Back to Journals » Journal of Inflammation Research » Volume 15
Mendelian Randomization Study Implies Causal Linkage Between Telomere Length and Juvenile Idiopathic Arthritis in a European Population
Authors Zhang J ![]()
Received 23 December 2021
Accepted for publication 2 February 2022
Published 15 February 2022 Volume 2022:15 Pages 977—986
DOI https://doi.org/10.2147/JIR.S354619
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Jun Zhang
The First Affiliated Hospital of Chongqing Medical University, Chongqing Key Laboratory of Ophthalmology, Chongqing Eye Institute, Chongqing Branch of National Clinical Research Center for Ocular Diseases, Chongqing, People’s Republic of China
Correspondence: Jun Zhang, Tel/Fax +86-23-89012851, Email [email protected]
Background: Telomere maintenance is increasingly being considered as fundamental to the progression of immune-mediated inflammatory diseases. However, the causality underlying the purported relationship has not been fully elucidated. In the present work, we applied Mendelian randomization (MR) analysis to obtain estimates of the causal effect of telomere length (TL) on the risk of juvenile idiopathic arthritis (JIA) and JIA-associated iridocyclitis.
Methods: Two-sample MR analysis was conducted using summary-level data from the largest genome-wide association studies concerning TL (78,592 individuals), JIA (6056 cases and 25,086 controls), and JIA-associated iridocyclitis (1430 cases and 9,2767 controls). All the participants were of European ancestry. The inverse variance weighted (IVW) method was applied to estimate the causal effects. Sensitivity analyses incorporating multiple complementary MR approaches were implemented to test the robustness of the association and examine potential bias from pleiotropy.
Results: In our MR analysis, genetically predicted shorter TL was associated with an increased risk of JIA (IVW: odds ratio=1.68, 95% CI: 1.13– 2.48, P=0.009), but not with the risk of JIA-associated iridocyclitis (IVW: odds ratio=1.75, 95% CI: 0.81– 3.79, P=0.155). The other MR methods produced consistent results. Besides, a leave-one-out sensitivity analysis yielded similar findings and validated the robustness of the causal relationship. MR-Egger regression revealed no notable horizontal pleiotropy (intercept=0.046, P=0.175).
Conclusion: This work provides evidence of a negative association between TL and JIA risk, but not for the association between TL and the risk of JIA-associated iridocyclitis, in a European population. Future studies with larger sample sizes are warranted to elucidate the underlying role of TL in these diseases.
Keywords: telomere length, Mendelian randomization, juvenile idiopathic arthritis, JIA-associated iridocyclitis, genetics
Introduction
Human telomeres are repetitive stretches of the hexameric nucleotide sequence TTAGGG located at the natural ends of linear chromosomes.1 Together with the associated protein complex capping the telomere, they are mainly dedicated to preserving DNA integrity and chromosomal stability in the human genome. In most somatic tissues, telomere attrition accompanied by the DNA damage response will ultimately contribute to cellular senescence or apoptosis,2,3 which is recognized as a primary hallmark of aging. Research suggests that telomeres may be inversely associated with the risk of immune-mediated inflammatory diseases, such as inflammatory bowel diseases, psoriasis, and rheumatoid arthritis.4–6 Although the etiology of these conditions remains poorly defined, advances in molecular research on telomere length (TL) indicate that their pathogenesis could be attributable to the expression of common inflammatory cytokines associated with cellular aging.7,8
Juvenile idiopathic arthritis (JIA) is a leading cause of rheumatic disease affecting children under the age of 16 years,9 with iridocyclitis as the most common extra-articular comorbidity, which involves the ocular iris and ciliary body.10 The clinical traits are recognized as complex immune-mediated inflammatory disorders, and are subject to a complicated association with the immune system dysfunction, and various environmental and genetic factors.11,12 Several population-based observational studies have found that JIA patients display significantly shorter TL compared to healthy controls.13–16 The relationship between JIA-associated iridocyclitis and TL has been seldom discussed. However, the case–control study design used in all retrieved research is prone to reverse causality and residual confounding. The difficult assays used for TL measurement, small sample sizes in observational studies, and lower statistical power are further limitations. Therefore, whether TL has a causal effect on the risk of JIA or JIA-associated iridocyclitis has not yet been reliably inferred.
