Back to Journals » Journal of Inflammation Research » Volume 18
Metabolic Reprogramming Intermediates of Glucose Regulate Macrophage Polarization: An Important Direction for Ameliorating Pulmonary Vascular Remodeling
Authors Wang J, Yuan R, Zhang S, Xu Z, Song L
Received 21 May 2025
Accepted for publication 3 November 2025
Published 17 November 2025 Volume 2025:18 Pages 16045—16062
DOI https://doi.org/10.2147/JIR.S541649
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Rongxue Wu
Junqi Wang,1,2 Rong Yuan,1,2 Shengkang Zhang,3 Zhaojun Xu,2,3 Lan Song1,2
1Medicine School, Hunan University of Chinese Medicine, Changsha, Hunan, 410208, People’s Republic of China; 2Hunan Provincial Key Laboratory of Vascular Biology and Translational Medicine, Hunan University of Chinese Medicine, Changsha, Hunan, 410208, People’s Republic of China; 3Cardiothoracic Surgery of the First Affiliated Hospital, Hunan University of Chinese Medicine, Changsha, Hunan, 410007, People’s Republic of China
Correspondence: Zhaojun Xu, Cardiothoracic Surgery of the First Affiliated Hospital, Hunan University of Chinese Medicine, Changsha, Hunan, 410007, People’s Republic of China, Email [email protected] Lan Song, Medicine School, Hunan University of Chinese Medicine, Changsha, Hunan, 410208, People’s Republic of China, Email [email protected]
Abstract: Pulmonary vascular remodeling (PVR) is a key pathological basis for various lung diseases and is centered on macrophage-driven pathological vascular remodeling. Macrophage functional polarization is closely related to metabolic reprogramming, a process that not only encompasses energy supply but also dictates cellular function through metabolic intermediates. To bridge the knowledge gap between metabolic regulation and clinical translation in PVR, this review focuses on key metabolites produced during glucose metabolism: pyruvate, citrate, succinate, and itaconate. These intermediates are not merely metabolic byproducts; rather, they directly influence the pathological processes of vascular endothelial cells, smooth muscle cells, and the extracellular matrix by modulating the polarization of macrophages. This review systematically elucidates the precise regulatory mechanisms of these metabolic signals, with the aim of providing new diagnostic and therapeutic targets for PVR. It emphasizes the immense potential of targeting metabolic intermediates for future precision medicine, ultimately promoting a paradigm shift in PVR therapy from traditional anti-proliferative interventions to an innovative model based on metabolic reprogramming.
Keywords: pulmonary vascular remodeling, macrophage polarization, glycolytic reprogramming, pyruvate, citrate, succinate, itaconate
Introduction
Pulmonary vascular remodeling (PVR), characterized by pathological alterations in the structure and function of pulmonary vessel walls, primarily manifests as vascular wall thickening and lumen narrowing. These changes lead to increased pulmonary vascular resistance and pulmonary artery hypertension (PAH), which can ultimately progress to right heart failure and death.1,2 As a common pathological feature in various pulmonary diseases, including PAH, acute respiratory distress syndrome (ARDS), and chronic obstructive pulmonary disease, PVR involves multiple processes such as endothelial cell dysfunction, abnormal proliferation of smooth muscle cells, and extracellular matrix (ECM) deposition.3,4 In recent years, macrophages have emerged as key immune effector cells driving the development of PVR, given their high plasticity and rapid responsiveness to microenvironmental changes.5,6
Depending on microenvironmental stimuli, macrophages can polarize into either pro-inflammatory M1 or anti-inflammatory/pro-repair M2 phenotypes. The balance between these polarization states is crucial for the pathogenesis of PVR. Research has demonstrated a strong link between macrophage polarization and their metabolic pathways, with metabolic reprogramming being particularly pronounced under hypoxic and inflammatory conditions.7,8 Specifically, M1 macrophages rely on glycolysis for a rapid energy supply to fuel their immune responses, whereas M2 macrophages predominantly utilize oxidative phosphorylation (OXPHOS) and fatty acid oxidation to support their anti-inflammatory and tissue repair functions.9–11 These findings highlight that metabolic reprogramming is not merely about energy supply but serves as a core determinant of macrophage functional phenotype conversion.
Furthermore, intermediate metabolites generated during glucose metabolic reprogramming, such as succinate, citrate, pyruvate, and itaconate, are no longer viewed simply as byproducts of energy production. Instead, they function as critical signaling molecules.12–14 These metabolites can directly influence macrophage polarization and function by modulating epigenetic modifications, such as histone acetylation, or by activating signaling pathways like HIF-1α. Their effects extend further to the vessel wall, promoting PVR by driving smooth muscle cell proliferation, endothelial cell phenotypic transformation, and abnormal ECM deposition6 (Figure 1). This review aims to systematically explore the precise regulatory role of glucose metabolism intermediates on macrophage polarization and their subsequent impact on PVR pathogenesis, in an effort to provide new theoretical support for developing targeted immunometabolic intervention strategies.
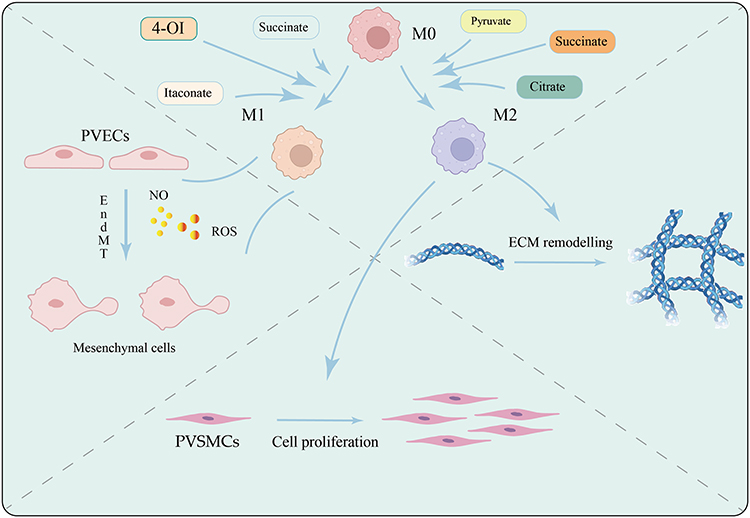 |
Figure 1 Metabolic intermediates of glycolysis in macrophages regulate their polarization states (M1 vs M2), orchestrating crosstalk among pulmonary vascular endothelial cells, smooth muscle cells, and immune cells, ultimately driving the progression of pulmonary. Created in BioRender. Junlan, T. (2025) https://BioRender.com/cqjmnah. |
Macrophage Polarization and Glycolytic Reprogramming: Metabolic Characteristics and Regulatory Mechanisms of M1/M2 Phenotypes
Glycolytic metabolic reprogramming refers to the process by which cells select and regulate their energy metabolism pathways in response to specific microenvironmental cues and external stimuli. This process not only involves changes in energy supply but also entails the generation of metabolic intermediates, the adjustment of metabolic pathways, and the reconstruction of regulatory networks. For macrophages, glucose metabolic reprogramming is fundamental to their polarization, functional execution, and participation in various physiological and pathological processes. Current research indicates that based on differences in their immune functions, polarized macrophage subtypes are broadly classified into classically activated M1 macrophages and alternatively activated M2 macrophages.15
Under physiological conditions, macrophages predominantly utilize the tricarboxylic acid (TCA) cycle and OXPHOS for energy production. However, under conditions of hypoxia or surging energy demand, such as in rapidly proliferating cells, they switch to glycolysis as the main energy source. In the context of PVR, abnormal blood flow, local hypoxia, and the continuous release of inflammatory mediators collectively alter the microenvironment, activating hypoxia-inducible factor (HIF-1α). Concurrently, inflammatory signals (eg, TNF-α, IL-1β) and oxidative stress signals also participate in regulating cellular metabolism. This results in a critical phenomenon: even in the presence of sufficient oxygen, macrophages preferentially utilize glycolysis over mitochondrial OXPHOS for energy production.6 This metabolic shift not only provides energy for the polarization process but also, through its metabolic products and associated signaling pathways, dictates the direction of macrophage polarization and their corresponding biological functions.16 Of note, macrophage polarization exhibits temporal dynamics. An animal model study by Fan et al demonstrated a transient increase in M1 macrophages during the early stage, which are subsequently replaced by M2 macrophages. The latter, by continuously secreting pro-proliferative factors such as IL-4 and IL-13, accelerate the progression of the disease. In summary, the polarization state of macrophages and their metabolic reprogramming dynamically interact throughout the disease course, with glucose metabolic reprogramming serving as both a consequence and a driver. Therefore, by exploring the distinct roles of glucose metabolic reprogramming in macrophage polarization, the following sections of this review will provide a detailed exposition of the core mechanisms and regulatory networks linking macrophage polarization and glucose metabolic reprogramming.
Metabolic Characteristics of M1 Macrophages
M1 macrophage polarization is triggered by various cytokine stimuli and accompanied by a core metabolic reprogramming that is characterized by significantly enhanced glycolysis and pentose phosphate pathway (PPP) activity.17 This metabolic shift involves a progressive transition of cellular energy metabolism from OXPHOS to glycolysis, alongside an interruption of TCA cycle. Glycolysis not only provides rapid energy for M1 macrophages but also regulates immune responses through intermediate metabolites such as citrate and succinate, which facilitates the rapid proliferation and pro-inflammatory cytokine secretion necessary for combating pathogens. This metabolic reprogramming is precisely regulated by both metabolic factors like HIF-1α and PKM2 and non-metabolic molecular signals. For instance, eRNA has been shown to act as a signaling molecule that participates in M1 macrophage activation and is closely associated with the expression of pro-inflammatory genes.18 This synergistic interplay between non-metabolic signals and core metabolic reprogramming is essential for maintaining the pro-inflammatory M1 phenotype. The reduced activity of isocitrate dehydrogenase and succinate dehydrogenase in the TCA cycle of M1 macrophages leads to the accumulation of metabolic intermediates, including succinate and itaconate, which act as important signaling molecules in addition to their metabolic roles.17 Notably, succinate accumulation can stabilize HIF-1α by inhibiting succinate dehydrogenase (SDH), thereby further promoting glycolysis and enhancing the expression of pro-inflammatory factors like IL-1β.
