")
Back to Journals » OncoTargets and Therapy » Volume 13
MET Oncogene in Non-Small Cell Lung Cancer: Mechanism of MET Dysregulation and Agents Targeting the HGF/c-Met Axis
Received 16 September 2019
Accepted for publication 16 February 2020
Published 25 March 2020 Volume 2020:13 Pages 2491—2510
DOI https://doi.org/10.2147/OTT.S231257
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Hongge Liang, Mengzhao Wang
Peking Union Medical College Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100730, People’s Republic of China
Correspondence: Mengzhao Wang
Peking Union Medical College Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, No. 1 Shuaifuyuan, Dongcheng District, Beijing 100730, People’s Republic of China
Tel +86 10-69155039
Email [email protected]
Abstract: Non-small cell lung cancer (NSCLC) is the leading cause of cancer-related death worldwide and has a poor prognosis. Current treatments for advanced NSCLC included traditional chemotherapy, radiotherapy, targeted therapy, and immunotherapy. The efficacy of targeted therapy relies on oncogene addiction. Mesenchymal-epithelial transition factor (MET) gene can encode unconventional receptor tyrosine kinases with pleiotropic functions, when signals are abnormally activated, it can initiate and maintain tumor transformation, promote cell proliferation, survival, tumor invasion and angiogenesis. Thus, it is a promising therapeutic target. Previous studies have shown that elevated levels of HGF and/or overexpression of c-Met are associated with poor prognosis in lung cancer. In preclinical and clinical trials, c-MET inhibitors have shown some antitumor activity in NSCLC. Although the efficacy results of MET inhibitors in Phase III clinical trials are disappointing, given the molecular heterogeneity of NSCLC, only subgroups of patients with MET gene alterations may benefit from c-MET inhibitors. The challenge for the future is to screen out the potential beneficiaries. To solve this problem, there is need for large data analysis for the detection methods and treatment effects, to establish standards that meet the MET activation status, and determine reliable thresholds to achieve effective patient stratification and clinical decision making. This article summarized the structure of the hepatocyte growth factor (HGF)/c-Met axis, the different mechanisms of MET addiction, as well as MET amplification as acquired resistance mechanism to epidermal growth factor receptor-tyrosine kinase inhibitors, the latest advances of MET inhibitors, and immuotherapy in the treatment of NSCLC with MET alterations.
Keywords: c-mesenchymal-epithelial transition, receptor tyrosine kinases, non-small cell lung cancer, treatment, oncogene addiction
Introduction
Non-small cell lung cancer (NSCLC) is the leading cause of cancer-related death worldwide.1 Most patients with NSCLC are diagnosed at an advanced stage, and traditional chemotherapy and radiotherapy have shown limited efficacy.2 Although immunotherapy has changed the current state of treatment for NSCLC, many patients do not respond to programmed cell death protein-1 (PD-1)/ programmed death-ligand 1 (PD-L1) inhibitors, especially in patients harboring driven mutations. Small molecule tyrosine kinase inhibitors (TKIs) are now approved for treatment in patients with NSCLC harboring epidermal growth factor receptor (EGFR) mutations, BRAF V600E mutations, anaplastic lymphoma kinase (ALK) rearrangements, and ROS1 rearrangements; however, all patients will inevitably develop progression of disease.3 Therefore, it is necessary to find new therapeutic targets that drive the pathogenesis of NSCLC and develop more effective targeted drugs.
C-mesenchymal-epithelial transition factor (c-MET), the hepatocyte growth factor (HGF) receptor, is an oncogene encoding tyrosine kinase receptor. It mainly exists in epithelial cells, and plays an important role in embryogenesis, tumor growth, and metastasis.4 Once tyrosine kinase receptors are activated by their ligands, mitosis is triggered. This can regulate a variety of cellular functions.5,6 The dysregulation of MET/HGF axis pathway is involved in the proliferation, survival, invasion, and metastasis of tumor cells.7,8 This can be found in NSCLC and other solid tumors such as breast cancer, cervical cancer, stomach cancer, and colon cancer.9,10 C-MET alterations in NSCLC include point mutations, amplification, fusion, and protein overexpression, which are associated with poor prognosis.11–15 Previous preclinical and clinical studies suggested that MET activation is both a primary oncogenic driver mutation and a secondary driver of acquired resistance to targeted therapy in other genomic subpopulations.16 Therefore, agents targeting c-MET are a promising treatment strategy for NSCLC. At present, a number of pre-clinical and clinical studies have been conducted on many drugs targeting MET (small molecule TKI, MET antibody, and HGF antibody).5,17,18 This article reviewed the mechanisms of MET gene addiction, and the clinical application of MET inhibitors in NSCLC.
Dysregulated c-MET Signaling in NSCLC
The c-MET gene is located on chromosome 7 q21-31 belonging to the HGF receptor family, which encodes a protein tyrosine kinase, and regulates important cellular processes including cell differentiation, proliferation, cell cycle, movement, and apoptosis. HGF is a paracrine-signaling molecule that is produced and secreted by mesenchymal cells, which is the only ligand for c-MET.19 The extracellular portion of c-MET consists of the immunoglobulin (Ig)-like, plexins, transcription factors (IPT) domain, the plexin-semaphorin-integrin (PSI) domain, and the Sema domain (homologous to semaphorin) responsible for binding to HGF. The intracellular portion of c-MET consists of the juxtamembrane (JM) domain, the Catalytic domain, and the Docking site responsible for signal transduction (Figure 1). HGF/c-MET binding leads to receptor dimerization, tyrosine residues autophosphorylate, substrate docking, and activation of downstream signaling pathways such as PI3K/AKT, RAS/ERK/MAPK, Wnt/β-catenin, SRC, and STAT320-29;20–29 thereby, inducing excessive cell proliferation, and is closely related to the occurrence and development of tumors. It is reported that the deregulation of the MET signaling in NSCLC can induce tumor invasion and metastasis,30 and can interact with other signaling pathways such as EGFR.31,32
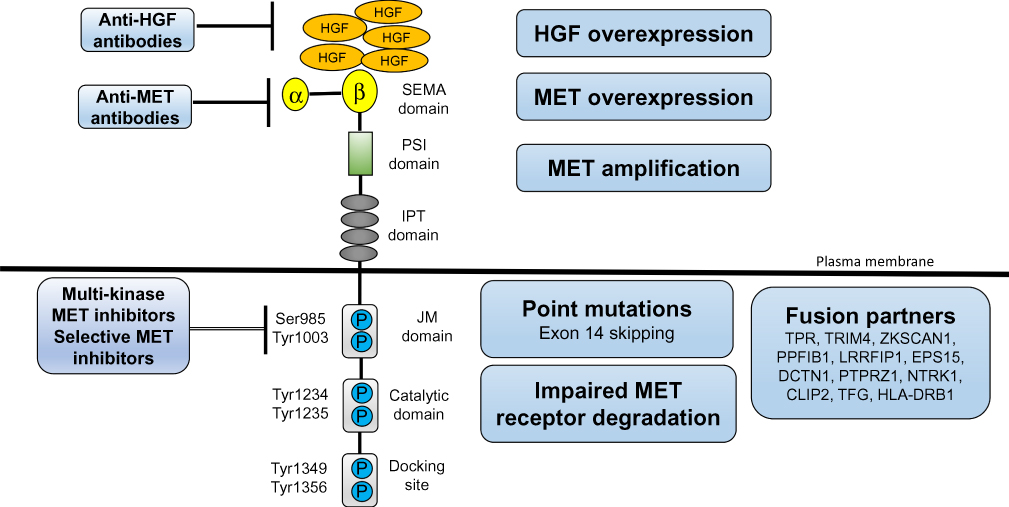 |
Figure 1 Major Mechanism of MET/HGF axis dysregulation. The extracellular portion of c-MET consists of a four immunoglobulin (Ig)-like modules, a cysteine-rich, MET-related sequence domain, and a Sema domain (homologous to semaphorin) responsible for binding to HGF. The intracellular portion of c-MET consists of the paramembrane domain, the Catalytic domain, and the Docking site responsible for signal transduction. Various mechanisms of MET/HGF axis addiction in NSCLC, including MET/HGF overexpression, and MET gene alterations (including point mutations, amplification, and fusion). A number of drugs targeting MET (small molecule TKIs, MET antibody, and HGF antibody) have been studied. MET, c-mesenchymal-epithelial transition factor; HGF, hepatocyte growth factor; NSCLC, non-small cell lung cancer. |
MET Exon 14 Skipping Mutations
METex14 alterations (point mutations, deletions, insertions, and complex mutations) lead to decreased degradation of MET receptor, resulting in the activation of MET signaling and the tumorigenesis.33–36 The evaluation methods for METex14 skipping mutations include differential MET exon expression,37,38 quantitative reverse transcription-polymerase chain reaction (qRT-PCR)39 or direct RNA sequencing.37,40 In addition, DNA and RNA sequencing can be used to detect Y1003, a “functional mimetic” of the METex14 skipping mutation, accounting for approximately 2% of all METex14 alterations.41
In 2015, Frampton et al performed a large-scale molecular profiling of METex14 alterations in 38,028 tumor samples. Of the 221 tumor samples containing METex14 alterations, 193 were found to be lung cancer, of which 131 were lung adenocarcinoma.42 METex14 skipping mutations have been reported in 2–4% of NSCLC.37,43–45 The mutation rate was closely related to the histological subtype of NSCLC, most commonly in sarcomatoid carcinoma (4.9% ~ 31%), followed by adenosquamous carcinoma (5%), adenocarcinoma (3%) and squamous cell carcinoma (2%).39,41,46–48 In addition, METex14 mutations are likely to be mutually exclusive of other genetic alterations in NSCLC and are more likely to occur in elderly, non-smoker patients.39,42 In a study of 933 patients with non-squamous NSCLC, no mutations in the KRAS, EGFR, erb-b2 receptor tyrosine kinase 2 gene (ERBB2), ALK, ROS1, or RET were found in 30 patients with METex14 alterations.39 In 687 Asian patients with NSCLC, METex14 skipping mutations are poor prognostic factors of overall survival.49 In NSCLC patients, METex14 skipping mutations are associated with the efficacy of MET protein TKIs, especially in patients with advanced lung sarcomatoid carcinomas.38,42,48,50–54
MET Amplification
MET amplification is the carcinogenic driver. Fluorescence in situ hybridization (FISH) can distinguish between the two based on the ratio of MET/CEP7 (centromeric chromosome 7). In polyploid, each copy of MET is associated with centromeres, and the MET/CEP7 ratio remains constant as the number of copies increases. In MET amplification, the MET/CEP7 ratio increased due to the increased copy number of the MET gene.55 Next generation sequencing can also be used to evaluate MET amplification and provide other potentially clinically relevant genomic alteration information.56
The MET amplification was reported in 2–5% of NSCLC.57,58 The high MET gene amplification (MET/CEP7 ≥ 5) is very rare, with an incidence of only 0.34%, and no other oncogenic driver genes were found in these patients compared to patients with low MET gene amplification (MET/CEP7 ratio < 5), which also suggests that high MET gene amplification may act as a carcinogenic driver.59 In addition, the incidence of MET amplification was higher in NSCLC patients treated with erlotinib/gefitinib, ranging from 5% to 22%.31,32,60 MET FISH-positive patients with advanced NSCLC have a poor overall survival.13,14,61 There is a correlation between high amplification of MET gene and high response rate of crizotinib.62 In a Phase I clinical trial (NCT00585195), a patient with advanced lung adenocarcinoma harboring high levels of MET amplification by FISH (MET/CEP7>5), and received crizotinib treatment, obtained a persistent partial response (PR).63 Preliminary results showed that 1 (16.7%) of 6 patients with moderate MET amplification (MET/CEP7 ratio >2.2 to <5) obtained PR; and 3 (50%) of 6 patients with high-level MET amplification (MET/CEP7 ratio >5) obtained PR. No PR was observed in patients with low MET amplification levels (MET/CEP7 ratio 1.8 to 2.2).64 Notably, Zhang et al reported a rapid response to crizotinib in a patient with lung adenocarcinoma, which showed an increase in MET copy number but a low MET/CEP7 ratio.65 Therefore, larger clinical trials are needed to assess the predictive value of MET gene amplification for targeted therapies.
