Back to Journals » Journal of Inflammation Research » Volume 14
Melatonin as a Potential Regulator of Oxidative Stress, and Neuroinflammation: Mechanisms and Implications for the Management of Brain Injury-Induced Neurodegeneration
Authors Ikram M ![]() , Park HY, Ali T
, Park HY, Ali T ![]() , Kim MO
, Kim MO ![]()
Received 25 August 2021
Accepted for publication 15 November 2021
Published 27 November 2021 Volume 2021:14 Pages 6251—6264
DOI https://doi.org/10.2147/JIR.S334423
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monika Sharma
Muhammad Ikram,1,* Hyun Young Park,2,3,* Tahir Ali,1 Myeong Ok Kim1,4
1Division of Life Science and Applied Life Science (BK21 Four), College of Natural Sciences, Gyeongsang National University, Jinju, 52828, Republic of Korea; 2Department of Pediatrics, Maastricht University Medical Center, Maastricht, 6202 AZ, the Netherlands; 3School for Mental Health and Neuroscience (MHeNS), Maastricht Medical Center, Maastricht, 6229 ER, the Netherlands; 4Alz-Dementia Korea Co., Jinju, 52828, Republic of Korea
*These authors contributed equally to this work
Correspondence: Myeong Ok Kim Tel +82-55-772-1345, 2655(Sec.)
Fax +82-55-772-2656
Email [email protected]
Abstract: This review covers the preclinical and clinical literature supporting the role of melatonin in the management of brain injury-induced oxidative stress, neuroinflammation, and neurodegeneration, and reviews the past and current therapeutic strategies. Traumatic brain injury (TBI) is a neurodegenerative condition, unpredictably and potentially progressing into chronic neurodegeneration, with permanent cognitive, neurologic, and motor dysfunction, having no standard therapies. Due to its complex and multi-faceted nature, the TBI has highly heterogeneous pathophysiology, characterized by the highest mortality and disability worldwide. Mounting evidence suggests that the TBI induces oxidative and nitrosative stress, which is involved in the progression of chronic and acute neurodegenerative diseases. Defenses against such conditions are mostly dependent on the usage of antioxidant compounds, the majority of whom are ingested as nutraceuticals or as dietary supplements. A large amount of literature is available regarding the efficacy of antioxidant compounds to counteract the TBI-associated damage in animal and cellular models of the TBI and several clinical studies. Collectively, the studies have suggested that TBI induces oxidative stress, by suppressing the endogenous antioxidant system, such as nuclear factor erythroid 2–related factor-2 (Nrf-2) increasing the lipid peroxidation and elevation of oxidative damage. Moreover, elevated oxidative stress may induce neuroinflammation by activating the microglial cells, releasing and activating the inflammatory cytokines and inflammatory mediators, and energy dyshomeostasis. Thus, melatonin has shown regulatory effects against the TBI-induced autophagic dysfunction, regulation of mitogen-activated protein kinases, such as ERK, activation of the NLRP-3 inflammasome, and release of the inflammatory cytokines. The collective findings strongly suggest that melatonin may regulate TBI-induced neurodegeneration, although further studies should be conducted to better facilitate future therapeutic windows.
Keywords: melatonin, antioxidants, brain injury, oxidative and nitrosative stress, neurodegeneration
Introduction
A review paper by Kaur and Sharma defined Traumatic Brain Injury (TBI) as “the injury resulting from mechanical energy to the brains from any sort of external physical stimulus”.1 It occurs from the absorption by the brain tissues of the energy linked with a mechanical force, which does not act directly to the head of an individual, which causes a myriad of metabolic, molecular, and biochemical effects, affecting the overall cell’s homeostasis causing permanent or temporary cognitive and motor dysfunction, consciousness, or psychological capabilities.2 Some of the well-known causes of TBI include fallings, assault, traffic accidents, and sport-associated concussions.3 Traumatic brain injury affects more than 10 million people yearly in the world, representing 30–40% of all injury-related disabilities and mortalities among individuals of all ages, causing tremendous economic and social impact.4 Epidemiological studies have suggested a 2–3-times higher incidence of TBI-associated consequences in the next ten years compared to Alzheimer’s disease or other neurodegenerative conditions.5 Interestingly, two of the most studied risk factors for the TBI are sex and age, as the males are three times more prone to suffer from a TBI compared to the females. While in case of age, the persons of 65 years or older, and kids under 14 are more likely to suffer from TBI. Annually, up to 2 million Americans suffer from a TBI, which accounts for around 1.4 million emergency visits, 2.75 million hospitalizations, and 5.2 thousand deaths annually in the US, contributing 30% of all deaths in the USA per year. A complete discussion and facts related to the epidemiology of TBI may be found somewhere else.6 Not different in East Asian countries. For example, according to the Lancet Neurology (2019), China has the most TBI patients than almost all countries of the world, making it a major public health concern. Population-based mortality of TBI is estimated to be 13 cases per 100 000 people, which is not less than reported in the other countries.7 Similarly, a retrospective longitudinal cohort study evaluated TBI trends from 2008 to 2017, acquired from Korean National Health Insurance Service–National Health Information Database (NHIS-NHID) (2020). Interestingly, mortality per 100,000 people was significantly higher among people aged ≥70 years than in the other age group.
It is also confirmed from a worldwide meta-analysis of the incidence of TBI that males have the most TBI among the adult population. Also, in the meta-analysis by continent showed in all age groups, there was no significant difference among continents including European and North American studies. The result obtained from this study is essential, as it is their first systematic review and meta-analysis that highlighted the heterogeneity of TBI incidence.
Although significant research has been made to develop therapeutic strategies against traumatic brain injury-induced neurodegeneration, it still represents the major global and public health challenge among all ages overall in the world regardless of the patient’s financial status and income level.8 Previously, a wide set of studies has been published on the role of melatonin in the management of brain injury-induced neurodegeneration.9 Therefore, the overall aim of this review is to summarize the role of melatonin against brain injury, including subarachnoid hemorrhage and cellular models of brain injury. In this review, broadly three broad thematic areas will be discussed, including the processes and etiology of TBI, evidence on therapeutic options from pre-clinical and clinical studies of TBI, and the use of melatonin as a potential regulator of TBI in multiple pathways will be discussed. The motivation behind the compilation of this review article was our previous study conducted on the role of melatonin against traumatic brain injury induced-neurodegeneration.10 For a collection of the articles, several independent databases, including but not limited to PubMed (https://pubmed.ncbi.nlm.nih.gov), Google Scholar (https://scholar.google.co.kr), and Web of Sciences (https://apps.webofknowledge.com). The papers were randomly selected, care was taken to avoid biases and repetition, and the main findings of the articles were collected from the abstracts, and some established protocols were followed.11 The paper has two major parts, one is traumatic brain-injury and the protective effects of melatonin against brain injury, the contents of the study has been shown in Figure 1.