Enhanced glycolysis necessitates an increased supply of glucose, which is typically achieved by upregulating the expression of glucose transporters. This funnels more glucose-6-phosphate (G-6-P) into the PPP. The robust activity of the PPP promotes the generation of NADPH and nucleotide precursors, which are essential for supporting antioxidant responses and rapid proliferation.19 Furthermore, M1 macrophages not only upregulate aerobic glycolysis but also stimulate glycogen synthesis. Carbon tracing experiments have shown an increase in PCK1-mediated phosphoenolpyruvate (PEP) synthesis, with a portion of this PEP flux being directed toward glycogen synthesis. The G-6-P derived from glycogenolysis then enters the PPP. As a crucial branch of glycolysis, the PPP is highly active in M1 macrophages, where it generates reactive oxygen species (ROS) via NADPH oxidase to maintain reduced glutathione (GSH) levels. This mechanism is critical for countering oxidative stress and plays a key role in the inflammatory response.20
Regarding the underlying regulatory mechanisms, inflammatory cytokine stimulation upregulates hexokinase mRNA levels, which in turn promotes glycolysis.21 Research indicates that the mammalian target of rapamycin complex 1 (mTORC1) mediates glycolysis-induced activation of the NLRP3 inflammasome via hexokinase, thereby regulating the secretion of IL-1β and IL-18 by M1 macrophages.22 Furthermore, the expression of pyruvate kinase M2 (PKM2) is elevated in M1 macrophages, where it tends to exist as a dimer. This dimer can translocate to the nucleus and interact with HIF-1α, promoting the expression of glycolytic enzymes such as PFKFB3 and LDHA while also upregulating the transcription of pro-inflammatory factors like IL-1β and IL-6.23 As a key regulator of glycolysis, hypoxia-inducible factor 1α (HIF-1α) demonstrates enhanced stability and activity upon activation of mTOR signaling. This in turn further promotes the expression of glycolysis-related proteins and the accumulation of succinate, thereby sustaining the pro-inflammatory phenotype of M1 macrophages.24,25
Metabolic Characteristics of M2 Macrophages
In contrast to M1 macrophages, M2 macrophages are more geared towards anti-inflammatory and tissue repair functions, with their primary energy sources being oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) and a lower dependence on glycolysis. Due to their metabolic flexibility, M2 cells can derive energy from both glucose and fatty acid metabolism, which maintains the integrity of the TCA cycle and allows pyruvate to enter the cycle to support energy metabolism for their anti-inflammatory and tissue repair functions. Studies have shown that while glucose metabolism is not completely suppressed in IL-4-induced M2 macrophages, a certain level of glycolytic activity is maintained, although their energy is predominantly reliant on fatty acid oxidation. It is widely believed that fatty acid oxidation is the main energy source during macrophage polarization. Interestingly, in the context of early IL-4 stimulation, glucose metabolism is more significant, suggesting that glycolysis also plays an important role in M2 polarization.26 Furthermore, OXPHOS generates abundant ATP to meet the energetic demands of M2 macrophages for maintaining cellular homeostasis and tissue repair. Research indicates that as long as OXPHOS maintains sufficient activity, macrophage M2 polarization may not require glycolytic stimulation.27
In M2 macrophages, glucose is metabolized via glycolysis to produce pyruvate, which enters the TCA cycle as a substrate for OXPHOS, thereby supporting mitochondrial respiration. Concurrently, IL-4 enhances glucose metabolism through the AKT and mTORC1 signaling pathways, promoting glucose uptake by macrophages. It also upregulates the activity of the pyruvate dehydrogenase complex (PDC), which boosts the conversion of pyruvate to acetyl-CoA, providing an ample energy source for the TCA cycle and OXPHOS.28 Moreover, the expression pattern of pyruvate kinase M2 (PKM2) in M2 macrophages differs from that in M1 cells; it predominantly exists in a tetrameric form. This tetrameric PKM2 is more likely to promote the entry of pyruvate into the TCA cycle rather than engaging in pro-inflammatory signaling pathways. However, the specific regulatory mechanism between glycolysis and OXPHOS during M2 macrophage activation remains to be fully elucidated.
Regarding FAO, it is a dominant pathway in M2 macrophages, promoting their function and polarization by regulating fatty acid metabolism and anti-inflammatory responses.27 In recent years, the relationship between FAO and inflammasomes has emerged as a new focal point in the field of immunometabolism. The inflammasome is a crucial intracellular multiprotein complex that is primarily involved in activating inflammatory responses, particularly in driving the release of pro-inflammatory cytokines.
Crosstalk Between Macrophages and Key Cells in Pulmonary Vascular Remodeling
PVR refers to the dynamic and pathological changes in the pulmonary artery vessel wall, encompassing alterations in both cellular components and the extracellular matrix. This process is a key pathological feature of numerous pulmonary vascular diseases, leading to increased vascular resistance and, ultimately, right heart failure.2 PVR involves multiple cell types and molecular mechanisms, including endothelial cell dysfunction, smooth muscle cell proliferation and migration, and extracellular matrix remodeling.1 n the early stages of pulmonary vascular injury, an inflammatory response characterized by the infiltration of immune cells (macrophages, T/B lymphocytes) and the release of pro-inflammatory cytokines (IL-6, IL-13, TNF-α) induces endothelial dysfunction and abnormal smooth muscle cell proliferation. This, in turn, triggers the vascular wall thickening and fibrosis associated with the remodeling process. As a crucial component of the innate immune system, macrophages not only play a pivotal role in inflammatory responses but also directly and indirectly influence PVR pathogenesis through metabolic regulation, cytokine secretion, and intercellular signaling. The lung contains two primary types of macrophages, which are categorized by their anatomical location: interstitial macrophages (IMs) and alveolar macrophages (AMs), distributed respectively in the lung interstitium (the connective tissue surrounding vessels and airways) and on the alveolar surface.29 Macrophage origin further distinguishes them as either tissue-resident or monocyte-derived. The former originates from the embryonic yolk sac or fetal liver and maintains pulmonary homeostasis via self-renewal, while the latter is recruited to the lungs from peripheral blood monocytes under specific stimuli and subsequently differentiates into macrophages.29,30 Notably, macrophages exhibit high plasticity, functionally polarizing in response to microenvironmental signals. This is particularly evident in IMs under pathological conditions. Single-cell transcriptome sequencing by Liu et al revealed that monocytes can replenish resident AMs that are depleted by inflammatory reactions, and then gradually mature into bone-marrow-derived macrophages.31 In their study, Kumar et al noted a significant increase in the number of abnormally proliferating perivascular IMs in pulmonary hypertension, whereas the number of AMs remained unchanged.32 Furthermore, Qiu et al used single-cell sequencing to demonstrate a significant increase in AMs following hypoxia and identified MHCIIhiLYVE1loCCR2hi IMs as the most prominent subpopulation. They further showed that inhibiting this subtype could alleviate the progression of PH.33 These results collectively suggest that IMs may play a more critical role in the pathological process of pulmonary vascular remodeling.
More importantly, these shifts in cell numbers are typically accompanied by a change in their functional phenotype. Under disease conditions, macrophages can be stimulated by bacterial lipopolysaccharide (LPS) and interferon-γ (IFN-γ) to undergo M1 activation.34 Activated M1 cells, in turn, can induce T helper 1 (Th1) lymphocytes to release more pro-inflammatory factors like IFN-γ,35 and further amplify the inflammatory cascade by secreting a variety of pro-inflammatory cytokines and chemokines (eg, IL-1β, IL-6, TNF-α, and IL-12). In contrast, M2 macrophages, activated by signals such as IL-4 and IL-13, primarily function to suppress inflammation and promote tissue repair.36 Throughout disease progression, the early stage is often dominated by M1 macrophages, but as the disease advances, the proportion of M2 macrophages gradually increases and they assume a critical role.37 Consequently, the polarization balance between M1 and M2 macrophages is central to regulating the inflammatory microenvironment in PVR, as it governs the initiation, maintenance, and resolution of the inflammatory response (Figure 2).
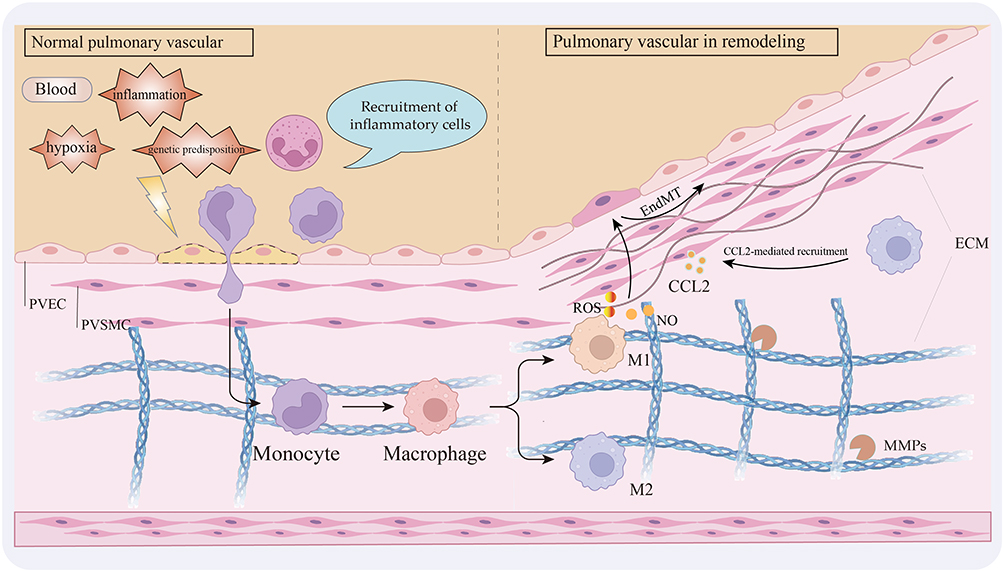 |
Figure 2 Under conditions such as hypoxia, inflammation, and genetic susceptibility, immune cells including monocytes are recruited and transmigrate across the pulmonary endothelium, where they differentiate into macrophages and polarize toward either the M1 or M2 phenotype. Both polarization states contribute to the progression of pulmonary vascular remodeling. Created in BioRender. Junlan, T. (2025) https://BioRender.com/j9l0pnb. |
Regulation of Endothelial Function by M1/M2 Macrophages
Endothelial cells, the primary cell type lining the pulmonary vasculature, are crucial for maintaining vascular homeostasis and regulating vessel tone. Under normal conditions, they maintain vascular permeability through tight junctions. However, inflammatory cytokines (eg, TNF-α, IL-1β) and oxidative stress can disrupt these junctions, leading to a loss of barrier function and increased vascular permeability,38,39 This compromised barrier allows monocytes to be recruited to the damaged vessel wall by chemokines (eg, CCL-2), which are typically released by vascular endothelial cells, smooth muscle cells, and other immune cells.40 Endothelial cell injury and apoptosis are key initiating factors throughout the pathological process of PVR. Upon their migration and activation into macrophages at the site of injury, a bidirectional regulatory crosstalk with endothelial cells emerges that continues to drive disease progression.