Impaired MET Receptor Degradation
Impaired MET receptor degradation seems to be a mechanism for ligand-independent aberrant MET signaling.66 The HGF/SF binding to MET induces dimerization of the c-MET receptor and activation of downstream signaling pathways. Tyrosine 1003 (Y1003), contained in c-MET juxtamembrane domain encoded by METex14, is a direct binding site for c-Cbl, an E3 ubiquitin ligase. After c-Cbl binds to Y1003, the MET receptor is internalized into endosomes and ubiquitinated, and degraded by the lysosomal pathway.35 However, in NSCLC, nearly all of the METex14 skipping can delete Y1003, c-Cbl binding site, in the juxtamembrane domain.42 This leads to MET ubiquitination abrogation, increased MET protein stability, and impaired MET degradation. This induces ligand-independent MET activation.35,42,66,67 Besides METex14 skipping, mutant cells of Casitas B-lineage lymphoma (CBL), an E3 ubiquitin ligase, show decreased MET ubiquitination, increased MET expression, and higher sensitivity to MET inhibitor SU11274 than CBL wild-type cells, suggesting that CBL status maybe a potential biomarker for MET-targeted therapeutics in NSCLC.68
MET Fusion
The MET fusion gene consists of METs that are lacking in juxtamembrane regulatory sequence and different N-terminal partners. The genes that are currently found to be fused with MET include TPR, TRIM4, ZKSCAN1, PPFIB1, LRRFIP1, EPS15, DCTN1, PTPRZ1, NTRK1, CLIP2, TFG, HLA-DRB1, etc. These chimeric proteins are constitutively phosphorylated in a xenograft animal model and induce tumorigenesis.69 A case report described a patient with lung cancer with HLA-DRB1-MET fusion who obtained PR for 8 months after receiving crizotinib.70 As with the fusions described above, this chimeric MET contains a kinase domain without a juxtamembrane regulatory sequence. Another kinase fusion, KIF5B-MET, was found in a patient with lung adenocarcinoma. This fusion-driven MET activation is probably due to constitutive dimerization and is likely an operational target which can be inhibited by agents. It is similar to other fusions in lung cancer, such as ALK, ROS1, and RET.71
Overexpression of MET/p-MET
In addition to gene amplification, mutation or fusion, the MET pathway can also activate protein overexpression by upregulating the secretion signal of MET or HGF, inducing tumor transformation.72,73 The MET overexpression rate in unselected NSCLC ranges from 15% to 70%, which depends on the antibody assay and positive threshold.10,74–76 In addition to the total level of protein, ligand-activated MET that induces phosphorylation of the juxtamembrane domain can also be detected by phosphorylation-MET (p-MET). Specific expression of p-MET has been observed in approximately two-thirds of lung cancer samples.10,36 Previous study has reported the expression and prognosis value of MET, p-MET, and HGF in lung cancer patients (n=129).77 The high expression of two specific forms of p-MET, cytoplasmic expression of Y1003 (P=0.016; HR =1.86; 95% CI: 1.12–3.07), and nuclear expression of Y1365 (P=0.034; HR=1.70; 95% CI: 1.04–2.78) were negative prognostic factors for overall survival. Thus, the particular type of p-MET may be biomarkers for selecting patients who are likely to benefit from MET inhibitors.
Although the overexpression of MET protein is associated with poor prognosis in lung cancer, it was less effective as a predictive biomarker for targeted therapeutic efficacy.78 Previous studies have found that MET overexpression has higher sensitivity and negative predictive value;49,79 however, due to the significant intratumoral heterogeneity, immunohistochemical evaluation of MET overexpression remains challenging, limiting its use as a biomarker in the clinic.80,81 This difficulty in screening patients for MET-targeted therapy may also partly explain the failure of MET inhibitors in recent phase III clinical trials of NSCLC.82,83
Role of MET in Induction of EGFR-TKIs Resistance
MET amplification is a potential resistance pattern of EGFR-TKIs in NSCLC, accounting for 50–60% of the first- and second-generation EGFR-TKIs acquired resistance,31,32,84 accounting for 15–19% of the third-generation EGFR-TKIs acquired resistance.85,86 In 2018, ESMO reported two important studies on the mechanisms of acquired resistance to osimertinib.85,86 FLAURA and AURA2 study provided patients treated with first-line and second-line osimertinib, respectively. Second-generation sequencing was performed on patient plasma samples at disease progression and/or treatment discontinuation. The results showed that MET amplification was the most common acquired resistance mechanism to first-line (15% MET amplification, n=91) and second-line (19% MET amplification, n=73) treatment. In addition, increased circulating levels of HGF in tumor and stromal cells can lead to dysregulation of the MET pathway,87,88 which can also confer EGFR-TKIs resistance.87,89 In vitro and in vivo preclinical studies have shown that MET-targeted agents can reverse the resistance to EGFR inhibitors and restore sensitivity in this particular genetic context.32,90,91