To validate these findings and avoid the potential impact from confounding factors, Mendelian randomization (MR) analysis is widely adopted to estimate the causal effects of genetically predicted exposures on one or more complex outcomes.17 Given that germline genetic information is fixed at conception in a random process according to Mendel’s laws of inheritance, the MR method utilizes genetic variants such as single-nucleotide polymorphisms (SNPs) as instrumental variables (IVs) for risk factors of interest,18 which can produce a more reliable association than before. To our knowledge, there has been no previous MR investigation on the causal link between TL and JIA. Therefore, in the current study, we performed a two-sample MR using summary-level statistics from large genome-wide association study (GWAS) databases to evaluate whether TL is causally associated with JIA risk. Furthermore, we also examined the potential causal sequence of TL on JIA-associated iridocyclitis.
Materials and Methods
Data Sources
We adopted a standard two-sample MR framework in which IV-exposure and IV-outcome interactions were derived from independent and non-overlapping individual datasets.
Summary statistics on leukocyte TL were obtained from the most recent, largest GWAS meta-analysis, which included EPIC-InterAct (n=19,799), EPIC-CVD (n=11,915), and ENGAGE consortium (n=46,898).19 All participants were of European ancestry. Mean leukocyte TL was measured using an established quantitative polymerase chain reaction technique and expressed as a ratio of the telomere repeat number (T) to a single-copy gene (S). The detailed procedure is described in a previous study.20 The TL measurements were standardized by quantifying the standard curve or using calibration samples. The final reported result for each sample was the average of three independently measured T/S values.21
JIA GWAS summary statistics were acquired from the recently updated meta-analysis of 6056 patients with all JIA subtypes and 25,086 controls of European descent.22 Detailed and clear descriptions, including the study procedure, were presented in the two involved studies.22,23 All eligible JIA patients must meet the criteria created and approved by the board-certified rheumatologists of the International League of Associations for Rheumatology (ILAR). The JIA GWAS data were systematically investigated regarding the specificity and sharing of JIA susceptibility loci across ILAR subtypes; that is, all subtypes of JIA were included in our cases without particular exploration of each subtype. Besides, to explore the causal inference between TL and JIA comorbidity (iridocyclitis), summary statistics from the MRC IEU Open GWAS Project (https://gwas.mrcieu.ac.uk/), with 1430 JIA-associated iridocyclitis cases and 92,767 controls of European ancestry from the FinnGen Study, were included in this work.
The study was approved by the ethical committee of each institutional review board, and written informed consent was provided by all participants.
Genetic Instruments for Telomere Length
Three essential model assumptions of MR analysis should be fulfilled to guarantee valid IVs (Figure 1), namely: 1) the selected IVs are robustly associated with the exposure (TL); 2) the eligible IVs for exposure are not associated with any confounders; and 3) the selected IVs only affect the outcome exclusively through the exposure (TL).
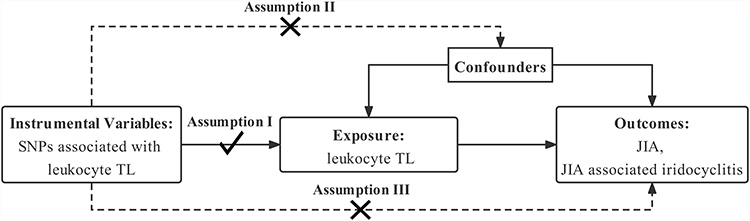 |
Figure 1 Mendelian randomization model of leukocyte TL and risk of JIA and JIA-associated iridocyclitis. The study design is under the assumption that genetic variants are directly associated with TL, but not indirectly with TL and not directly with confounders, that is, the instrumental variables influence JIA and JIA-associated iridocyclitis only directly through TL. Abbreviations: SNP, single-nucleotide polymorphism; TL, telomere length; JIA, juvenile idiopathic arthritis. |
Based on the three MR core assumptions, the top 20 SNPs (shown in Table 1) regarded as IVs for leukocyte TL were chosen at the established threshold of GWAS significance (P<5×10−8) and assessed through additive models adjusted for age, gender, and cohort-specific covariates.19 These SNPs were mainly located close to the genes coding for proteins involved in telomere homeostasis. Proxy SNPs identified through the online platform LDlink (https://ldlink.nci.nih.gov/) were applied when the SNPs were unavailable for the outcome in the GWAS. Besides, palindromic SNPs were not included in our research. The strength of the eligible IVs was calculated by R2 and the F-statistic.