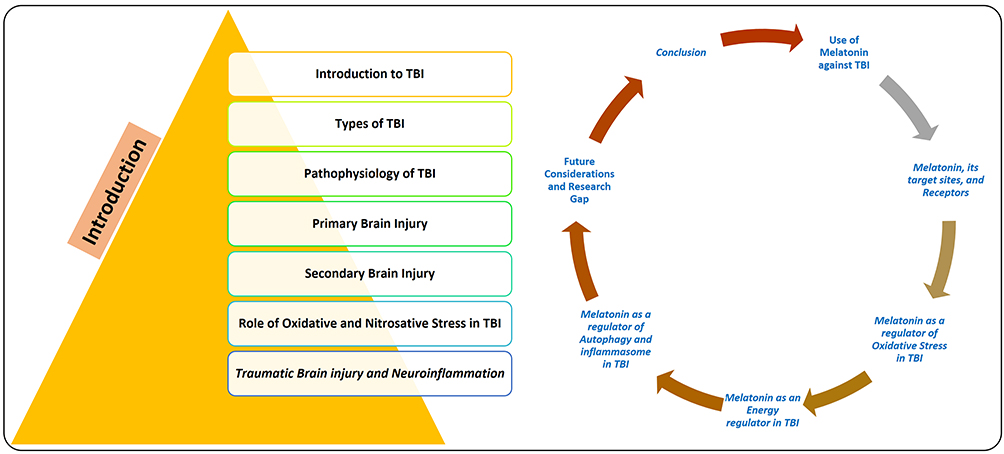 |
Figure 1 Diagram, showing the overall contents of the manuscript covering the introduction and the contents related to melatonin. |
Types of TBI
There are several systems to analyze the severity of TBI, the most common one is the Glasgow Coma Scale (GCS). The GCS classifies TBI into mild (GCS range 13–15), moderate (GCS range 9–12), and severe (GCS range 3–8), which are based on scores obtained from specific clinical evaluations, such as eye-opening, verbal communication and motor functions, which has been given elsewhere.12 The TBI can be divided into three categories based on its distinctive physical insult: closed head, penetrating, and explosive blast TBI. The closed-head TBI is the highest occurring incident among civilians caused by blunt objects, such as falling, vehicle accidents, and sports injuries. A brain injury in which a foreign body penetrates the brain parenchyma, causing focal damages, intracranial hemorrhage, edema, and ischemic conditions is called penetrating TBI.13 Explosive TBI is a form of TBI caused by the explosion, introduced in the 20th century, which is war-related TBI.14 Here, in explosion blast TBI, the brain is badly affected by rapid pressure shock waves, transmitting a significant amount of energy from the skull into the brain parenchyma.15
Pathophysiology of TBI
The pathophysiological consequences of TBI can be divided into the following two categories: primary brain injury, and secondary brain injury.
Primary Brain Injury
The primary brain injury comprises the direct physical insult to the neuronal, vascular, and glial cells.16 It may cause deformation, compression, displacement, shearing, stretching, tearing, and crushing of brain cells and the blood circulatory system. The direct head injury can cause injury in adjacent (coup) brain cells or may damage the opposite (contrecoup) sides of the cells. The concussion involves the primary injury, which is not severe and is associated with rapid loss of consciousness (for more than several minutes) and occurs as a result of coup and contrecoup lesions. The contusion causes glial and vascular cell injury, while in severe cases it may cause neuronal injury. A primary brain insult is an irreversible event that causes neuro-electro-chemical events (secondary injury) that ends in neurodegeneration.2
Secondary Brain Injury
The molecular and biochemical changes caused by the primary neuronal injury cause secondary brain injury. Several factors may contribute to secondary brain injury, including mitochondrial dysfunction, excitotoxicity, oxidative stress and nitrosative stress, neuroinflammation, lipid peroxidation, and axonal degeneration.17,18 A sketch (Figure 2) shows the pathophysiology of brain injury.
 |
Figure 2 Pathophysiology of traumatic brain injury. A brief illustration of the primary brain injury, which is consisted of stretching, compression, and tearing followed by secondary brain injury which leads to further induce oxidative stress, neuroinflammation, excitotoxicity, apoptotic cell death, blood-brain barrier disruption, dendritic degeneration, synaptic loss, and ultimately progress towards neurodegeneration and cognitive dysfunctions. |
Role of Oxidative and Nitrosative Stress in TBI
Studies related to TBI have extensively highlighted the role of Reactive Oxygen Species (ROS) and free radicals in brain injury, facilitating lipid peroxidation, protein oxidation, and DNA damage, causing excitotoxicity (glutamates), ionic dysregulation, and mitochondrial apoptosis.19 The excitotoxicity and release of glutamate cause an influx of Ca2+ into cells through the N-methyl D-aspartate (NMDA) receptors,20 causing mitochondrial dysfunction with oxidative phosphorylation, and energy dyshomeostasis.21 Among ROS, superoxides are produced from the malfunctioning of the mitochondrial electron transport chain and activation of microglia and macrophages.22 The superoxide anion is converted into H2O2 + O2 by Superoxide dismutase (SOD), through the glutathione peroxidase. The overproduction of superoxide coupled with acidosis favors the H2O2 to react with extravasated hemoglobin, producing hydroxyl radicals, which react with several biological molecules (capable of donating an electron).23 The nuclear factor (erythroid-derived 2)-like 2 (Nrf-2), encoded by the NFE2L2 gene, is an important transcription factor acting as antioxidant defense system.24 Nrf-2 is known as the molecular switch turning on/off the Nrf-2 signaling acting as an integral component of the oxidative-stress pathway, which is activated in response to the oxidative damage.25 Under normal physiological conditions, Nrf2 is found in the cytoplasm bound to Kelch-like erythroid cell-derived protein with CNC homology- (ECH-) associated protein 1 (Keap-1), acting as a primary inhibitor of Nrf-2, where it undergoes proteasomal degradation and ubiquitination. Under physiological stress, the Nrf-2 is separated from the Keap-1, permeated into the nucleus, where it attaches with Antioxidant responsive element (ARE) and induces the transcription of ARE-associated antioxidant enzymes, such as catalases, superoxide dismutase (SOD), glutathione S-transferase (GSTP),26 heme oxygenase-1 (HO-1).27
Apart from oxidative stress, nitrosative stress also plays a pivotal role in the execution of neuronal cell loss in TBI28 and other neurodegenerative conditions.29 It affects the level of nitric oxide (NO), affecting the extracellular and intracellular signaling, which is crucial for cell survival and functioning, and is produced by the activity of nitric oxide synthase (NOS).30 The elevated level of peroxynitrite and RNS actively suppresses the endogenous antioxidant mechanisms, causing lipid peroxidation of mitochondrial membranes,31 affecting the Ca2+ homeostasis,32 formation of the membrane permeability transition pores,33 and neurodegeneration.29 Figure 3 has been given, showing the role of oxidative and nitrosative stress and its consequences.
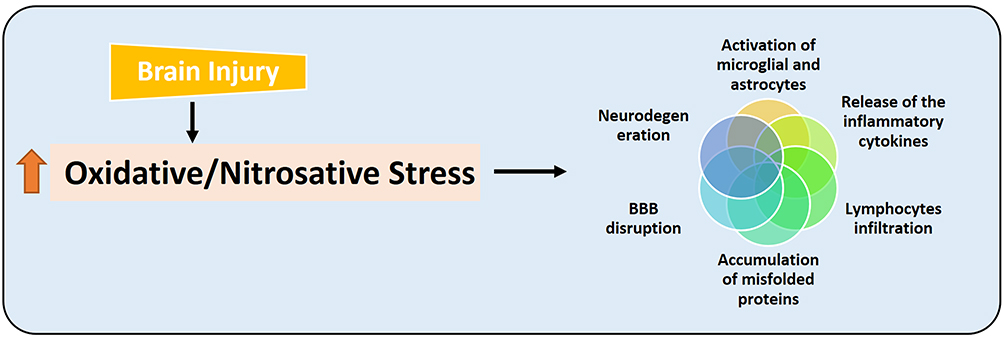 |
Figure 3 Brain injury-induced oxidative and nitrosative stress and its consequences. Diagrammatic representation, showing that brain injury induces oxidative and nitrosative stress, which causes further deterioration by disrupting the blood-brain barrier, activating the astrocytes and microglial cells, releasing the inflammatory cytokines, infiltration of the lymphocytes, and may cause the accumulation of misfolded proteins and neurodegeneration. |
Traumatic Brain Injury and Neuroinflammation
Within 24 hours of TBI, the permeability of the blood-brain barrier (BBB) increases the infiltration of circulating monocytes, neutrophils, and lymphocytes into the brain parenchyma,34 which activates the complement system and release inflammatory cytokines such as tissue necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6).35,36 The release of these inflammatory cytokines is associated with BBB dysfunction and brain edema. It was established that upon injury the activated microglia and macrophages reach the site of insult to mitigate the deleterious impacts of the brain injury and provide a safe environment.37 The activated microglia may cause the release of inflammatory cytokines, reactive oxygen species, and release of glutamate, which exacerbates the injury and neuronal cell death.38 The maturation of IL-1β and its release into the systemic circulation is mainly regulated by nucleotide oligomerization-like receptor protein 3 (NLRP3) inflammasome, which is localized in immune cells.39 The NLRP3 inflammasome is made of NLRP3, oligomers apoptosis-associated speck-like (ASC) adapter protein, and pro-caspase-1, cleaving caspase-1 and maturation of IL-1β, and neuroinflammation.40 Figure 4 shows the overall brain injury associated with neuroinflammation and its consequences.