In fact, synergistic effects of epidermal growth factor (EGF) and HGF on cell proliferation have been demonstrated in preclinical studies of NSCLC. When cells are stimulated by HGF and EGF, respectively, an increase in cell wrinkles can be observed, to form on the mobile cell surface. When these growth factors are combined, an additive effect can be observed,92 indicating that the combination of MET inhibitors and EGFR-TKIs may have synergistic anti-tumor effects. This provide a theoretical basis for the clinical application of combination targeted therapy in the treatment of patients with NSCLC, especially of patients with EGFR-TKIs resistance.
Therapeutics to Inhibit the HGF/c-Met Axis
Only patients with MET gene alterations may have an objective response (tumor regression) after receiving MET-targeted therapy, whereas targeting the HGF/MET axis in MET wild-type patients has little effect on cancer cell growth or on the efficacy of chemotherapy agents.93 In many cases, MET inhibitors have been reported to be effective in patients with NSCLC, who have high levels of MET amplification63,64 or METex14 skipping mutations.38,42,51,54,94–96 There are a variety of MET inhibitors, including small molecule TKIs and monoclonal antibodies (mAbs) against MET or its ligand HGF, for clinical studies of NSCLC. The results of clinical studies of c-MET inhibitors for NSCLC are shown in Table 1. Table 2 summarizes ongoing clinical studies on c-MET inhibitors for NSCLC.
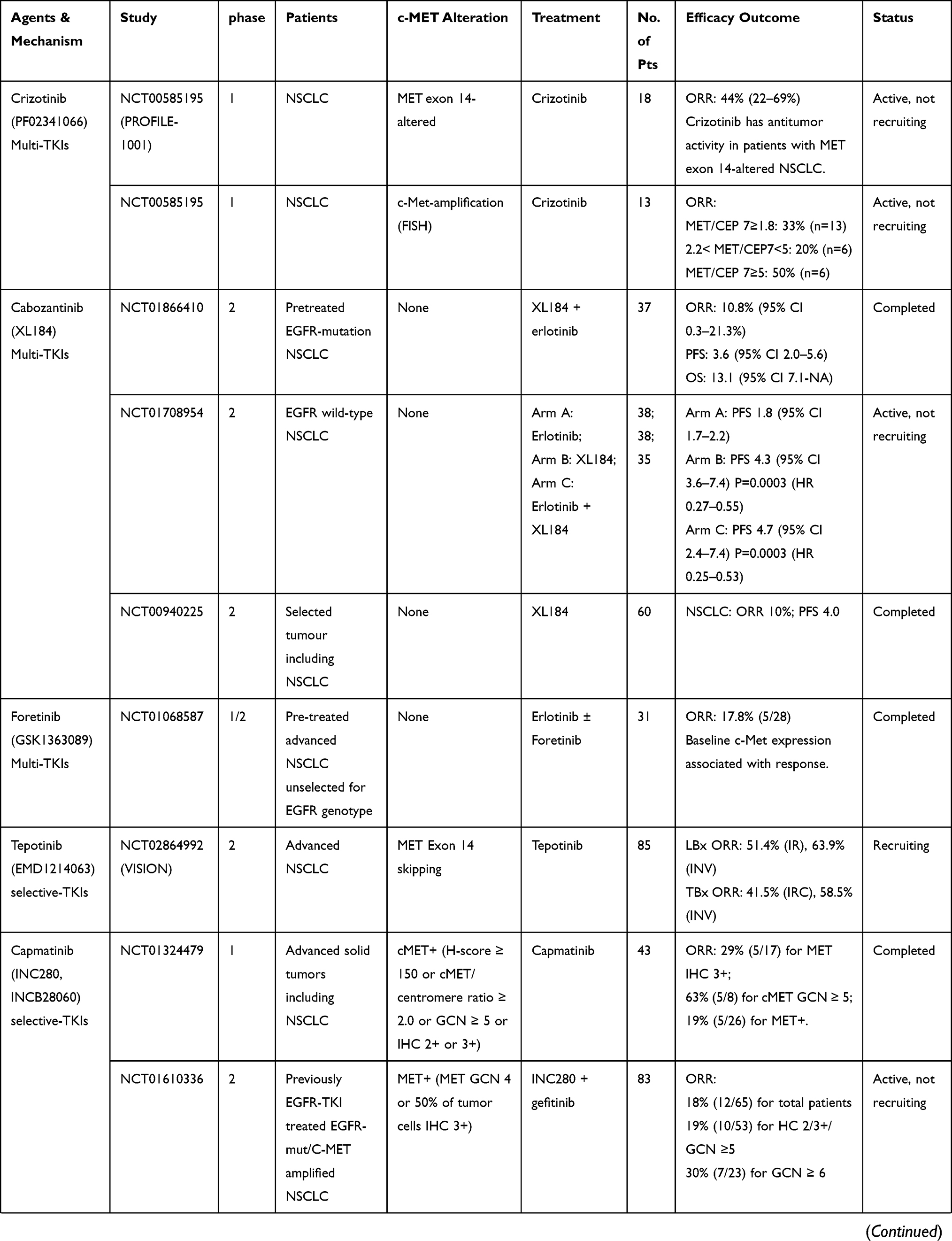 | 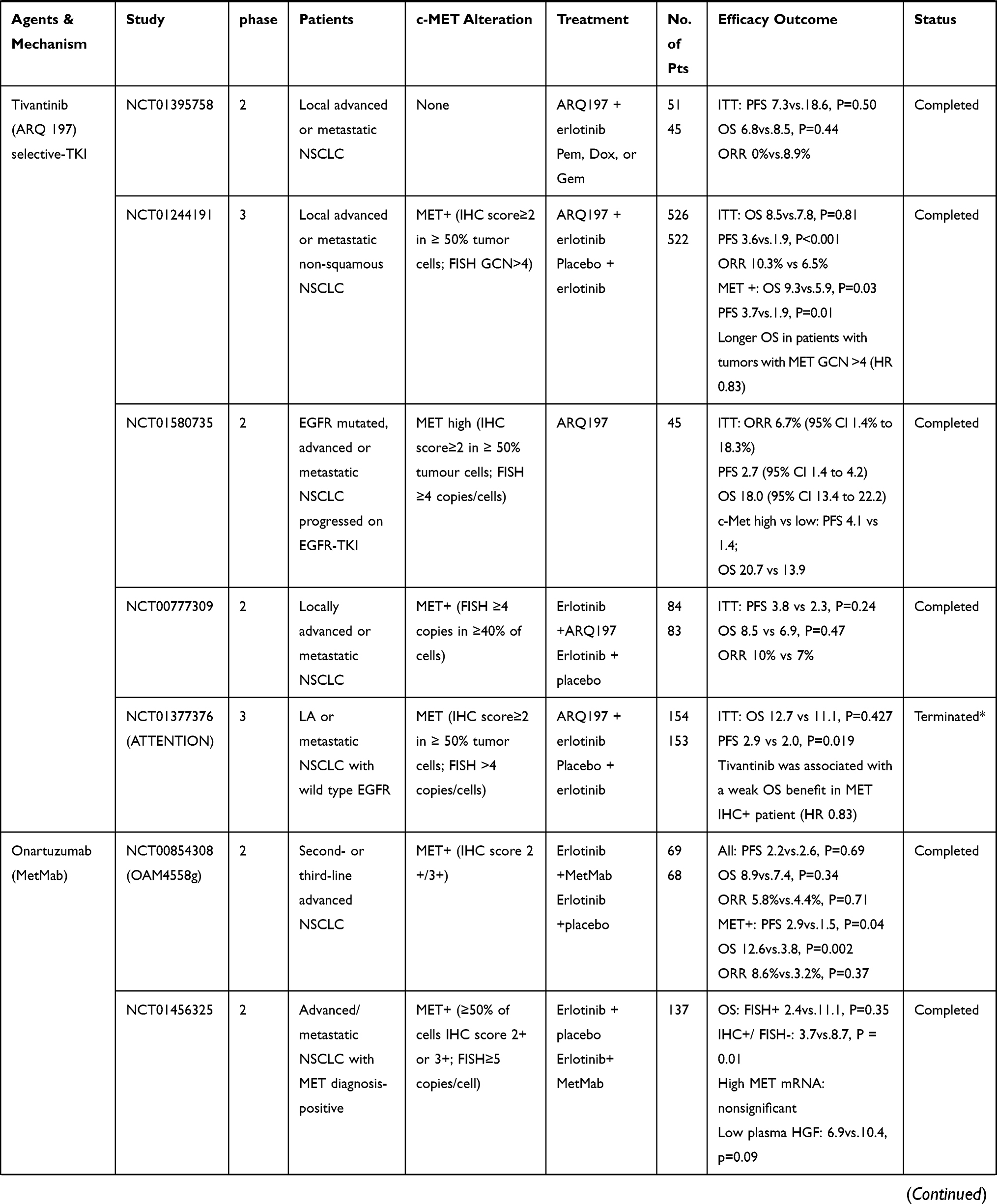 | 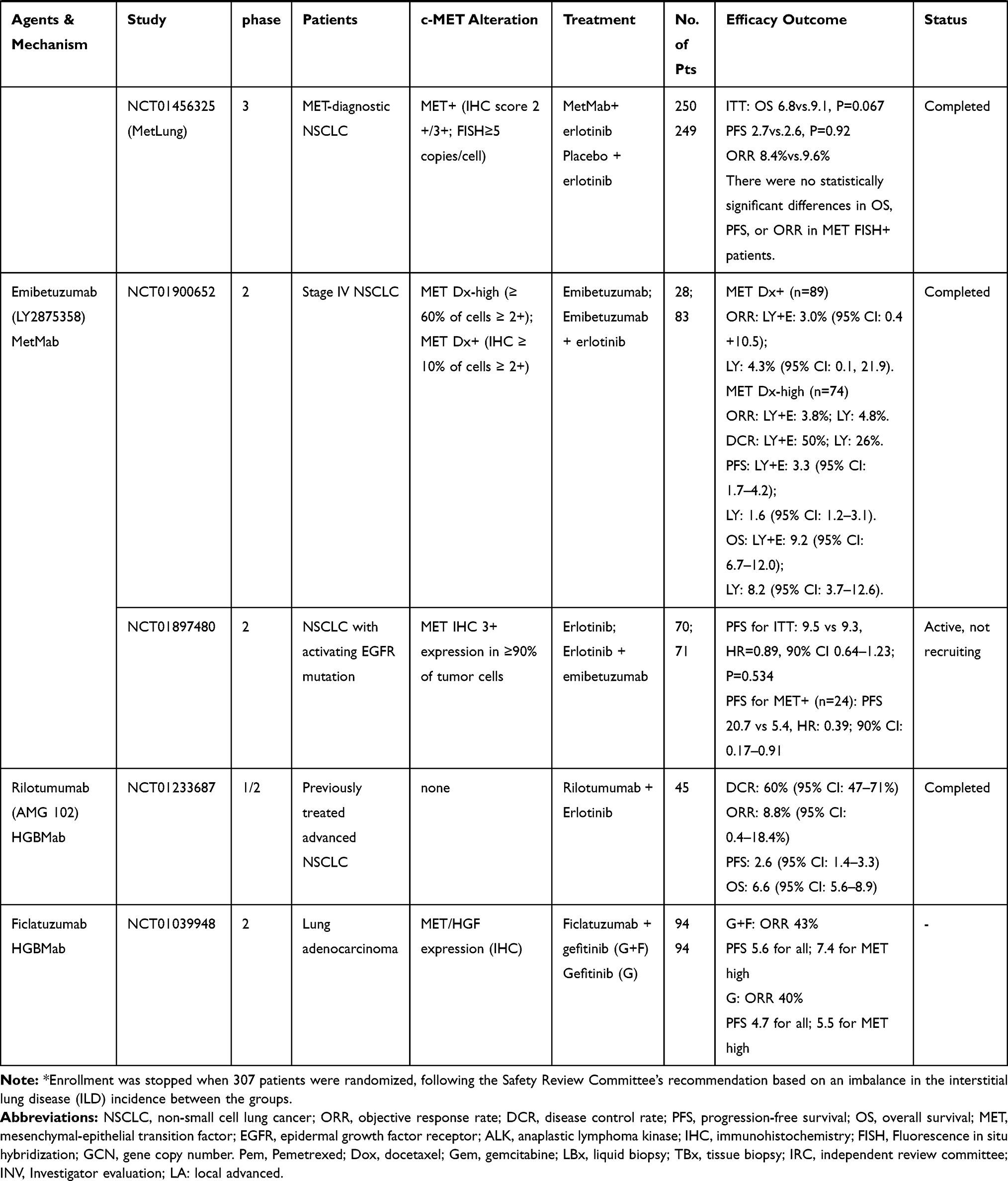 |  |
Table 1 Results of Clinical Trials with Targeted MET Inhibitors in NSCLC |
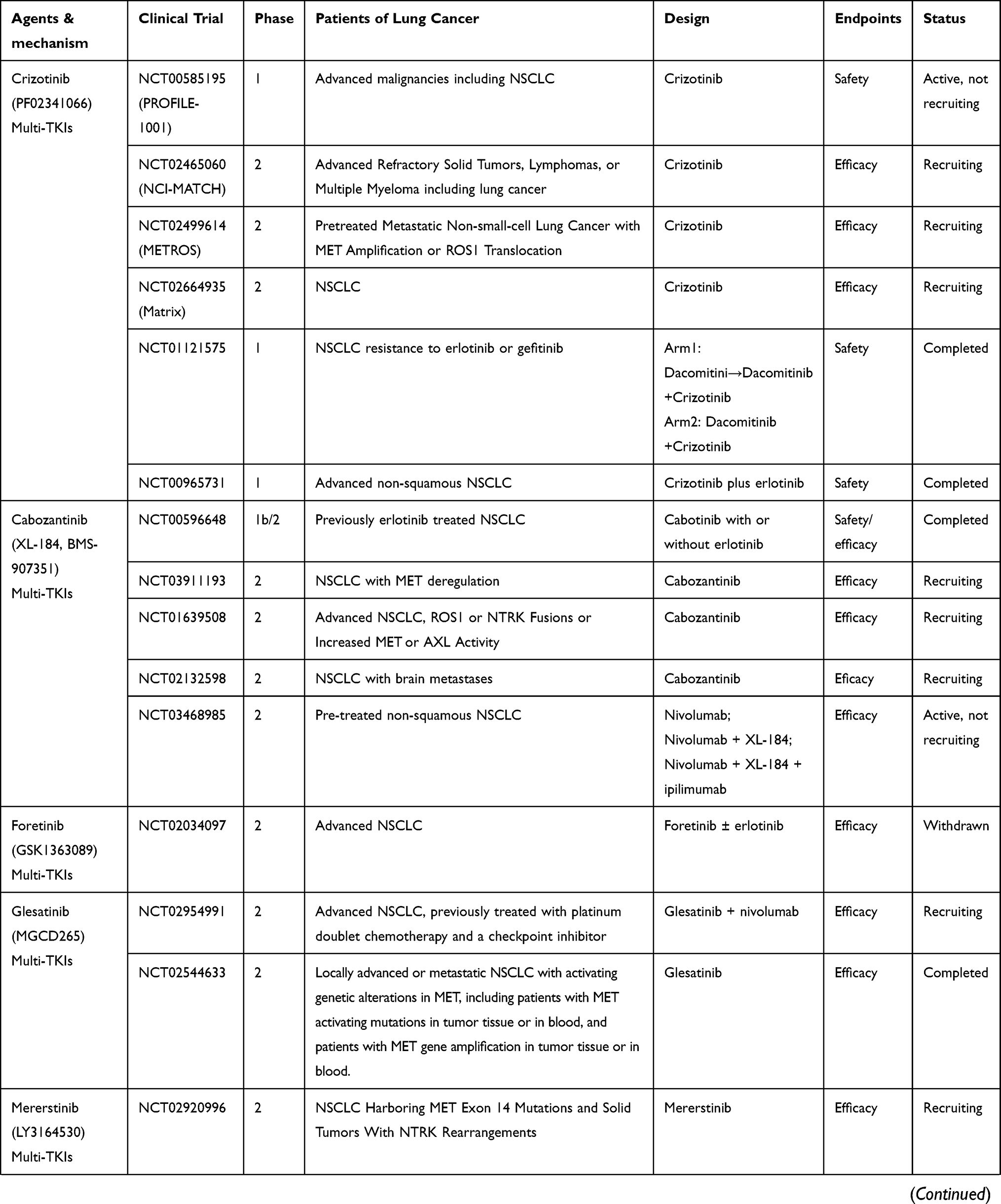 | 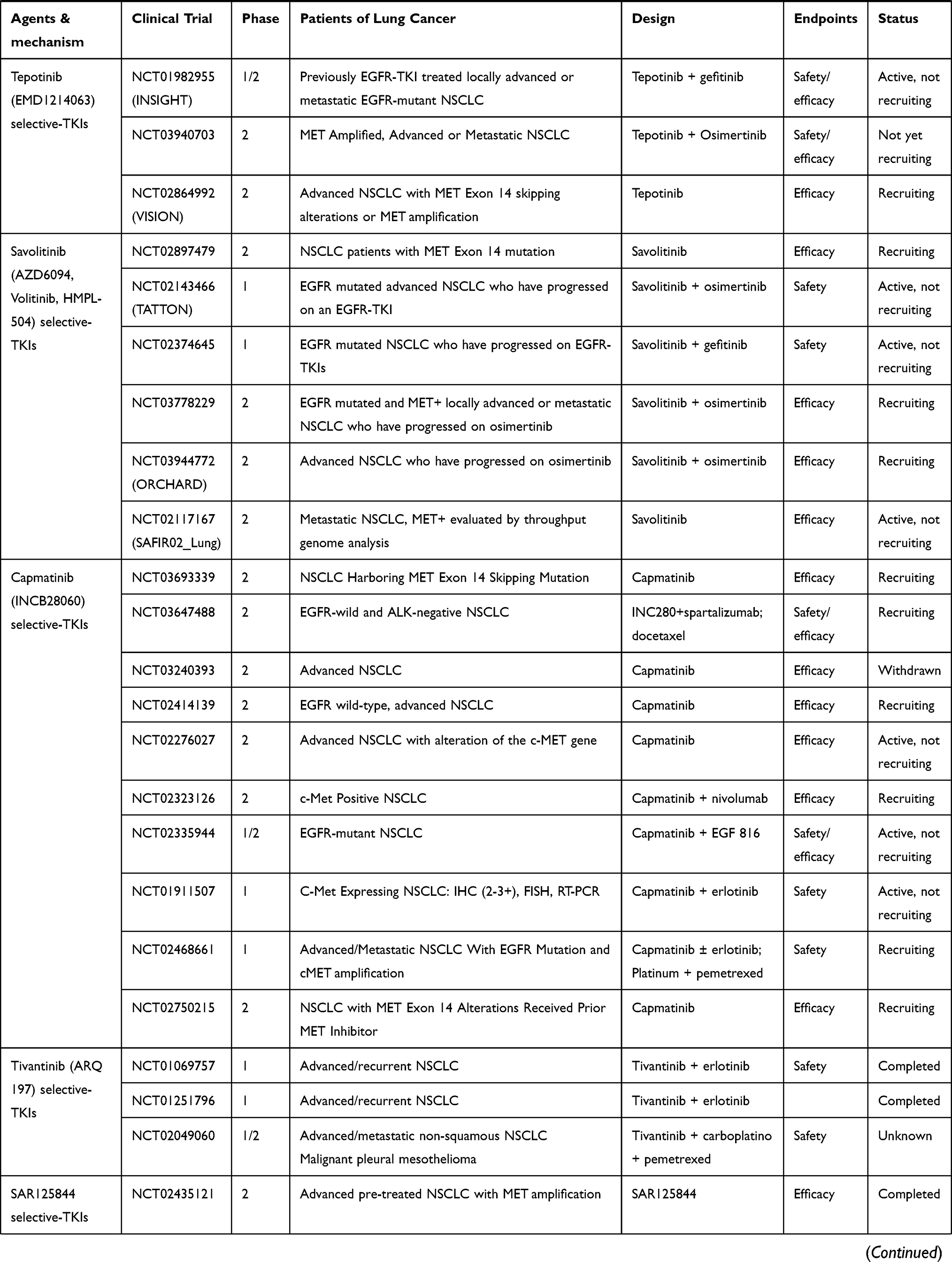 | 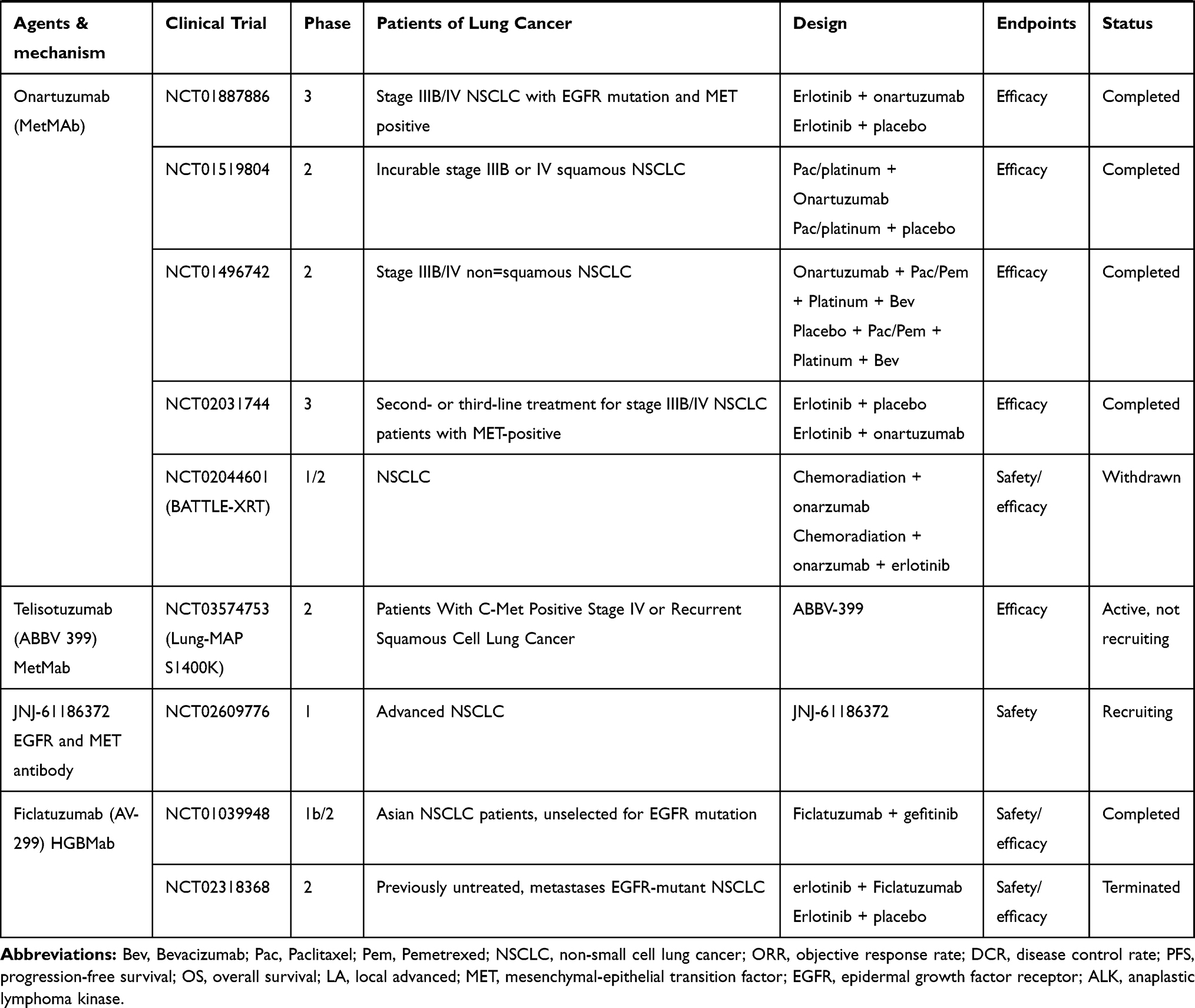 |
Table 2 Ongoing Clinical Studies with MET Inhibitors in Advanced NSCLC |
Multi-Kinase MET Inhibitors
Crizotinib (PF-02341066)
Crizotinib is an oral multi-target TKI that competes with c-Met tyrosine kinase domain for binding the adenosine triphosphate (ATP) site,30 preventing receptor activation and downstream signaling transmission, as well as competing for ATP binding sites with other kinases, including vascular endothelial growth factor receptor (VEGFR), ROS1, and EML4-ALK, etc.64,97,98 Crizotinib has been approved by the FDA for patients with advanced NSCLC harboring ALK or ROS1 fusion.99,100
In PROFILE-1001 study, of 18 patients with NSCLC harbored METex14 skipping mutations who received crizotinib, 8 patients obtained an objective response (44%; 95% CI: 22–69%);50 13 patients with c-MET amplification were divided into low (1.8–2.2), medium (>2.2 - <5), and high (≥5) FISH ratio grades. The results showed that 4 patients obtained an objective response (33%; 95% CI: 10–65%). One of the four was mid-level while three were of high-level amplification group64 A multicenter retrospective analysis showed that for patients with METex14 NSCLC, treatment with a MET inhibitor (including crizotinib) is associated with an improvement in overall survival. The median overall survival (mOS) of 34 metastatic patients, who had never received MET inhibitor therapy, was 8.1 months, while mOS of 27 metastatic patients who had received at least one MET inhibitor (including crizotinib, glesatinib, capmatinib, and ABBV-399), was 24.6 months. Treatment with MET inhibitors significantly prolonged these patients’ mOS (HR=0.11; 95% CI: 0.01–0.92; p = 0.04). The median progression-free survival (PFS) of 22 patients receiving crizotinib was 7.36 months.101 In addition, previous studies have also evaluated the efficacy and safety of crizotinib in combination with other targeted therapies in patients with NSCLC, including erlotinib102 and dacomitinib.103 However, combination therapy leads to many adverse events (43% of patients had grade 3 or 4 treatment-related adverse events [AEs]) and the efficacy was poor. With these poor results, there are no plans for further research.
Crizotinib has clinical activity in patients with NSCLC with METex14 and MET amplification (FISH ratio grades > 5), indicating that METex14 evaluated by NGS and MET amplification evaluated by FISH appears to be biomarkers for crizotinib treatment in patients with NSCLC. This needs further confirmation. Besides, due to the adverse effects and limited efficacy, combination of crizotinib with other targeted agents is not recommended.