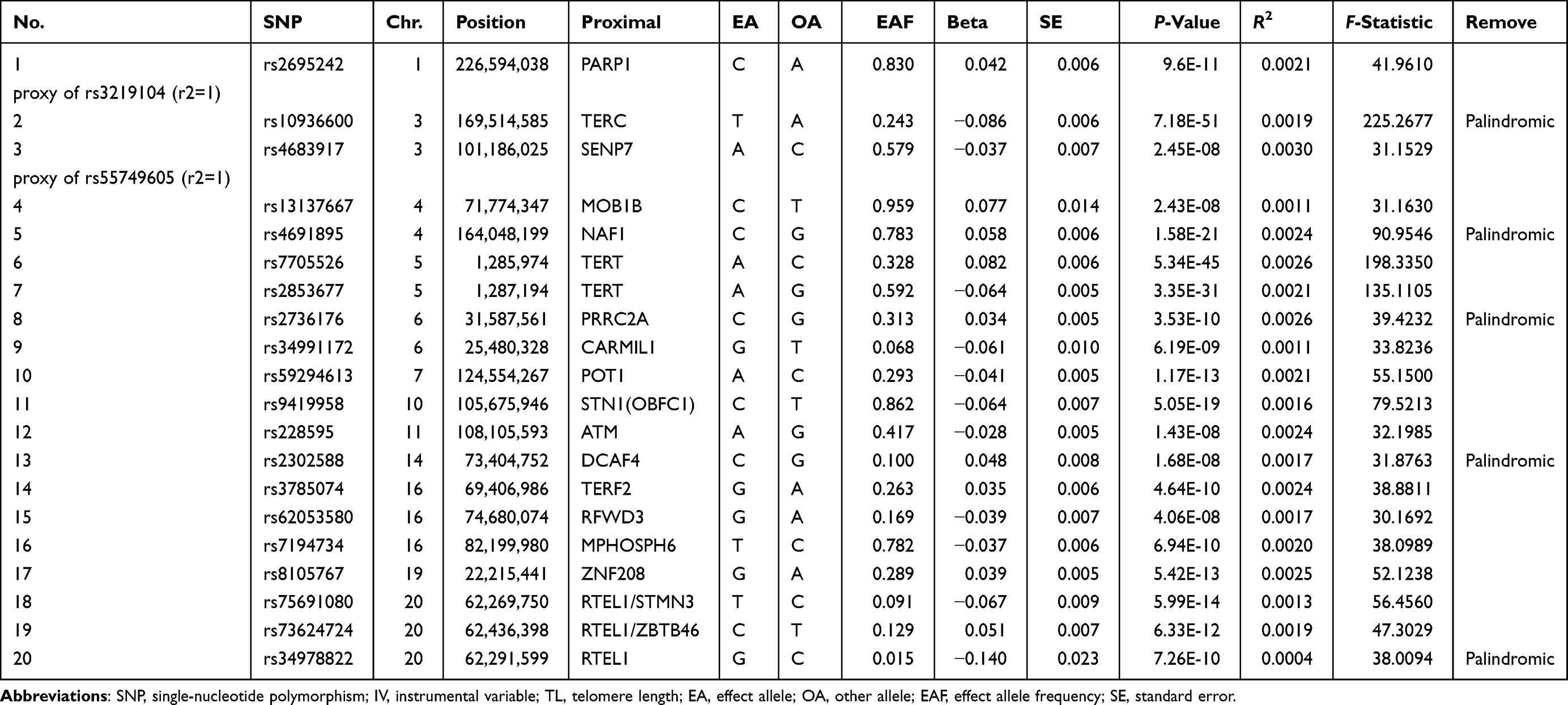 |
Table 1 Characteristics of SNPs Selected as the IV and Association with Leukocyte TL |
Statistical Analysis
Mendelian Randomization Analysis
Our two-sample MR analysis was conducted using the “TwoSampleMR” R package.24 The inverse variance weighted (IVW) method18 was applied to obtain the combined estimate of the causal effect. The conventional IVW method relies on the assumption that all of the SNPs obtained in the analysis are valid instruments. Besides, the maximum likelihood-based method25 was utilized to provide appropriately estimated confidence intervals (CIs) when weak IVs exist. For each SNP, causal effect estimates were generated for relative outcomes as odds ratios (ORs) per one SD unit increase in the putative risk factor using the Wald ratio.