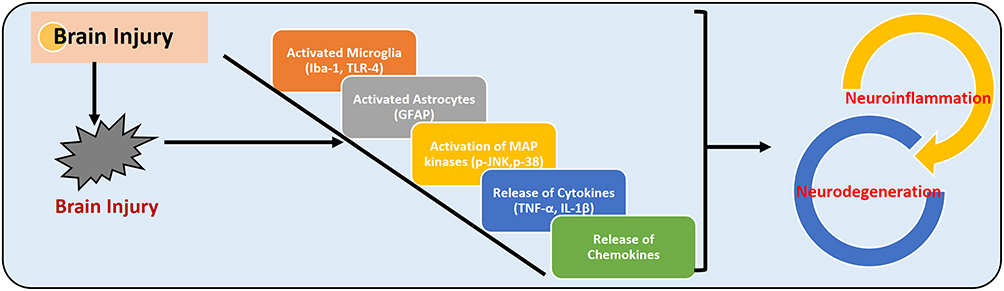 |
Figure 4 Traumatic brain injury and neuroinflammation. The figure illustrates the activation of microglia (Iba-1 and microglial receptor TLR-4), astrocytes (GFAP), MAP kinases (such as p-JNK and p-38), the release of the inflammatory cytokines (TNF-α and IL-1β), and release of chemokines, which may induce neuroinflammation and neurodegeneration. |
Use of Melatonin Against Traumatic Brain Injury-Induced Neurodegeneration
The therapeutic approaches deeply vary among the individuals because of the complex nature, limited knowledge of the pathophysiological mechanisms of TBI, and the response of the patient to the drugs. The most frequently used drugs in the management of TBI include osmotic therapy such as mannitol, which regulates osmotic pressure, decreases cerebral edema, and cerebral blood flow. Other drugs used to reduce secondary brain injury include opioids (morphine, to reduce the pain), and sedatives (benzodiazepines, barbiturates) which reduce neuronal activity.41 Table 1 depicts a summarized chart on the therapeutic drugs or approaches to treating TBI in the pre-clinical and clinical trial stage. As mentioned, there is no therapeutic drug available against TBI-induced neurodegeneration, although several pre-clinical drugs, such as melatonin and plant-based polyphenols have drawn much attention.42 Failure to develop effective drugs against brain injury-induced neurodegeneration may be partly because most drugs target a single aspect of the disease. So, current efforts are being made to develop drugs having multi-targeted beneficial effects against injury. In this regard, melatonin, a hormone from the pineal gland has drawn much attention, due to its multi-targeted effects against injuries. Here, we give a list of previously used compounds and drugs against TBI. Table 1.
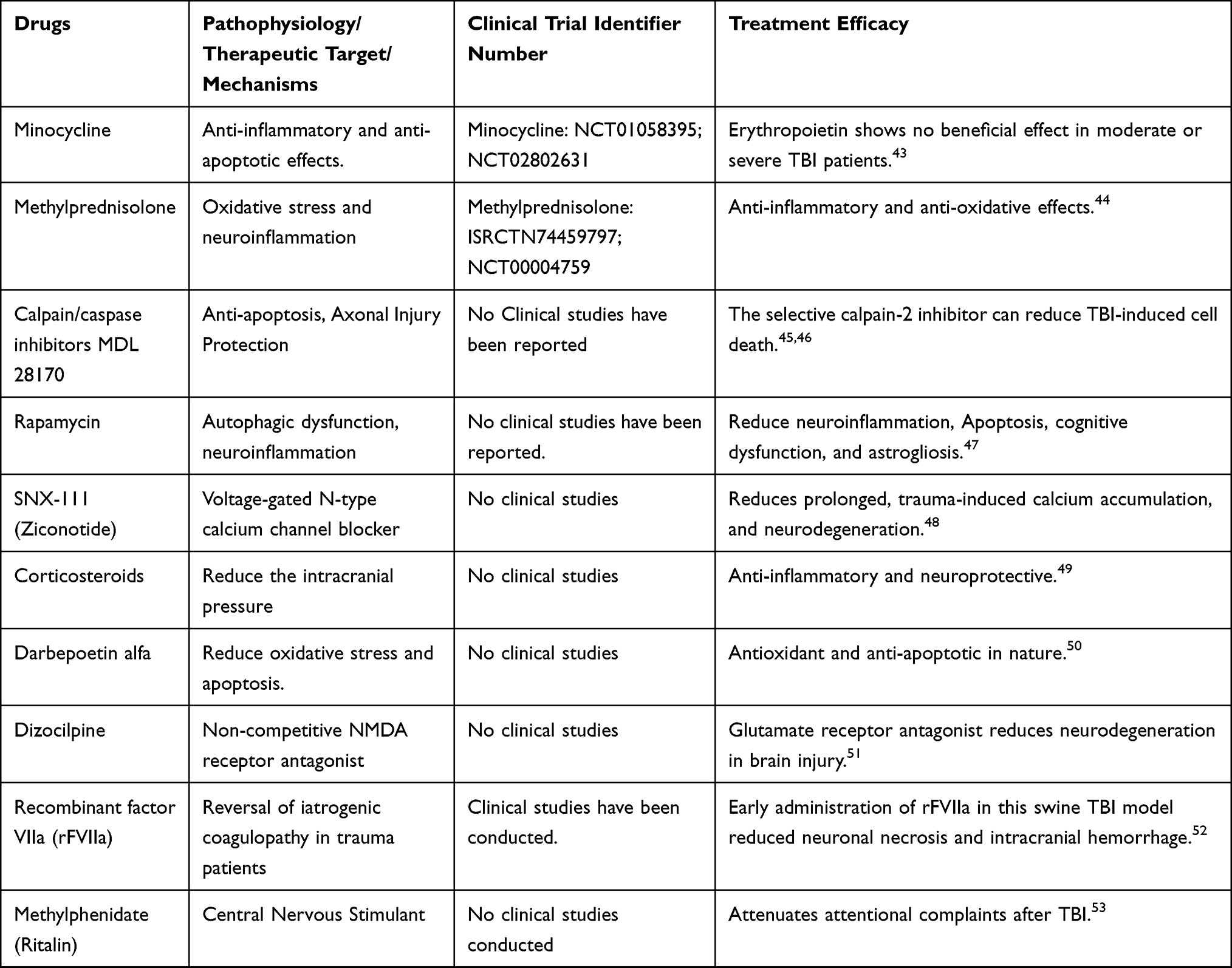 |
Table 1 Summary of the Drugs Used Against Traumatic Brain Injury-Induced Neurodegeneration, Therapeutic Targets, and Implications |
Melatonin, Its Target Sites, and Receptors
Melatonin (N-acetyl-5-hydroxytryptamine), a hormone of darkness synthesized within the pinealocytes from tryptophan, once synthesized it is released into the peripheral system and cerebrospinal fluid (CSF). Melatonin acts both centrally and peripherally and the binding sites for melatonin are found in several areas of the brain including the hypothalamus, pars tuberalis, immune cells, gonads, kidneys, and heart.54 Melatonin mediates its effects through receptor-dependent and independent mechanisms. The non-receptor-mediated effects are due to amphipathic properties, meaning that it can easily cross the cell and nuclear membranes, as it can be easily found in the nuclei of several cells in the brain and other body tissues.55 A prominent example of the non-receptor-mediated effects of melatonin includes antioxidant action against different models of oxidative stress. Melatonin acts as a free radical scavenger and potent anti-oxidant in vivo and in vitro models, independent of the presence of the receptor.56 Previously, several studies have suggested strong antioxidant, anti-inflammatory, and neuroprotective effects against different models of neurodegeneration.10 In this review, we have collected data from different sources concerning the use of melatonin in the post-injury period to counteract the effects of TBI-associated neurodegeneration.
