Back to Journals » Journal of Inflammation Research » Volume 16
Metabolic Reprogramming and Its Regulatory Mechanism in Sepsis-Mediated Inflammation
Authors Liu W ![]() , Liu T
, Liu T ![]() , Zheng Y, Xia Z
, Zheng Y, Xia Z
Received 6 January 2023
Accepted for publication 8 March 2023
Published 20 March 2023 Volume 2023:16 Pages 1195—1207
DOI https://doi.org/10.2147/JIR.S403778
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Wenzhang Liu,1,* Tianyi Liu,1,* Yongjun Zheng,1 Zhaofan Xia1,2
1Department of Burn Surgery, Changhai Hospital, Naval Medical University, Shanghai, 200433, People’s Republic of China; 2Research Unit of Key Techniques for Treatment of burns and Combined Burns and Trauma Injury, Chinese Academy of Medical Sciences, Shanghai, 200433, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yongjun Zheng; Zhaofan Xia, Email [email protected]; [email protected]
Abstract: Sepsis is a systemic inflammatory disease caused by an infection that can lead to multiple organ failure. Sepsis alters energy metabolism, leading to metabolic reprogramming of immune cells, which consequently disrupts innate and adaptive immune responses, triggering hyperinflammation and immunosuppression. This review summarizes metabolic reprogramming and its regulatory mechanism in sepsis-induced hyperinflammation and immunosuppression, highlights the significance and intricacies of immune cell metabolic reprogramming, and emphasizes the pivotal role of mitochondria in metabolic regulation and treatment of sepsis. This review provides an up-to-date overview of the relevant literature to inform future research directions in understanding the regulation of sepsis immunometabolism. Metabolic reprogramming has great promise as a new target for sepsis treatment in the future.
Keywords: immune cell, hyperinflammation, immunosuppression, immunometabolism, mitochondria
Introduction
Sepsis is a systemic inflammatory disease caused by pathogens, such as bacteria, viruses, or fungi, which can lead to multiple organ dysfunction and even death in severe cases. Sepsis is the leading cause of patient death in intensive care units.1 In 2017, an estimated incidence of 48.9 million cases of sepsis had been recorded worldwide, and 11.0 million sepsis-related deaths were reported, accounting for 19.7% of all global deaths.2
The metabolism of immune and non-immune cells undergoes substantial changes during sepsis, ultimately leading to organ dysfunction that involves the heart, lungs, kidneys, liver, and brain. The pathophysiology of organ dysfunction is characterized by disturbances in the host response to infection, including hyperinflammation and immunosuppression, which is a pathological alteration also known as immune paralysis.3 During sepsis, an alteration in the pathway of cellular energy metabolism occurs. To meet the increased energy demands that accompany sepsis, the cells promote cell proliferation and growth by changing their metabolic pattern, which helps the cells resist external pressure as well as confers novel functions to the cells. During the hyperinflammatory stage of sepsis, energy is preferentially generated through glycolysis instead of through the more efficient process of oxidative phosphorylation, OXPHOS) despite an adequate supply of oxygen.4,5 This phenomenon is termed “metabolic reprogramming”, which is similar to the Warburg effect discovered in tumor cells, also known as aerobic glycolysis.6 Glycolysis may be beneficial during the initial stage of inflammation by increasing the production of metabolic intermediates to meet cell biosynthesis and energy requirements, thus facilitating processes such as cell growth, proliferation, and differentiation.7 However, cellular metabolism fails to restore oxidative phosphorylation and metabolic homeostasis owing to mitochondrial dysfunction that occurs in later stages, leading to organ dysfunction.8
This review aims to summarize the changes in the metabolic pathways of immune cells during sepsis with hyperinflammation and immune paralysis, with an emphasis on the significance and intricacies of metabolic reprogramming during sepsis and the dominant role of mitochondria in the regulation of sepsis metabolism. We believe this summary would inform future studies with regard to the current body of knowledge in the sepsis immunometabolism.
Metabolic Reprogramming During Sepsis
Metabolic Reprogramming Associated with Hyperinflammation
Pathogen recognition receptors (PRRs) are a class of germ line-encoded receptors that recognize pathogen-associated molecular patterns (PAMPs). The activation of PRRs is vital for the activation of innate immunity, which plays a dominant role in the first-line immune defense until the more specific adaptive immunity is mounted. In addition, PRRs can interact with damage-associated molecular patterns (DAMPs) released from damaged or dying cells. The most studied PRRs are Toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), and DNA sensors.9
During the immune activation stage of sepsis, the metabolic pathway of immune cells changes from oxidative phosphorylation to glycolysis.11,12 After infection, PRRs activate immune cells by recognizing PAMPs or DAMPs before mounting an inflammatory response. However, abnormal activation of innate immune cells can lead to hyperinflammation, which is characterized by elevated secretion of inflammatory mediators.10,11 Hyperinflammation during sepsis leads to energy deficiency, which further promotes changes in cellular metabolism. This metabolic reprogramming ensures a rapid ATP supply; however, the resulting metabolic intermediates can also serve as nutrients for immune cell proliferation and biosynthesis. For example, reduced nicotinamide adenine dinucleotide phosphate (NADPH) produced during glycolysis is an essential cofactor for various metabolic processes such as lipid synthesis.12
Metabolic intermediates generated by glycolytic enzymes supply oxidative and non-oxidative branches of the pentose phosphate pathway (PPP). The non-oxidative PPP branch converts glycolytic intermediates into ribose-5-phosphate, which is required to synthesize nucleic acids and sugar-phosphate precursors necessary for amino acid synthesis.13 Dihydroxyacetone phosphate and glyceraldehyde 3-phosphate, produced by glycolysis, are the primary substrates for lipid synthesis and amino acid synthesis, respectively.14 In addition, citrate is transformed into acetyl-CoA via the Krebs cycle, which can be used to synthesize cholesterol and fatty acids.15
In neutrophil granulocytes, enhanced glycolysis promotes the establishment of neutrophil extracellular traps (NETs),16 cell-trapping nets composed of depolymerized chromatin, and intracellular granular proteins that capture and kill pathogens.17 Neutrophils die during the establishment of NETs; this process is termed NETosis. NETosis is a process of cell death distinct from apoptosis and cell necrosis. Neutrophil glycolysis contributes to the establishment of NETs. However, owing to limited bacterial clearance, increased neutrophil glycolysis may suppress their transport to infection sites, thus retaining inflammation during sepsis.18 The metabolic shift to the PPP in neutrophils is vital for releasing NADPH oxidase (NOX)-dependent NETs. NADPH produced through the PPP fuels massive NOX-induced reactive oxygen species (ROS) generation and induces the formation of NOX-dependent NETs.16
Macrophages are critical components of the innate immune system. Immunosuppressive (M2) and proinflammatory (M1) macrophages represent two distinct polarization phenotypes. M1 macrophages are induced by lipopolysaccharide (LPS), TLR ligands, or interferon-gamma (IFN-γ). Lipid biosynthesis is essential for membrane remodeling and the synthesis of inflammatory mediators in M1 macrophages.15 Lipogenesis is regulated at the transcriptional level by the sterol regulatory element-binding proteins (SREBPs). Srebp1-a is abundantly expressed in macrophages and positively regulates their inflammatory response. Im et al19 show that mice with a targeted deficiency in SREBP-1a are resistant to endotoxic shock and sepsis. Fatty acid synthase (FAS) is a key enzyme for fatty acid biosynthesis in M1 macrophages. Wei et al20 showed that FAS deletion in macrophages prevented adipose macrophage recruitment and inflammation in mice. Moon et al21 suggest that uncoupling protein 2 (UCP2) regulates NLRP3-mediated caspase-1 activation through lipid synthesis mediated by FAS. Their results identify UCP2 as a potential therapeutic target in inflammatory diseases such as sepsis. However, Bidault et al22 showed that lipid accumulation in pro-inflammatory macrophages is likely to come from uptake or reduced FAO rather than increased fatty acid synthesis.