Sensitivity Analysis
After MR analysis, Cochran’s Q test was applied to assess heterogeneity among the effects of individual TL-associated SNPs on JIA or JIA-associated iridocyclitis. Besides, several important sensitivity analyses were performed to verify MR model assumptions and evaluate the MR results. The MR-Egger regression was used to identify potential directional pleiotropy26 and an intercept P-value >0.05 indicated a lack of horizontal pleiotropy. The MR-RAPS (robust adjusted profile score) approach was used to validate the robustness of systemic and idiosyncratic pleiotropy.27 For each outlying SNP, the MR-PRESSO (MR pleiotropy residual sum and outlier) test was applied to assess the causal estimates.28 Besides, we applied radial variants of the IVW model for a better visualization of the causal estimate. Leave-one-SNP-out analysis was also used to assess to identify potentially influential SNPs and verify the reliability of the results. With the PhenoScanner database (http://www.phenoscaner.medschl.cam.ac.uk/), those SNPs that correlated with confounding factors were manually screened and omitted.
All statistical analyses in this study were conducted using the “TwoSampleMR” package in R software (version 4.1.0). All presented P-values were two sided and statistical significance was set at the 5% level.
Results
In our MR analysis, 20 leukocyte TL-related SNPs were described as IV models. Two SNPs that failed the identification in the JIA GWAS were replaced by their corresponding proxy SNPs. We searched the selected SNPs in the PhenoScanner dataset and did not identify any potential SNPs in association with other confounding factors. After harmonization and removal of palindromic SNPs with intermediate allele frequencies, the remaining 13 sentinel SNPs were taken as IVs for TL. The F-statistics for these IVs were greater than 10 (range: 30.17–198.34), indicating that these SNPs were sufficiently strong for MR analysis. These variants explained around 2.98% of the variance in leukocyte TL.
Genetically Determined TL Was Associated with JIA Risk
No obvious evidence was observed for the heterogeneity of these TL-associated SNPs in Cochran’s Q test (P=0.577). The IVW method under a fixed-effects model was implemented for primary estimation. As presented in Table 2, primary MR analysis indicated that genetically predicted shorter TL was associated with increased risk of JIA (OR=1.68, 95% CI: 1.13–2.48, P=0.009) (Figure 2). Similar effect estimates were observed through the maximum likelihood-based method and MR-PRESSO method. Notably, we used the MR-RAPS estimator and found that the results remained largely consistent with our primary findings (OR=1.69, 95% CI: 1.13–2.51, P=0.01). This indicates the robustness of the causal association between TL and JIA. Furthermore, we tried to find the causal effect between genetically determined TL and JIA-associated iridocyclitis. No strong evidence was yielded based on IVW, MR-PRESSO, MR-RAPS, or maximum likelihood-based methods (IVW, OR=1.75, 95% CI: 0.81–3.79, P=0.155). The detailed data from these methods are presented in the Supplementary Material, Table S1.