Melatonin as a Regulator of Oxidative Stress in Traumatic Brain Injury
Oxidative stress plays a pivotal role in the pathophysiology of TBI, which suggests that antioxidant therapeutic approaches may provide a rescuing effect against TBI-induced oxidative damage.57,58 Several natural compounds have shown beneficial effects against brain injury-induced oxidative stress-mediated neurodegeneration, such as melatonin, quercetin, and other plyphenols10,59 that. A study conducted on melatonin against the controlled weight‐drop TBI model has shown rescuing effects of melatonin against the elevated oxidative stress (as shown by reduced expression of total glutathione, an enhanced ratio of oxidized glutathione to the total glutathione). Moreover, there was a decrease in STAT1 DNA‐binding activity, and neuronal cells. There was a marked upregulation in the mRNA level of the inhibitor of cytokine signaling (SOCS3), interleukin‐6 (IL‐6), inducible nitric oxide synthase (iNOS), and a reduced expression of protein inhibitor of activated STAT (PIAS1), after 24 hours of the brain contusion. As SOCS3 and PIAS1 are the endogenous inhibitors of STAT1. Interestingly, they have suggested that activation of SOCS‐3 and elevated oxidative stress in the TBI may aid in the activation of STAT1 inactivation, and melatonin may reduce the TBI-induced oxidative stress, STAT1 inhibition, and increase in the expression of SOCS‐3 and inflammatory mediators.60
To evaluate the effect of melatonin against subarachnoid hemorrhage (SAH)-induced neurodegeneration and oxidative stress, the SAH model was developed by injection of arterial fresh autologous blood (non-heparinized) into the chiasmatic cistern. To the SAH + melatonin co-treated group, melatonin was injected at a dose of 150 mg/kg 2 and 24 hours after the SAH, and brain samples were collected after 48 hours of SAH. Their findings suggested that melatonin significantly enhanced the levels of Nrf-2‐ARE signaling and associated genes, such as Nrf-2, NAD (P) H: quinone oxidoreductase-1, heme oxygenase‐1 (HO-1), and glutathione S‐transferase α‐1. Moreover, melatonin regulated the blood-brain barrier (BBB) dysfunction, cortical apoptosis, brain-edema, and neurological impairment. The findings suggest that melatonin may rescue the SAH-induced neurodegeneration and oxidative stress by regulating the endogenous antioxidant regulators.61
A similar study was conducted on melatonin against the mouse model of traumatic brain injury. Here, the Melatonin (10mg/kg/intraperitoneal injection) was administered at 0, 1, 2, 3, and 4 hours post-TBI. For confirmatory purposes, a second experiment was conducted where the Nrf2 wild-type (Nrf2 (+/+) group) and Nrf2-knockout mice were used, and the TBI was induced in these groups and injected with melatonin (10mg/kg, i.p) at the corresponding time points. The findings have suggested that melatonin after TBI markedly regulated the effects of brain injury-induced oxidative stress, cortical neuronal degeneration, and brain edema. Furthermore, melatonin boosted the nuclear translocation of Nrf2 from the cytoplasm; thereby enhancing the expression of Nrf2-ARE signaling and related proteins, such as heme oxygenase-1 and NAD(p)h: Quinone oxidoreductase-1; and reduced the antioxidant enzymes, such as glutathione peroxidase and superoxide dismutase. Interestingly, in Nrf2 knockout mice, the neuroprotective effects of melatonin were partly reduced,62 showing that melatonin through Nrf2 rescued the mice brains against TBI-induced neurodegeneration.
A study conducted on closed head injury (CHI) has suggested that melatonin (5 mg/kg) had pronounced effects on neurobehavioral recovery, suppression of the redox‐sensitive transcription factors nuclear factor‐kappa-B (NF‐κB), and AP‐1 in mice subjected to closed head injury. After one hour of CHI, melatonin facilitated recovery for at least 1 week and decreased lesion size by two-fold. Neuroprotection against closed head injury-induced neurodegeneration was achieved with melatonin. The findings have indicated that melatonin is neuroprotective, via potentiation of brain antioxidants and regulation of NF‐κB and AP‐1 activation.63 Overall findings have suggested that melatonin may reduce brain injury-induced oxidative stress and its consequences. Figure 5 shows a summary of the protective effect of melatonin against brain-injury associated neuroinflammation and neurodegeneration, by counteracting the oxidative and nitrosative stress, boosting the endogenous antioxidant mechanism (Nrf-2, HO-1 and GSH), and lipid peroxidation.
 |
Figure 5 Melatonin is a potent antioxidant in brain injury-induced neurodegeneration. |
Melatonin as an Energy Regulator in Traumatic Brain Injury
Apart from the elevated oxidative stress, energy imbalance and dyshomeostasis also play a pivotal role in the progression of brain injury-induced neurodegeneration.64 Both preclinical and clinical studies have suggested that brain injury causes a prolonged suppression in glucose metabolism that affects brain physiology.65 Most of the metabolic dysfunctions occur in the cortical and hippocampal regions of the brain,66 highlighting the roles of metabolic dysfunction in brain injury-induced neurodegeneration and its pathological consequences.67 Melatonin boosts the energy sources (such as glucose and lactate) or provides alternative energy sources (glyceryl triacetate, acetyl-L-carnitine),68,69 which may confer neuroprotection against brain injury-induced neurodegeneration. Several studies have indicated that boosting the energy sensors through its kinases may confer neuroprotection after TBI-induced neurodegeneration. A study conducted on the role of phospho-5′AMP-activated protein kinase (p-AMPK) has shown that pharmacological enhancement of p-AMPK expression is neuroprotective.70 To unveil the role of melatonin against the TBI-induced downregulated expression of p-AMPK, we conducted a study on TBI, which suggested that TBI-evoked oxidative stress, induced neuroinflammation, dysregulated the brain energy homeostasis, and contributed to neurodegeneration and memory dysfunction in mice. As TBI reduced the expression of phospho-5′AMP-activated protein kinase (p-AMPK) and phospho- cAMP-response element-binding (p-CREB), and activated the phosphorylation of NF-κB in TBI mice brains, interestingly melatonin enhanced the expression of p-AMPK, p-CREB, and reduced the p-NF-κB in the TBI-induced mice. Moreover, melatonin regulated mitochondrial dysfunction, amyloidogenic markers, and behavioral dysfunctions in TBI-induced mice.10
A study conducted on 7-day-old rat pups, which were subjected to contusion injury and were examined after 24 hours of TBI, showed elevated expression of thiobarbituric acid reactive substances (TBARS), and there was no increase in the antioxidant enzymes such as superoxide dismutase (SOD) and glutathione peroxidase (GPx). Contrarily, Melatonin at a single dose (5 mg/kg) reduced the level of TBARS levels in brain hemispheres, showing that melatonin reduced brain injury-induced oxidative stress in rat pups.71
Surgical brain injury (SBI) is another form of brain injury, which occurs as an inevitable consequence of neurosurgical procedures. SBI may cause postoperative complications such as brain edema and inflammation following blood-brain barrier (BBB) leakage and neurological disorders. They have suggested that administration of melatonin (5 mg/kg, 15 mg/kg) to the rodent models (before the surgical procedures) markedly reduced brain edema, reduced lipid peroxidation, and may protect the brains against SBI-induced neurodegeneration. They have shown that a higher dose of melatonin 150mg/kg is deleterious, as it may induce lipid peroxidation and brain edema.72