Cabozantinib (XL-184, BMS-907351)
Cabozantinib is another multi-kinase inhibitor targeting c-Met, VEGFR1, VEGFR2, VEGFR3, RET, TIE2, FLT-3 and KIT, which blocks a variety of precancerous signaling pathways.104 It has significant oral bioavailability and blood-brain barrier penetration.
A phase I clinical trial analyzed the efficacy and safety of cabozantinib in patients with various solid tumors including NSCLC.105 The results showed that in NSCLC patients (n=60), the objective response rate was 10%, and the median PFS was 4.0 months. Another study evaluated the efficacy of cabozantinib in combination with erlotinib in patients with EGFR-mutant NSCLC who were resistant to EGFR-TKIs.106 The results showed that the objective response rate was 10.8% (95% CI 0.3–21.3%), median PFS was 3.6 months (95% CI: 2.0–5.6), and the mOS was 13.1 months (95% CI 7.1-NA). Recently, a randomized Phase II trial evaluated the efficacy of erlotinib monotherapy, cabozantinib monotherapy, and erlotinib, in combination with cabozantinib in patients with EGFR wild-type NSCLC.107 There were 111 patients (38, 38, 35) included in the preliminary analysis. The results showed a significant improvement in the median PFS between the cabozantinib group (4.3 vs 1.8; HR: 0.39, p = 0.0003) and the combination treatment group (4.7 vs 1.8; HR: 0.37, p = 0.0003) compared with the erlotinib group, with tolerable safety.
In clinical trials related to cabozantinib, MET alterations have not been evaluated, but mainly focused on the EGFR status. The theoretical basis is that MET alterations, especially MET amplification may be a potential acquired resistance mechanism to EGFR-TKIs. All these results indicate that cabozantinib alone or in combination with EGFR-TKIs has a potential research prospect for NSCLC patients with EGFR wild-type or resistance to EGFR-TKIs.
Foretinib (XL-880)
Foretinib is a multi-target kinase inhibitor whose targets include c-Met, ROS-1, RON, VEGFR2, KIT, AXL, TIE2, and PDGFR.108 It can also block proliferation and anti-angiogenesis. Recently, a Phase I clinical trial evaluated the efficacy and safety of foretinib in combination with erlotinib in patients with advanced NSCLC who progressed after chemotherapy.108 The results showed that the objective response rate (ORR) of 28 evaluable patients (including wild-type EGFR patients) was 17.8% (5/28). Among the 18 samples, regardless of the EGFR gene status, the baseline MET immunohistochemistry (IHC) expression correlated with the ORR.
MET status evaluated by IHC may be a biomarker for foretinib combined with erlotinib treatment in NSCLC patients who have received chemotherapy. This need further confirmation.
Glesatinib (MGCD265)
Glesatinib (MGCD265) is an oral type II kinase inhibitor targeting c-Met, VEGFR1, VEGFR2, VEGFR3, TIE2, and RON.4 In an ongoing phase I study,109 glesatinib showed initial anti-tumor activity in patients with advanced solid tumors with MET positive or AXL rearrangements. All three NSCLC patients with MET alterations (two METex14 mutations, one gene copy number [GCN] increase) showed significant tumor shrinkage at the first assessment.
For NSCLC patients with METex14, glasetinib has shown very promising prospects in phase I clinical trials. This also indicates that METex14 is a potential biomarker. This need to be further confirmed by larger samples.
Selective MET Inhibitors
Capmatinib (INCB28060)
Capmatinib is a highly selective ATP-competitive c-MET inhibitor. It is 10,000 times more selective for c-Met than other kinases, and can inhibits MET activity at picomolar concentrations.110 A phase I clinical trial have evaluated the efficacy and safety of capmatinib in NSCLC patients with c-MET positive (H-score ≥ 150; or c-MET/centromere ratio ≥ 2.0; or GCN ≥ 5; or IHC 2+ or 3+) and EGFR wild type.111 A total of 43 patients were included in the NSCLC expansion group. The results showed that in c-MET-positive, cMET IHC 3+, and cMET GCN ≥ 5 patients, the ORR were 19% (5/26), 29% (5/17), and 63% (5/8), respectively. Another Phase I study evaluated the efficacy and safety of capmatinib as a single-agent therapy in patients with advanced solid tumors (including a group of EGFR wild-type MET+ NSCLC patients; n = 55).111 Preliminary antitumor activity was observed in NSCLC patients with high MET gene copy number (GCN ≥ 6; ORR=47%) or MET overexpression (IHC 3+; ORR=24%). A phase II study evaluated the efficacy and safety of a combination of capmatinib and gefitinib in patients with EGFR-mutant NSCLC who progressed after gefitinib treatment.112 The results showed that in 65 evaluable patients, the ORR was 18% and the disease control rate (DCR) was 80%. Patients with MET amplification (GCN ≥ 6) obtained an ORR of up to 30%.