Numerous studies have revealed the possible mechanisms of metabolic reprogramming associated with hyperinflammation. For instance, four main mechanisms of metabolic reprogramming occur in macrophages or dendritic cells (DC) following LPS-induced sepsis (Figure 1): (1) LPS upregulates the expression of inducible nitric oxide synthase (iNOS) and increases the production of nitric oxide (NO), which inhibits mitochondrial respiration through nitrosylation of cytochrome C oxidase and complex I in the electron transport chain23,24 and increases glycolytic metabolism; (2) LPS activates mammalian target of rapamycin (mTOR) and increases the expression of hypoxia-inducible factor-1α (HIF-1α). HIF-1α promotes glycolytic metabolism through the expression of glucose transporter 1 (GLUT1) and lactate dehydrogenase (LDH);25 (3) LPS increases the expression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), which increases the level of the glycolytic intermediate, fructose 2,6-biphosphate (F-2,6-BP) that allosterically activates 6-phosphofructokinase-1 and increases glycolytic metabolism; (4) LPS inhibits the AMP-dependent protein kinase, adenosine 5’-monophosphate-activated protein kinase (AMPK), resulting in decreased β-oxidation and the mitochondrial biosynthesis of fatty acids.4
 |
Figure 1 Mechanisms of metabolic reprogramming occur in macrophages or DC following LPS-induced sepsis. Red arrows, up regulation; green arrows, down regulation. |
Metabolic Reprogramming Associated with Immunosuppression
Anti-inflammation alleviates inflammation and initiates tissue reconstruction, whereas excessive suppression of inflammation may result in immune paralysis. Under this circumstance, immune cells cannot respond appropriately to inflammatory stimulation, thus rendering the host susceptible to “secondary infection”. Some of the patients who survived the “inflammatory storm” died of uncontrollable infection as a result of immunosuppression, which is usually distinguished by the decreased expression of genes encoding pro-inflammatory factors (such as TNF-α, IL-6, and CCL2), the recruitment of T cells, and the increased expression of anti-inflammatory factors (such as IL-4, IL-10, IL-35).26,27
Changes in cellular metabolic pathways are the basis of immunosuppression, mainly from glycolysis to fatty acid oxidation (FAO) (Figure 2). During immunosuppression, HIF-1α pathway is inhibited, promoting the transformation of metabolic pathway, and enhancing anti-inflammatory response. The process is regulated by Toll-like receptors (TLR), transcription factors, metabolites et al.28 In addition, the expression of the fatty acid transporter CD36 and carnitine palmitoyl transferase 1 (CPT1) increases, allowing more fatty acids to enter the mitochondria for β-oxidation. Sirtuins are a family of highly conserved NAD(+)-dependent deacetylases that serve as sensors to detect energy availability, regulate metabolic procedures, and guide the occurrence and development of sepsis. SIRT1 and SIRT6 support the metabolic transition from glycolysis to FAO by regulating metabolic reprogramming through the SIRT1–RELB–SIRT6 axis, SIR6 reduced HIF-1α-dependent glycolysis and SIRT1 enhanced PGC-1-dependent fatty acid flux and oxidation. This transition from glucose metabolism to fatty acid metabolism constitutes metabolic reprogramming associated with the early and late stages of sepsis.29
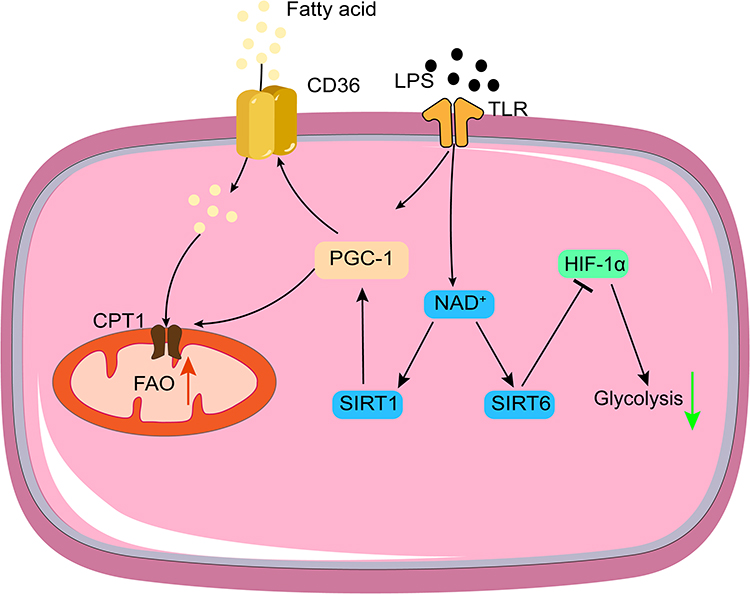 |
Figure 2 Mechanisms of metabolic reprogramming during immunosuppression. Red arrows, up regulation; green arrows, down regulation. |
Glycolysis of immune cells in the early phase of sepsis is an integral part of the host defense response initiation. However, high glycolytic levels can also lead to immunosuppression. Therefore, in addition to promoting the anti-inflammatory phenotype of immune cells by FAO, lactic acid produced during hyperinflammation also plays a strong immunosuppressive role. In healthy volunteers, artificially increased blood lactate levels induced by breathing exercises are involved in the protection against subsequent LPS-induced inflammatory responses.30 Specifically, lactate inhibits the TLR/NF-κB pathway and pro-inflammatory cytokine production by stimulating macrophage polarization.31 Zhang et al32 reported that elevated intracellular lactate levels suppress RLR-mediated IFN activation in M1 macrophages. Efferocytosis-induced lactate acts in a paracrine manner and contributes to an anti-inflammatory environment by increasing the expression of the anti-inflammatory genes, including TGF-β and IL-10, as well as M2-like genes, such as Vegfa, Mgl1, and Cd206.33 Liu et al34 reported that increased intracellular lactate concentration in macrophages promotes an M2-like phenotype via HIF-2α by inducing the expression of its target genes, including Vegf and the M2-like homeostatic genes Mrc1, Arg1, and Retnla. In addition, the lactylation of histones acts as a mechanism to trigger the expression of homeostatic genes, which have traditionally been associated with M2-type macrophages.35 Lactate suppresses the activation of TLR-induced monocyte macrophages and the secretion of the pro-inflammatory cytokines TNF-a and IL-23, as well as the chemokines CCL2 and CCL7 in vitro.35 High lactate concentrations can also prevent monocytes from differentiating into DC and reduce the accumulation of DC.36 The PD-1/PD-L1 pathway activated by lactate can also cause immunosuppression in septic acute kidney injury by inducing apoptosis in lymphocytes.37
The intricacy of the metabolic relationship between hyperinflammation and immunosuppression impedes the development of effective therapies for sepsis.38 Self-regulation is a highly delicate process, and timely regulation of immune cell metabolism is crucial for maintaining immune homeostasis. Once immune cells are widely activated, timely regulation of the level of glycolysis to limit hyperinflammation, avoid immunosuppression caused by excessive lactate levels, and reverse the immunosuppressive state are aspects that should be the focus of future research.39
Metabolic Regulation During Sepsis
Metabolic Regulation in Hyperinflammation
Hyperinflammation occurs during the early stage of sepsis, damaging host organs, tissues, and cells. This process is closely related to metabolic reprogramming. Glycolysis ensures sufficient energy and nutrient supply to activate the immune system and achieve a more effective and rapid immune response. Therefore, limiting the glycolysis pathway can reduce organ damage caused by an “inflammation storm” in the early stage of sepsis.