To evaluate the effects of melatonin on the regeneration of motor axons in the TBI-induced model, two types of injury models were developed; reversible and acute presynaptic degeneration induced by α‐Latrotoxin and compression/transection of the sciatic nerves. The findings have suggested that melatonin accelerated the process of nerve repair via melatonin receptor-1 (MT1) and is partly due to sustained activation of the ERK1/2 signalings. These findings have suggested that the regenerative effects of melatonin are receptor-mediated and may offer a potential application for the management of several peripheral neurodegenerative conditions.73
The neuronal‐specific K+–Cl− cotransporter‐2 (KCC2) is the main Cl− extruder in neuronal cells, playing a role in the Cl- homeostasis and physiological functions of the neurons. A study conducted on the role of melatonin on the expression of KCC2 in the TBI-induced rat model has suggested reduced expression of KCC2 (protein and mRNA) in the ipsilateral peri‐core parietal cortex. On the other hand, with the administration of melatonin (10 mg/kg intraperitoneally at 1, 2, 3, and 4 hours after TBI), the expression of KCC2 was significantly enhanced. Moreover, with the administration of melatonin, there was a significant increase in the expression of brain‐derived neurotrophic factor (BDNF) and phospho-extracellular signal‐regulated kinase (p‐ERK), reduced neuronal apoptosis, brain edema, and neurological dysfunction in the brain injury-induced rats. The overall findings suggest that melatonin may enhance the expression of KCC2, BDNF, and p-ERK, thereby reducing neurodegeneration and neurological deficits in TBI-induced animals.74 Similarly, a study conducted by Nilgün Senol has also investigated the anti-oxidant effects of melatonin in the cerebral cortex and blood of traumatic brain injury-induced rats. The findings have suggested that TBI reduced the β-carotene, vitamin C, vitamin E, glutathione, and erythrocyte, glutathione levels, and plasma vitamin C level in the cerebral cortex of TBI-induced mice, which were enhanced with the administration of melatonin, suggesting that melatonin may regulate the level of anti-oxidant enzymes thereby regulate the oxidative stress associated with TBI. The overall findings support the anti-oxidant potentials of melatonin against brain injury-induced neurodegeneration.75
Melatonin as a Regulator of Autophagic Dysfunction and Activated NLRP-3 Inflammasome in TBI-Induced Mice
Autophagy is a self-catabolic physiological mechanism by which cells recycle and conserve their organelles in a nutrient-deprived or stressed state, which is a lysosomal degradation mechanism responsible for survival, development, differentiation, and homeostasis. It serves as a safeguard protecting the organisms against diverse pathogenic events including cancer, infections, aging, neurodegeneration, and heart diseases.76 Also, autophagic dysfunction is involved in several pathological conditions, including TBI and other brain injuries,77 and boosting autophagy may counteract the pathological consequences of TBI. To explore the effects of melatonin on the autophagy-related markers in animal models of brain injury, several studies have been conducted on melatonin. A study has suggested that melatonin activates mitophagy via the mTOR signaling, thereby reducing the TBI‐induced inflammation. Moreover, melatonin markedly regulated neuronal cell death and behavioral dysfunctions in TBI-induced mice. The collective findings have suggested that melatonin may protect the mice’s brain against TBI‐induced neuroinflammation by activating the autophagy and inhibiting the IL-1β secretion via regulating the autophagy of damaged mitochondria.78 Another study suggested that melatonin markedly regulated the secondary brain injury induced by TBI upregulated autophagy, accompanied by a reduction in translocation of Bax to mitochondria and the release of cytochrome C into the cytoplasm. Collectively, they have suggested that melatonin regulates autophagy and mitochondrial apoptosis, thereby protecting the mice’s brains against TBI-induced secondary brain injury.79 In addition to oxidative stress, the innate immune response is also playing a critical role in the pathophysiology of brain-injury-induced neurodegeneration. One key mechanism of the innate immune system is NLRP-3 inflammasome activation, which is an important component of the innate immune response to inflammasome activities and tissue damage, and the abnormal activation of this system has been extensively reported in several neurological conditions, including AD, PD and TBI.40 Several studies have suggested that inhibition of the NLRP-3 inflammasome activation in the TBI may reduce the activation of the inflammatory cytokines and neuroinflammation.80 To analyze the effects of melatonin against the subarachnoid hemorrhage (SAH)-induced activated inflammasome, a comprehensive study has been conducted. The findings have indicated that melatonin through upregulation of autophagy (LC3-II/LC3-I and Atg 5), and mitophagy markers (Parkin and PINK-1) suppressed the NLRP-3 inflammasome activation. Moreover, melatonin regulated the pathological changes in mitochondria, reduced oxidative stress, and reduced the pro-inflammatory cytokine after SAH-induction. Interestingly, the inhibition of autophagy (through 3-MA) reversed the protective effects of melatonin against SAH-induced NLRP-3 inflammasome activation and release of the inflammatory mediators, showing the protective effects of melatonin are autophagy-dependent.81 Figure 6 shows the effects of melatonin against the activated NLRP-3 inflammasomes and subsequent neurodegeneration.
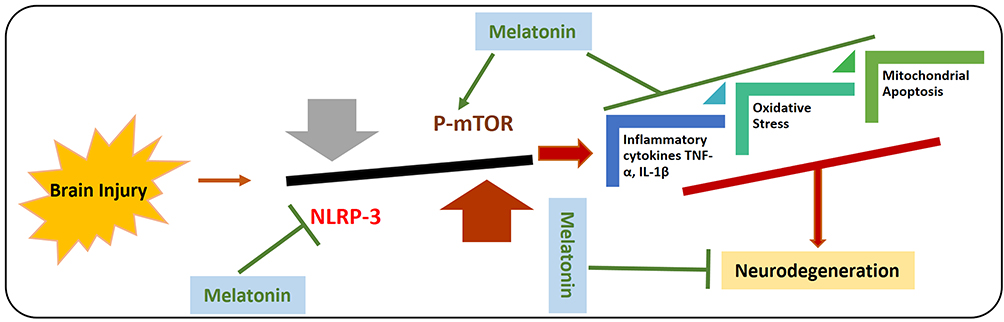 |
Figure 6 Effects of melatonin against Brain injury-induced activated NLRP-3 inflammasome and autophagic dysfunction. Graphical representation of effects of melatonin against brain injury-induced the activation of NLRP-3 inflammasome and inhibits the phosphorylation of mTOR. As the activated NLRP-3 inflammasome are responsible for the release of inflammatory cytokines, mitochondrial apoptosis, and elevated oxidative stress. Moreover, it has been suggested that melatonin has regulatory effects on the mTOR-mediated autophagy and NLRP-3 inflammasome activation. |
Future Considerations, Research Gap, and Conclusion
The overall findings strongly support the notion that melatonin may counteract the brain-injury associated neurodegenerations by regulating oxidative stress, neuroinflammation, and autophagic dysfunction. Melatonin has shown neuroprotective effects in receptor dependent and independent manners.10 The receptor-independent effects are achieved through activating the endogenous antioxidant mechanisms, reducing lipid peroxidation, and reducing the reactive oxygen species. For example, melatonin attenuates oxidative insult by activating specific activator of transcription factor Nrf2 [REF]. The neuroprotective effects of melatonin on TBI were verified using Nrf2 knockout mice, which caused a loss in the neuroprotective effects of melatonin, indicating that the neuroprotective effects of melatonin are Nrf2 dependent.62 In addition, our study conducted on brain injury has suggested that melatonin may regulate the expression of p-AMPK and p-CREB, thereby reducing neuroinflammation and neurodegeneration [REF]. Also, our previous study spanned on the role of melatonin against ethanol-induced neurodegenerations in 7-day-old rat pups. Our finding is in agreement with the above studies that revealed related mechanisms of Nrf2 and melatonin significantly reduced neuroinflammation and neurodegeneration via the Nrf2 and its associated genes, as Nrf2 knocked down in HT-22 cells resulted in significant loss in the neuroprotective effects of melatonin [REF].