A subgroup of a phase II study evaluated the efficacy of capmatinib in METex14+ and EGFR/ALK-negative NSCLC patients (GEOMETRY mono-1), which included a total of six cohorts. Preliminary results showed that for pre-treatment patients with NSCLC with METex4 skipping mutation (cohort 4), capmatinib has a certain ORR and tolerable toxicity, whereas for newly diagnosed patients (cohort 5b) ORR was higher. In 2019, ASCO reported updated data. As of April 15, 2019, a total of 97 patients (cohort 4: n=69; cohort 5b: n=28) with NSCLC with METex14 skipping mutations were enrolled. The results showed that for patients in cohort 4, the ORR was 40.6% (95% CI: 28.9–53.1%), and the median PFS was 5.42 months (95% CI: 4.17–6.97). For patients in cohort 5b, the ORR was 67.9% (95% CI: 47.6–84.1), and the median PFS was 9.69 months (95% CI: 5.52–13.86). Toxcity was tolerable. In this study, 13 patients had baseline brain metastases, and the intracranial ORR was 54% (7/13), of these, 4 patients achieved complete response (CR); the intracranial DCR was 92% (12/13). The response time of internal lesion was consistent with the response time of the extracranial lesion.
These results indicate that MET amplification and METex14 may be potential biomarkers for the treatment of capmatinib in patients with NSCLC. Capmatinib appears to be more effective in newly diagnosis NSCLC patients with METex14.
Tivantinib (ARQ197)
Tivantinib is a non-ATP competitive c-MET inhibitor. A randomized, double-blind, open-label phase II trial evaluated the efficacy and safety of erlotinib in combination with tivantinib versus erlotinib in combination with placebo in patients with advanced NSCLC. A total of 167 patients were enrolled: the proportion of patients harboring EGFR mutations in the combination and standard treatment groups was 10% and 18%, respectively, and the proportion of patients with MET GCN ≥ 4 was 26% and 26.5%, respectively. Compared to standard treatment group, the combination treatment group had a value-advantaged ORR (10% vs 7%), median PFS (3.8 vs 2.3 months; HR = 0.81; p = 0.24), and mOS (8.5 vs 6.9 months; hazard ratio [HR] = 0.87; p = 0.47), but there was no statistical difference. An exploratory analysis of non-squamous NSCLC showed a benefit trend for PFS (HR = 0.71) and overall survival (HR = 0.72) in the combination group compared with the standard treatment group. Subgroup analysis showed that patients with EGFR wild-type (HR = 0.70), KRAS mutation (HR = 0.76), and MET FISH-positive (>5, HR = 0.45) had an advantage with PFS. In the two groups, both were safely tolerated.113
The MARQUEE phase III study evaluated the efficacy and safety of erlotinib in combination with tivantinib vs erlotinib in combination with placebo in patients with pre-treated NSCLC.114 A total of 1048 patients were enrolled. Interim analysis showed that combination therapy improved median PFS (3.6 vs 1.9 months; HR-0.74; 95% CI: 0.62–0.89; p < 0.001) but did not improve mOS (8.5 vs 7.8 months; HR=0.98; 95% CI: 0.84–1.15) in intention-to-treat (ITT) patients. Exploratory subgroup analysis showed that the combination therapy prolonged mOS (HR=0.70; 95% CI: 0.49–1.01) in patients with high MET expression (MET GCN ≥ 4). This result was confirmed by a phase III study in Asian patients with previously treated stage IIIB/IV non-squamous NSCLC harboring wild-type EGFR (ATTENTION study).115 This study began enrollment with a target of 460 patients. Due to an imbalance in the incidence of interstitial lung disease (ILD) between the 2 groups, 307 patients with NSCLC were finally randomized to receive either erlotinib in combination with tivantinib or erlotinib in combination with placebo. ILD occurred in 6 patients (0 deaths) and 14 patients (3 deaths) in the placebo and tivantinib group, respectively. In ITT population, compared to placebo group, the tivantinib group had significantly prolonged PFS (2.9 months vs 2.0 months; HR = 0.719; p = 0.019), and had a value-advantaged overall survival (12.7 months and 11.1 months; HR = 0.891; p = 0.427).
These results indicate that for patients with NSCLC, MET amplification evaluated by FISH appears to be a potential biomarker for the treatment of tivantinib in combination of erlotinib.
Savolitinib (Volitinib, AZD6094, HMPL-504)
Savolitinib is a selective small molecule MET inhibitor that blocks c-Met activity in an ATP-dependent way. It has high selectivity for MET.116 A phase II clinical trial evaluated the efficacy and safety of savolitinib in combination with osimertinib in patients with NSCLC with positive T790M mutations resistance to osimertinib.117 Preliminary results showed that of the seven patients who received combination therapy, two obtained PR, with tolerable safety.
Osimertinib combined with savolitinib may be a therapy strategy for patients with NSCLC who are resistant to osimertinib. These may show better clinical efficacy in patients with MET alterations after osimertinib resistance. However, further confirmation is needed.
Tepotinib (EMD1214063)
Tepotinib is an oral, ATP-competitive, highly selective c-MET TKI. Its selectivity to c-Met is 1000 times that of other kinases.118 A phase II study evaluated the efficacy and safety of epotinib in patients with NSCLC harboring METEx14 mutations and EGFR/ALK wild-type. The primary end point was the ORR assessed by the independent review committee (IRC) while the secondary endpoint was ORR and safety evaluated by the investigator (INV). As at the time of the data analysis, 85 patients were enrolled (55 and 52 patients in the liquid biopsy [LBx] and biopsy group [TBx], respectively). Thirty-five patients in the LBx group were evaluated for efficacy, the ORR, evaluated by IRC, was 51.4% and 63.9% evaluated by INV. Forty-one patients in the TBx group underwent efficacy evaluation, the ORR was 41.5% evaluated by IRC and 58.5% evaluated by INV. The toxicity was tolerable.
These results indicate that for patients with NSCLC, METex14 maybe a biomarker for tepotinib treatment.
Anti-MET Antibodies
Onartuzumab (MetMAb)
Onartuzumab is a fully humanized, c-MET monoclonal antibody that blocks the alpha chain binding of the HGF to the c-MET ligand binding domain.119 In a phase II study,120 pre-treated NSCLC patients were randomized into two groups, one receiving onartuzumab in combination with erlotinib and the other receiving placebo plus erlotinib. A total of 137 patients were enrolled in this study, of which 26 patients (23%) harbored KRAS mutations while 13 patients (12%) harbored EGFR mutations. The results showed that compared with placebo group, the onartuzumab group showed no prolonged PFS (HR=1.09; P=0.69) and overall survival (HR=0.80; P=0.34) in the ITT population. In MET-positive patients (n=66), PFS (HR=0.53; p = 0.04) and overall survival (HR=0.37; p = 0.002) were significantly prolonged in the onartuzumab group. However, in MET-negative patients (n=62), PFS (HR=1.82, p=0.05) and overall survival (HR=1.78, p=0.16) were poor in the onartuzumab group. MET-positive patients in the placebo group had a worse PFS (HR=1.71; p=0.06) and overall survival (HR=2.61; p=0.004) than MET-negative patients. Thus, in MET-positive people, onartuzumab plus erlotinib can improve PFS and overall survival, while in MET-negative patients, the combination therapy is even worse. Therefore, a larger randomized trial included 499 pre-treated patients with MET-positive NSCLC.121 They were randomized (1:1) to receive erlotinib in combination with placebo or erlotinib in combination with onartuzumab. The results showed that patients in the onartuzumab group did not have improved overall survival (6.8 vs 9.1 months; HR=1.27; p = 0.067), PFS (2.7 vs 2.6 months; HR=0.99; p=0.92), and overall RR (8.4% vs 9.6%; p = 0.63). The trial has been terminated.
Larger clinical trials have not confirmed better efficacy of onartuzumab combined with erlotinib in patients with MET-positive NSCLC. The screening of biomarkers may need to be explored.
Emibetuzumab (LY2875358)
Emibetuzumab is a c-MET monoclonal antibody that blocks the binding of HGF and c-MET, leading to internalization and degradation of c-MET, and prevention of signal transmission. A phase II clinical trial (NCT01900652) evaluated the efficacy and safety of emibetuzumab with or without erlotinib in patients with MET-positive stage IV NSCLC.122 A total of 111 patients were enrolled in this study, of which 28 patients received emibetuzumab monotherapy, and 83 patients received emibetuzumab plus erlotinib. The results showed that in patients with MET Dx+ (≥ 10% of cells ≥ 2+ by IHC, n=89) the ORR of monotherapy group was 4.3% (95% CI: 0.1–21.9%), and the ORR of the combination treatment group was 3.0% (95% CI: 0.4–10.5%). In MET Dx-high population (≥ 60% of cells ≥ 2+; n = 74), the combination treatment group had longer PFS (3.3 vs 1.6 months) and overall survival (9.2 vs 8.2 months), and lower ORR (3.8% vs 4.8%) than the monotherapy group. Another phase II clinical trial (NCT01897480) evaluated the efficacy and safety of erlotinib with or without emibetuzumab in patients with advanced NSCLC harboring EGFR mutations.123 A total of 141 patients were enrolled, of which 71 patients were treated with combination therapy and 70 patients received erlotinib monotherapy. The results showed that in the ITT population, the combination treatment group had advantaged value PFS (9.5 vs 9.3 months; HR=0.89; 90% CI 0.64–1.23; P=0.534), with no statistical difference. Exploratory analysis showed that in patients with MET high expression (≥ 90% of cells MET IHC 3+; n = 24), the combination treatment significantly prolonged PFS (20.7 vs 5.4 months; HR=0.39; 90% CI: 0.17–0.91) compared to emibetuzumab monotherapy. In the rest of the population, PFS was not statistically different (HR=1.1; 90% CI: 0.7–1.7).
These results indicate that MET IHC status may be a potential biomarker for the treatment of emibetuzumab in patients with NSCLC.
Telisotuzumab (ABBV 399)
Telisotuzumab is a novel anti-c-MET antibody drug conjugate that can be coupled to monomethyl staphylococin E (MMAE), which mediates killing requiring a threshold level of c-MET expression.124 In a phase I study, 16 patients with MET-positive (IHC H score ≥ 150) NSCLC were treated with ABBV-399.125 The results showed that 3 patients (18.8%; 95% CI: 4.1%-45.7%) obtained PR. The median response duration time was 4.8 months, and the median PFS was 5.7 months (95% CI, 1.2–15.4 months). The toxicity was tolerable.
Telisotumumab has certain effect on NSCLC patients with MET IHC positive, further confirmation is needed.
JNJ-61186372
JNJ-372 can block ligand binding by binding to EGFR and MET, promote receptor degradation, and trigger antibody-dependent cytotoxicity in the EGFR mutant (EGFRm) NSCLC model.126 A phase I study evaluated the efficacy and safety of JIN-61186372 in patients with advanced NSCLC harboring EGFR mutations. In 2019, ASCO congress reported the interim results. Up to the cut-off data analysis, a total of 116 patients were enrolled in the study, of which 97% harbored EGFR mutations. In 88 evaluable patients, 28% (25/88) achieved PR. In 47 patients who were resistant to third generation EGFR-TKI, 21.3% (10/47) achieved PR (6 confirmed), 4 of whom were C797S, 1 was MET amplification, and 5 had no identifiable EGFR/MET gene alterations. In 20 patients with EGFR 20ins, 30% (6/20) achived PR (3 confirmed). The toxicity was tolerable. Patients with resistance to third generation EGFR-TKI, including C797S, MET amplification, and exon 20ins, all had initial responses, and research on expanded doses are ongoing.