Upon migration to the injury site, monocytes differentiate into polarized M1 or M2 macrophages, establishing a complex regulatory network with endothelial cells. In the early stages of PVR, M1 macrophages produce a significant amount of cytotoxic mediators, including nitric oxide (NO) and reactive oxygen species (ROS),41 While these mediators play a protective role in clearing pathogens, their over-activation can cause oxidative stress and apoptosis in vascular endothelial cells, compromising the endothelial barrier and thereby exacerbating tissue damage and vascular remodeling. Concurrently, M1 macrophages can activate and upregulate the expression of adhesion molecules (eg, ICAM-1 and VCAM-1), enhancing the adhesion and infiltration of monocytes to the damaged area and further amplifying the inflammatory response.42 n addition to secreted cytokines, macrophages can also regulate endothelial function by releasing non-metabolic molecules such as extracellular eRNA. Studies have demonstrated that macrophage-derived eRNA can be taken up by vascular endothelial cells, inducing apoptosis and barrier dysfunction.43 Furthermore, during the pathogenesis of PVR, the role of macrophages extends beyond initiating inflammation and damaging endothelial cells; they can also induce endothelial cells to undergo a phenotypic transition.44 When exposed to stimuli such as inflammation, oxidative stress, or hypoxia, endothelial cells are induced to undergo Endothelial-to-Mesenchymal Transition (EndMT).45 During this transition, endothelial cells lose their characteristic markers (eg, VE-cadherin, CD31) and begin to express mesenchymal cell markers like α-SMA and fibronectin. This phenotypic shift not only compromises the endothelial barrier and promotes the abnormal proliferation and migration of smooth muscle cells, but also provides a new pathological cell population that drives pulmonary vascular remodeling. Notably, EndMT may directly contribute to PAH by transforming endothelial cells into smooth muscle-like cells with higher proliferative and migratory potential. This notion is supported by clinical and experimental findings, including the observation of EndMT markers in both PAH patients and the vascular lesions of BMPR2 mutant rats.46
The interaction between endothelial cells and macrophages during EndMT is especially crucial. On one hand, EndMT cells can regulate macrophage polarization by secreting specific factors. Multiple studies, including those in mouse models and in vitro cell culture experiments, have demonstrated that in pancreatic ductal adenocarcinoma, EndMT cells secrete the chaperone HSP90α, which induces M2 macrophage polarization and further HSP90α secretion to promote tumor growth,47 Conversely, other studies in mice, both in vitro and in vivo, have shown that CD163+ macrophages release pro-inflammatory cytokines rather than mediating an anti-inflammatory response. These macrophages activate intracellular signaling pathways within endothelial cells, leading to an apoptosis-prone EndMT phenotype and identifying NF-κB as a key pathway responsible for CD163+ macrophage-induced EndMT formation.44,48,49
In summary, the interaction between endothelial cells and macrophages in PVR forms a complex bidirectional regulatory network. Endothelial injury attracts and activates macrophages via the secretion of chemokines and the expression of adhesion molecules. In a reciprocal manner, macrophages modulate endothelial cell function and phenotypic transformation by secreting bioactive molecules and metabolic products. Mechanistic studies of this interaction not only enhance our understanding of PVR pathogenesis but also provide a theoretical basis for exploring novel therapeutic targets.
The Effect of Macrophage Polarization on Smooth Muscle Cell Proliferation and Phenotype
The interaction between smooth muscle cells (SMCs) and macrophages is a critical factor in the pathological progression of PVR. These two cell types not only directly participate in vascular structural changes but also play significant roles in the development of the disease.
Under normal physiological conditions, SMCs are located in the tunica media of blood vessels, where they provide mechanical strength and stability to the vasculature through interactions with the extracellular matrix. Concurrently, their contractile and relaxation capabilities regulate blood flow and pressure. However, under pathological conditions, inflammatory factors (eg, IL-6) and growth factors (eg, platelet-derived growth factor, PDGF) induce excessive proliferation of vascular SMCs by activating signaling pathways such as MAPK and PI3K/Akt,50,51 These cells then migrate to the intimal layer, initiating a phenotypic switch from a contractile phenotype with low proliferative capacity to a synthetic phenotype characterized by significantly enhanced proliferation and migration. This transformation further exacerbates vascular lumen narrowing.52
During PVR, SMCs not only undergo their own phenotypic transformation but also recruit macrophages to local inflammatory or injury sites by secreting chemokines like CCL2, thereby amplifying the local immune response. The link between chemokines and macrophage recruitment and activation has been demonstrated in clinical patients and animal models: models deficient in specific chemokines show a significant reduction in PVR, suggesting a core role for the SMC-macrophage interaction in disease progression.53 The relationship between M2 macrophages and SMCs is particularly prominent, as M2 macrophages promote the proliferation of pulmonary artery SMCs (PASMCs) and vascular remodeling by secreting pro-angiogenic factors such as VEGF and PDGF-β.37 Research by Abid et al has shown that M2 macrophages cooperate with PASMCs via CCR2/CCR5-mediated signaling pathways to promote PASMC proliferation and migration.54 The pro-proliferative effect of M2 macrophage culture supernatant on PASMC proliferation was significantly attenuated by blocking CCR2 and CCR5, indicating that the CCR2/CCR5 signaling pathway plays a key role in this process.54 Furthermore, CCR5 is expressed in endothelial cells, SMCs, and macrophages of PH patients and is also upregulated in rodent models of chronic hypoxia. Conversely, mice lacking CCR5 and exposed to hypoxia exhibit reduced PASMC proliferation and do not develop pulmonary hypertension.55 These findings suggest that M2 macrophages not only function by secreting anti-inflammatory factors but also likely participate in the pathological progression of PVR through direct communication with PASMCs.
In summary, the interaction between PASMCs and macrophages is a key mechanism driving disease progression. PASMCs secrete chemokines that recruit macrophages to the lesion site. M2 macrophages then further promote PASMC proliferation and migration through the secretion of pro-angiogenic factors and the activation of the CCR2/CCR5 signaling pathway. This series of intercellular interactions highlights the critical pathological basis for the cooperative roles of PASMCs and M2 macrophages in PVR progression.
Macrophage Polarization and Extracellular Matrix Remodeling
The extracellular matrix (ECM), composed of various matrix proteins such as collagen, elastin, and glycosaminoglycans, plays a crucial role in maintaining vascular structure, elasticity, and stability. In PVR, macrophages interact with the ECM to regulate local inflammation and fibrosis, thereby driving vascular remodeling. ECM metabolic dysregulation also plays a key role in this process, with excessive deposition of matrix proteins, dysregulated degradation, and increased stiffness. These changes can regulate cell behavior via mechanotransduction signals, forming a pathological positive feedback loop.56,57
Macrophages regulate local inflammation and fibrosis progression through their interaction with the ECM. On one hand, ECM components such as fibronectin, laminin, and glycosaminoglycans can act as chemoattractants for macrophages, promoting their migration to injury sites. Upon migration to these areas, macrophages exacerbate local inflammatory responses by secreting various inflammatory factors (eg, TNF-α, IL-1β, IL-6),58 On the other hand, macrophages regulate the dynamic balance of the ECM during remodeling by secreting matrix metalloproteinases (MMPs) and their inhibitors (TIMPs).59 The dynamic regulation of the ECM is critically dependent on the balance between MMPs and TIMPs. Excessive MMP activity coupled with insufficient TIMP levels results in abnormal ECM degradation. This process also releases growth factors stored within the matrix (eg, VEGF, TGF-β), further exacerbating vascular pathological changes.60
In regulating vascular remodeling, different macrophage polarization phenotypes have distinct effects on ECM metabolism. M2 macrophages interact with T helper 2 (Th2) cells to secrete anti-inflammatory and pro-repair factors such as IL-10, TGF-β, CD206, Arg-1, and CCL-18. This action both suppresses M1 pro-inflammatory factor production and mitigates tissue damage while also promoting ECM deposition and vascular wall repair, thereby alleviating abnormal vascular permeability.15,61 However, the over-activation of M2 macrophages can lead to fibrosis and abnormal ECM deposition. Through their regulation of TGF-β and other ECM-related factors, these cells promote the deposition of Type I and Type III collagen, increasing vascular wall stiffness and ultimately driving pathological vascular remodeling.62 Additionally, MMP-2 and MMP-9 secreted by M1 macrophages can degrade collagen and fibronectin. This action provides space for smooth muscle cell migration and releases matrix-stored growth factors (eg, VEGF), further accelerating the remodeling process.63
In summary, macrophages regulate endothelial cell function, smooth muscle cell proliferation, migration, and phenotypic changes, and also influence ECM metabolic balance. This intricate pathological network exacerbates vascular wall thickening and stiffness while driving a positive feedback loop of pathological inflammation and tissue remodeling that accelerates PVR progression. Single-cell RNA sequencing to investigate the mechanisms by which different polarized macrophage phenotypes contribute to PVR not only offers a new perspective for understanding the pathogenesis of diseases like pulmonary hypertension but also presents potential therapeutic strategies for the targeted regulation of macrophage, smooth muscle cell, and endothelial cell phenotypic transitions.
