifestations without determined cause. Sao Paulo Med J. 2001;119(5):160–164. | ||
Selim LA, Hassan SA, Salem F, et al. Selective screening for inborn errors of metabolism by tandem mass spectrometry in Egyptian children: a 5 year report. Clin Biochem. 2014;47(9):823–828. | ||
Kumta NB. Inborn errors of metabolism (IEM) – an Indian perspective. Indian J Pediatr. 2005;72(4):325–332. | ||
Hyánek J. [Diagnostic significance of the test with ferric chloride in the urine in phenylketonuria]. Cesk Pediatr. 1969;24:159–165. | ||
Kuhara T. Diagnosis and monitoring of inborn errors of metabolism using urease-pretreatment of urine, isotope dilution, and gas chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;781(1–2):497–517. | ||
Kuhara T. Diagnosis of inborn errors of metabolism using filter paper urine, urease treatment, isotope dilution and gas chromatography-mass spectrometry. J Chromatogr B Biomed Sci Appl. 2001;758(1):3–25. | ||
Daily DK, Ardinger HH, Holmes GE. Identification and evaluation of mental retardation. Am Fam Physician. 2000;61(4):1059–1067. | ||
Dimauro S, Moraes CT. Mitochondrial encephalomyopathies. Arch Neurol. 1993;50(11):1197–1208. | ||
Ezgu F. Inborn errors of metabolism. Adv Clin Chem. 2016;73:195–250. | ||
Hao H, Li S, Zhou W, et al. Metabolic products in urine of preterm infants characterized via gas chromatography-mass spectrometry. Int J Clin Exp Med. 2015;8(9):16454–16462. | ||
Sheffali G, Vaswani M, Kalra V. An approach to neurometabolic disorders by a simple metabolic screen. Indian Pediatr. 2000;37(1):63–69. | ||
Alfadhel M, Benmeakel M, Hossain MA, et al. Thirteen year retrospective review of the spectrum of inborn errors of metabolism presenting in a tertiary center in Saudi Arabia. Orphanet J Rare Dis. 2016;11(1):126. | ||
Temtamy SA, Kandil MR, Demerdash AM, Hassan WA, Meguid NA, Afifi HH. An epidemiological/genetic study of mental subnormality in Assiut Governorate, Egypt. Clin Genet. 1994;46(5):347–351. | ||
Wasant P, Vatanavicharn N, Srisomsap C, Sawangareetrakul P, Liammongkolkul S, Svasti J. Retrospective study of patients with suspected inborn errors of metabolism at Siriraj Hospital, Bangkok, Thailand (1997–2001). J Med Assoc Thai. 2005;88(6):746–753. | ||
Munir KM. The co-occurrence of mental disorders in children and adolescents with intellectual disability/intellectual developmental disorder. Curr Opin Psychiatry. 2016;29(2):95–102. | ||
van Ool JS, Snoeijen-Schouwenaars FM, Schelhaas HJ, Tan IY, Aldenkamp AP, Hendriksen JG. A systematic review of neuropsychiatric comorbidities in patients with both epilepsy and intellectual disability. Epilepsy Behav. 2016;60:130–137. | ||
Blau N, Blaskovics ME, Duran M. Simple test in urine and blood. In: Blau N, Duran M, Blaskovics ME, editors. Physician’s Guide to the Laboratory Diagnosis of Metabolic Diseases. 1st ed. Oxford: Chapman & Hall Medical; 1996:3–11. | ||
Multicentric study on genetic causes of mental retardation in India. ICMR Collaborating Centres & Central Co-ordinating Unit. Indian J Med Res. 1991;94:161–169. | ||
Jeste SS. Neurodevelopmental behavioral and cognitive disorders. Continuum (Minneap Minn). 2015;21(3 Behavioral Neurology and Neuropsychiatry):690–714. | ||
Yap S, Rushe H, Howard PM, Naughten ER. The intellectual abilities of early-treated individuals with pyridoxine-nonresponsive homocystinuria due to cystathionine beta-synthase deficiency. J Inherit Metab Dis. 2000;24(4):437–447. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.