As one of the critical factors regulating immune cell metabolism during sepsis, HIF-1α can promote the expression of GLUT140 and LDH, increase the glycolytic metabolism of inflammatory cells, promote the secretion of pro-inflammatory cytokines in macrophages, and generate “inflammation storm” in the early period of sepsis, making HIF-1α a new target for sepsis therapy. The inhibition of glycolysis contributed to neutrophil immunosuppression during sepsis and might be controlled by PI3K/Akt-HIF-1α pathway-mediated decrease in LDHA expression, which could provide a scientific theoretical basis for the management and treatment of patients with sepsis.41 The suppressor of cytokine signaling 1 (SOCS1) is a negative regulator of metabolic reprogramming in sepsis.42 It can negatively regulate the expression of hexokinase 1 (HK1) induced by HIF-1α in macrophages. HK1 increases macrophage-mediated inflammatory cytokine production. Moreover, HK1 mitochondrial dissociation can shift glucose metabolism by inhibiting GAPDH and increasing PPP.43 Pyruvate kinase M2 (PKM2) can also interact with HIF-1α and activate the HIF-1α-dependent transcription of enzymes necessary for aerobic glycolysis in macrophages.44 Deoxycholagonin weakens the interaction between PKM2 and HIF-1α by regulating the PKM2/HIF-1α axis, thus affecting the expression of glycolytic genes downstream of HIF-1α.45 Lidocaine can significantly inhibit LPS-induced secretion of inflammatory cytokines by inhibiting the HIF-1α-mediated glycolysis pathway, resulting in anti-inflammatory effects.46 The microRNA miR-223 can induce M2 polarization of macrophages by downregulating the HIF-1α interference glycolysis pathway, resulting in an anti-inflammatory state.47
In addition, inducing macrophages to undergo anti-inflammatory M2 polarization by interfering with glycolysis is an effective method in the treatment of sepsis.47 Methylsulfonylmethane promotes the expression of Arg1 via the lactate–H3K18la pathway to control the M2 polarization of macrophages, thereby protecting against lethal doses of methicillin-resistant Staphylococcus aureus infection.48 Adipose-derived mesenchymal stromal cells regulate lipid metabolism and lipid droplet biogenesis through the AKT/mTOR peroxisome proliferator-activated receptor γ (PPAR-γ) signaling in macrophages.49 Sema7A induces metabolic reprogramming of macrophages through the activation of mTOR and AKT2 signaling pathways, which is characterized by decreased FAO and oxidative phosphorylation, increased glycolysis and PPP, disrupted Krebs cycle, and enhanced M2-type polarization in macrophages.50 Exosomes originating from human bone marrow mesenchymal stem cells have also been reported to be capable of effectively downregulating sepsis-induced glycolysis in macrophages and promoting M2-type polarization.51 Fatty acid metabolism is found to be up-regulated in the induction of macrophage M2 polarization. Prostaglandin E2 (PGE2), metabolic product of arachidonic acid, can enhance mitochondrial oxidative phosphorylation through suppressing PPAR-γ, resulting in the M2 polarization of macrophages.52
Metabolic Regulation in Immunosuppression
The excessive anti-inflammatory response can trigger immune paralysis or compensatory anti-inflammatory response syndrome (CARS), increasing the susceptibility of patients to infection and aggravating the development of sepsis.53 Lymphocyte apoptosis is one of the causes of immunosuppression during sepsis and is associated with poor prognosis. EGFR regulates GLUT1 transport to the cell surface through the TBK1/Exo84/RalA protein system, which is a crucial process by which the Warburg effect and the subsequent cellular activation and apoptosis of CD4 T lymphocytes are induced, and this regulation by EGFR may eventually affect immune function and induce immune cell exhaustion in sepsis.54 PPAR-γ inhibits the pro-inflammatory response of immune cells such as macrophages by promoting the β-oxidative pathway of fatty acids.38 PPAR-γ induces T-cell apoptosis by inhibiting the PI3K/Akt signaling pathway in human and mouse sepsis.55 In the recovery stage of inflammation, SIRT4 antagonizes the effect of SIRT1–SIRT3 on mitochondrial function, reverses the activities of pyruvate dehydrogenase, glutamate dehydrogenase, and malonyl-CoA decarboxylase, restores the flexibility of mitochondria to select metabolic substrates, and promotes the rebalance of cell metabolism and mitochondrial energy metabolism. Mitochondrial SIRT4 reverses fatty acid metabolism to glucose metabolism by controlling the expression and function of the nuclear SIRT1 and the mitochondrial pyruvate dehydrogenase complex (PDHC)/pyruvate dehydrogenase kinase isoenzyme 1 (PDK1) axis.56 SIRT1 plays a dominant role in immune metabolism during sepsis. Inhibition of SIRT1 activity during immunosuppression can restore the homeostasis of immune metabolism and significantly increase the survival rate of patients during sepsis. Collectively, sirtuins mutually interact to regulate the outcome of sepsis. Precise metabolic targeting therapy can improve the survival rate of patients with sepsis.
Metabolic reprogramming is vital in inducing and maintaining immune tolerance during sepsis. In LPS-activated macrophages, mitochondrial glycerol 3-phosphate dehydrogenase (GPD2) regulates glucose oxidation to drive inflammatory responses. Drawn-out exposure to LPS triggers LPS tolerance, in which macrophages induce immunosuppression to limit the detrimental effects of sustained inflammation.57 In an in vitro model that imitated sepsis-induced immunosuppression, metabolic defects in human monocytes were partially restored by IFN-γ by enhancing glycolysis.58 Xu et al59 identified NLRC3 as a driver of monocyte and macrophage tolerance. NLRC3 deficiency activates cellular glycolytic pathways and cytokines by regulating the p300–NF-κB–NFAT5 pathway and improves lung host defense responses by regulating macrophage function in a sepsis-induced immunosuppression model in response to secondary infection. Additionally, NLRC3 deficiency is associated with improved survival rates in patients with sepsis.
As mentioned earlier, the lactate produced by glycolysis plays a potent immunosuppressive role. Lactate has been suggested to induce M2 polarization of macrophages through the mTORC1/ATP6V0D2 and HIF-1α pathway.60,61 The immunosuppressive effects of myeloid-derived suppressor cells (MDSCs) in sepsis have also been well documented. When lactate was added to bone marrow cells cultured in vitro, the proportion and number of MDSCs increased significantly.62 In addition, lactate can activate the G protein-coupled receptor GPR81 (also known as HCA1). Lactate negatively regulates TLR induction of the NLRP3 inflammasome and production of IL-1β via ARRB2 and GPR81.63 Yang et al demonstrated a new mechanism by which lactate plays a previously unknown role in inhibiting pro-inflammatory cytokine production via GPR81-mediated Yes-associated protein (YAP) inactivation, leading to the interruption of YAP and NF-κB interactions and nuclear translocation in macrophages.64 Schenz et al65 suggested that extracellular lactate acts as a metabolic checkpoint independent of the already-known GPR81 signaling-dependent mechanism and shapes monocyte function via distinct acute and long-term effects. Temporary elevation of systemic lactate may be considered a mechanism to withstand excessive inflammation. When lactate is trafficked out of the cell through the monocarboxylic acid transporter MCT4, it can regulate immune cell migration and cytokine production, producing a T-cell “stop migration” signal mediated by the lactate transporter SLC5A12 and SLC16A1.66 MCT1 and MCT4, which transport lactate into and out of cells, have become new targets for tumor therapy; however, there are still few studies on sepsis. Therefore, intensive research into the roles of MCT1 and MCT4 in immune paralysis during sepsis may provide a new target for the treatment of sepsis. An overview of immune metabolism regulation mechanism can be found in Table 1.