The Regulatory Role of Glucose Metabolism Intermediates in Macrophage Polarization States
Macrophages play a crucial role in inflammation, immune regulation, and tissue repair, and the reprogramming of glucose metabolism (including pathways such as glycolysis and OXPHOS) is closely linked to their polarization states, which further influence the progression of PVR. The metabolites of glucose metabolism not only provide energy for cells but also play significant roles in cell signaling, immune responses, and tissue repair (Figure 3 and Table 1)
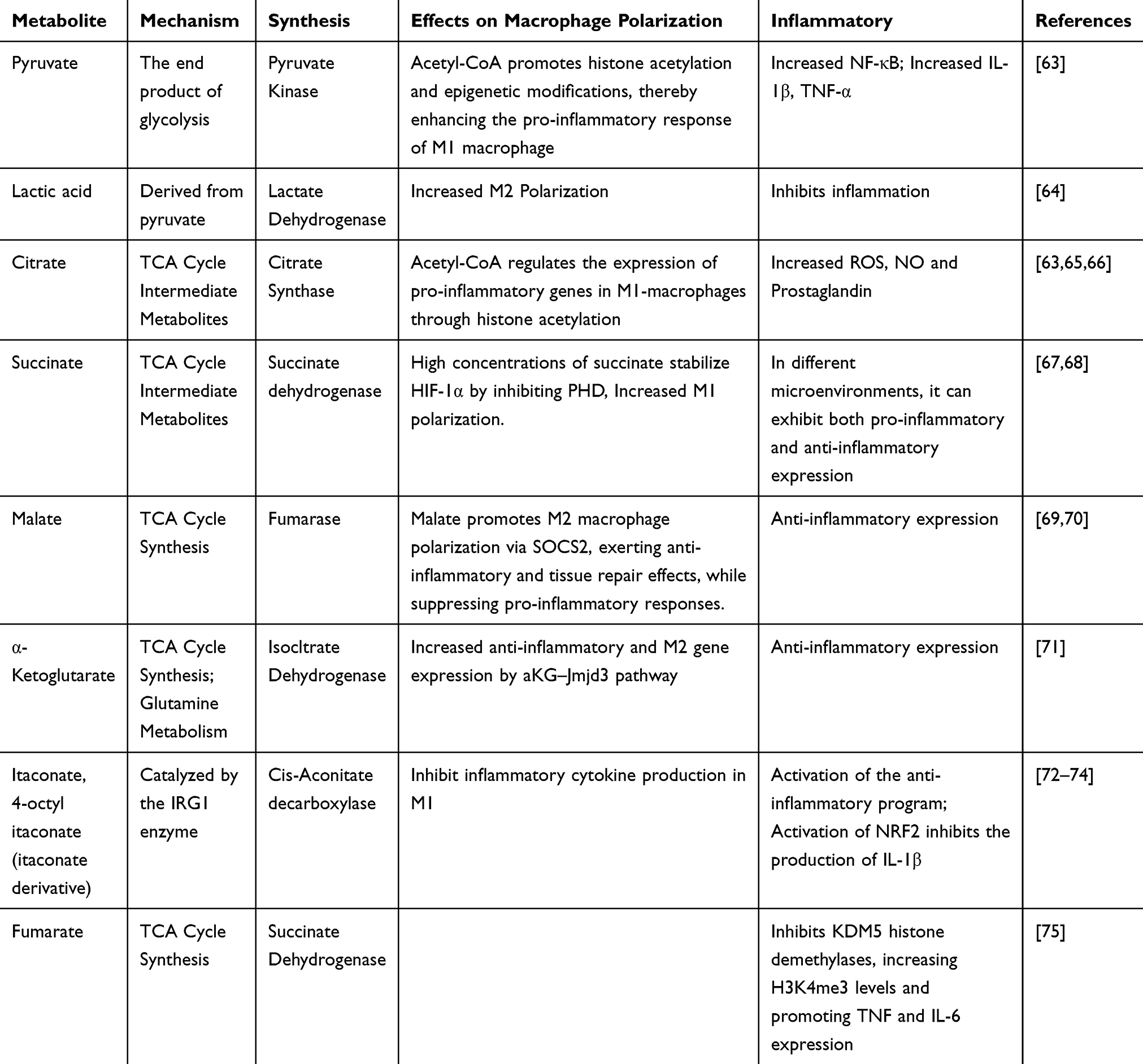 |
Table 1 The Regulatory Role of Metabolic Intermediates in Macrophage Polarization and Inflammation |
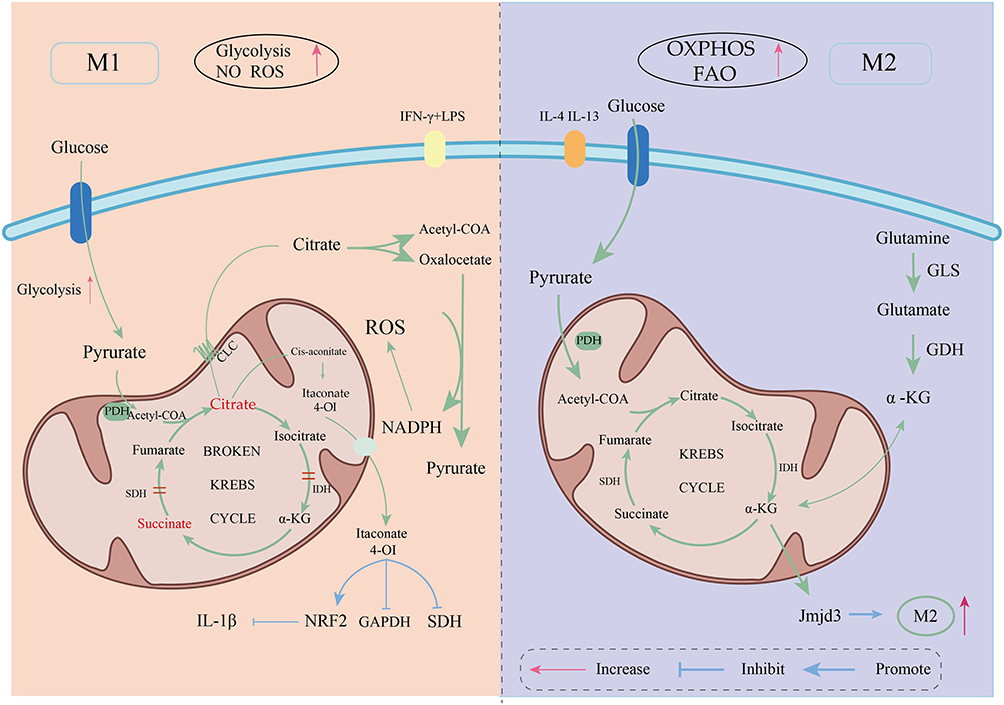 |
Figure 3 The metabolic differences between M1 and M2 macrophages. |
Pyruvate
Pyruvate, the final product of glycolysis, is also a key intersection point for multiple biosynthetic pathways, and its metabolic fate profoundly impacts cellular function. In the pathological state of pulmonary vascular remodeling PVR, local hypoxia and inflammatory stimuli significantly enhance the glycolytic rate in macrophages, leading to the production of large amounts of pyruvate.
Pyruvate primarily follows two metabolic fates. On one hand, it can enter the mitochondria and be catalyzed by the pyruvate dehydrogenase complex (PDC) to enter the TCA cycle, generating citrate and subsequent metabolites. This process increases the level of acetyl-CoA in the cytoplasm, promoting histone acetylation and related epigenetic modifications. These epigenetic changes help activate the transcription of pro-inflammatory genes, thereby enhancing the inflammatory response of M1 macrophages;76 On the other hand, under the condition of enhanced aerobic glycolysis in M1 macrophages, pyruvate is converted into lactate in the cytoplasm through the catalysis of lactate dehydrogenase (LDH). Lactate not only serves as a signaling molecule, playing a role in intracellular and extracellular signal transduction with key immune regulatory functions, but increasing evidence also suggests that lactate acts as a metabolic regulator of macrophages, closely linked to their polarization. A 2019 study demonstrated that lactate can induce histone lactylation, which enhances the expression of M2 genes during the M1 polarization phase of macrophages.64,77 This finding offers a new perspective for intervening in the reciprocal transformation of macrophage polarization states in disease conditions. In the context of PVR, enhancing the expression of M1-associated genes and suppressing the proportion and activity of M2 macrophages during the later stages of disease progression may represent a viable strategy to attenuate vascular remodeling. This metabolic diversion mechanism determines the energy utilization patterns and functional phenotypes of macrophages under distinct microenvironmental conditions, underscoring the pivotal role of glucose metabolism reprogramming in immune regulation.
Notably, the balance and regulation of pyruvate metabolism involve multiple key catalytic enzymes and regulatory factors. For instance, pyruvate dehydrogenase kinase (PDK) inhibits the activity of the PDC via phosphorylation, thereby directing pyruvate toward lactate production. In mammals, the PDK family comprises four main isoenzymes (PDK1–PDK4), each exhibiting distinct regulatory properties and tissue-specific expression patterns—features that are also relevant to the regulation of macrophage polarization,78 PDK1 deficiency has been shown to attenuate LPS-induced glycolysis, reduce the number of M1 macrophages, and suppress the production of associated pro-inflammatory mediators such as IL-6, IL-12, IL-1β, and iNOS. Concurrently, it promotes the activation of M2 macrophages;79,80 PDK2 suppresses macrophage activation and M1 polarization through the VSIG4–PI3K/Akt/STAT3 signaling pathway;81 PDK4 promotes M1 macrophage polarization through the HIF-1α–PDK4 signaling axis;82 The combined activity of PDK4 and PDK2 synergistically promotes M1 macrophage polarization83 (Table 2).
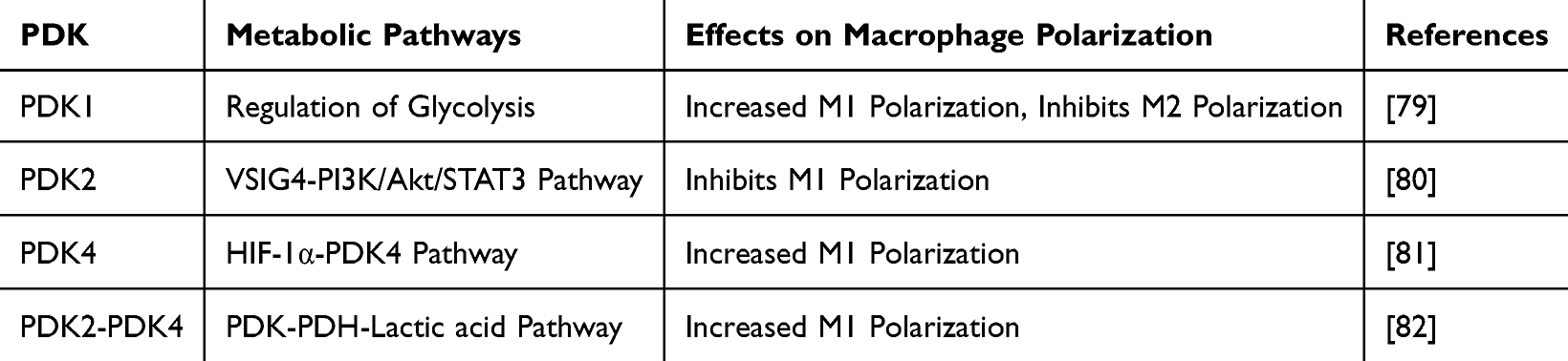 |
Table 2 The Impact of Pyruvate Dehydrogenase Kinase (PDK) on Macrophage Polarization |
In summary, under pro-inflammatory stimulation and upregulation of the Warburg effect, pyruvate—as the end product of glycolysis and a central node in cellular energy metabolism—undergoes metabolic branching toward either mitochondrial oxidation via the TCA cycle or cytosolic conversion into lactate. These divergent pathways not only directly influence cellular energy supply but also modulate macrophage polarization through mechanisms such as acetylation-dependent epigenetic regulation, signaling pathway activation, and microenvironmental acidification. Targeting key regulatory enzymes such as PDK and LDH thus presents a promising therapeutic strategy for modulating macrophage function.