Another main mechanism is the regulation of autophagic dysfunction in brain injury, as the regulation of autophagy is responsible for counteracting neuroinflammation and neurodegeneration.78 A study conducted on the role of melatonin against the TBI-induced neuroinflammation has suggested that melatonin reduced the expression of IL-1β level in the melatonin-treated groups compared to the TBI-induced mice. Similarly, melatonin enhanced the expression of IL-6, TNF-α IL-10 compared to the TBI model group (p < 0.001). These findings suggested that melatonin reversed the inflammatory cytokines involved in the brain injury-induced edema and neurodegeneration after TBI.82 Besides the receptor-independent mechanisms, several studies show that melatonin regulates ERK phosphorylation, thereby modulating the axonal regeneration via its melatonin receptor (MT1).73 The available literatures strongly highlighted that the protective effects of melatonin against brain injury-induced neurodegeneration, but several aspects are left unexplored. For example, still there are limited studies available on whether regulation of sleep via melatonin in the TBI-induced model. In mammals and humans, the melatonin is secreted at night and the peak plasma concentration reaches at night (3–4 am) with a robust circadian rhythm. The rise of melatonin secretion is strongly associated with sleep propensity about 2 hours before the person’s regular bedtime.83 Several studies have suggested that in TBI, there is a reduction in sleep quality, as melatonin has the sleep-promoting quality and is involved in the circadian regulation of the sleep-wake cycle. Abrogated melatonin concentration may contribute to sleep-related issues after TBI.84 Studies have shown that TBI patients showed reduced (42% less) melatonin production overnight compared to healthy participants. Similarly, the timing of dim light melatonin onset (DLMO) was delayed by 1.5 hours in patients with TBI compared to the controls, proving that circadian regulation of melatonin synthesis is a feature of TBI, contributing to the sleep deprivation.85 Another study has demonstrated a clear disruption of the diurnal rhythm in the TBI in the intensive care unit (ICU) patients. These patients with TBI peaked maximal endogenous serum melatonin level compared to control or patients with trauma without TBI, which is due to the stress caused by ICU. These findings widen the therapeutic window of melatonin, which might improve TBI patient recovery by balancing the circadian rhythm, strengthening the notion that administration of melatonin may counteract the TBI-associated sleep complications.86
Few other shortcomings are in the timing of administration of melatonin, as melatonin is specifically activated in response to the darkness, and more detailed studies tackling whether administration or endogenous melatonin has therapeutic implication needs to be further investigated. Also, more mechanistic studies may be focused to unveil the specific dose, duration of treatment, pre- and post-treatment of melatonin in different disease states, and effects of sex differences on the efficacy of melatonin. One study has suggested that compared to the female rats, melatonin exhibited more significant neuroprotective effects (including improved locomotor and exploratory activities, elevated neuronal amount, and reduced neuronal apoptosis) in male rats exposed to TBI.87
Conclusively, the overall findings suggest that melatonin may reduce brain injury-induced neurodegeneration by regulating oxidative stress, neuroinflammation, autophagy, and apoptotic cell death. The exact mechanisms behind the neuroprotective effects of melatonin are still not clear as mentioned previously, more efforts should be focused on unveiling the neuroprotective effects of melatonin. Current therapeutic approaches based on the regulation of oxidative stress, neuroinflammation, and neurodegeneration are quite generalized. Due to the heterogeneity of TBI mechanisms, current review focuses on oxidative and nitrosative stress after TBI. On the other hand, still very little attention has been given to underlying pathology on the association of melatonin and immune activation such as microglia and its receptors. Since immune system is very plastic, meaning that it can not only remain over a long period after injury, but immune activation also drive both injury and repair. This calls for future research, particularly to the long-term outcomes on immune responses after brain injury-induced neurodegeneration. Based on available studies, we suggest that melatonin may be further evaluated for its efficacy against brain injury-associated degenerative conditions.
Abbreviations
ICU, intensive care unit; ARE, antioxidant responsive element; TBI, traumatic brain injury; ROS, reactive oxygen species; SOD, superoxide dismutase; RNS, reactive nitrosative stress; Nrf-2, nuclear factor erythroid 2-related factor 2; HO-1, heme oxygenase-1; NOS, nitric oxide synthase; IL-1β, interleukin-1β; Aβ, amyloid beta; TNF-α, tissue necrosis factor-alpha; NF-kB, nuclear factor-kappa-B; DLMO, dim light melatonin onset.
Author Contributions
All authors contributed to data analysis, drafting, or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This research was supported by the Neurological Disorder Research Program of the National Research Foundation (NRF) funded by the Korean Government (MSIT) (2020M3E5D9080660).
Disclosure
The authors declare no conflict of interest.
References
1. Kaur P, Sharma S. Recent advances in pathophysiology of traumatic brain injury. Curr Neuropharmacol. 2018;16(8):1224–1238. doi:10.2174/1570159X15666170613083606
2. McKee AC, Daneshvar DH. The neuropathology of traumatic brain injury. Handb Clin Neurol. 2015;127:45–66.
3. Ahmed S, Venigalla H, Mekala HM, Dar S, Hassan M, Ayub S. Traumatic brain injury and neuropsychiatric complications. Indian J Psychol Med. 2017;39(2):114–121. doi:10.4103/0253-7176.203129
4. Maas AIR, Menon DK, Adelson PD, et al. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017;16(12):987–1048. doi:10.1016/S1474-4422(17)30371-X
5. Dams-O’Connor K, Guetta G, Hahn-Ketter AE, Fedor A. Traumatic brain injury as a risk factor for Alzheimer’s disease: current knowledge and future directions. Neurodegener Dis Manag. 2016;6(5):417–429. doi:10.2217/nmt-2016-0017
6. Gardner AJ, Zafonte R. Neuroepidemiology of traumatic brain injury. Handb Clin Neurol. 2016;138:207–223.
7. Jiang JY, Gao GY, Feng JF, et al. Traumatic brain injury in China. Lancet Neurol. 2019;18(3):286–295. doi:10.1016/S1474-4422(18)30469-1
8. Ng SY, Lee AYW. Traumatic brain injuries: pathophysiology and potential therapeutic targets. Front Cell Neurosci. 2019;13:528. doi:10.3389/fncel.2019.00528
9. Osier N, McGreevy E, Pham L, et al. Melatonin as a therapy for traumatic brain injury: a review of published evidence. Int J Mol Sci. 2018;19(5):1539. doi:10.3390/ijms19051539
10. Rehman SU, Ikram M, Ullah N, et al. Neurological enhancement effects of melatonin against brain injury-induced oxidative stress, neuroinflammation, and neurodegeneration via AMPK/CREB signaling. Cells. 2019;8(7):760. doi:10.3390/cells8070760
11. Gulpinar O, Guclu AG. How to write a review article? Turk J Urol. 2013;39(Suppl 1):44–48. doi:10.5152/tud.2013.054
12. Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2(7872):81–84. doi:10.1016/S0140-6736(74)91639-0
13. Black KL, Hanks RA, Wood DL, et al. Blunt versus penetrating violent traumatic brain injury: frequency and factors associated with secondary conditions and complications. J Head Trauma Rehabil. 2002;17(6):489–496. doi:10.1097/00001199-200212000-00001
14. Warden D. Military TBI during the Iraq and Afghanistan wars. J Head Trauma Rehabil. 2006;21(5):398–402. doi:10.1097/00001199-200609000-00004
15. Ling GS, Ecklund JM. Traumatic brain injury in modern war. Curr Opin Anaesthesiol. 2011;24(2):124–130. doi:10.1097/ACO.0b013e32834458da
16. Hemphill MA, Dauth S, Yu CJ, Dabiri BE, Parker KK. Traumatic brain injury and the neuronal microenvironment: a potential role for neuropathological mechanotransduction. Neuron. 2015;85(6):1177–1192. doi:10.1016/j.neuron.2015.02.041
17. Ray SK, Dixon CE, Banik NL. Molecular mechanisms in the pathogenesis of traumatic brain injury. Histol Histopathol. 2002;17(4):1137–1152. doi:10.14670/HH-17.1137
18. Kumar Sahel D, Kaira M, Raj K, Sharma S, Singh S. Mitochondrial dysfunctioning and neuroinflammation: recent highlights on the possible mechanisms involved in traumatic brain injury. Neurosci Lett. 2019;710:134347. doi:10.1016/j.neulet.2019.134347
19. Bullock R, Fujisawa H. The role of glutamate antagonists for the treatment of CNS injury. J Neurotrauma. 1992;9(Suppl 2):S443–462.
20. Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci. 2004;61(6):657–668. doi:10.1007/s00018-003-3319-x
21. Tavazzi B, Signoretti S, Lazzarino G, et al. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery. 2005;56(3):582–589. doi:10.1227/01.NEU.0000156715.04900.E6
22. Battelli MG, Polito L, Bortolotti M, Bolognesi A. Xanthine oxidoreductase-derived reactive species: physiological and pathological effects. Oxid Med Cell Longev. 2016;2016:3527579. doi:10.1155/2016/3527579
23. Collin F. Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int J Mol Sci. 2019;20(10):2407. doi:10.3390/ijms20102407
24. Ikram M, Ullah R, Khan A, Kim MO. Ongoing research on the role of gintonin in the management of neurodegenerative disorders. Cells. 2020;9(6):1464. doi:10.3390/cells9061464
25. Liu XF, Zhou DD, Xie T, et al. The Nrf2 signaling in retinal ganglion cells under oxidative stress in ocular neurodegenerative diseases. Int J Biol Sci. 2018;14(9):1090–1098. doi:10.7150/ijbs.25996
26. Zhou T, Zong R, Zhang Z, et al. SERPINA3K protects against oxidative stress via modulating ROS generation/degradation and KEAP1-NRF2 pathway in the corneal epithelium. Invest Ophthalmol Vis Sci. 2012;53(8):5033–5043. doi:10.1167/iovs.12-9729
27. David JA, Rifkin WJ, Rabbani PS, Ceradini DJ. The Nrf2/Keap1/ARE pathway and oxidative stress as a therapeutic target in type II diabetes mellitus. J Diabetes Res. 2017;2017:4826724. doi:10.1155/2017/4826724
28. Wang CC, Wee HY, Hu CY, Chio CC, Kuo JR. The effects of memantine on glutamic receptor-associated nitrosative stress in a traumatic brain injury rat model. World Neurosurg. 2018;112:e719–e731. doi:10.1016/j.wneu.2018.01.140
29. Ikram M, Park TJ, Ali T, Kim MO. Antioxidant and neuroprotective effects of caffeine against Alzheimer’s and Parkinson’s disease: insight into the role of Nrf-2 and A2AR signaling. Antioxidants (Basel). 2020;9(9):902.
30. Fresta CG, Chakraborty A, Wijesinghe MB, et al. Non-toxic engineered carbon nanodiamond concentrations induce oxidative/nitrosative stress, imbalance of energy metabolism, and mitochondrial dysfunction in microglial and alveolar basal epithelial cells. Cell Death Dis. 2018;9(2):245. doi:10.1038/s41419-018-0280-z
31. Bains M, Hall ED. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim Biophys Acta. 2012;1822(5):675–684. doi:10.1016/j.bbadis.2011.10.017
32. Hall ED, Andrus PK, Oostveen JA, Fleck TJ, Gurney ME. Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. J Neurosci Res. 1998;53(1):66–77. doi:10.1002/(SICI)1097-4547(19980701)53:1<66::AID-JNR7>3.0.CO;2-H
33. Castilho RF, Kowaltowski AJ, Meinicke AR, Bechara EJ, Vercesi AE. Permeabilization of the inner mitochondrial membrane by Ca2+ ions is stimulated by t-butyl hydroperoxide and mediated by reactive oxygen species generated by mitochondria. Free Radic Biol Med. 1995;18(3):479–486. doi:10.1016/0891-5849(94)00166-H
34. Lotocki G, de Rivero Vaccari JP, Perez ER, et al. Alterations in blood-brain barrier permeability to large and small molecules and leukocyte accumulation after traumatic brain injury: effects of post-traumatic hypothermia. J Neurotrauma. 2009;26(7):1123–1134. doi:10.1089/neu.2008.0802
35. Buttram SD, Wisniewski SR, Jackson EK, et al. Multiplex assessment of cytokine and chemokine levels in cerebrospinal fluid following severe pediatric traumatic brain injury: effects of moderate hypothermia. J Neurotrauma. 2007;24(11):1707–1717. doi:10.1089/neu.2007.0349
36. Frugier T, Morganti-Kossmann MC, O’Reilly D, McLean CA. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J Neurotrauma. 2010;27(3):497–507. doi:10.1089/neu.2009.1120
37. Faden AI, Wu J, Stoica BA, Loane DJ. Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br J Pharmacol. 2016;173(4):681–691. doi:10.1111/bph.13179
38. Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19(8):312–318. doi:10.1016/0166-2236(96)10049-7
39. Henao-Mejia J, Elinav E, Strowig T, Flavell RA. Inflammasomes: far beyond inflammation. Nat Immunol. 2012;13(4):321–324. doi:10.1038/ni.2257
40. O’Brien WT, Pham L, Symons GF, Monif M, Shultz SR, McDonald SJ. The NLRP3 inflammasome in traumatic brain injury: potential as a biomarker and therapeutic target. J Neuroinflammation. 2020;17(1):104. doi:10.1186/s12974-020-01778-5
41. Helmy A, Vizcaychipi M, Gupta AK. Traumatic brain injury: intensive care management. Br J Anaesth. 2007;99(1):32–42. doi:10.1093/bja/aem139
42. Pontifex MG, Malik M, Connell E, Muller M, Vauzour D. Citrus polyphenols in brain health and disease: current perspectives. Front Neurosci. 2021;15:640648. doi:10.3389/fnins.2021.640648
43. Scott G, Zetterberg H, Jolly A, et al. Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain. 2018;141(2):459–471. doi:10.1093/brain/awx339
44. Thompson HJ, Bakshi A. Methylprednisolone was associated with an increase in death after head injury. Evid Based Nurs. 2005;8(2):51. doi:10.1136/ebn.8.2.51
45. Thompson SN, Carrico KM, Mustafa AG, Bains M, Hall ED. A pharmacological analysis of the neuroprotective efficacy of the brain- and cell-permeable calpain inhibitor MDL-28170 in the mouse controlled cortical impact traumatic brain injury model. J Neurotrauma. 2010;27(12):2233–2243. doi:10.1089/neu.2010.1474
46. Buki A, Farkas O, Doczi T, Povlishock JT. Preinjury administration of the calpain inhibitor MDL-28170 attenuates traumatically induced axonal injury. J Neurotrauma. 2003;20(3):261–268. doi:10.1089/089771503321532842
47. Campolo M, Casili G, Lanza M, et al. The inhibition of mammalian target of rapamycin (mTOR) in improving inflammatory response after traumatic brain injury. J Cell Mol Med. 2021;25(16):7855–7866. doi:10.1111/jcmm.16702
48. Samii A, Badie H, Fu K, Luther RR, Hovda DA. Effects of an N-type calcium channel antagonist (SNX 111; Ziconotide) on calcium-45 accumulation following fluid-percussion injury. J Neurotrauma. 1999;16(10):879–892. doi:10.1089/neu.1999.16.879
49. Alderson P, Roberts I. Corticosteroids for acute traumatic brain injury. Cochrane Database Syst Rev. 2005;1:CD000196.