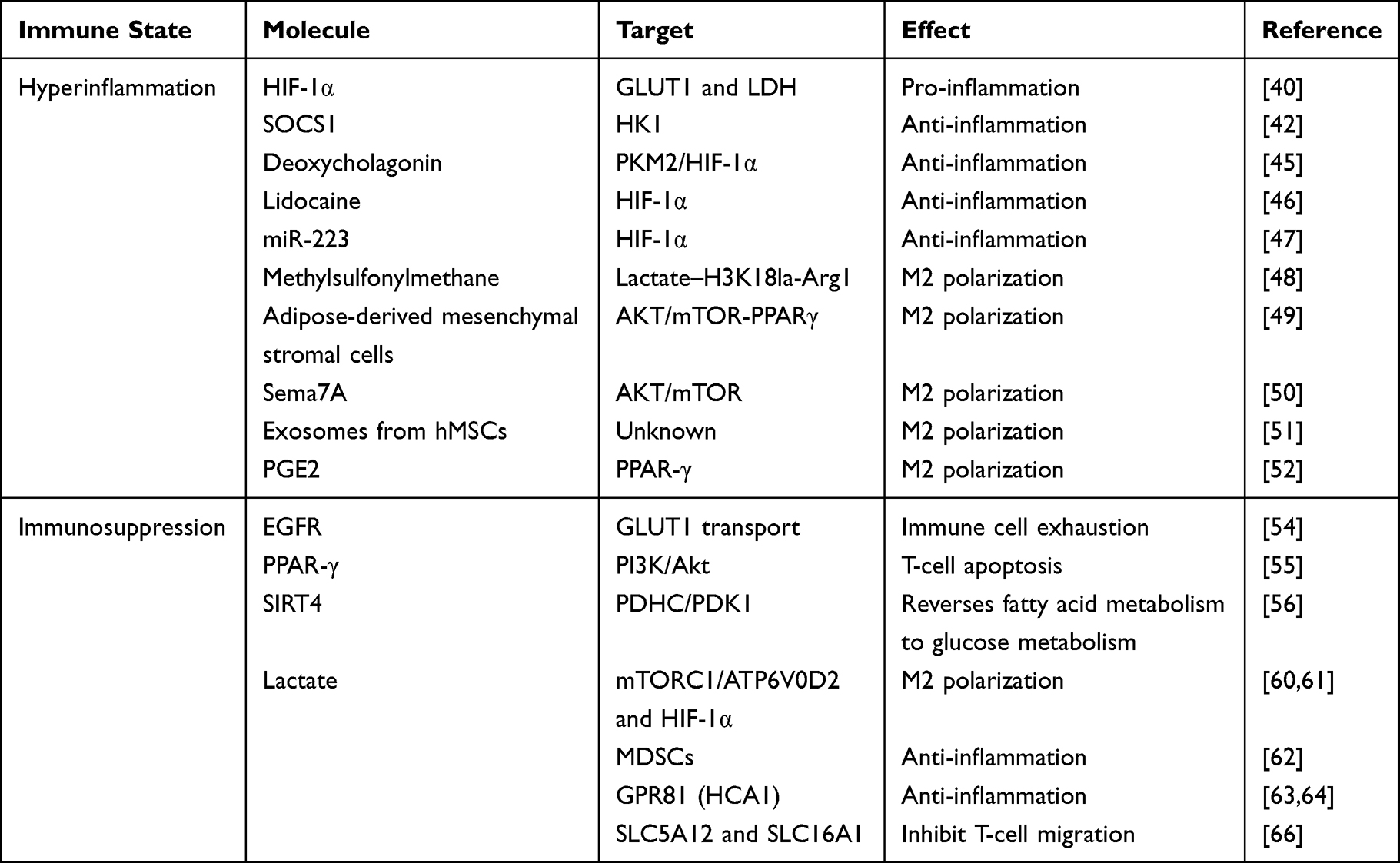 |
Table 1 Overview of Immune Metabolism Regulation Mechanism |
Characteristics of Mitochondria in the Metabolic Modulation of Sepsis
Sepsis-Induced Factors Contributing to Mitochondrial Dysfunction
Mitochondrial dysfunction is an essential factor that leads to septic multiple organ dysfunction syndrome and is known to be the leading cause of death in patients with severe sepsis. The main manifestations of mitochondrial dysfunction are electron transport chain damage and metabolic disorders. Abnormalities in energy metabolism lead to continuous inflammation, cause organ dysfunction, and affect the prognosis of sepsis.
Sepsis mainly affects mitochondrial function in the following ways. First, sufficient oxygen supply is a prerequisite for ATP synthesis in the mitochondria. A mismatch between systemic circulation and microcirculation during sepsis,67,68 abnormal volume distribution, and cardiac insufficiency69 may result in inadequate oxygen supply to the tissues. The metabolic pathway of the affected tissues changes to anaerobic glycolysis under hypoxic conditions, which does not produce ATP effectively. This results in ATP production below a certain level, triggering apoptosis and necrosis pathways.70 Second, PDHC is the main mitochondrial metabolic enzyme that enables energy generation via oxidative phosphorylation in the mitochondria. Its primary role is to catalyze the irreversible conversion of pyruvate to acetyl-CoA while reducing NAD+ to NADH, which enters the respiratory chain to produce ATP. At the same time, PDHC exerts its function at the branching node of oxidative phosphorylation and glycolysis, and its role in metabolic reprogramming should not be neglected. During sepsis, PDHC activity decreases, glucose metabolism changes from aerobic oxidation to anaerobic glycolysis, and a large amount of lactic acid accumulates, resulting in intracellular Ca2+ overload, mitochondrial membrane damage, and, consequently, mitochondrial dysfunction.71 PDHC reverses sepsis-induced anaerobic glycolysis, thus promoting aerobic oxidation, reducing lactate production, and conditioning LPS-induced barrier function failure of vascular endothelial cells.72 Briana et al showed extensive protein remodeling in mitochondria, inactivation of PDHC, and impaired pyruvate-fueled oxidative phosphorylation during sepsis-induced cardiomyopathy.73 During sepsis, PDHC is regulated by HIF-1α,74 glucocorticoid receptor,74 and PPAR-α pathways, which also directly affect oxidative phosphorylation. Dichloroacetate (DCA) is a PDK inhibitor that can increase the activity of PDHC and the pyruvate flux into mitochondria, promoting oxidative phosphorylation. In a septic mouse model, DCA treatment was found to reverse shock, reduce hepatocyte injury, significantly improve hyperlactatemia and immune paralysis, and significantly improve septic survival.75
Mitochondrial homeostasis plays a decisive role in regulating cellular energy metabolism and signaling pathways. PPAR-γ coactivator 1 alpha (PGC-1α) is one of the most important transcription factors that can drive mitochondrial biogenesis to respond to external stimuli and maintain mitochondrial homeostasis. The activity of PGC-1α is regulated through post-translational modifications. AMPK-induced phosphorylation and SIRT1-induced deacetylation activate PGC-1α.76 As a critical regulatory molecule in cell metabolism, AMPK regulates various aspects of mitochondrial homeostasis and promotes the mutual effect between mitochondrial division and autophagy by regulating dynamin-related protein 1 (Drp1), mitochondrial fission factor (Mff), PTEN-induced putative kinase (Pink), and parkin. Excessive free radicals produced during sepsis lead to oxidative stress and mitochondrial DNA damage, consequently leading to impaired mitochondrial protein synthesis and respiratory function. In response to these injuries, mitochondrial autophagy (mitophagy) is initiated to clear damaged mitochondria and activate mitochondrial biosynthesis.77 TEPP-46-induced activation of PKM2 promotes PGC-1α-mediated mitochondrial biogenesis by inhibiting the PI3K/Akt signaling pathway and promotes endotoxin tolerance by inhibiting pro-inflammatory cytokine release, namely, TNF-α and IL-6, in vitro and in vivo.78 Mitochondrial proteins containing the Asn-Glu-Glu-Thr sequence (MitoNEET) control the establishment of inter-mitochondrial junctions and promote autophagy resistance. The inhibition of mitoNEET promotes the accumulation of mitophagy-associated proteins (Pink1 and parkin) and activates the expression of PGC-1α to regulate mitochondrial biogenesis.79 Zhang et al80 reported that Pink1/parkin-mediated mitochondrial autophagy is activated during sepsis and has an anti-apoptotic effect on DC, which modulates the functions of the immune system. Furthermore, the APPL1–Rab5 axis restricts NLRP3 inflammasome activation through early endosomal-dependent mitophagy in macrophages. During sepsis, Ca/calmodulin-dependent protein kinase IV (CaMK-IV) regulates mitochondrial division by directly phosphorylating mitogen Drp1 and decreasing the expression of fusion proteins Mfn1/2 and OPA1. At the same time, CaMK-IV is also a regulator of PINK1 kinase and parkin expression, which regulates mitochondrial autophagy.81 Mdivi-1 inhibits mitochondrial division by targeting Drp1, which can restore mitochondrial morphology, reduce LPS-induced metabolic reprogramming, and reduce the production of ROS and inflammatory factors.82
Treatment of Sepsis by Regulating Mitochondrial Function
Mitochondria play a crucial role in disease progression by influencing ATP production, mainly by regulating cellular metabolism. In addition, they are responsible for regulating ROS production and redox signals as well as maintaining immune cell function. Through timely and effective regulation of mitochondrial function, prognosis during sepsis can be improved, and the mortality rate of sepsis can be significantly reduced.