JNJ-61186372 has clinical activity in NSCLC patients who harbor EGFR mutation or who are resistant to the third-generation EGFR-TKI, regardless of whether or not the cause of resistance is due to MET amplification.
Anti-HGF Antibodies
Rilotumumab (AMG-102)
Rilotumumab is a fully humanized IgG2 monoclonal antibody that binds to the HGFβ chain and inhibits HGF binding to c-MET.127,128 A Phase 1/2 study evaluated the efficacy and safety of rilotumumab in combination with erlotinib in patients with NSCLC regardless of the EGFR status.129 In all evaluable patients (n=45), the ORR was 8.8% (90% CI: 0.4–18.4%), the DCR was 60% (95% CI: 47–71%), the median PFS was 2.6 months (90% CI: 1.4–3.3 months) and the mOS was 6.6 months (90% CI: 5.6–8.9 months). For patients with wild-type EGFR (n=33), the DCR was 60.6% (90% CI: 46.3–73.3%), the median PFS was 2.6 months (90% CI: 1.4–2.7 months), and the mOS was 7.0 months (90% CI: 5.6–13.4 months).
The results suggested that blocking HGF combined with EGFR-TKI may increase the efficacy of NSCLC patients with EGFR wild-type.
Ficlatuzumab (AV-299)
Ficlatuzumab is a high-affinity humanized IgG1 monoclonal antibody directed against HGF that inhibits HGF-induced c-MET signaling pathway by blocking HGF/cMET binding.130 A randomized phase II clinical trial evaluated the efficacy of gefitinib with or without ficlatuzumab in patients with NSCLC.131 In the EGFR mutations and low c-MET expression subgroup, patients receiving ficlatuzumab plus gefitinib had an improvement in ORR (41% vs 22%) and median PFS (11 vs 5.5 months). However, in the ITT population, the combination therapy did not significantly improve ORR (40% vs 38%, P=0.77), PFS (5.6 vs 4.7 months, HR=0.94, P=0.67) and overall survival (24.7 vs 21.8 months, HR=0.98, P=0.91) compared with the gefitinib monotherapy group.
Ficlatuzumab plus gefitinib can improve the clinical efficacy in NSCLC patients with EGFR mutations and low c-MET expression, indicating that MET expression maybe a biomarker for the ficlatuzumab treatment.
Immunotherapy
For patients with NSCLC with MET alterations, immunotherapy can be administered after exhaustion of the targeted therapy and chemotherapy. When to start immunotherapy for these patients; however, remains to be explored. A study retrospectively analyzed the clinical activity of immunotherapy in patients with NSCLC with oncogenic driver alterations, including 36 patients with MET alterations (MET amplification n=13, METex14 skipping mutation n=23).132 They were all received immunotherapy in different treatment lines. In MET alteration subgroup analysis, ORR, median PFS and OS were 16%, 3.4 months and 18.4 months, respectively. Another study retrospectively included 147 patients to evaluate the PD-L1 expression, tumor mutational burden (TMB), and clinical activity of immunotherapy in patients with METex14 lung cancers. The PD-L1 positive (PD-L1 ≥ 1%) rate was 63% (57/111), while TMB was lower in tumors with METex14 alterations compared with unselected NSCLCs. In 24 patients who received immunotherapy, the ORR was 17%, and the median OS was 18.2 months. The median PFS was 1.9 months for the 21 patients assessable for this end point.133 Therefore, compared with the targeted agents and chemotherapy, immunotherapy had a poor clinical activity in MET alteration patients with NSCLC. Single-agent immunotherapy should not be considered before receiving targeted therapies and chemotherapy. The role of combination treatment (ie immunotherapy and chemotherapy, immunotherapy and targeted therapy, immunotherapy and antiangiogenic agents, or combination immunotherapy) in MET alteration patients with NSCLC need further investigation.
Future Challenges
In recent phase III clinical trials, the efficacy of MET inhibitors to treat NSCLC was disappointed.114,115,134 Whether the target agents were inhibiting the c-MET receptor itself or inhibiting its ligand HGF, they were not effective as monotherapy for patients with unselected NSCLC. From the current research results, the subgroups of patients with NSCLC harboring MET gene alterations, especially MET amplification or METex14 skipping mutation, respond to MET inhibitors. Therefore, it is important to find appropriate biomarkers for selecting patients. In order to improve the applicability of biomarkers, it is important to set two thresholds that distinguish between MET positive and negative or between high and low HGF, and to choose the appropriate method to achieve this. To solve this problem, there is need for large data analysis for the detection methods and treatment effects, to establish standards that meet the MET activation status, and determine reliable thresholds to achieve effective patient stratification and clinical decision making.
In fact, MET inhibitors cannot block the growth of tumors without MET gene alterations, which does not mean that MET inhibitors are not effective in this case. When MET inhibitors are combined with kinase inhibitors in the upstream or downstream signaling pathways, they can inhibit parallel kinase signaling from other receptors, which may improve the clinical outcomes. However, the toxicity of targeted combination therapies could be considerable. In addition, since MET is related to the properties of activated dendritic cell tolerogenic, MET inhibitors may act synergistically with immunological checkpoint inhibitors such as PD-1 antibodies to restore the immune stimulating microenvironment, and release the tumoricidal activity of cytotoxic T cells. Other combination therapies include HGF/c-Met inhibitors plus inhibitors that inhibit the vitality or function of stromal cells such as tumor-associated fibroblasts, endothelial cells, or macrophages.
Conclusion
The MET/HGF axis is a promising therapeutic target in advanced NSCLC. A variety of MET inhibitors have been developed, some of which have entered phase III clinical trials. However, the results of phase III clinical trials to date have been disappointing. Various mechanisms of MET activation in lung cancer, including MET/HGF overexpression, MET gene alterations (such as mutations, amplification, translocation, or transcriptional disorders), and impaired MET degradation provide a range of potential biomarkers. The challenge now is to use a range of agents currently under development to determine which biomarkers are most likely to select the appropriate patient for MET-targeted therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. Ca-Cancer J Clin. 2017;67:7–30. doi:10.3322/caac.21387
2. Burdett S, Stephens R, Stewart L, et al. Chemotherapy in addition to supportive care improves survival in advanced non-small-cell lung cancer: a systematic review and meta-analysis of individual patient data from 16 randomized controlled trials - NSCLC meta-analyses collaborative group. J Clin Oncol. 2008;26:4617–4625.
3. Tsao AS, Scagliotti GV, Bunn PA, et al. Scientific advances in lung cancer 2015. J Thoracic Oncol. 2016;11:613–638. doi:10.1016/j.jtho.2016.03.012
4. Birchmeier C, Birchmeier W, Gherardi E, et al. Met, metastasis, motility and more. Nature Rev Mol Cell Biol. 2003;4:915–925. doi:10.1038/nrm1261
5. Blumenschein GR, Mills GB, Gonzalez-angulo AM. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J Clin Oncol. 2012;30:3287–3296. doi:10.1200/JCO.2011.40.3774
6. Rosario M, Birchmeier W. How to make tubes: signaling by the Met receptor tyrosine kinase. Trends Cell Biol. 2003;13:328–335. doi:10.1016/S0962-8924(03)00104-1
7. Gentile A, Trusolino L, Comoglio PM. The Met tyrosine kinase receptor in development and cancer. Cancer Metast Rev. 2008;27:85–94. doi:10.1007/s10555-007-9107-6
8. Zhang YW, Su YL, Volpert OV, et al. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc Natl Acad Sci USA. 2003;100:12718–12723. doi:10.1073/pnas.2135113100
9. Olivero M, Rizzo M, Madeddu R, et al. Overexpression and activation of hepatocyte growth factor scatter factor in human non-small-cell lung carcinomas. Brit J Cancer. 1996;74:1862–1868. doi:10.1038/bjc.1996.646
10. Ma PC, Tretiakova MS, MacKinnon AC, et al. Expression and mutational analysis of MET in human solid cancers. Gene Chromosome Canc. 2008;47:1025–1037. doi:10.1002/gcc.20604
11. Beau-faller M, Ruppert AM, Voegeli AC, et al. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naive cohort. J Thoracic Oncol. 2008;3:331–339. doi:10.1097/JTO.0b013e318168d9d4
12. Masuya D, Huang C, Liu D, et al. The tumour-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non-small-cell lung cancer patients. Brit J Cancer. 2004;90:1555–1562. doi:10.1038/sj.bjc.6601718
13. Cappuzzo F, Marchetti A, Skokan M, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009;27:1667–1674. doi:10.1200/JCO.2008.19.1635
14. Dimou A, Non L, Chae YK, et al. MET gene copy number predicts worse overall survival in patients with Non-Small Cell Lung Cancer (NSCLC); A systematic review and meta-analysis. PLoS One. 2014;9.
15. Guo BP, Cen H, Tan XH, et al. Prognostic value of MET gene copy number and protein expression in patients with surgically resected non-small cell lung cancer: a meta-analysis of published literatures. PLoS One. 2014;9.
16. Christensen JG, Zou HY, Arango ME, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6:3314–3322. doi:10.1158/1535-7163.MCT-07-0365
17. Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol. 2013;31:1089–1096. doi:10.1200/JCO.2012.43.9422
18. Peters S, Adjei AA. MET: a promising anticancer therapeutic target. Nat Rev Clin Oncol. 2012;9:314–326. doi:10.1038/nrclinonc.2012.71
19. Basile JR, Afkhami T, Gutkind JS. Semaphorin 4D/Plexin-B1 induces endothelial cell migration through the activation of PYK2, Src, and the phosphatidylinositol 3-kinase-Akt pathway. Mol Cell Biol. 2005;25:6889–6898. doi:10.1128/MCB.25.16.6889-6898.2005
20. Maroun CR, Naujokas MA, Holgado-madruga M, et al. The tyrosine phosphatase SHP-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream from the Met receptor tyrosine kinase. Mol Cell Biol. 2000;20:8513–8525. doi:10.1128/MCB.20.22.8513-8525.2000
21. Lee YY, Kim HP, Kang MJ, et al. Phosphoproteomic analysis identifies activated MET-axis PI3K/AKT and MAPK/ERK in lapatinib-resistant cancer cell line. Exp Mol Med. 2013;45:e64. doi:10.1038/emm.2013.115
22. Scagliotti GV, Novello S, von Pawel J. The emerging role of MET/HGF inhibitors in oncology. Cancer Treat Rev. 2013;39:793–801. doi:10.1016/j.ctrv.2013.02.001
23. Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011;3:S7–S19. doi:10.1177/1758834011422556
24. Finocchiaro G, Toschi L, Gianoncelli L, et al. Prognostic and predictive value of MET deregulation in non-small cell lung cancer. Ann Transl Med. 2015;3:83.