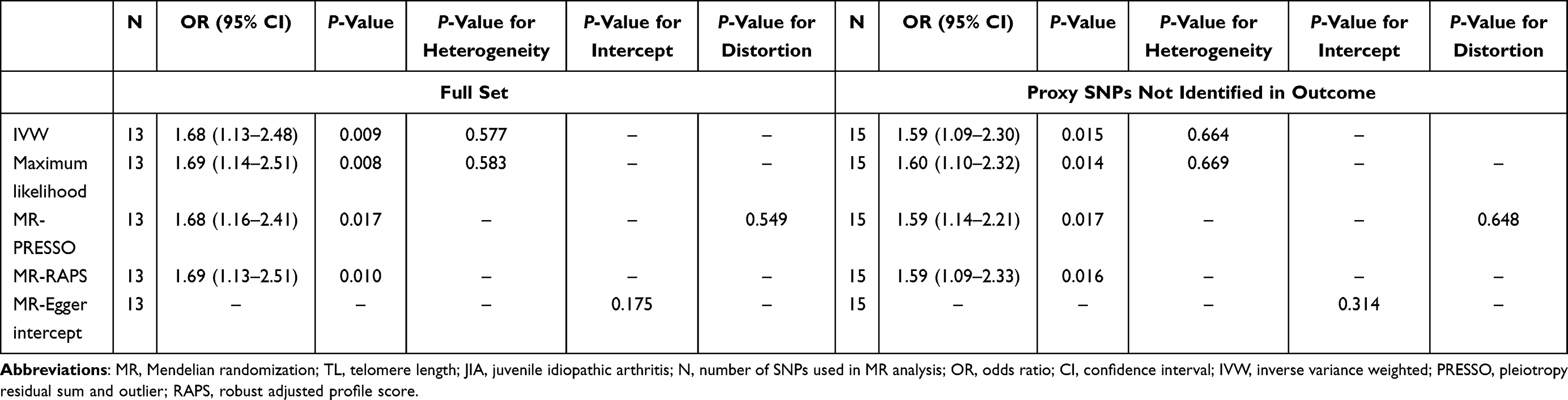 |
Table 2 MR Results of the Association Between Leukocyte TL and the Risk of JIA |
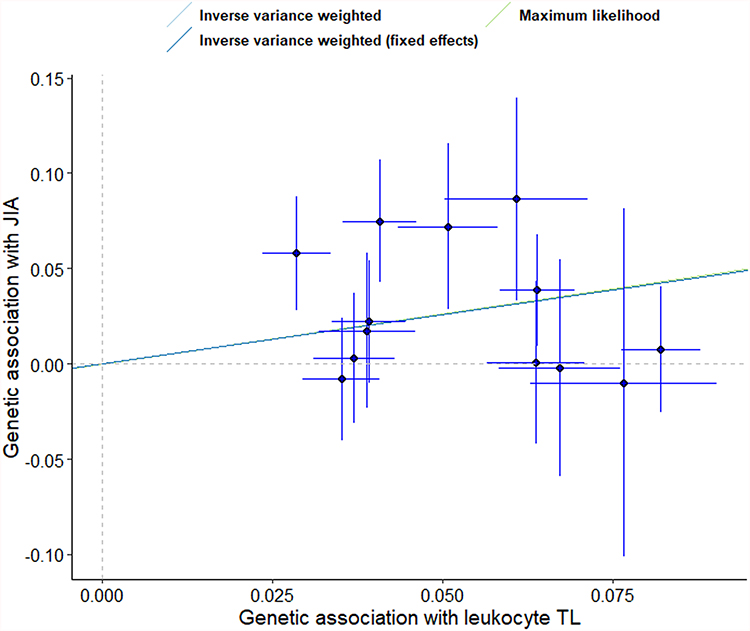 |
Figure 2 Scatter plot of the effect size and 95% CIs of each SNP on leukocyte TL and JIA risk. The horizontal axis represents genetic associations of each variant on leukocyte TL. The vertical axis indicates the genetic association of each SNP with risk of JIA.Abbreviations: SNP, single-nucleotide polymorphism; TL, telomere length; JIA, juvenile idiopathic arthritis. |
In an alternative approach, we utilized the proxy package to explore the potential causal effects of SNPs that were not identified in the outcome factors. Our MR analysis also yielded very comparable point estimates through multiple MR methods (IVW, OR=1.59, 95% CI: 1.09–2.30, P=0.015) (Table 2). Thus, genetic predisposition towards shorter TL was found to be a causative factor for a higher JIA risk. However, no significant estimates were discovered between TL and JIA-associated iridocyclitis (IVW, OR=1.65, 95% CI: 0.84–3.27, P=0.147). The detailed data are presented in the Supplementary Material, Table S1.
Pleiotropy and Sensitivity Analysis
The MR-Egger regression did not suggest any evidence of directional pleiotropy (intercept=0.046, SE=0.032, P=0.175). The result of MR-PRESSO analysis did not highlight any outliers for any IVs (P-distortion=0.549) (Table 2). In addition, the IVW radial MR approach did not delineate any evidence of outlying genetic variants (Figure 3 and the Supplementary Material, Figure S1). In the sensitivity analyses, the leave-one-out method demonstrated that the causal links between genetically determined TL and JIA or JIA-associated iridocyclitis were not driven by a single SNP, suggesting the stability of our MR analysis (shown in the Supplementary Material, Figures S2 and S3).
 |
Figure 3 Radial plots to visualize individual outlier SNPs in the MR estimates for the effect of leukocyte TL and risk of JIA. The radial curve displays the ratio estimate for each SNP, as well as the overall IVW estimate (in blue). Black dots show valid SNPs. Abbreviation: IVW, inverse variance weighted method; SNP, single-nucleotide polymorphism; MR, Mendelian randomization; TL, telomere length; JIA, juvenile idiopathic arthritis.. |
As suggested by Brion et al (https://shiny.cnsgenomics.com/mRnd/), we also calculated the sample power to validate our analysis. Under the current situation, our study had 99% power to detect a causal effect of a 68% increase in JIA risk at a significance level of 0.05.
Discussion
To the best of our knowledge, this is the first two-sample MR study to evaluate the relationships between leukocyte TL and JIA risk, as well as between leukocyte TL and the risk of JIA-associated iridocyclitis. Having made an effort to fulfill stringent MR assumptions and utilize multiple complementary MR methodologies, our results demonstrated that shorter genetically predicted TL could increase the risk of JIA in populations of European ancestry. However, there was no evidence supporting the causal linkage between TL and JIA-associated iridocyclitis.