Free radicals and ROS produced in the mitochondria can cause mitochondrial damage. During sepsis, hypoxia can induce glycolysis, reduce the oxidative phosphorylation pathway, and subsequently reduce the production of mitochondrial ROS, thus reducing mitochondrial damage to mitochondria.83 Therefore, by reducing the production of ROS to protect mitochondrial function, damage to target organs can be alleviated. MitoNEET is also a vital regulator of mitochondrial oxidative capacity. Using NL-1 or mitoNEET shRNA to inhibit mitoNEET can abolish LPS-induced ROS formation and mitochondrial dysfunction.84 The mitochondrial UCP2 can reduce the production of mitochondrial ROS by triggering proton leakage.85 UCP2 prevents oxidative stress and apoptosis of cardiomyocytes induced by endotoxins during septic cardiomyopathy.86 UCP2 can prevent LPS-induced acute renal injury by protecting the morphology and function of the mitochondria and reducing ROS production of ROS.87 The upregulation of SIRT3 to inhibit oxidative stress can protect mitochondrial function and induce intestinal epithelial cell autophagy, which alleviates intestinal injury caused by sepsis to some extent.88 In addition, mitochondrial homeostasis, which includes the balance between mitochondrial fusion and division and mitochondrial biogenesis, protects the lungs and kidneys from persistent oxidative stress.89
Mitochondria are also involved in maintaining immune cell functions. Kraft et al90 revealed that the early activation of mitochondrial biogenesis can effectively reverse leukocyte mitochondrial damage in the early stages of sepsis and improve clinical outcomes. Moreover, the antiviral signal protein on the mitochondrial membrane serves as a signal bridge to participate in the RLR pathway against the RNA virus.32 Mitochondrial DAMPs are released from damaged or stressed mitochondria, which include mitochondrial DNA (mtDNA). MtDNA activates the innate immune responses and mediates inflammation through the TLR-9, NLRP3 inflammasome, and cGAS-STING signaling pathways.91 In addition, the regulation of mitochondria in innate immunity is affected by intermediate products of the metabolic pathway. Itaconate (ITA) is a Krebs cycle metabolite with anti-inflammatory activity. Under LPS stimulation, macrophages undergo metabolic reprogramming, and the mitochondrial metabolic enzyme IRG1 protein is rapidly induced to catalyze the decarboxylation of cis-aconitine to produce a high concentration of ITA. ITA alkylates the transcription factor TFEB in the cytoplasm, resulting in its translocation from the cytoplasm to the nucleus. TFEB activates lysosome biosynthesis in the nucleus and enhances the role of macrophages in scavenging invasive bacteria in innate immunity.92 ITA inhibits the DNA dioxygenase TET and regulates inflammatory gene expression by competitively binding with α-ketoglutarate, confirming that TET2 is a crucial anti-inflammatory target of ITA.93 Therefore, as the main organelles of cell metabolism, mitochondria play an important role in immune metabolism. Regulating metabolic reprogramming of immune cells in sepsis by regulating mitochondrial function will provide a new direction for the regulation of immune homeostasis in sepsis.
Significance of Metabolic Reprogramming in the Clinical Treatment of Sepsis
Wagner et al94 investigated the association between mitochondrial respiratory states and inflammatory biomarkers in patients with septic shock, found that metabolic change observed in mitochondrial complex I and complex II of lymphocytes is associated with a decrease in IL-6 levels, which can signal a decrease in global inflammatory activity. Fatmi et al95 have shown that miR-26a represents a promising biomarker for neonatal sepsis, and miR-23b can be used as a tool for predicting early and late neonatal sepsis progression and fatal outcomes. A review of the currently available data on miRNAs involved in the major target organs (heart, lung, liver, kidney and blood) during sepsis found at least 122 miRNAs and signaling pathways involved in sepsis-related organ dysfunction, which may help clinicians to detect, prevent, and treat sepsis-related organ failures early.96 In addition, precise detection of metabolite types and levels is expected to be a potential method to assess the immune status of sepsis. During hyperinflammatory stage, small molecule drugs, microRNA targeting HIF-α-related signaling pathways and promoting M2-type polarization by metabolic regulation can be promising ways to avoid excessive inflammatory response causing multiple organ damage. During immunosuppression, correcting excessive lactic acid production can not only reduce the immunosuppressive effect of lactic acid, but also avoid the intracellular Ca2+ overload and mitochondrial membrane damage caused by a large amount of lactate accumulation, leading to mitochondrial dysfunction.
In recent years, the concept of immune metabolism has attracted more and more attention to the nutritional treatment of sepsis. Adequate clinical nutrition requires us to be aware of the dynamics and diversity of metabolic disorders during sepsis. Both insufficient and excessive energy supply are associated with worse treatment outcomes.97 As mentioned earlier, metabolic reprogramming during sepsis contribute to blood lactate concentrations involved in glycolysis, pyruvate dehydrogenase activity, and reduced lactate clearance.98 A systemic review conclude that Thiamin supplementation may improve lactate clearance when administered in the first 24 h, which can alleviate the immunosuppressive effect and damage to mitochondria. Ω-3 lipid is involved in arachidonic acid metabolism, which can induce macrophage M2 polarization. Ω-3 lipid emulsions enhance pro-resolving lipid mediator biosynthesis, which recessed neutrophil infiltration, reduced pro-inflammatory mediators, reduced the classical monocyte and enhanced the non-classical monocytes/macrophages recruitment and finally increased the efferocytosis in sepsis.99 A systematic review showed that parenteral nutrition containing fish oil, which is full of Ω-3 lipid, could significantly decrease mortality in sepsis patients.100 In addition, Due to both energetic and metabolic consequences, mitochondrial dysfunction appears to play a significant role in the pathogenesis of sepsis.101 This makes them a valuable subject for further research and a potential target for future therapies.
Conclusions
In summary, the balance of inflammatory responses during sepsis is inextricably linked to metabolic reprogramming. During hyperinflammatory response, cellular metabolism alters from OXPHOS to aerobic glycolysis; however, excessive glycolysis can trigger immunosuppression. Metabolic imbalance and mitochondrial dysfunction play vital roles in the occurrence, development, and recovery from sepsis. A comprehensive study on the metabolic relationship between hyperinflammation and immunosuppression, timely regulation of immune cell metabolism by studying the biomarkers of cell metabolic status, suppression of organ damage caused by excessive inflammatory reactions, and prevention of immune paralysis will significantly help inform future clinical management in improving the survival rate of patients with sepsis. As the core organelle of cell metabolism, further understanding of the roles of mitochondria in metabolic reprogramming during sepsis will offer novel and valuable insights into the treatment and prognosis of sepsis. Over the years, more and more researches pay attention to the nutritional treatment of sepsis. More evidence-based medical evidences are needed to decide how to assess nutritional status of an ICU patient, how to define the amount of energy to provide and how to adapt according to various clinical conditions.