Citrate Metabolism
Citrate is not only a critical intermediate in TCA cycle but also serves as a key signaling molecule involved in the regulation of cellular functions and epigenetic modifications.84 In normal cells, citrate participates in the TCA cycle to support ATP production. However, in macrophages activated by pro-inflammatory stimuli, disruption of the TCA cycle leads to intracellular accumulation of citrate.17,85
The citrate accumulated in the mitochondria must be transported to the cytoplasm through the citrate transporter (SLC25A1, also known as CIC) located on the mitochondrial inner membrane. This transport process is dependent on the concentration gradient.86 Once in the cytoplasm, citrate is cleaved by ATP citrate lyase (ACLY) into acetyl-CoA and oxaloacetate.87 This metabolic pathway is a critical hub for metabolic epigenetics and can regulate the polarization of M1/M2 macrophages.88 Studies have shown that silencing CIC or ACLY significantly reduces the production of ROS, NO, and prostaglandins, suggesting that citrate efflux plays an important role in pro-inflammatory responses.84 During the activation of M1 macrophages, both the mRNA and protein expression levels of CIC and ACLY are significantly increased,89 Both enzymes, as key regulators of macrophage-mediated inflammatory responses, are crucial for the production of inflammatory mediators. They influence immune cell recruitment, the progression of inflammation, and even vascular dilation.90 In summary, regulating the CIC/ACLY axis may offer a potential therapeutic strategy for alleviating pulmonary vascular remodeling by influencing macrophage polarization. However, ACLY plays crucial roles in various tissues, and targeting ACLY expression specifically in macrophages within the lung tissue remains a significant challenge. This represents a major obstacle for the further clinical translation of this strategy.
Acetyl-CoA participates in the reactions of histone acetyltransferases, leading to increased histone acetylation levels. This modification alters chromatin structure, thereby regulating the expression of pro-inflammatory or anti-inflammatory genes.91 This mechanism plays a crucial role in regulating macrophage polarization, particularly the balance between M1 and M2 phenotypes. Under pro-inflammatory conditions, the increase in cytosolic acetyl-CoA levels is often associated with enhanced histone acetylation, which facilitates the activation of a range of pro-inflammatory genes (such as IL-1β, TNF-α, etc), promoting M1 macrophage polarization and its inflammatory response. Conversely, in anti-inflammatory or repair environments, relatively lower acetyl-CoA availability favors the maintenance of M2 macrophage function.92
Similarly, oxaloacetate, produced from citrate breakdown, plays multiple roles. It can be converted into malate through intracellular enzymatic reactions, and under the action of malate dehydrogenase, it generates pyruvate and NADPH. This provides the cell with reducing power, supporting fatty acid synthesis and maintaining redox balance.93 NADPH, as a crucial reducing cofactor, not only participates in biosynthetic reactions but also plays a key role in combating oxidative stress. This is essential for maintaining normal cell function and regulating macrophage phenotypes. Oxaloacetate, by supporting NADPH production, helps regulate ROS levels and intracellular signaling, indirectly influencing macrophage polarization.94
Therefore, in the context of PVR, metabolic reprogramming triggered by local hypoxia and inflammatory stimuli leads to citrate efflux into the cytoplasm, where it is broken down by ACLY to generate acetyl-CoA and oxaloacetate. Together, these metabolites form a dual regulatory system. Acetyl-CoA promotes the expression of pro-inflammatory phenotypes by regulating histone acetylation and lipid metabolism, while oxaloacetate regulates NADPH levels and the redox state, influencing cellular energy balance and signaling. This system jointly determines the direction of macrophage polarization towards M1 or M2 phenotypes. This mechanism plays a crucial role in local inflammation, tissue repair, and the progression of PVR, providing new theoretical insights and therapeutic targets for metabolic interventions in related diseases.
Itaconate and Its Derivatives
Itaconate was first synthesized in 1836, and at that time, it was primarily used for industrial purposes,95 With ongoing research, it has been discovered that itaconate plays a significant role in immune regulation. Subsequently, researchers identified that the enzyme encoded by the Irg1 gene is responsible for the synthesis of itaconate in macrophages. This gene was recognized as encoding the enzyme that catalyzes the decarboxylation of cis-aconitate (an intermediate of the TCA cycle) to produce itaconate.96 Researchers have used techniques such as gene knockout models (Irg1−/−) to analyze the role of itaconate and its derivatives in macrophages, investigating their regulatory mechanisms in inflammatory responses.12,72
Itaconate exerts anti-inflammatory effects through various mechanisms, one of which is by inhibiting SDH to reduce ROS generation in mitochondria. Studies have shown that itaconate shares a similar structure with maleic acid and can competitively inhibit SDH, thereby reducing LPS-induced ROS production. This effect has been validated in mouse bone marrow-derived macrophages (BMDMs).97 However, itaconate’s effectiveness as an SDH inhibitor is relatively weak, as experimental results show that only at high concentrations does itaconate significantly inhibit SDH. This suggests that SDH inhibition is not the sole mechanism underlying itaconate’s anti-inflammatory effects.73 In addition to its inhibition of SDH, studies have shown that due to the structural similarity between itaconate and malate, itaconate can be transported from the mitochondrial inner membrane to the cytoplasm via common carriers.73 This process subsequently activates the NRF2 pathway to regulate the antioxidant response.74 This process subsequently activates the NRF2 pathway to regulate the antioxidant response,98 thereby further inhibiting ROS production and the generation of the pro-inflammatory cytokine IL-1β. -octylitaconate (4-OI), a derivative of itaconate, alkylates the cysteine residues of GAPDH, thereby inhibiting its enzymatic activity and activating anti-inflammatory programs.99 This mechanism further underscores the role of itaconate and its derivatives in regulating macrophage inflammatory responses. Specifically, 4-OI not only reduces ROS levels in macrophages stimulated by LPS but also inhibits IL-1β production through NRF2 activation, thereby highlighting its potential in mitigating inflammatory responses.74 Notably, itaconate also exhibits a negative feedback regulation with the type IFN signaling pathway. IFN-β induces the expression of Irg1 and the production of itaconate, while the itaconate derivative 4-OI can attenuate the expression of IFN-dependent genes by inhibiting the IFN signaling pathway.100 Interestingly, although itaconate accumulates in macrophages after M1 polarization and retains its anti-inflammatory effects, studies have shown that the reduction of IRG1 and itaconate is essential for M2 macrophage polarization. This finding further supports the inhibitory role of itaconate when it is excessively activated during immune responses,101 It also provides a novel approach to alleviating vascular remodeling by modulating M2 macrophage polarization.
Based on the aforementioned studies, itaconate alleviates inflammation by inhibiting SDH, activating NRF2-mediated antioxidant pathways, and negatively regulating IFN signaling, thereby reducing inflammation-associated ROS production and the expression of pro-inflammatory cytokines. Given the close association between the development of pulmonary vascular remodeling (PVR) and inflammation, oxidative stress, and cellular metabolic abnormalities, these mechanisms suggest that itaconate and its derivatives may mitigate endothelial cell damage and SMCs hyperproliferation by modulating the local immune microenvironment in the lungs and balancing M1 and M2 macrophage polarization. This, in turn, may inhibit or reverse pathological vascular remodeling. Although most studies to date have focused on the effects of itaconate in macrophages, the precise mechanisms underlying its role in the interplay between lung inflammation and vascular remodeling remain incompletely understood. Further in vitro and in vivo experiments are needed to explore the therapeutic potential and mechanisms of itaconate in diseases associated with PVR.
Succinate and α-Ketoglutarate
Within the context of PVR, the significance of succinate in the reprogramming of macrophage glucose metabolism is increasingly recognized. Upon macrophage activation, glycolysis is upregulated, leading to enhanced succinate production, which subsequently inhibits succinate dehydrogenase activity and suppresses succinate-mediated inflammation. Traditionally regarded as an intermediate in the TCA cycle and a substrate for the mitochondrial electron transport chain, succinate has now been identified as a signaling molecule. It exerts its effects by binding and activating its specific receptor, SUCNR1 (G protein-coupled receptor 91), thereby participating in processes such as inflammation and immune activation.102,103 Meanwhile, in the TCA cycle, succinate is derived from the conversion of α-ketoglutarate (α-KG), and these two metabolites are closely linked in cellular metabolic pathways. During macrophage activation, the concentrations of succinate and α-KG influence each other, collectively regulating cellular energy metabolism and the inflammatory response.