25. Birchmeier C, Birchmeier W, Gherardi E, et al. Met, metastasis, motility and more. Nat Rev Mol Cell Bio. 2003;4:915–925. doi:10.1038/nrm1261
26. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Bio. 2010;11:834–848. doi:10.1038/nrm3012
27. Appleman LJ. MET signaling pathway: a rational target for cancer therapy. J Clin Oncol. 2011;29:4837–4838. doi:10.1200/JCO.2011.37.7929
28. Gherardi E, Sandin S, Petoukhov MV, et al. Structural basis of hepatocyte growth factor/scatter factor and MET signalling. Proc Natl Acad Sci USA. 2006;103:4046–4051. doi:10.1073/pnas.0509040103
29. Zhang YW, Woude GFV. HGF/SF-Met signaling in the control of branching morphogenesis and invasion. J Cell Biochem. 2003;88:408–417. doi:10.1002/jcb.10358
30. Christensen JG, Burrows J, Salgia R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005;225:1–26. doi:10.1016/j.canlet.2004.09.044
31. Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–20937. doi:10.1073/pnas.0710370104
32. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi:10.1126/science.1141478
33. Kong-beltran M, Seshagiri S, Zha JP, et al. Somatic mutations lead to an oncogenic deletion of Met in lung cancer. Cancer Res. 2006;66:283–289. doi:10.1158/0008-5472.CAN-05-2749
34. Weidner KM, Sachs M, Riethmacher D, et al. Mutation of juxtamembrane tyrosine residue-1001 suppresses loss-of-function mutations of the met receptor in epithelial-cells. Proc Natl Acad Sci USA. 1995;92:2597–2601. doi:10.1073/pnas.92.7.2597
35. Peschard P, Fournier TM, Lamorte L, et al. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol Cell. 2001;8:995–1004. doi:10.1016/S1097-2765(01)00378-1
36. Ma PC, Jagadeeswaran R, Jagadeesh S, et al. Functional expression and mutations of c-met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65:1479–1488. doi:10.1158/0008-5472.CAN-04-2650
37. Collisson EA, Campbell JD, Brooks AN, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550.
38. Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5:842–849. doi:10.1158/2159-8290.CD-14-1467
39. Awad MM, Oxnard GR, Jackman DM, et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-met overexpression. J Clin Oncol. 2016;34:721. doi:10.1200/JCO.2015.63.4600
40. Heist RS, Shim HS, Gingipally S, et al. MET exon 14 skipping in non-small cell lung cancer. Oncologist. 2016;21:481–486. doi:10.1634/theoncologist.2015-0510
41. Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol. 2016;11:1493–1502. doi:10.1016/j.jtho.2016.06.004
42. Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5:850–859. doi:10.1158/2159-8290.CD-15-0285
43. Kalluri R. The biology and function of fibroblasts in cancer. Nature Rev Cancer. 2016;16:582–598. doi:10.1038/nrc.2016.73
44. Onozato R, Kosaka T, Kuwano H, et al. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thoracic Oncol. 2009;4:5–11. doi:10.1097/JTO.0b013e3181913e0e
45. Seo JS, Ju YS, Lee WC, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22:2109–2119. doi:10.1101/gr.145144.112
46. Saffroy R, Fallet V, Girard N, et al. MET exon 14 mutations as targets in routine molecular analysis of primary sarcomatoid carcinoma of the lung. Oncotarget. 2017;8:42428–42437. doi:10.18632/oncotarget.16403
47. Liu XW, Jia YX, Stoopler MB, et al. Next-generation sequencing of pulmonary sarcomatoid carcinoma reveals high frequency of actionable MET gene mutations. J Clin Oncol. 2016;34:794. doi:10.1200/JCO.2015.62.0674
48. Vuong HG, Ho ATN, Altibi AMA, et al. Clinicopathological implications of MET exon 14 mutations in non-small cell lung cancer - A systematic review and meta-analysis. Lung Cancer. 2018;123:76–82. doi:10.1016/j.lungcan.2018.07.006
49. Tong JH, Yeung SF, Chan AWH, et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin Cancer Res. 2016;22:3048–3056. doi:10.1158/1078-0432.CCR-15-2061
50. Drilon AE, Camidge DR, Ou SHI, et al. Efficacy and safety of crizotinib in patients (pts) with advanced MET exon 14-altered non-small cell lung cancer (NSCLC). J Clin Oncol. 2016;34. doi:10.1200/JCO.2016.34.15_suppl.108
51. Lee C, Usenko D, Frampton GM, et al. MET 14 deletion in sarcomatoid non-small-cell lung cancer detected by next-generation sequencing and successfully treated with a MET inhibitor. J Thoracic Oncol. 2015;10:E113–E114. doi:10.1097/JTO.0000000000000645
52. Waqar SN, Morgensztern D, Sehn J. MET mutation associated with responsiveness to crizotinib. J Thoracic Oncol. 2015;10:E29–E31. doi:10.1097/JTO.0000000000000478
53. Mendenhall MA, Goldman JW. MET-mutated NSCLC with major response to crizotinib. J Thoracic Oncol. 2015;10:E33–E34. doi:10.1097/JTO.0000000000000491
54. Jenkins RW, Oxnard GR, Elkin S, et al. Response to crizotinib in a patient with lung adenocarcinoma harboring a MET splice site mutation. Clin Lung Cancer. 2015;16:E101–E104. doi:10.1016/j.cllc.2015.01.009
55. Drilon A, Cappuzzo F, Ou S-HI, et al. Targeting MET in lung cancer: will expectations finally be MET? J Thoracic Oncol. 2017;12:15–26. doi:10.1016/j.jtho.2016.10.014
56. Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–1484. doi:10.1038/nm.3729
57. Rodgers K, Network CGAR. Comprehensive molecular profiling of lung adenocarcinoma (vol 511, pg 543, 2014). Nature. 2014;514.
58. Kawakami H, Okamoto I, Okamoto W, et al. Targeting MET amplification as a new oncogenic driver. Cancers. 2014;6:1540–1552. doi:10.3390/cancers6031540
59. Noonan SA, Berry L, Lu X, et al. Identifying the appropriate FISH criteria for defining MET copy number-driven lung adenocarcinoma through oncogene overlap analysis. J Thoracic Oncol. 2016;11:1293–1304. doi:10.1016/j.jtho.2016.04.033
60. Chen HJ, Mok TS, Chen ZH, et al. Clinicopathologic and molecular features of epidermal growth factor receptor T790M mutation and c-MET amplification in tyrosine kinase inhibitor-resistant Chinese non-small cell lung cancer. Pathol Oncol Res. 2009;15:651–658. doi:10.1007/s12253-009-9167-8
61. Go H, Jeon YK, Park HJ, et al. High MET gene copy number leads to shorter survival in patients with non-small cell lung cancer. J Thoracic Oncol. 2010;5:305–313. doi:10.1097/JTO.0b013e3181ce3d1d
62. Caparica R, Yen CT, Coudry R, et al. Responses to crizotinib can occur in high-level MET-amplified non-small cell lung cancer independent of MET exon 14 alterations. J Thorac Oncol. 2017;12:141–144. doi:10.1016/j.jtho.2016.09.116
63. Ou SHI, Kwak EL, Siwak-tapp C, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thoracic Oncol. 2011;6:942–946. doi:10.1097/JTO.0b013e31821528d3
64. Camidge DR, Ou SHI, Shapiro G, et al. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC). J Clin Oncol. 2014;32:8001. doi:10.1200/jco.2014.32.15_suppl.8001
65. Zhang Y, Wang WY, Wang Y, et al. Response to crizotinib observed in lung adenocarcinoma with MET Copy number gain but without a high-level MET/CEP7 ratio, MET overexpression, or exon 14 splicing mutations. J Thoracic Oncol. 2016;11:E59–E62. doi:10.1016/j.jtho.2015.12.102
66. Gherardi E, Birchmeier W, Birchmeier C, et al. Targeting MET in cancer: rationale and progress. Nature Rev Cancer. 2012;12:89–103. doi:10.1038/nrc3205
67. Abella JV, Peschard P, Naujokas MA, et al. Met/hepatocyte growth factor receptor ubiquitination suppresses transformation and is required for Hrs phosphorylation. Mol Cell Biol. 2005;25:9632–9645. doi:10.1128/MCB.25.21.9632-9645.2005
68. Tan YHC, Mirzapoiazova T, Won BM, et al. Differential responsiveness of MET inhibition in non-small-cell lung cancer with altered CBL. Sci Rep. 2017;7:1–3.
69. Yeh I, Botton T, Talevich E, et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat Commun. 2015;6. doi:10.1038/ncomms8174
70. Davies KD, Ng TL, Estrada-bernal A, et al. Dramatic response to crizotinib in a patient with lung cancer positive for an HLA-DRB1-MET gene fusion. JCO Precis Oncol. 2017;2017.
71. Stransky N, Cerami E, Schalm S, et al. The landscape of kinase fusions in cancer. Nat Commun. 2014;5. doi:10.1038/ncomms5846
72. Kentsis A, Reed C, Rice KL, et al. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat Med. 2012;18:1118. doi:10.1038/nm.2819
73. Danilkovitch-miagkova A, Zbar B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J Clin Invest. 2002;109:863–867. doi:10.1172/JCI0215418
74. Park S, Choi YL, Sung CO, et al. High MET copy number and MET overexpression: poor outcome in non-small cell lung cancer patients. Histol Histopathol. 2012;27:197–207. doi:10.14670/HH-27.197
75. Tsao M-S, Yang Y, Marcus A, et al. Hepatocyte growth factor is predominantly expressed by the carcinoma cells in non-small-cell lung cancer. Hum Pathol. 2001;32:57–65. doi:10.1053/hupa.2001.21133
76. Tsuta K, Kozu Y, Mimae T, et al. c-MET/phospho-MET protein expression and MET gene copy number in non-small cell lung carcinomas. J Thorac Oncol. 2012;7:331–339. doi:10.1097/JTO.0b013e318241655f
77. Tretiakova M, Salama AKS, Karrison T, et al. MET and phosphorylated MET as potential biomarkers in lung cancer. J Environ Pathol Tox. 2011;30:341–354. doi:10.1615/JEnvironPatholToxicolOncol.v30.i4.70
78. Ye S, Li JK, Hao K, et al. The efficacy and risk profile of c-Met inhibitors in non-small cell lung cancer: a meta-analysis. Sci Rep. 2016;6. doi:10.1038/srep35770
79. Lee GD, Lee SE, Oh DY, et al. MET exon 14 skipping mutations in lung adenocarcinoma: clinicopathologic implications and prognostic values. J Thoracic Oncol. 2017;12:1233–1246. doi:10.1016/j.jtho.2017.04.031
80. Kwon D, Koh J, Kim S, et al. MET exon 14 skipping mutation in triple-negative pulmonary adenocarcinomas and pleomorphic carcinomas: an analysis of intratumoral MET status heterogeneity and clinicopathological characteristics. Lung Cancer. 2017;106:131–137. doi:10.1016/j.lungcan.2017.02.008
81. Casadevall D, Gimeno J, Clave S, et al. MET expression and copy number heterogeneity in nonsquamous non-small cell lung cancer (nsNSCLC). Oncotarget. 2015;6:16215–16226. doi:10.18632/oncotarget.v6i18
82. Scagliotti GV, Novello S, Schiller JH, et al. Rationale and design of MARQUEE: a phase III, randomized, double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic, nonsquamous, non-small-cell lung cancer. Clin Lung Cancer. 2012;13:391–395. doi:10.1016/j.cllc.2012.01.003
83. Spigel DR, Edelman MJ, Mok T, et al. Treatment rationale study design for the metlung trial: a randomized, double-blind phase III study of onartuzumab (MetMAb) in combination with erlotinib versus erlotinib alone in patients who have received standard chemotherapy for stage IIIB or IV met-positive non-small-cell lung cancer. Clin Lung Cancer. 2012;13:500–504. doi:10.1016/j.cllc.2012.05.009
84. Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi:10.1016/j.ccr.2009.11.022
85. Papadimitrakopoulou VA, Wu YL, Han JY, et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol. 2018;29:741. doi:10.1093/annonc/mdy424.064
86. Ramalingam SS, Cheng Y, Zhou C, et al. Mechanisms of acquired resistance to first-line osimertinib: preliminary data from the phase III FLAURA study. Ann Oncol. 2018;29:viii740. doi:10.1093/annonc/mdy424.063
87. Yano S, Wang W, Li Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68:9479–9487. doi:10.1158/0008-5472.CAN-08-1643