Abbreviations
PRR, pathogen recognition receptor; PAMP, pathogen-associated molecular pattern; DAMP, damage-associated molecular pattern; TLR, Toll-like receptor; NLR, NOD-like receptor; RLR, RIG-I-like receptor; PPP, pentose phosphate pathway; NADPH, reduced nicotinamide adenine dinucleotide phosphate; NET, neutrophil extracellular traps; NOX, NADPH oxidase; ROS, reactive oxygen species; DC, dendritic cells; LPS, lipopolysaccharide; mTOR, mammalian target of rapamycin; HIF-1α, hypoxia-inducible factor-1α; GLUT1, glucose transporter 1; LDH, lactate dehydrogenase; PFKFB3, fructose-2,6-bisphosphatase 3; AMPK, adenosine 5’-monophosphate-activated protein kinase; FAO, fatty acid oxidation; OXPHOS, oxidative phosphorylation; IFN-γ, interferon-gamma; HK1, hexokinase 1; PKM2, pyruvate kinase M2; PPAR-γ, peroxisome proliferator-activated receptor γ; PDHC, pyruvate dehydrogenase complex; MDSCs, myeloid-derived suppressor cells; YAP, Yes-associated protein; PGC-1α, PPAR-γ coactivator 1 alpha; CaMK-IV, Ca/calmodulin-dependent protein kinase IV; UCP2, uncoupling protein 2; ITA, Itaconate.
Acknowledgments
This work was supported by the National Nature Science Foundation of China (81930057, 82072170, 82172201); Shanghai Rising Star Program (22QA1411700); Youth Medical Talents-Specialist Program; CAMS Innovation Fund for Medical Sciences (2019-I2M-5-076), Achievements Supportive Fund (2018-CGPZ-B03).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol. 2011;22(6):999–1006. doi:10.1681/asn.2010050484
2. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the global burden of disease study. Lancet. 2020;395(10219):200–211. doi:10.1016/s0140-6736(19)32989-7
3. Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers. 2016;2:16045. doi:10.1038/nrdp.2016.45
4. Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25(7):771–784. doi:10.1038/cr.2015.68
5. Zheng Z, Ma H, Zhang X, et al. Enhanced glycolytic metabolism contributes to cardiac dysfunction in polymicrobial sepsis. J Infect Dis. 2017;215(9):1396–1406. doi:10.1093/infdis/jix138
6. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi:10.1126/science.123.3191.309
7. Soto-Heredero G, Gómez de Las Heras MM, Gabandé-Rodríguez E, Oller J, Mittelbrunn M. Glycolysis - a key player in the inflammatory response. Febs j. 2020;287(16):3350–3369. doi:10.1111/febs.15327
8. Owen AM, Patel SP, Smith JD, et al. Chronic muscle weakness and mitochondrial dysfunction in the absence of sustained atrophy in a preclinical sepsis model. Elife. 2019;8. doi:10.7554/eLife.49920
9. Kumar S, Ingle H, Prasad DV, Kumar H. Recognition of bacterial infection by innate immune sensors. Crit Rev Microbiol. 2013;39(3):229–246. doi:10.3109/1040841x.2012.706249
10. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi:10.1016/j.cell.2010.01.022
11. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249(1):158–175. doi:10.1111/j.1600-065X.2012.01146.x
12. Donnelly RP, Finlay DK. Glucose, glycolysis and lymphocyte responses. Mol Immunol. 2015;68(2Pt C):513–519. doi:10.1016/j.molimm.2015.07.034
13. Stincone A, Prigione A, Cramer T, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. 2015;90(3):927–963. doi:10.1111/brv.12140
14. Kim JA, Yeom YI. Metabolic signaling to epigenetic alterations in cancer. Biomol Ther. 2018;26(1):69–80. doi:10.4062/biomolther.2017.185
15. Batista-Gonzalez A, Vidal R, Criollo A, Carreño LJ. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front Immunol. 2019;10:2993. doi:10.3389/fimmu.2019.02993
16. Awasthi D, Nagarkoti S, Sadaf S, Chandra T, Kumar S, Dikshit M. Glycolysis dependent lactate formation in neutrophils: a metabolic link between NOX-dependent and independent NETosis. Biochim Biophys Acta Mol Basis Dis. 2019;1865(12):165542. doi:10.1016/j.bbadis.2019.165542
17. Denning NL, Aziz M, Gurien SD, Wang P. DAMPs and NETs in sepsis. Front Immunol. 2019;10:2536. doi:10.3389/fimmu.2019.02536
18. Tan C, Gu J, Chen H, et al. Inhibition of aerobic glycolysis promotes neutrophil to influx to the infectious site via CXCR2 in sepsis. Shock. 2020;53(1):114–123. doi:10.1097/shk.0000000000001334
19. Im SS, Yousef L, Blaschitz C, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. 2011;13(5):540–549. doi:10.1016/j.cmet.2011.04.001
20. Wei X, Song H, Yin L, et al. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature. 2016;539(7628):294–298. doi:10.1038/nature20117
21. Moon JS, Lee S, Park MA, et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest. 2015;125(2):665–680. doi:10.1172/jci78253
22. Bidault G, Virtue S, Petkevicius K, et al. SREBP1-induced fatty acid synthesis depletes macrophages antioxidant defences to promote their alternative activation. Nat Metab. 2021;3(9):1150–1162. doi:10.1038/s42255-021-00440-5
23. Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345(1):50–54. doi:10.1016/0014-5793(94)00424-2
24. Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci U S A. 1998;95(13):7631–7636. doi:10.1073/pnas.95.13.7631
25. Mole DR, Blancher C, Copley RR, et al. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284(25):16767–16775. doi:10.1074/jbc.M901790200
26. Zhang Q, Hu Y, Zhang J, Deng C. iTRAQ‑based proteomic analysis of endotoxin tolerance induced by lipopolysaccharide. Mol Med Rep. 2019;20(1):584–592. doi:10.3892/mmr.2019.10264
27. Morris M, Li L. Molecular mechanisms and pathological consequences of endotoxin tolerance and priming. Arch Immunol Ther Exp. 2012;60(1):13–18. doi:10.1007/s00005-011-0155-9
28. Rubio I, Osuchowski MF, Shankar-Hari M, et al. Current gaps in sepsis immunology: new opportunities for translational research. Lancet Infect Dis. 2019;19(12):e422–e436. doi:10.1016/s1473-3099(19)30567-5
29. Liu TF, Vachharajani VT, Yoza BK, McCall CE. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J Biol Chem. 2012;287(31):25758–25769. doi:10.1074/jbc.M112.362343
30. Zwaag J, Ter Horst R, Blazenovic I, et al. Involvement of lactate and pyruvate in the anti-inflammatory effects exerted by voluntary activation of the sympathetic nervous system. Metabolites. 2020;10(4). doi:10.3390/metabo10040148
31. Zhou HC, Yu WW, Yan XY, et al. Lactate-driven macrophage polarization in the inflammatory microenvironment alleviates intestinal inflammation. Front Immunol. 2022;13:1013686. doi:10.3389/fimmu.2022.1013686
32. Zhang W, Wang G, Xu ZG, et al. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell. 2019;178(1):176–189.e15. doi:10.1016/j.cell.2019.05.003
33. Ivashkiv LB. The hypoxia-lactate axis tempers inflammation. Nat Rev Immunol. 2020;20(2):85–86. doi:10.1038/s41577-019-0259-8
34. Liu N, Luo J, Kuang D, et al. Lactate inhibits ATP6V0d2 expression in tumor-associated macrophages to promote HIF-2α-mediated tumor progression. J Clin Invest. 2019;129(2):631–646. doi:10.1172/jci123027
35. Zhang D, Tang Z, Huang H, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–580. doi:10.1038/s41586-019-1678-1