TL, a recognized complex heritable trait, is influenced by oxidative damage and replicative stress caused by genetic and epigenetic factors, as well as extrinsic factors such as socioeconomic status, working conditions, and stress.29,30 Longer TL in offspring could be attributed to higher paternal age at conception.31 Besides, an increase in telomerase activity was inversely correlated with improvement in depressive symptoms and disease severity.32 It has been reported that shortened TL is associated with a high level of psychosocial stress, which could be attributed to increased oxidative stress and reduced telomerase activity.33 Moreover, in an observational study on rheumatoid arthritis, the levels of oxidative stress biomarkers rose as disease activity increased and thereafter led to significant telomere shortening.34 In our MR study, we disregarded genetic instruments associated with the above traits and solely explored the relationship between TL and diseases (JIA and JIA-associated iridocyclitis) from a genetic perspective.
Our findings were consistent with the evidence from limited observational studies; that is, genetically predicted TL was associated with the risk of JIA. JIA patients showed significantly shorter TL than healthy controls.13,16 A previous study demonstrated that shorter TL was measured in naive CD45RA+CD4+T cells from JIA patients with persistent immune activation.15 However, the exact biological mechanisms of telomere shortening in JIA remain to be elucidated. Several possible explanations may be advanced. First, some studies have indicated that the loss of the telomere would lead to functional consequences after infection due to autoimmune deficiency.13,35 Second, a change in TL may drive the premature immunosenescence of CD4+ CD28− T cells, which have been implicated in immune system diseases and chronic immune-mediated diseases owing to their pro-inflammatory and cytotoxic characteristics.36,37 In addition, some studies have shown that shortened or dysfunctional telomeres are associated with hyperactivity of the transcription factor NF-kB and overexpression of inflammatory cytokines such as TNF-alpha and IL-6 in circulating macrophages.38,39 Shortened TL has also been linked to the upregulation of NLRP3 inflammasome and the ATM-YAP1-pro-IL-18 pathway in inflammatory bowel diseases.4,40 These findings imply that TL may be involved in the pathogenesis of immune-mediated inflammatory diseases.
Contrary to the inevitable bias in observational research, our two-sample MR study, with a large sample size and sufficient statistical power, can address the concern of unmeasured or uncontrolled confounding factors by reanalyzing GWAS summary data. Furthermore, our results are in keeping with a causal role of shortened TL in increasing the risk of multiple immune-mediated disorders such as rheumatoid arthritis and hip osteoarthritis, as discovered in a growing number of MR studies.6,41 Although the exact role of TL in JIA has not yet been clearly defined, these findings suggest that telomere attrition seems to be a common trait of these disorders. Thus, the modulation of TL, except for a better understanding of various TL analyses, may represent a potential therapeutic approach for taking control of a disease risk.
In the MR study, we discovered that TL was related to JIA risk, rather than to the risk of JIA-associated iridocyclitis, suggesting the specificity of its comorbidity. Unfortunately, no studies have been published to evaluate the role of TL in JIA-associated iridocyclitis until now. One study revealed longer TL in ocular birdshot uveitis patients compared to unrelated healthy controls.42 These contradictory phenomena may be attributed to deleterious lifestyle, distinct cellular turnover due to chronic inflammation, and other determinants in the disease process.3 Furthermore, the relatively small populations and low incidence of JIA-associated iridocyclitis would unquestionably result in the inconsistency of the results. Further studies are warranted to explore the biological mechanisms of TL in JIA-associated iridocyclitis.
Major strengths of this MR study include the robust genetic instruments identified from the largest GWAS, explaining 2.98% of the variance in TL. In order to reduce bias in the causal estimate, we conducted the two-sample MR, where IVs associated with JIA or JIA-associated iridocyclitis and the estimation of the IVs were derived from two independent individuals. Moreover, we used stringent criteria for the inclusion of SNPs as IVs in our analyses and undertook comprehensive assessment for confounding. The intercept of the MR-Egger regression test was close to zero, suggesting no strong evidence for directional pleiotropy. Besides, no outliers were highlighted by the MR-PRESSO test, which is often used to detect and correct the outliers in IVW linear regression. Therefore, the consistent results observed across multiple complementary approaches lend further support to our findings.