88. Jiang W, Hiscox S, Matsumoto K, et al. Hepatocyte growth factor scatter factor, its molecular, cellular and clinical implications in cancer. Crit Rev Oncol Hemat. 1999;29:209–248. doi:10.1016/S1040-8428(98)00019-5
89. Kosaka T, Yamaki E, Mogi A, et al. Mechanisms of resistance to EGFR TKIs and development of a new generation of drugs in non-small-cell lung cancer. J Biomed Biotechnol. 2011;2011:1–7. doi:10.1155/2011/165214
90. Bardelli A, Corso S, Bertotti A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658–673. doi:10.1158/2159-8290.CD-12-0558
91. Pietrantonio F, Oddo D, Gloghini A, et al. MET-driven resistance to dual EGFR and BRAF blockade may be overcome by switching from EGFR to MET inhibition in BRAF-mutated colorectal cancer. Cancer Discov. 2016;6:963–971. doi:10.1158/2159-8290.CD-16-0297
92. Puri N, Salgia R. Synergism of EGFR and c-Met pathways, cross-talk and inhibition, in non-small cell lung cancer. J Carcinog. 2008;7:9. doi:10.4103/1477-3163.44372
93. Benvenuti S, Gentile A, Lazzari L, et al. An ‘in-cell trial’ to assess the efficacy of a monovalent anti-MET antibody as monotherapy and in association with standard cytotoxics. Mol Oncol. 2014;8:378–388. doi:10.1016/j.molonc.2013.12.006
94. Awad MM, Leonardi GC, Kravets S, et al. Impact of MET inhibitors on survival among patients with non-small cell lung cancer harboring MET exon 14 mutations: a retrospective analysis. Lung Cancer. 2019;133:96–102. doi:10.1016/j.lungcan.2019.05.011
95. Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thoracic Oncol. 2016;11:1493–1502. doi:10.1016/j.jtho.2016.06.004
96. Liu SY, Gou LY, Li AN, et al. The unique characteristics of MET exon 14 mutation in chinese patients with NSCLC. J Thoracic Oncol. 2016;11:1503–1510. doi:10.1016/j.jtho.2016.05.016
97. Zou HY, Li QH, Lee JH, et al. An orally available small-molecule inhibitor of c-met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67:4408–4417. doi:10.1158/0008-5472.CAN-06-4443
98. Cui JJ, Tran-dube M, Shen H, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem. 2011;54:6342–6363. doi:10.1021/jm2007613
99. Malik SM, Maher VE, Bijwaard KE, et al. US food and drug administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20:2029–2034. doi:10.1158/1078-0432.CCR-13-3077
100. Kazandjian D, Blumenthal GM, Luo L, et al. Benefit-risk summary of crizotinib for the treatment of patients with ROS1 alteration-positive, metastatic non-small cell lung cancer. Oncologist. 2016;21:974–980. doi:10.1634/theoncologist.2016-0101
101. Awad MM, Leonardi GC, Kravets S, et al. Impact of MET inhibitors on survival among patients (pts) with MET exon 14 mutant (METdel14) non-small cell lung cancer (NSCLC). J Clin Oncol. 2017;35:8511. doi:10.1200/JCO.2017.35.15_suppl.8511
102. Ou SHI, Govindan R, Eaton KD, et al. Phase I results from a study of crizotinib in combination with erlotinib in patients with advanced nonsquamous non-small cell lung cancer. J Thoracic Oncol. 2017;12:145–151. doi:10.1016/j.jtho.2016.09.131
103. Janne PA, Shaw AT, Camidge DR, et al. Combined pan-HER and ALK/ROS1/MET inhibition with dacomitinib and crizotinib in advanced non-small cell lung cancer: results of a phase I study. J Thoracic Oncol. 2016;11:737–747. doi:10.1016/j.jtho.2016.01.022
104. Taniguchi H, Yamada T, Takeuchi S, et al. Impact of MET inhibition on small-cell lung cancer cells showing aberrant activation of the hepatocyte growth factor/MET pathway. Cancer Sci. 2017;108:1378–1385. doi:10.1111/cas.2017.108.issue-7
105. Schoffski P, Gordon M, Smith DC, et al. Phase II randomised discontinuation trial of cabozantinib in patients with advanced solid tumours. Eur J Cancer. 2017;86:296–304. doi:10.1016/j.ejca.2017.09.011
106. Reckamp KL, Mack PC, Ruel N, et al. Biomarker analysis of a phase II trial of cabozantinib and erlotinib in patients (pts) with EGFR-mutant NSCLC with epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) resistance: a California Cancer Consortium phase II trial (NCI 9303). J Clin Oncol. 2015;33:8087. doi:10.1200/jco.2015.33.15_suppl.8087
107. Neal JW, Dahlberg SE, Wakelee HA, et al. Erlotinib, cabozantinib, or erlotinib plus cabozantinib as second-line or third-line treatment of patients with EGFR wild-type advanced non-small-cell lung cancer (ECOG-ACRIN 1512): a randomised, controlled, open-label, multicentre, Phase 2 trial. Lancet Oncol. 2016;17:1661–1671. doi:10.1016/S1470-2045(16)30561-7
108. Leighl NB, Tsao MS, Liu G, et al. A phase I study of foretinib plus erlotinib in patients with previously treated advanced non-small cell lung cancer: Canadian cancer trials group IND.196. Oncotarget. 2017;8:69651–69662. doi:10.18632/oncotarget.18753
109. Kollmannsberger CK, Sharma S, Shapiro G, et al. Phase I study of receptor tyrosine kinase (RTK) inhibitor, MGCD265, in patients (pts) with advanced solid tumors. J Clin Oncol. 2015;33:2589. doi:10.1200/jco.2015.33.15_suppl.2589
110. Liu XD, Wang Q, Yang GJ, et al. A novel kinase inhibitor, INCB28060, blocks c-MET-dependent signaling, neoplastic activities, and cross-talk with EGFR and HER-3. Clin Cancer Res. 2011;17:7127–7138. doi:10.1158/1078-0432.CCR-11-1157
111. Schuler MH, Berardi R, Lim WT, et al. Phase (Ph) I study of the safety and efficacy of the cMET inhibitor capmatinib (INC280) in patients (pts) with advanced cMET plus non-small cell lung cancer (NSCLC). J Clin Oncol. 2016;34:9067. doi:10.1200/JCO.2016.34.15_suppl.9067
112. Wu YL, Kim DW, Felip E, et al. Phase (Ph) II safety and efficacy results of a single-arm ph ib/II study of capmatinib (INC280) + gefitinib in patients (pts) with EGFR-mutated (mut) cMET-positive (cMET plus) non-small cell lung cancer (NSCLC). J Clin Oncol. 2016;34:9020. doi:10.1200/JCO.2016.34.15_suppl.9020
113. Sequist LV, von Pawel J, Garmey EG, et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol. 2011;29:3307–3315. doi:10.1200/JCO.2010.34.0570
114. Scagliotti G, von Pawel J, Novello S, et al. Phase III multinational, randomized, double-blind, placebo-controlled study of tivantinib (ARQ 197) plus erlotinib versus erlotinib alone in previously treated patients with locally advanced or metastatic nonsquamous non-small-cell lung cancer. J Clin Oncol. 2015;33:2667. doi:10.1200/JCO.2014.60.7317
115. Yoshioka H, Azuma K, Yamamoto N, et al. A randomized, double-blind, placebo-controlled, phase III trial of erlotinib with or without a c-Met inhibitor tivantinib (ARQ 197) in Asian patients with previously treated stage IIIB/IV nonsquamous nonsmall-cell lung cancer harboring wild-type epidermal growth factor receptor (ATTENTION study). Ann Oncol. 2015;26:2066–2072. doi:10.1093/annonc/mdv288
116. Jia H, Dai GX, Weng JY, et al. Discovery of (S)-1-(1-(Imidazo[1,2-a]pyridin-6-yl)ethyl)-6-(1-methyl-1H-pyrazol-4-yl)-1H-[1,2,3]triazolo[4,5-b]pyrazine (Volitinib) as a highly potent and selective mesenchymal-epithelial transition factor (c-Met) inhibitor in clinical development for treatment of cancer. J Med Chem. 2014;57:7577–7589. doi:10.1021/jm500510f
117. Oxnard GR, Ramalingam SS, Ahn MJ, et al. Preliminary results of TATTON, a multi-arm phase Ib trial of AZD9291 combined with MEDI4736, AZD6094 or selumetinib in EGFR-mutant lung cancer. J Clin Oncol. 2015;33:2509. doi:10.1200/jco.2015.33.15_suppl.2509
118. Bladt F, Faden B, Friese-hamim M, et al. EMD 1214063 and EMD 1204831 constitute a new class of potent and highly selective c-met inhibitors. Clin Cancer Res. 2013;19:2941–2951. doi:10.1158/1078-0432.CCR-12-3247
119. Merchant M, Ma XL, Maun HR, et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc Natl Acad Sci USA. 2013;110:E2987–E2996. doi:10.1073/pnas.1302725110
120. Spigel DR, Ervin TJ, Ramlau RA, et al. Randomized phase II trial of onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2013;31:4105. doi:10.1200/JCO.2012.47.4189
121. Spigel DR, Edelman MJ, O’Byrne K, et al. Results from the phase III randomized trial of onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIB or IV non-small-cell lung cancer: mETLung. J Clin Oncol. 2017;35:412. doi:10.1200/JCO.2016.69.2160
122. Camidge DR, Moran T, Demedts I, et al. A randomized, open-label, phase 2 study of emibetuzumab plus erlotinib (LY plus E) and emibetuzumab monotherapy (LY) in patients with acquired resistance to erlotinib and MET diagnostic positive (MET Dx plus) metastatic NSCLC. J Clin Oncol. 2016;34:9070. doi:10.1200/JCO.2016.34.15_suppl.9070
123. Scagliotti GV, Moro-sibilot D, Kollmeier J, et al. A randomized, controlled, open label phase II study of erlotinib (E) with or without the MET antibody emibetuzumab (Emi) as first-line treatment for EGFRmt non-small cell lung cancer (NSCLC) patients who have disease control after an 8-week lead-in treatment with erlotinib. J Clin Oncol. 2017;35.
124. Wang JY, Anderson MG, Oleksijew A, et al. ABBV-399, a c-met antibody-drug conjugate that targets both MET-amplified and c-met-overexpressing tumors, irrespective of MET pathway dependence. Clin Cancer Res. 2017;23:992–1000. doi:10.1158/1078-0432.CCR-16-1568
125. Strickler JH, Weekes CD, Nemunaitis J. First-in-human phase I, dose-escalation and -expansion study of telisotuzumab vedotin, an antibody-drug conjugate targeting c-met, in patients with advanced solid tumors (vol 36, pg 3298, 2018). J Clin Oncol. 2019;37:261.
126. Emdal KB, Dittmann A, Reddy RJ, et al. Characterization of in vivo resistance to osimertinib and JNJ-61186372, an EGFR/met bispecific antibody, reveals unique and consensus mechanisms of resistance. Mol Cancer Ther. 2017;16:2572–2585. doi:10.1158/1535-7163.MCT-17-0413
127. Burgess TL, Sun J, Meyer S, et al. Biochemical characterization of AMG 102: a neutralizing, fully human monoclonal antibody to human and nonhuman primate hepatocyte growth factor. Mol Cancer Ther. 2010;9:400–409. doi:10.1158/1535-7163.MCT-09-0824
128. Giordano S. Rilotumumab, a mAb against human hepatocyte growth factor for the treatment of cancer. Curr Opin Mol Ther. 2009;11:448–455.
129. Tarhini AA, Rafique I, Floros T, et al. Phase 1/2 study of rilotumumab (AMG 102), a hepatocyte growth factor inhibitor, and erlotinib in patients with advanced non-small cell lung cancer. Cancer. 2017;123:2936–2944. doi:10.1002/cncr.v123.15
130. D’arcangelo M, Cappuzzo F. Focus on the potential role of ficlatuzumab in the treatment of non-small cell lung cancer. Biologics. 2013;7:61–68.
131. Mok TSK, Geater SL, Su WC, et al. A randomized phase 2 study comparing the combination of ficlatuzumab and gefitinib with gefitinib alone in asian patients with advanced stage pulmonary adenocarcinoma. J Thoracic Oncol. 2016;11:1736–1744. doi:10.1016/j.jtho.2016.05.038
132. Mazieres J, Drilon A, Lusque A, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol. 2019;30:1321–1328. doi:10.1093/annonc/mdz167
133. Sabari JK, Leonardi GC, Shu CA, et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann Oncol. 2018;29:2085–2091. doi:10.1093/annonc/mdy334
134. Spigel DR, Edelman MJ, O’byrne K, et al. Onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIb or IV NSCLC: results from the pivotal phase III randomized, multicenter, placebo-controlled METLung (OAM4971g) global trial. J Clin Oncol. 2014;32:8000. doi:10.1200/jco.2014.32.15_suppl.8000
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.