36. Gottfried E, Kunz-Schughart LA, Ebner S, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood. 2006;107(5):2013–2021. doi:10.1182/blood-2005-05-1795
37. Xu J, Ma X, Yu K, et al. Lactate up-regulates the expression of PD-L1 in kidney and causes immunosuppression in septic Acute Renal Injury. J Microbiol Immunol Infect. 2021;54(3):404–410. doi:10.1016/j.jmii.2019.10.006
38. Koutroulis I, Batabyal R, McNamara B, Ledda M, Hoptay C, Freishtat RJ. Sepsis immunometabolism: from defining sepsis to understanding how energy production affects immune response. Crit Care Explor. 2019;1(11):e0061. doi:10.1097/cce.0000000000000061
39. Liu J, Zhou G, Wang X, Liu D. Metabolic reprogramming consequences of sepsis: adaptations and contradictions. Cell Mol Life Sci. 2022;79(8):456. doi:10.1007/s00018-022-04490-0
40. Shao C, Lin S, Liu S, et al. HIF1α-induced glycolysis in macrophage is essential for the protective effect of ouabain during endotoxemia. Oxid Med Cell Longev. 2019;2019:7136585. doi:10.1155/2019/7136585
41. Pan T, Sun S, Chen Y, et al. Immune effects of PI3K/Akt/HIF-1α-regulated glycolysis in polymorphonuclear neutrophils during sepsis. Crit Care. 2022;26(1):29. doi:10.1186/s13054-022-03893-6
42. Pineros Alvarez AR, Glosson-Byers N, Brandt S, et al. SOCS1 is a negative regulator of metabolic reprogramming during sepsis. JCI Insight. 2017;2(13). doi:10.1172/jci.insight.92530
43. De Jesus A, Keyhani-Nejad F, Pusec CM, et al. Hexokinase 1 cellular localization regulates the metabolic fate of glucose. Mol Cell. 2022;82(7):1261–1277.e9. doi:10.1016/j.molcel.2022.02.028
44. Yang L, Xie M, Yang M, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. 2014;5:4436. doi:10.1038/ncomms5436
45. Pan L, Hu L, Zhang L, et al. Deoxyelephantopin decreases the release of inflammatory cytokines in macrophage associated with attenuation of aerobic glycolysis via modulation of PKM2. Int Immunopharmacol. 2020;79:106048. doi:10.1016/j.intimp.2019.106048
46. Lin S, Jin P, Shao C, et al. Lidocaine attenuates lipopolysaccharide-induced inflammatory responses and protects against endotoxemia in mice by suppressing HIF1alpha-induced glycolysis. Int Immunopharmacol. 2020;80:106150. doi:10.1016/j.intimp.2019.106150
47. Dang CP, Leelahavanichkul A. Over-expression of miR-223 induces M2 macrophage through glycolysis alteration and attenuates LPS-induced sepsis mouse model, the cell-based therapy in sepsis. PLoS One. 2020;15(7):e0236038. doi:10.1371/journal.pone.0236038
48. Ma W, Ao S, Zhou J, et al. Methylsulfonylmethane protects against lethal dose MRSA-induced sepsis through promoting M2 macrophage polarization. Mol Immunol. 2022;146:69–77. doi:10.1016/j.molimm.2022.04.001
49. Souza-Moreira L, Soares VC, Dias S, Bozza PT. Adipose-derived mesenchymal stromal cells modulate lipid metabolism and lipid droplet biogenesis via AKT/mTOR -PPARgamma signalling in macrophages. Sci Rep. 2019;9(1):20304. doi:10.1038/s41598-019-56835-8
50. Körner A, Bernard A, Fitzgerald JC, et al. Sema7A is crucial for resolution of severe inflammation. Proc Natl Acad Sci U S A. 2021;118(9). doi:10.1073/pnas.2017527118
51. Deng H, Zhu L, Zhang Y, et al. Differential lung protective capacity of exosomes derived from human adipose tissue, bone marrow, and umbilical cord mesenchymal stem cells in sepsis-induced acute lung injury. Oxid Med Cell Longev. 2022;2022:7837837. doi:10.1155/2022/7837837
52. Xu M, Wang X, Li Y, et al. Arachidonic acid metabolism controls macrophage alternative activation through regulating oxidative phosphorylation in PPARγ dependent manner. Front Immunol. 2021;12:618501. doi:10.3389/fimmu.2021.618501
53. Cohen J, Vincent JL, Adhikari NK, et al. Sepsis: a roadmap for future research. Lancet Infect Dis. 2015;15(5):581–614. doi:10.1016/s1473-3099(15)70112-x
54. Huang L, Zhang X, Fan J, et al. EGFR promotes the apoptosis of CD4(+) T lymphocytes through TBK1/Glut1 induced Warburg effect in sepsis. J Adv Res. 2022. doi:10.1016/j.jare.2022.04.010
55. Schmidt MV, Paulus P, Kuhn AM, et al. Peroxisome proliferator-activated receptor γ-induced T cell apoptosis reduces survival during polymicrobial sepsis. Am J Respir Crit Care Med. 2011;184(1):64–74. doi:10.1164/rccm.201010-1585OC
56. Tao J, Zhang J, Ling Y, McCall CE, TF Liu. Mitochondrial sirtuin 4 resolves immune tolerance in monocytes by rebalancing glycolysis and glucose oxidation homeostasis. Front Immunol. 2018;9:419. doi:10.3389/fimmu.2018.00419
57. Langston PK, Nambu A, Jung J, et al. Glycerol phosphate shuttle enzyme GPD2 regulates macrophage inflammatory responses. Nat Immunol. 2019;20(9):1186–1195. doi:10.1038/s41590-019-0453-7
58. Cheng SC, Scicluna BP, Arts RJ, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol. 2016;17(4):406–413. doi:10.1038/ni.3398
59. Xu J, Gao C, He Y, et al. NLRC3 expression in macrophage impairs glycolysis and host immune defense by modulating the NF-κB-NFAT5 complex during septic immunosuppression. Mol Ther. 2022. doi:10.1016/j.ymthe.2022.08.023
60. Choi SY, Collins CC, Gout PW, Wang Y. Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite? J Pathol. 2013;230(4):350–355. doi:10.1002/path.4218
61. Zhao Y, Zhao B, Wang X, et al. Macrophage transcriptome modification induced by hypoxia and lactate. Exp Ther Med. 2019;18(6):4811–4819. doi:10.3892/etm.2019.8164
62. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. 2013;191(3):1486–1495. doi:10.4049/jimmunol.1202702
63. Hoque R, Farooq A, Ghani A, Gorelick F, Mehal WZ. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology. 2014;146(7):1763–1774. doi:10.1053/j.gastro.2014.03.014
64. Yang K, Xu J, Fan M, et al. Lactate suppresses macrophage pro-inflammatory response to LPS stimulation by inhibition of YAP and NF-κB activation via GPR81-mediated signaling. Front Immunol. 2020;11:587913. doi:10.3389/fimmu.2020.587913
65. Schenz J, Heilig L, Lohse T, et al. Extracellular lactate acts as a metabolic checkpoint and shapes monocyte function time dependently. Front Immunol. 2021;12:729209. doi:10.3389/fimmu.2021.729209
66. Haas R, Smith J, Rocher-Ros V, et al. Lactate regulates metabolic and pro-inflammatory circuits in control of t cell migration and effector functions. PLoS Biol. 2015;13(7):e1002202. doi:10.1371/journal.pbio.1002202
67. De Backer D, Ricottilli F, Ospina-Tascón GA. Septic shock: a microcirculation disease. Curr Opin Anaesthesiol. 2021;34(2):85–91. doi:10.1097/aco.0000000000000957
68. Inkinen N, Pettilä V, Lakkisto P, et al. Association of endothelial and glycocalyx injury biomarkers with fluid administration, development of acute kidney injury, and 90-day mortality: data from the FINNAKI observational study. Ann Intensive Care. 2019;9(1):103. doi:10.1186/s13613-019-0575-y
69. Lin Y, Xu Y, Zhang Z. Sepsis-Induced Myocardial Dysfunction (SIMD): the pathophysiological mechanisms and therapeutic strategies targeting mitochondria. Inflammation. 2020;43(4):1184–1200. doi:10.1007/s10753-020-01233-w