Despite multiple advantages of the stringent MR assumptions, several limitations of the current study are worthy of mention. First, since the included participants in this study were of European ancestry, our findings may not be extrapolated to other populations, and it is urgent that our results are verified in various races. Second, the full individual-level statistics of the most recent GWAS were not available, which made it difficult to perform further functional validation. Third, the effects of age, sex, and other confounding environmental factors on TL cannot be ignored. Only summary-level data was applied in our MR analysis, which limited further stratified analyses on these specific factors. Fourth, the uncertain incompatibility in sample size between JIA and JIA-associated iridocyclitis may lead to some bias in our MR analysis. Fifth, though the genetic effects on the exposure of interest are independent and fixed throughout the life course, the age incompatibility between TL in the adult population and JIA in pediatric individuals should be noticed in our MR study. Whereas various JIA subtypes were taken together into the evaluation of their causal inference with TL, the exploration on the association of genetically predicted TL with a specific JIA subtype is still a challenge in current MR analysis. Therefore, subgroup analysis based on a set of JIA subtypes is recommended in the further GWAS study. Finally, JIA and its comorbidities are considered complex clinical traits involving various components, including genetic, immunological, environmental, and socioeconomic factors, suggesting that genetic variants could temptingly and partially explain the results.
Conclusions
To our knowledge, our results provide novel evidence to support a potential effect of leukocyte TL on JIA risk. The relationship between genetically predicted leukocyte TL and JIA-associated iridocyclitis could not be confirmed in the present work. Further large-scale studies among different populations are warranted to validate our findings and elucidate the causal role of TL in these disorders.
Data Sharing Statement
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval
Ethical approval for this study was not required as these analyses were based on summary statistics from published GWAS or the data were publicly accessible and no individual-level data were used.
Acknowledgments
I thank all the investigators of the UK Biobank and the MRC IEU Open GWAS Project for providing the data publicly.
Disclosure
The author declares that there is no conflict of interest in this work.
References
1. Wood AM, Laster K, Rice EL, et al. A beginning of the end: new insights into the functional organization of telomeres. Nucleus. 2015;6:172–178.
2. Barnes PJ. Mechanisms of development of multimorbidity in the elderly. Eur Respir J. 2015;45:790–806.
3. Heba AC, Toupance S, Arnone D, et al. Telomeres: new players in immune-mediated inflammatory diseases? J Autoimmun. 2021;123:102699.
4. Kang Y, Zhang H, Zhao Y, et al. Telomere dysfunction disturbs macrophage mitochondrial metabolism and the NLRP3 inflammasome through the PGC-1alpha/TNFAIP3 axis. Cell Rep. 2018;22:3493–3506.
5. Wu K, Higashi N, Hansen ER, et al. Telomerase activity is increased and telomere length shortened in T cells from blood of patients with atopic dermatitis and psoriasis. J Immunol. 2000;165:4742–4747.
6. Zeng Z, Zhang W, Qian Y, et al. Association of telomere length with risk of rheumatoid arthritis: a meta-analysis and Mendelian randomization. Rheumatology (Oxford). 2020;59:940–947.
7. Pont AR, Sadri N, Hsiao SJ, et al. mRNA decay factor AUF1 maintains normal aging, telomere maintenance, and suppression of senescence by activation of telomerase transcription. Mol Cell. 2012;47:5–15.
8. Zota AR, Geller RJ, Romano LE, et al. Association between persistent endocrine-disrupting chemicals (PBDEs, OH-PBDEs, PCBs, and PFASs) and biomarkers of inflammation and cellular aging during pregnancy and postpartum. Environ Int. 2018;115:9–20.
9. Thierry S, Fautrel B, Lemelle I, et al. Prevalence and incidence of juvenile idiopathic arthritis: a systematic review. Joint Bone Spine. 2014;81:112–117.
10. Hayworth JL, Turk MA, Nevskaya T, et al. The frequency of uveitis in patients with juvenile inflammatory rheumatic diseases. Joint Bone Spine. 2019;86:685–690.
11. Nigrovic PA, Colbert RA, Holers VM, et al. Biological classification of childhood arthritis: roadmap to a molecular nomenclature. Nat Rev Rheumatol. 2021;17:257–269.
12. Sen ES, Dick AD, Ramanan AV. Uveitis associated with juvenile idiopathic arthritis. Nat Rev Rheumatol. 2015;11:338–348.
13. Prelog M, Schwarzenbrunner N, Sailer-Hoeck M, et al. Indications for a disturbed peripheral T-cell homeostasis in juvenile idiopathic arthritis (JIA): absent expansion of CD28 T-cells and no decrease of naive T-cells in cytomegalovirus-positive patients with JIA. J Rheumatol. 2008;35:520–527.
14. Almanzar G, Zlamy M, Koppelstaetter C, et al. Increased replication of CD4+ naive T cells and changes in T cell homeostasis in a case of acute exacerbation of juvenile idiopathic arthritis: a case comparison study. J Med Case Rep. 2013;7:135.