50. Kertmen H, Gurer B, Yilmaz ER, et al. Antioxidant and antiapoptotic effects of darbepoetin-alpha against traumatic brain injury in rats. Arch Med Sci. 2015;11(5):1119–1128. doi:10.5114/aoms.2015.54869
51. O’Neil DA, Nicholas MA, Lajud N, Kline AE, Bondi CO. Preclinical models of traumatic brain injury: emerging role of glutamate in the pathophysiology of depression. Front Pharmacol. 2018;9:579. doi:10.3389/fphar.2018.00579
52. Kim B, Haque A, Arnaud FG, et al. Use of recombinant factor VIIa (rFVIIa) as pre-hospital treatment in a swine model of fluid percussion traumatic brain injury. J Emerg Trauma Shock. 2014;7(2):102–111. doi:10.4103/0974-2700.130880
53. Whyte J, Hart T, Vaccaro M, et al. Effects of methylphenidate on attention deficits after traumatic brain injury: a multidimensional, randomized, controlled trial. Am J Phys Med Rehabil. 2004;83(6):401–420. doi:10.1097/01.PHM.0000128789.75375.D3
54. Reppert SM, Weaver DR, Godson C. Melatonin receptors step into the light: cloning and classification of subtypes. Trends Pharmacol Sci. 1996;17(3):100–102. doi:10.1016/0165-6147(96)10005-5
55. Costa EJ, Lopes RH, Lamy-Freund MT. Permeability of pure lipid bilayers to melatonin. J Pineal Res. 1995;19(3):123–126. doi:10.1111/j.1600-079X.1995.tb00180.x
56. Tan DX, Reiter RJ, Manchester LC, et al. Chemical and physical properties and potential mechanisms: melatonin as a broad spectrum antioxidant and free radical scavenger. Curr Top Med Chem. 2002;2(2):181–197. doi:10.2174/1568026023394443
57. Ikram M, Jo MG, Park TJ, et al. Oral administration of gintonin protects the brains of mice against abeta-induced Alzheimer disease pathology: antioxidant and anti-inflammatory effects. Oxid Med Cell Longev. 2021;2021:6635552. doi:10.1155/2021/6635552
58. Khan A, Park TJ, Ikram M, et al. Antioxidative and anti-inflammatory effects of Kojic acid in abeta-induced mouse model of Alzheimer’s disease. Mol Neurobiol. 2021;58(10):5127–5140. doi:10.1007/s12035-021-02460-4
59. Du G, Zhao Z, Chen Y, et al. Quercetin protects rat cortical neurons against traumatic brain injury. Mol Med Rep. 2018;17(6):7859–7865. doi:10.3892/mmr.2018.8801
60. Tsai MC, Chen WJ, Tsai MS, Ching CH, Chuang JI. Melatonin attenuates brain contusion-induced oxidative insult, inactivation of signal transducers and activators of transcription 1, and upregulation of suppressor of cytokine signaling-3 in rats. J Pineal Res. 2011;51(2):233–245. doi:10.1111/j.1600-079X.2011.00885.x
61. Wang Z, Ma C, Meng CJ, et al. Melatonin activates the Nrf2-ARE pathway when it protects against early brain injury in a subarachnoid hemorrhage model. J Pineal Res. 2012;53(2):129–137. doi:10.1111/j.1600-079X.2012.00978.x
62. Ding K, Wang H, Xu J, et al. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2-ARE signaling pathway as a potential mechanism. Free Radic Biol Med. 2014;73:1–11. doi:10.1016/j.freeradbiomed.2014.04.031
63. Beni SM, Kohen R, Reiter RJ, Tan DX, Shohami E. Melatonin-induced neuroprotection after closed head injury is associated with increased brain antioxidants and attenuated late-phase activation of NF-kappaB and AP-1. FASEB J. 2004;18(1):149–151. doi:10.1096/fj.03-0323fje
64. Brooks GA, Martin NA. Cerebral metabolism following traumatic brain injury: new discoveries with implications for treatment. Front Neurosci. 2014;8:408.
65. Hovda DA, Yoshino A, Kawamata T, Katayama Y, Becker DP. Diffuse prolonged depression of cerebral oxidative metabolism following concussive brain injury in the rat: a cytochrome oxidase histochemistry study. Brain Res. 1991;567(1):1–10. doi:10.1016/0006-8993(91)91429-5
66. Yoshino A, Hovda DA, Kawamata T, Katayama Y, Becker DP. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res. 1991;561(1):106–119. doi:10.1016/0006-8993(91)90755-K
67. Dietrich WD, Alonso O, Busto R, Ginsberg MD. Widespread metabolic depression and reduced somatosensory circuit activation following traumatic brain injury in rats. J Neurotrauma. 1994;11(6):629–640. doi:10.1089/neu.1994.11.629
68. Prins ML, Fujima LS, Hovda DA. Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J Neurosci Res. 2005;82(3):413–420. doi:10.1002/jnr.20633
69. Davis LM, Pauly JR, Readnower RD, Rho JM, Sullivan PG. Fasting is neuroprotective following traumatic brain injury. J Neurosci Res. 2008;86(8):1812–1822. doi:10.1002/jnr.21628
70. Hill JL, Kobori N, Zhao J, et al. Traumatic brain injury decreases AMP-activated protein kinase activity and pharmacological enhancement of its activity improves cognitive outcome. J Neurochem. 2016;139(1):106–119. doi:10.1111/jnc.13726
71. Ozdemir D, Uysal N, Gonenc S, et al. Effect of melatonin on brain oxidative damage induced by traumatic brain injury in immature rats. Physiol Res. 2005;54(6):631–637.
72. Lee S, Jadhav V, Ayer R, et al. The antioxidant effects of melatonin in surgical brain injury in rats. Acta Neurochir Suppl. 2008;102:367–371.
73. Stazi M, Negro S, Megighian A, et al. Melatonin promotes regeneration of injured motor axons via MT1 receptors. J Pineal Res. 2021;70:e12695.
74. Wu H, Shao A, Zhao M, et al. Melatonin attenuates neuronal apoptosis through up-regulation of K(+) -Cl(-) cotransporter KCC2 expression following traumatic brain injury in rats. J Pineal Res. 2016;61(2):241–250. doi:10.1111/jpi.12344
75. Senol N, Naziroglu M. Melatonin reduces traumatic brain injury-induced oxidative stress in the cerebral cortex and blood of rats. Neural Regen Res. 2014;9(11):1112–1116. doi:10.4103/1673-5374.135312
76. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi:10.1016/j.cell.2007.12.018
77. Lipinski MM, Wu J, Faden AI, Sarkar C. Function and mechanisms of autophagy in brain and spinal cord trauma. Antioxid Redox Signal. 2015;23(6):565–577. doi:10.1089/ars.2015.6306
78. Lin C, Chao H, Li Z, et al. Melatonin attenuates traumatic brain injury-induced inflammation: a possible role for mitophagy. J Pineal Res. 2016;61(2):177–186. doi:10.1111/jpi.12337
79. Ding K, Xu J, Wang H, Zhang L, Wu Y, Li T. Melatonin protects the brain from apoptosis by enhancement of autophagy after traumatic brain injury in mice. Neurochem Int. 2015;91:46–54. doi:10.1016/j.neuint.2015.10.008
80. Kuwar R, Rolfe A, Di L, et al. A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J Neuroinflammation. 2019;16(1):81. doi:10.1186/s12974-019-1471-y
81. Cao S, Shrestha S, Li J, et al. Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation. Sci Rep. 2017;7(1):2417. doi:10.1038/s41598-017-02679-z
82. Dehghan F, Shahrokhi N, Khaksari M, et al. Does the administration of melatonin during post-traumatic brain injury affect cytokine levels? Inflammopharmacology. 2018;26(4):1017–1023. doi:10.1007/s10787-017-0417-1
83. Grivas TB, Savvidou OD. Melatonin the “light of night” in human biology and adolescent idiopathic scoliosis. Scoliosis. 2007;2:6. doi:10.1186/1748-7161-2-6
84. Grima NA, Rajaratnam SMW, Mansfield D, Sletten TL, Spitz G, Ponsford JL. Efficacy of melatonin for sleep disturbance following traumatic brain injury: a randomised controlled trial. BMC Med. 2018;16(1):8. doi:10.1186/s12916-017-0995-1
85. Grima NA, Ponsford JL, St Hilaire MA, Mansfield D, Rajaratnam SM. Circadian melatonin rhythm following traumatic brain injury. Neurorehabil Neural Repair. 2016;30(10):972–977.
86. Seifman MA, Gomes K, Nguyen PN, et al. Measurement of serum melatonin in intensive care unit patients: changes in traumatic brain injury, trauma, and medical conditions. Front Neurol. 2014;5:237. doi:10.3389/fneur.2014.00237
87. Li SS, Xie LL, Li ZZ, Fan YJ, Qi MM, Xi YG. Androgen is responsible for enhanced susceptibility of melatonin against traumatic brain injury in females. Neurosci Lett. 2021;752:135842. doi:10.1016/j.neulet.2021.135842
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.