Intracellular succinate oxidation is closely linked to the increase in mitochondrial membrane potential. In LPS-activated macrophages, the shift towards glycolysis reduces mitochondrial ATP synthesis, leading to a significant increase in mitochondrial membrane potential. This elevated membrane potential, in conjunction with succinate oxidation, promotes the generation of mitochondrial ROS. As signaling molecules, ROS activate HIF-1α, thereby enhancing the expression of pro-inflammatory genes.104 α-ketoglutarate (α-KG) inhibits M1 macrophage activation while promoting M2 macrophage activation. It exerts anti-inflammatory effects by mediating both metabolic and epigenetic reprogramming,71 Studies have shown that measuring the α-KG/succinate ratio reveals that macrophages stimulated with IL-4 exhibit a higher α-KG/succinate ratio compared to those treated with LPS. Manipulating this ratio allows for the customization of macrophage immune responses.105 Additionally, when macrophages are stimulated by pro-inflammatory factors such as LPS, partial reprogramming of the TCA cycle leads to succinate accumulation within the cells. High concentrations of succinate can inhibit PHDs, which depend on α-KG and oxygen for catalysis. As succinate levels rise, PHD activity is competitively inhibited, resulting in the stabilization of HIF-1α. This stabilized HIF-1α translocates to the nucleus, where it forms heterodimers and binds to hypoxia response elements (HREs), thereby initiating the transcription of a series of downstream genes.67,104,106 The stabilization of HIF-1α further promotes the expression of pro-inflammatory cytokines, which can be considered a key function of succinate as a signaling marker.21 SUCNR1 is a G protein-coupled receptor and serves as the specific receptor for extracellular succinate,103 Succinate primarily mediates its signaling through the Di and Gq signaling pathways. Upon binding to SUCNR1, succinate activates downstream signaling cascades, such as the cAMP/PKA and PLC/IP3/Ca2+ pathways, thereby modulating macrophage inflammatory responses.68 SUCNR1 exhibits a markedly paradoxical role in pro- and anti-inflammatory signaling, with its effects being significantly influenced by environmental factors. During early inflammation or hypoxia, the activation of SUCNR1 can induce the production of inflammatory factors (eg, IL-1β and TNF-α), thereby enhancing the macrophage inflammatory response and exacerbating M1 macrophage-mediated vascular damage. This suggests a pro-inflammatory role for SUCNR1 in the early stages of PVR, which drives vascular inflammation.68,107 Conversely, other studies have shown that extracellular succinate, through binding to this receptor, activates the PKA–CREB–KLF4 signaling pathway. This enhances IL-4-induced anti-inflammatory gene expression, such as Arg1, Mrc1, Fizz1, and IL-10, and consequently suppresses the expression of pro-inflammatory factors like IL-1β, IL-6, Tnf, and Nos2.108 This mechanism suggests that under specific conditions, SUCNR1 may promote M2 polarization or mediate the resolution of inflammation. Furthermore, LysM-Cre SUCNR1fl/fl mice (with myeloid-specific SUCNR1 deletion) exhibit a stronger inflammatory response and metabolic dysfunction under both steady-state and high-fat diet conditions. This implies that the absence of SUCNR1 leads to a predominant pro-inflammatory macrophage phenotype,16,108 The high expression of SUCNR1 in M2 macrophages, which are generated from primary human monocyte cultures with a combination of IL-4 and IL-13, in stark contrast to M1 macrophages, strongly suggests a critical role for SUCNR1 in the M2 polarization process.109
In summary, succinate’s role is multifaceted: it promotes an increase in mitochondrial membrane potential, induces ROS generation, and stabilizes HIF-1α. This, in turn, activates a cascade of downstream pro-inflammatory gene expression, thereby aggravating the local inflammatory environment. The changing balance between succinate and α-KG also participates in regulating macrophage phenotypic transformation, offering a new regulatory perspective for maintaining metabolic homeostasis and immune balance. The dual role of SUCNR1 consequently makes its function in PVR more intricate. The specific functional tendencies of SUCNR1 at different stages of PVR (eg, from early inflammation to late fibrosis), and the mechanisms by which it is regulated in a given microenvironment to mediate either pro- or anti-inflammatory responses, remain to be fully elucidated. This complexity makes SUCNR1 a multifaceted regulatory node. A precise understanding of its role in PVR will be instrumental in developing more stage-specific intervention strategies.
Conclusion and Outlook
PVR is a central pathological process in various pulmonary vascular diseases, including pulmonary arterial hypertension. Its essence lies in the dynamic reconstruction of the vascular wall structure, involving endothelial cell dysfunction, SMCs proliferation and phenotypic transformation, as well as abnormal deposition and degradation of the ECM. According to the 2022 pulmonary hypertension diagnostic guidelines, pulmonary hypertension is a global health issue, with a prevalence of 1% in the global population, particularly higher in individuals over 65 years of age.2 However, the treatment of PVR-related diseases is still in its early stages, with no specific therapeutic approaches currently available, Existing medications—such as oral endothelin receptor antagonists (bosentan, ambrisentan, macitentan);110,111 oral type 5 phosphodiesterase inhibitors (sildenafil and avanafil, which exert vasodilatory and antiproliferative effects on pulmonary artery smooth muscle cells);112 and the oral soluble guanylate cyclase stimulator Riociguat (which has direct vasodilatory effects)65 —primarily target endothelial dysfunction and abnormal SMC proliferation.
This review systematically elucidates how intermediate metabolites from glucose metabolic pathways are no longer merely byproducts of energy metabolism but serve as crucial signaling molecules that directly determine macrophage polarization and, in turn, influence PVR progression. Our key findings include: (1) The accumulation of succinate and citrate is a hallmark of M1 polarization. Succinate promotes inflammatory gene expression by stabilizing HIF-1α. Citrate efflux into the cytoplasm is converted into acetyl-CoA by ACLY, which then directly enhances the transcription of pro-inflammatory genes via histone acetylation, aggravating endothelial damage and vascular inflammation. (2) Pyruvate and its downstream product, lactate, serve as key metabolic hubs that regulate M1/M2 conversion and inflammation resolution by modulating PDK family activity and histone lactylation. (3) Itaconate, an endogenous metabolite, possesses potent anti-inflammatory effects. It negatively regulates M1 polarization and inflammatory responses by inhibiting SDH or activating the NRF2 signaling pathway, suggesting its potential value in attenuating PVR.
Despite the significant progress made in understanding these metabolic regulatory mechanisms, translating these findings into precision therapies for PVR faces formidable challenges. Future research should prioritize addressing the following knowledge gaps and translational barriers:
- The Challenge of Tissue- and Cell-Specific Delivery: Many metabolic enzymes (eg, ACLY) are widely expressed across systemic tissues. The primary clinical translational barrier is how to specifically deliver metabolites or metabolic enzyme inhibitors to IMs while avoiding off-target effects on other organs or on Ams within the lung. Therefore, there is an urgent need to develop novel targeted delivery systems (such as nanocarriers or conjugated antibodies) that target ACLY, PDK, or itaconate derivatives to enable precise intervention on macrophages at the PVR lesion site.
- The Mechanisms of Dynamic Pathology and Cellular Crosstalk: Most existing research is based on static models, and a systematic analysis of macrophage polarization and metabolic features throughout the dynamic progression of PVR is lacking. Concurrently, the mechanism by which macrophages paracrinely regulate endothelial cells and PASMCs via metabolites remains unclear. To address this, it is crucial to combine single-cell RNA sequencing and metabolomics techniques to analyze the metabolic profiles of macrophages at different disease stages. Furthermore, developing more physiologically relevant pulmonary vascular organoid or microfluidic models is necessary to simulate and verify the direct paracrine effects of macrophage metabolites on vascular wall cells.
- The Validation and Translation of Clinical Biomarkers: The changes in metabolic intermediates (eg, succinate, itaconate) in the plasma or lung tissue of PVR patients have not been validated on a large scale as biomarkers for disease diagnosis or prognostic assessment. Therefore, in clinical cohorts, it is imperative to perform metabolomic analysis of plasma and exhaled breath condensate (EBC) from PVR patients to validate the biomarker potential of intermediates like succinate and citrate. This will promote precision phenotyping and personalized therapy based on immunometabolic dysregulation.
In conclusion, a deeper understanding of the regulatory mechanisms of glucose metabolic intermediates on macrophage polarization will open new directions for PVR treatment. This will drive a paradigm shift in the field, moving from traditional anti-proliferative interventions to an innovative model of targeted immunometabolic regulation.
Acknowledgment
We gratefully acknowledge the Bioinformatics Core, Medical School, Hunan University of Chinese Medicine for their expert advice and assistance throughout the data analysis, manuscript writing, and submission process.
Funding
Hunan University of Chinese Medicine Liu Liang Research Station Project, Grant/Award Number: 24YS001. Project of Education Department of Hunan Province, Grant/Award Number: 22A0243. Natural Science Foundation of Hunan Province, Grant/Award Numbers: 2024JJ5299, 2025JJ80944. Open Fund for National Key Laboratory Cultivation Base of Chinese Medicinal Powder & Innovative Medicinal Jointly Established by Province and Ministry, Grant/Award Numbers: 23PTKF1005. Open Fund for Hunan Provincial Key Laboratory of Vascular Biology and Translational Medicine, Grant/Award Numbers: 2024XG0033. Ningxia Autonomous Region Key Research and Development Program,Grant/Award Number:2023BEG02033.Hunan University of Chinese Medicine graduate student scientific research innovation project, Grant/Award Number: 2024CX198,LXBZZ2024168,2025CX103.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jia Z, Wang S, Yan H, et al. Pulmonary vascular remodeling in pulmonary hypertension. J Pers Med. 2023;13(2):366. doi:10.3390/jpm13020366
2. Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43(38):3618–3731.