15. Dvergsten JA, Mueller RG, Griffin P, et al. Premature cell senescence and T cell receptor-independent activation of CD8+ T cells in juvenile idiopathic arthritis. Arthritis Rheum. 2013;65:2201–2210.
16. Picarelli MM, Danzmann LC, Grun LK, et al. Arterial stiffness by oscillometric device and telomere length in juvenile idiopathic arthritis with no cardiovascular risk factors: a cross-sectional study. Pediatr Rheumatol Online J. 2017;15:34.
17. Davies NM, Holmes MV, Davey smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601.
18. Burgess S, Scott RA, Timpson NJ, et al. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. 2015;30:543–552.
19. Li C, Stoma S, Lotta LA, et al. Genome-wide association analysis in humans links nucleotide metabolism to leukocyte telomere length. Am J Hum Genet. 2020;106:389–404.
20. Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002;30:e47.
21. Cawthon RM. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res. 2009;37:e21.
22. Lopez-Isac E, Smith SL, Marion MC, et al. Combined genetic analysis of juvenile idiopathic arthritis clinical subtypes identifies novel risk loci, target genes and key regulatory mechanisms. Ann Rheum Dis. 2020;80(3):321.
23. McIntosh LA, Marion MC, Sudman M, et al. Genome-wide association meta-analysis reveals novel juvenile idiopathic arthritis susceptibility loci. Arthritis Rheumatol. 2017;69:2222–2232.
24. Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408.
25. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37:658–665.
26. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32:377–389.
27. Zhao Q, Chen Y, Wang J, et al. Powerful three-sample genome-wide design and robust statistical inference in summary-data Mendelian randomization. Int J Epidemiol. 2019;48:1478–1492.
28. Verbanck M, Chen CY, Neale B, et al. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–698.
29. Fasching CL. Telomere length measurement as a clinical biomarker of aging and disease. Crit Rev Clin Lab Sci. 2018;55:443–465.
30. Srinivas N, Rachakonda S, Kumar R. Telomeres and telomere length: a general overview. Cancers (Basel). 2020;12:558.
31. Hjelmborg JB, Dalgard C, Mangino M, et al. Paternal age and telomere length in twins: the germ stem cell selection paradigm. Aging Cell. 2015;14:701–703.
32. Pawelczyk T, Grancow-Grabka M, Trafalska E, et al. Telomerase level increase is related to n-3 polyunsaturated fatty acid efficacy in first episode schizophrenia: secondary outcome analysis of the OFFER randomized clinical trial. Prog Neuropsychopharmacol Biol Psychiatry. 2018;83:142–148.
33. Simon NM, Smoller JW, McNamara KL, et al. Telomere shortening and mood disorders: preliminary support for a chronic stress model of accelerated aging. Biol Psychiatry. 2006;60:432–435.
34. Gamal RM, Hammam N, Zakary MM, et al. Telomere dysfunction-related serological markers and oxidative stress markers in rheumatoid arthritis patients: correlation with diseases activity. Clin Rheumatol. 2018;37:3239–3246.
35. Georgin-Lavialle S, Aouba A, Mouthon L, et al. The telomere/telomerase system in autoimmune and systemic immune-mediated diseases. Autoimmun Rev. 2010;9:646–651.
36. Thewissen M, Somers V, Venken K, et al. Analyses of immunosenescent markers in patients with autoimmune disease. Clin Immunol. 2007;123:209–218.
37. Maly K, Schirmer M. The story of CD4+ CD28- T cells revisited: solved or still ongoing? J Immunol Res. 2015;2015:348746.
38. Zhang J, Rane G, Dai X, et al. Ageing and the telomere connection: an intimate relationship with inflammation. Ageing Res Rev. 2016;25:55–69.
39. Prasad KN, Wu M, Bondy SC. Telomere shortening during aging: attenuation by antioxidants and anti-inflammatory agents. Mech Ageing Dev. 2017;164:61–66.
40. Chakravarti D, Hu B, Mao X, et al. Telomere dysfunction activates YAP1 to drive tissue inflammation. Nat Commun. 2020;11:4766.
41. Yang J, Xu H, Cai B, et al. Genetically predicted longer telomere length may reduce risk of hip osteoarthritis. Front Genet. 2021;12:718890.
42. Vazirpanah N, Verhagen FH, Rothova A, et al. Aberrant leukocyte telomere length in Birdshot Uveitis. PLoS One. 2017;12:e0176175.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.