70. Bartolák-Suki E, Imsirovic J, Nishibori Y, Krishnan R, Suki B. Regulation of mitochondrial structure and dynamics by the cytoskeleton and mechanical factors. Int J Mol Sci. 2017;18(8). doi:10.3390/ijms18081812
71. Arulkumaran N, Deutschman CS, Pinsky MR, et al. Mitochondrial function in sepsis. Shock. 2016;45(3):271–281. doi:10.1097/shk.0000000000000463
72. Mao L, Sun M, Chen Z, et al. The pyruvate dehydrogenase complex mitigates LPS-induced endothelial barrier dysfunction by metabolic regulation. Shock. 2022;57(6):308–317. doi:10.1097/shk.0000000000001931
73. Shimada BK, Boyman L, Huang W, et al. Pyruvate-driven oxidative phosphorylation is downregulated in sepsis-induced cardiomyopathy: a study of mitochondrial proteome. Shock. 2022;57(4):553–564. doi:10.1097/shk.0000000000001858
74. Dasgupta A, Wu D, Tian L, et al. Mitochondria in the pulmonary vasculature in health and disease: oxygen-sensing, metabolism, and dynamics. Compr Physiol. 2020;10(2):713–765. doi:10.1002/cphy.c190027
75. McCall CE, Zhu X, Zabalawi M, et al. Sepsis, pyruvate, and mitochondria energy supply chain shortage. J Leukoc Biol. 2022. doi:10.1002/JLB.3MR0322-692RR
76. Gureev AP, Shaforostova EA, Popov VN. Regulation of mitochondrial biogenesis as a way for active longevity: interaction between the Nrf2 and PGC-1α signaling pathways. Front Genet. 2019;10:435. doi:10.3389/fgene.2019.00435
77. Zhao Y, Huang S, Liu J, et al. Mitophagy contributes to the pathogenesis of inflammatory diseases. Inflammation. 2018;41(5):1590–1600. doi:10.1007/s10753-018-0835-2
78. Yi Z, Wu Y, Zhang W, et al. Activator-mediated pyruvate kinase M2 activation contributes to endotoxin tolerance by promoting mitochondrial biogenesis. Front Immunol. 2020;11:595316. doi:10.3389/fimmu.2020.595316
79. Lee S, Lee S, Lee SJ, Chung SW. Inhibition of mitoNEET induces Pink1-Parkin-mediated mitophagy. BMB Rep. 2022;55(7):354–359. doi:10.5483/BMBRep.2022.55.7.040
80. Zhang Y, Chen L, Luo Y, et al. Pink1/parkin-mediated mitophagy regulated the apoptosis of dendritic cells in sepsis. Inflammation. 2022;45(3):1374–1387. doi:10.1007/s10753-022-01628-x
81. Zhang X, Griepentrog JE, Zou B, et al. CaMKIV regulates mitochondrial dynamics during sepsis. Cell Calcium. 2020;92:102286. doi:10.1016/j.ceca.2020.102286
82. Yu W, Wang X, Zhao J, et al. Stat2-Drp1 mediated mitochondrial mass increase is necessary for pro-inflammatory differentiation of macrophages. Redox Biol. 2020;37:101761. doi:10.1016/j.redox.2020.101761
83. Yang H, Zhang Z. Sepsis-induced myocardial dysfunction: the role of mitochondrial dysfunction. Inflamm Res. 2021;70(4):379–387. doi:10.1007/s00011-021-01447-0
84. Lee S, Seok BG, Lee SJ, Chung SW. Inhibition of mitoNEET attenuates LPS-induced inflammation and oxidative stress. Cell Death Dis. 2022;13(2):127. doi:10.1038/s41419-022-04586-2
85. Donadelli M, Dando I, Fiorini C, Palmieri M. UCP2, a mitochondrial protein regulated at multiple levels. Cell Mol Life Sci. 2014;71(7):1171–1190. doi:10.1007/s00018-013-1407-0
86. Huang J, Peng W, Zheng Y, et al. Upregulation of UCP2 expression protects against LPS-induced oxidative stress and apoptosis in cardiomyocytes. Oxid Med Cell Longev. 2019;2019:2758262. doi:10.1155/2019/2758262
87. Ding Y, Zheng Y, Huang J, et al. UCP2 ameliorates mitochondrial dysfunction, inflammation, and oxidative stress in lipopolysaccharide-induced acute kidney injury. Int Immunopharmacol. 2019;71:336–349. doi:10.1016/j.intimp.2019.03.043
88. Xu S, Li L, Wu J, et al. Melatonin attenuates sepsis-induced small-intestine injury by upregulating SIRT3-mediated oxidative-stress inhibition, mitochondrial protection, and autophagy induction. Front Immunol. 2021;12:625627. doi:10.3389/fimmu.2021.625627
89. Shi J, Yu J, Zhang Y, et al. PI3K/Akt pathway-mediated HO-1 induction regulates mitochondrial quality control and attenuates endotoxin-induced acute lung injury. Lab Invest. 2019;99(12):1795–1809. doi:10.1038/s41374-019-0286-x
90. Kraft BD, Chen L, Suliman HB, Piantadosi CA, Welty-Wolf KE. Peripheral blood mononuclear cells demonstrate mitochondrial damage clearance during sepsis. Crit Care Med. 2019;47(5):651–658. doi:10.1097/ccm.0000000000003681
91. Liao S, Luo J, Kadier T, Ding K, Chen R, Meng Q. Mitochondrial DNA release contributes to intestinal ischemia/reperfusion injury. Front Pharmacol. 2022;13:854994. doi:10.3389/fphar.2022.854994
92. Zhang Z, Chen C, Yang F, et al. Itaconate is a lysosomal inducer that promotes antibacterial innate immunity. Mol Cell. 2022;82(15):2844–2857.e10. doi:10.1016/j.molcel.2022.05.009
93. Chen LL, Morcelle C, Cheng ZL, et al. Itaconate inhibits TET DNA dioxygenases to dampen inflammatory responses. Nat Cell Biol. 2022;24(3):353–363. doi:10.1038/s41556-022-00853-8
94. Nedel WL, Strogulski NR, Rodolphi MS, Kopczynski A, Montes TH, Portela LV. Short-term inflammatory biomarkers profile are associated with deficient mitochondrial bioenergetics in lymphocytes of septic shock patients–a prospective cohort study. Shock. 2022;8:10–97. doi:10.1097/SHK.0000000000002055
95. Fatmi A, Chabni N, Cernada M, et al. Clinical and immunological aspects of microRNAs in neonatal sepsis. Biomed Pharmacother. 2022;145:112444. doi:10.1016/j.biopha.2021.112444
96. Maiese A, Scatena A, Costantino A, et al. Expression of MicroRNAs in sepsis-related organ dysfunction: a systematic review. Int J Mol Sci. 2022;23(16). doi:10.3390/ijms23169354
97. Singer P, Blaser AR, Berger MM, et al. ESPEN guideline on clinical nutrition in the intensive care unit. Clin Nutr. 2019;38(1):48–79. doi:10.1016/j.clnu.2018.08.037
98. Nolt B, Tu F, Wang X, et al. Lactate and immunosuppression in sepsis. Shock. 2018;49(2):120–125. doi:10.1097/shk.0000000000000958
99. Körner A, Schlegel M, Theurer J, et al. Resolution of inflammation and sepsis survival are improved by dietary Ω-3 fatty acids. Cell Death Differ. 2018;25(2):421–431. doi:10.1038/cdd.2017.177
100. Wang H, Su S, Wang C, Hu J, Dan W, Peng X. Effects of fish oil-containing nutrition supplementation in adult sepsis patients: a systematic review and meta-analysis. Burns Trauma. 2022;10:tkac012. doi:10.1093/burnst/tkac012
101. Wasyluk W, Zwolak A. Metabolic alterations in sepsis. J Clin Med. 2021;10(11). doi:10.3390/jcm10112412
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Advances in the Study of Immunosuppressive Mechanisms in Sepsis
Fu X, Liu Z, Wang Y
Journal of Inflammation Research 2023, 16:3967-3981
Published Date: 8 September 2023
Clinical Outcomes of Hospitalized Immunocompromised Patients With COVID-19 and the Impact of Hyperinflammation: A Retrospective Cohort Study
Zhang X, Han X, Li C, Cui J, Yuan X, Meng J, Han Z, Han X, Chen W, Xiong J, Xie W, Xie L
Journal of Inflammation Research 2025, 18:3385-3397
Published Date: 7 March 2025