3. Tejwani V, Yun X, Sikka G, et al. Airway epithelial genomic signatures in steroid-resistant COPD; role for SMAD3 in vascular remodeling in pulmonary hypertension; regulation of lung endothelial cell function by VEGFR3. Am J Respir Cell Mol Biol. 2019;61(3):392–394. doi:10.1165/rcmb.2019-0075RO
4. Borek I, Birnhuber A, Voelkel NF, et al. The vascular perspective on acute and chronic lung disease. J Clin Investig. 2023;133(16):e170502. doi:10.1172/JCI170502
5. Qiu H, Zhang Y, Li Z, et al. Donepezil ameliorates pulmonary arterial hypertension by inhibiting M2-macrophage activation. Front Cardiovasc Med. 2021;8:639541. doi:10.3389/fcvm.2021.639541
6. Stenmark KR, Tuder RM, El Kasmi KC. Metabolic reprogramming and inflammation act in concert to control vascular remodeling in hypoxic pulmonary hypertension. J Appl Physiol. 2015;119(10):1164–1172. doi:10.1152/japplphysiol.00283.2015
7. Blagih J, Jones RG. Polarizing macrophages through reprogramming of glucose metabolism. Cell Metab. 2012;15(6):793–795. doi:10.1016/j.cmet.2012.05.008
8. Wang S, Liu G, Li Y, et al. Metabolic reprogramming induces macrophage polarization in the tumor microenvironment. Front Immunol. 2022;13:840029. doi:10.3389/fimmu.2022.840029
9. Huang SCC, Everts B, Ivanova Y, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15(9):846–855. doi:10.1038/ni.2956
10. Huangfu N, Zheng W, Xu Z, et al. RBM4 regulates M1 macrophages polarization through targeting STAT1-mediated glycolysis. Int Immunopharmacol. 2020;83:106432. doi:10.1016/j.intimp.2020.106432
11. Ma H, Lin J, Li L, et al. Formaldehyde reinforces pro-inflammatory responses of macrophages through induction of glycolysis. Chemosphere. 2021;282:131149. doi:10.1016/j.chemosphere.2021.131149
12. Hooftman A, O’Neill LAJ. The immunomodulatory potential of the metabolite itaconate. Trends Immunol. 2019;40(8):687–698. doi:10.1016/j.it.2019.05.007
13. Harber KJ, De Goede KE, Verberk SGS, et al. Succinate is an inflammation-induced immunoregulatory metabolite in macrophages. Metabolites. 2020;10(9):372. doi:10.3390/metabo10090372
14. Huang L, Wang C, Xu H, et al. Targeting citrate as a novel therapeutic strategy in cancer treatment. Biochim Biophys Acta Rev Cancer. 2020;1873(1):188332. doi:10.1016/j.bbcan.2019.188332
15. Shapouri‐Moghaddam A, Mohammadian S, Vazini H, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425–6440. doi:10.1002/jcp.26429
16. Trauelsen M, Hiron TK, Lin D, et al. Extracellular succinate hyperpolarizes M2 macrophages through SUCNR1/GPR91-mediated gq signaling. Cell Rep. 2021;35(11):109246. doi:10.1016/j.celrep.2021.109246
17. O’Neill LAJ. A broken krebs cycle in macrophages. Immunity. 2015;42(3):393–394. doi:10.1016/j.immuni.2015.02.017
18. Cabrera-Fuentes H, Lopez M, McCurdy S, et al. Regulation of monocyte/macrophage polarisation by extracellular RNA. Thromb Haemost. 2015;113(03):473–481. doi:10.1160/TH14-06-0507
19. Faas M, Ipseiz N, Ackermann J, et al. IL-33-induced metabolic reprogramming controls the differentiation of alternatively activated macrophages and the resolution of inflammation. Immunity. 2021;54(11):2531–2546.e5. doi:10.1016/j.immuni.2021.09.010
20. Thwe PM, Pelgrom LR, Cooper R, et al. Cell-intrinsic glycogen metabolism supports early glycolytic reprogramming required for dendritic cell immune responses. Cell Metab. 2017;26(3):558–567.e5. doi:10.1016/j.cmet.2017.08.012
21. Liu L, Lu Y, Martinez J, et al. Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1 α-dependent. Proc Natl Acad Sci. 2016;113(6):1564–1569. doi:10.1073/pnas.1518000113
22. Chen G, Phan V, Luo X, et al. The mechanistic target of rapamycin complex 1 critically regulates the function of mononuclear phagocytes and promotes cardiac remodeling in acute ischemia. J Mol Cell Cardiol. 2021;159:62–79. doi:10.1016/j.yjmcc.2021.06.004
23. Certo M, Tsai CH, Pucino V, et al. Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat Rev Immunol. 2021;21(3):151–161. doi:10.1038/s41577-020-0406-2
24. Wang T, Liu H, Lian G, et al. HIF1 α -induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediat Inflamm. 2017;2017:1–10. doi:10.1155/2017/3102737
25. Dang B, Gao Q, Zhang L, et al. The glycolysis/HIF-1α axis defines the inflammatory role of IL-4-primed macrophages. Cell Rep. 2023;42(5):112471. doi:10.1016/j.celrep.2023.112471
26. Chiba S, Hisamatsu T, Suzuki H, et al. Glycolysis regulates LPS-induced cytokine production in M2 polarized human macrophages. Immunol Lett. 2017;183:17–23. doi:10.1016/j.imlet.2017.01.012
27. Wang F, Zhang S, Vuckovic I, et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab. 2018;28(3):463–475.e4. doi:10.1016/j.cmet.2018.08.012
28. Covarrubias AJ, Aksoylar HI, Yu J, et al. Akt-mTORC1 signaling regulates acly to integrate metabolic input to control of macrophage activation. eLife. 2016;5:e11612. doi:10.7554/eLife.11612
29. Evren E, Ringqvist E, Willinger T. Origin and ontogeny of lung macrophages: from mice to humans. Immunology. 2020;160(2):126–138. doi:10.1111/imm.13154
30. McQuattie-Pimentel AC, Ren Z, Joshi N, et al. The lung microenvironment shapes a dysfunctional response of alveolar macrophages in aging. J Clin Investig. 2021;131(4):e140299. doi:10.1172/JCI140299
31. Liu Z, Gu Y, Chakarov S, et al. Fate mapping via Ms4a3-expression history traces monocyte-derived cells. Cell. 2019;178(6):1509–1525.e19. doi:10.1016/j.cell.2019.08.009
32. Kumar R, Nolan K, Kassa B, et al. Monocytes and interstitial macrophages contribute to hypoxic pulmonary hypertension. J Clin Investig. 2025;135(6):e176865. doi:10.1172/JCI176865
33. Qiu F, Miao H, Hui H, et al. MHCIIhi LYVE1lo CCR2hi interstitial macrophages promote medial fibrosis in pulmonary arterioles and contribute to pulmonary hypertension. Circ Res. 2025;137(1):46–66. doi:10.1161/CIRCRESAHA.125.326173
34. Jeong S, Kim MB, Baek S, et al. Suppression of pro-inflammatory M1 polarization of LPS-stimulated RAW 264.7 macrophage cells by fucoxanthin-rich sargassum hemiphyllum. Mar Drugs. 2023;21(10):533. doi:10.3390/md21100533
35. Zhang Z, Xiong Y, Yao Y. Sinomenine hydrochloride alleviates autoimmune myocarditis via suppressing Th1 cell induced-M1 macrophage pyroptosis. Eur Heart J. 2024;45(Supplement_1):
36. Gao S, Zhou J, Liu N, et al. Curcumin induces M2 macrophage polarization by secretion IL-4 and/or IL-13. J Mol Cell Cardiol. 2015;85:131–139. doi:10.1016/j.yjmcc.2015.04.025
37. Ye Y, Xu Q, Wuren T. Inflammation and immunity in the pathogenesis of hypoxic pulmonary hypertension. Front Immunol. 2023;14:1162556. doi:10.3389/fimmu.2023.1162556
38. Evans CE, Cober ND, Dai Z, et al. Endothelial cells in the pathogenesis of pulmonary arterial hypertension. Eur Respir J. 2021;58(3):2003957. doi:10.1183/13993003.03957-2020
39. Shen YH, Ding D, Lian TY, et al. Panorama of artery endothelial cell dysfunction in pulmonary arterial hypertension. J Mol Cell Cardiol. 2024;197:61–77. doi:10.1016/j.yjmcc.2024.10.004
40. Medrano-Bosch M, Simón-Codina B, Jiménez W, et al. Monocyte-endothelial cell interactions in vascular and tissue remodeling. Front Immunol. 2023;14:1196033. doi:10.3389/fimmu.2023.1196033
41. Tsai CF, Chen GW, Chen YC, et al. Regulatory effects of quercetin on M1/M2 macrophage polarization and oxidative/antioxidative balance. Nutrients. 2021;14(1):67.
42. Turner CT, Zeglinski MR, Richardson KC, et al. Granzyme K expressed by classically activated macrophages contributes to inflammation and impaired remodeling. J Invest Dermatol. 2019;139(4):930–939.
43. Simsekyilmaz S, Cabrera-Fuentes HA, Meiler S, et al. Role of extracellular RNA in atherosclerotic plaque formation in mice. Circulation. 2014;129(5):598–606.
44. Mori M, Sakamoto A, Kawakami R, et al. CD163+ macrophages induce endothelial-to-mesenchymal transition in atheroma. Circ Res. 2024;135(2):e4–23. doi:10.1161/CIRCRESAHA.123.324082
45. Gaikwad AV, Eapen MS, McAlinden KD, et al. Endothelial to mesenchymal transition (EndMT) and vascular remodeling in pulmonary hypertension and idiopathic pulmonary fibrosis. Expert Rev Respir Med. 2020;14(10):1027–1043. doi:10.1080/17476348.2020.1795832
46. Gorelova A, Berman M, Al Ghouleh I. Endothelial-to-mesenchymal transition in pulmonary arterial hypertension. Antioxid Redox Signal. 2021;34(12):891–914. doi:10.1089/ars.2020.8169
47. Fan CS, Chen LL, Hsu TA, et al. Endothelial-mesenchymal transition harnesses HSP90α-secreting M2-macrophages to exacerbate pancreatic ductal adenocarcinoma. J Hematol Oncol. 2019;12(1):138. doi:10.1186/s13045-019-0826-2
48. Dai X, Liu Y, Wu Y, et al. DYZY01 alleviates pulmonary hypertension via inhibiting endothelial cell pyroptosis and rescuing endothelial dysfunction. Eur J Pharmacol. 2024;978:176785. doi:10.1016/j.ejphar.2024.176785
49. Hey J, Paulsen M, Toth R, et al. Epigenetic reprogramming of airway macrophages promotes polarization and inflammation in muco-obstructive lung disease. Nat Commun. 2021;12(1):6520. doi:10.1038/s41467-021-26777-9
50. Chang Y, Li JY, Jayakumar T, et al. Ketamine, a clinically used anesthetic, inhibits vascular smooth muscle cell proliferation via PP2A-activated PI3K/akt/ERK inhibition. Int J Mol Sci. 2017;18(12):2545. doi:10.3390/ijms18122545
51. Pantan R, Tocharus J, Phatsara M, et al. Synergistic effect of atorvastatin and cyanidin-3-glucoside against angiotensin II-mediated vascular smooth muscle cell proliferation and migration through MAPK and PI3K/akt pathways. Arch Pharmacal Res. 2016:1–2. doi:10.1007/s12272-016-0836-3
52. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84(3):767–801. doi:10.1152/physrev.00041.2003
53. Yan Q, Liu S, Sun Y, et al. CC chemokines modulate immune responses in pulmonary hypertension. J Adv Res. 2024;63:171–186. doi:10.1016/j.jare.2023.10.015
54. Abid S, Marcos E, Parpaleix A, et al. CCR2/CCR5-mediated macrophage–smooth muscle cell crosstalk in pulmonary hypertension. Eur Respir J. 2019;54(4):1802308. doi:10.1183/13993003.02308-2018
55. Amsellem V, Lipskaia L, Abid S, et al. CCR5 as a treatment target in pulmonary arterial hypertension. Circulation. 2014;130(11):880–891. doi:10.1161/CIRCULATIONAHA.114.010757
56. Thakker-Varia S, Tozzi CA, Poiani GJ, et al. Expression of matrix-degrading enzymes in pulmonary vascular remodeling in the rat. Am J Physiol Lung Cell Mol Physiol. 1998;275(2):L398–L406. doi:10.1152/ajplung.1998.275.2.L398
57. Ambade AS, Hassoun PM, Damico RL. Basement membrane extracellular matrix proteins in pulmonary vascular and right ventricular remodeling in pulmonary hypertension. Am J Respir Cell Mol Biol. 2021;65(3):245–258. doi:10.1165/rcmb.2021-0091TR
58. Sutherland TE, Dyer DP, Allen JE. The extracellular matrix and the immune system: a mutually dependent relationship. Science. 2023;379(6633):eabp8964. doi:10.1126/science.abp8964
59. Reel B, Sala‐Newby GB, Huang W, et al. Diverse patterns of cyclooxygenase‐independent metalloproteinase gene regulation in human monocytes. Br J Pharmacol. 2011;163(8):1679–1690. doi:10.1111/j.1476-5381.2011.01298.x
60. Kucherenko MM, Sang P, Yao J, et al. Elastin stabilization prevents impaired biomechanics in human pulmonary arteries and pulmonary hypertension in rats with left heart disease. Nat Commun. 2023;14(1):4416. doi:10.1038/s41467-023-39934-z
61. Li X, Hou R, Ding H, et al. Mollugin ameliorates murine allergic airway inflammation by inhibiting TH2 response and M2 macrophage activation. Eur J Pharmacol. 2023;946:175630. doi:10.1016/j.ejphar.2023.175630
62. Madsen DH, Bugge TH. Imaging collagen degradation in vivo highlights a key role for M2-polarized macrophages in extracellular matrix degradation. OncoImmunology. 2013;2(12):e27127. doi:10.4161/onci.27127
63. Butoi ED, Gan AM, Manduteanu I, et al. Cross talk between smooth muscle cells and monocytes/activated monocytes via CX3CL1/CX3CR1 axis augments expression of pro-atherogenic molecules. Biochim Biophys Acta Mol Cell Res. 2011;1813(12):2026–2035. doi:10.1016/j.bbamcr.2011.08.009
64. Zhang D, Tang Z, Huang H, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–580. doi:10.1038/s41586-019-1678-1
65. Ghofrani HA, Galiè N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369(4):330–340. doi:10.1056/NEJMoa1209655
66. Infantino V, Convertini P, Cucci L, et al. The mitochondrial citrate carrier: a new player in inflammation. Biochem J. 2011;438(3):433–436. doi:10.1042/BJ20111275
67. Stothers CL, Luan L, Fensterheim BA, et al. Hypoxia-inducible factor-1α regulation of myeloid cells. J Mol Med. 2018;96(12):1293–1306.
68. Ryan DG, O’Neill LAJ. Krebs cycle reborn in macrophage immunometabolism. Ann Rev Immunol. 2020;38(1):289–313.
69. O’Neill LAJ, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016;213(1):15–23. doi:10.1084/jem.20151570
70. Zhang FL, Hu Z, Wang YF, et al. Organoids transplantation attenuates intestinal ischemia/reperfusion injury in mice through L-malic acid-mediated M2 macrophage polarization. Nat Commun. 2023;14(1):6779.
71. Liu S, Yang J, Wu Z. The regulatory role of α-ketoglutarate metabolism in macrophages. Mediat Inflamm. 2021;2021:1–7. doi:10.1155/2021/5577577
72. Yang W, Wang Y, Tao K, et al. Metabolite itaconate in host immunoregulation and defense. Cell Mol Biol Lett. 2023;28(1):100.
73. Beloborodova N, Pautova A, Sergeev A, et al. Serum levels of mitochondrial and microbial metabolites reflect mitochondrial dysfunction in different stages of sepsis. Metabolites. 2019;9(10):196. doi:10.3390/metabo9100196
74. Mills EL, Ryan DG, Prag HA, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018;556(7699):113–117. doi:10.1038/nature25986
75. Chen Y, Shi R, Xiang Y, et al. Malate initiates a proton-sensing pathway essential for pH regulation of inflammation. Signal Transduct Target Ther. 2024;9(1):367.
76. Wellen KE, Hatzivassiliou G, Sachdeva UM, et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324(5930):1076–1080. doi:10.1126/science.1164097
77. Shi W, Cassmann TJ, Bhagwate AV, et al. Lactic acid induces transcriptional repression of macrophage inflammatory response via histone acetylation. Cell Rep. 2024;43(2):113746. doi:10.1016/j.celrep.2024.113746
78. Li C, Liu C, Zhang J, et al. Pyruvate dehydrogenase kinase regulates macrophage polarization in metabolic and inflammatory diseases. Front Immunol. 2023;14:1296687. doi:10.3389/fimmu.2023.1296687
79. Semba H, Takeda N, Isagawa T, et al. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun. 2016;7(1):11635. doi:10.1038/ncomms11635
80. Tan Z, Xie N, Cui H, et al. Pyruvate dehydrogenase kinase 1 participates in macrophage polarization via regulating glucose metabolism. J Immunol. 2015;194(12):6082–6089. doi:10.4049/jimmunol.1402469
81. Li J, Diao B, Guo S, et al. VSIG4 inhibits proinflammatory macrophage activation by reprogramming mitochondrial pyruvate metabolism. Nat Commun. 2017;8(1):1322. doi:10.1038/s41467-017-01327-4
82. Han X, Ma W, Zhu Y, et al. Advanced glycation end products enhance macrophage polarization to the M1 phenotype via the HIF-1α/PDK4 pathway. Mol Cell Endocrinol. 2020;514:110878. doi:10.1016/j.mce.2020.110878
83. Jha MK, Song GJ, Lee MG, et al. Metabolic connection of inflammatory pain: pivotal role of a pyruvate dehydrogenase kinase-pyruvate dehydrogenase-lactic acid axis. J Neurosci. 2015;35(42):14353–14369. doi:10.1523/JNEUROSCI.1910-15.2015
84. Iacobazzi V, Infantino V. Citrate – new functions for an old metabolite. Biol Chem. 2014;395(4):387–399. doi:10.1515/hsz-2013-0271
85. Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25(7):771–784. doi:10.1038/cr.2015.68
86. Zhao Y, Shen Y, Wen Y, et al. High-performance intensiometric direct- and inverse-response genetically encoded biosensors for citrate. ACS Cent Sci. 2020;6(8):1441–1450. doi:10.1021/acscentsci.0c00518
87. Baardman J, Verberk SGS, Van Der Velden S, et al. Macrophage ATP citrate lyase deficiency stabilizes atherosclerotic plaques. Nat Commun. 2020;11(1):6296. doi:10.1038/s41467-020-20141-z
88. Lauterbach MA, Hanke JE, Serefidou M, et al. Toll-like receptor signaling rewires macrophage metabolism and promotes histone acetylation via ATP-citrate lyase. Immunity. 2019;51(6):997–1011.e7. doi:10.1016/j.immuni.2019.11.009
89. Infantino V, Iacobazzi V, Palmieri F, et al. ATP-citrate lyase is essential for macrophage inflammatory response. Biochem Biophys Res Commun. 2013;440(1):105–111. doi:10.1016/j.bbrc.2013.09.037
90. Biju AK, Chennam lakshmikumar RR, Rengasamy KR. ATP-citrate lyase (ACLY): an extensive investigation from molecular insight to therapeutic implications. Nat Resour Hum Health. 2024;4(3):208–229. doi:10.53365/nrfhh/189500
91. Noe JT, Rendon BE, Geller AE, et al. Lactate supports a metabolic-epigenetic link in macrophage polarization. Sci Adv. 2021;7(46):eabi8602. doi:10.1126/sciadv.abi8602
92. Williams NC, O’Neill LAJ. A role for the krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation. Front Immunol. 2018;9:141. doi:10.3389/fimmu.2018.00141
93. Palmieri EM, Spera I, Menga A, et al. Acetylation of human mitochondrial citrate carrier modulates mitochondrial citrate/malate exchange activity to sustain NADPH production during macrophage activation. Biochim Biophys Acta Bioenergy. 2015;1847(8):729–738. doi:10.1016/j.bbabio.2015.04.009
94. Jha AK, Huang SCC, Sergushichev A, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42(3):419–430. doi:10.1016/j.immuni.2015.02.005
95. Baup S. Ueber eine neue pyrogen‐citronensäure, und über benennung der pyrogen‐säuren überhaupt. Ann Pharm. 1836;19(1):29–38. doi:10.1002/jlac.18360190107
96. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11(1):102. doi:10.1038/s41467-019-13668-3
97. Lampropoulou V, Sergushichev A, Bambouskova M, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016;24(1):158–166. doi:10.1016/j.cmet.2016.06.004
98. Bambouskova M, Gorvel L, Lampropoulou V, et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ–ATF3 inflammatory axis. Nature. 2018;556(7702):501–504. doi:10.1038/s41586-018-0052-z
99. Liao ST, Han C, Xu DQ, et al. 4-octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to exert anti-inflammatory effects. Nat Commun. 2019;10(1):5091. doi:10.1038/s41467-019-13078-5
100. Ryan TAJ, Hooftman A, Rehill AM, et al. Dimethyl fumarate and 4-octyl itaconate are anticoagulants that suppress tissue factor in macrophages via inhibition of type I interferon. Nat Commun. 2023;14(1):3513. doi:10.1038/s41467-023-39174-1
101. Ganta VC, Choi MH, Kutateladze A, et al. A MicroRNA93–interferon regulatory factor-9–immunoresponsive gene-1–itaconic acid pathway modulates M2-like macrophage polarization to revascularize ischemic muscle. Circulation. 2017;135(24):2403–2425. doi:10.1161/CIRCULATIONAHA.116.025490
102. Mills EL, Kelly B, Logan A, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167(2):457–470.e13. doi:10.1016/j.cell.2016.08.064
103. He W, Miao FJP, Lin DCH, et al. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature. 2004;429(6988):188–193. doi:10.1038/nature02488
104. Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi:10.1038/nature11986
105. Liu PS, Wang H, Li X, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18(9):985–994.
106. Mills E, O’Neill LAJ. Succinate: a metabolic signal in inflammation. Trends Cell Biol. 2014;24(5):313–320. doi:10.1016/j.tcb.2013.11.008
107. Qiu M, Geng H, Zou C, et al. Intestinal inflammation exacerbates endometritis through succinate production by gut microbiota and SUCNR1-mediated proinflammatory response. Int Immunopharmacol. 2025;146:113919.
108. Keiran N, Ceperuelo-Mallafré V, Calvo E, et al. SUCNR1 controls an anti-inflammatory program in macrophages to regulate the metabolic response to obesity. Nat Immunol. 2019;20(5):581–592. doi:10.1038/s41590-019-0372-7
109. Trauelsen M, Rexen Ulven E, Hjorth SA, et al. Receptor structure-based discovery of non-metabolite agonists for the succinate receptor GPR91. Mol Metab. 2017;6(12):1585–1596. doi:10.1016/j.molmet.2017.09.005
110. Galié N, Badesch D, Oudiz R, et al. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2005;46(3):529–535. doi:10.1016/j.jacc.2005.04.050
111. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809–818. doi:10.1056/NEJMoa1213917
112. Grünig E, Jansa P, Fan F, et al. Randomized trial of macitentan/tadalafil single-tablet combination therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2024;83(4):473–484. doi:10.1016/j.jacc.2023.10.045
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.