")
Back to Journals » Drug, Healthcare and Patient Safety » Volume 12
Meeting Regulatory Requirements for Drugs with a Narrow Therapeutic Index: Bioequivalence Studies of Generic Once-Daily Tacrolimus
Authors Gantar K, Škerget K, Mochkin I, Bajc A
Received 9 April 2020
Accepted for publication 15 August 2020
Published 8 September 2020 Volume 2020:12 Pages 151—160
DOI https://doi.org/10.2147/DHPS.S256455
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Siew Siang Chua
Kaja Gantar,1 Katja Škerget,1 Ilya Mochkin,2 Aleksander Bajc1
1Sandoz Development Center Slovenia, Lek Pharmaceuticals d.d, Ljubljana 1526, Slovenia; 2Sandoz International GmbH, Holzkirchen 83607, Germany
Correspondence: Ilya Mochkin
Sandoz International GmbH, Industriestraße 18, Holzkirchen 83607, Germany
Email [email protected]
Abstract: Despite growing clinical confidence in generics and their potential to reduce long-term healthcare costs, the transplant community have had real concerns about the use of generic immunosuppressants. One such immunosuppressant is tacrolimus, a cornerstone of lifelong treatment for patients who have undergone a solid organ transplant. Tacrolimus has a narrow therapeutic index (NTI), giving rise to questions about the potential for clinically relevant altered drug exposure. Its use in transplant patients also gives rise to questions about the most discriminative subject population for bioequivalence studies. The recognised need for stringent criteria to support approval of generic drugs with an NTI led the European Medicines Association and Health Canada to provide detailed information on requirements for bioequivalence studies and introduce tighter bioequivalence limits for these drugs, including tacrolimus. The aim of this article is to illustrate how regulatory guidance is implemented during the clinical development of generic immunosuppressants, using a generic, once-daily prolonged-release formulation of tacrolimus as an example.
Keywords: tacrolimus, bioequivalence, narrow therapeutic index, once-daily, generic
Plain Language Summary
A generic drug is a medication that has been developed to have the same active ingredient, dosing and route of administration, as another medication that is already on the market. Generics increase treatment options, and can be less expensive than their branded counterparts (the “reference product”). However, physicians and patients often have concerns about whether a generic truly works as well as its reference product, and without unanticipated side effects. For this reason, strict measures are in place to test whether or not a generic drug is “bioequivalent”, ie, can be switched in for the reference product. Tacrolimus is a drug used to subdue the immune systems of patients who have received a solid organ transplant, to prevent their bodies rejecting the transplant. Generic versions of tacrolimus have been developed; this paper describes the strict processes that were followed in the EU and Canada in order to obtain approval for one of these generic versions, a once-daily formulation. By describing the requirements that were met, our aim is to reassure physicians and patients that the generic formulation is indeed bioequivalent to the reference product and has met various strict criteria to be determined as such. This means that the generic version can be used in the same way as the reference product.
Introduction
Generics: Opportunities and Challenges
The foundation for the global generics market was laid with the Hatch-Waxman Act of 1984, which established an abbreviated new drug application process for generics in the United States (US).1 One of the advantages of generics is that they are less expensive to bring to market than the original, “branded” product, providing an alternative to the reference product (as long as both products are of similar quality).2 Often, this provides the opportunity for substantial cost savings for payers and patients. However, in practice, the uptake of available generics is not as high as it could be. In one study covering 13 European countries, generics filled less than 40% of reported prescriptions in 6 countries; the study authors noted that this compared poorly with the proportion of prescriptions filled with generics in the US (>80%).3 Despite growing confidence in generics, about one-third of 718 physicians in a US survey were categorised as ‘generic sceptics’ based on their responses to a short email survey.4 In a survey of US patients, 46% (95% confidence interval [CI]: 42–49%) of 737 respondents reported requesting a prescription for a brand-name drug over a generic in the preceding year.5
A number of questions remain about how to improve the costing and uptake of generic drugs. Reference, index and maximum pricing strategies have had an inconsistent impact on the uptake of generics in different countries, and these strategies have their own limitations including the potential to dampen competitive drug development and pricing. These issues have been recently and comprehensively reviewed in other publications,6,7 and will not be considered in detail here. However, demand-side policies aimed at physicians, pharmacists and even patients cannot succeed if stakeholders continue to lack trust in the quality of generics, or have concerns about possible side effects or risk of switching to a generic from a reference product. We hope to address some of these concerns in this paper, by using a specific example to shed some light on the process of regulatory approval for generic tacrolimus.
Tacrolimus
In patients who have undergone solid organ transplantation and require lifelong immunosuppression, generic immunosuppressants have potential to broaden treatment options and reduce costs. This could facilitate equitable access to healthcare in countries where patients face high out-of-pocket drug costs and are therefore at risk of cost-related non-adherence.8 Tacrolimus, used in combination with adrenal corticosteroids and other immunosuppressive agents, is a cornerstone of treatment for these patients. It is a calcineurin inhibitor that blocks interleukin-2 dependent T-cell activation and T-helper-cell dependent B-cell proliferation, which are key features of graft rejection. Tacrolimus is indicated for prophylaxis of transplant rejection in adult patients receiving allogeneic kidney, liver or heart transplants, and also to treat refractory rejection after allogeneic liver or kidney transplants.9 In clinical practice, physicians need to consider the intra-patient variability that has been reported with tacrolimus and could potentially affect outcomes.10–13 This variability has been attributed to a number of possible causes including diarrhoea, liver dysfunction, corticosteroid tapering, altered haematocrit levels, and poor adherence.14 For this reason, therapeutic drug monitoring is used routinely to maintain systemic concentrations and is also recommended if patients are converted to different formulations of tacrolimus.9 The first generic tacrolimus product (Sandoz, Holzkirchen, Germany) was approved by the US Food and Drug Administration (FDA) in 2009.15
Regulatory Approval of Generic Products
Strict standards for the quality and approval of generic drugs have been set by the FDA, the European Medicines Agency (EMA), Health Canada, and other regulatory bodies, and the requirements for approval of generics have been updated after stringent scientific consideration. Most notably, demonstration of bioequivalence is the central requirement. Generics are required to match their reference (“branded”) product with regard to active substance, dosage, administration, quality, and intended use. For example, the EMA defines a generic medicinal product as a product with
the same qualitative and quantitative composition in active substances and the same pharmaceutical form as the reference medicinal product, and whose bioequivalence with the reference medicinal product has been demonstrated by appropriate bioavailability studies.16
Once these criteria are met, the efficacy and safety established for the reference product can be extrapolated to its generic.16
Concerns About Generic Immunosuppressants Used Post-Transplant
Despite these strict regulatory standards, there have been concerns among the transplant community that substitution of a branded immunosuppressant for its generic counterpart could pose a clinically relevant risk of altered drug exposure.17 In addition, there is uncertainty about whether the bioequivalence demonstrated in studies with healthy volunteers provides sufficient assurance of therapeutic equivalence in transplant patients, who present distinct clinical challenges; this is a problem of inter-patient variability.17 Some transplant physicians have suggested that bioequivalence studies should be performed not only in healthy volunteers but also in transplant recipients, because drug pharmacokinetics may be affected in kidney or liver transplant patients, possibly to the detriment of patient outcomes.18
Some retrospective studies in transplant patients lend support to this concern, appearing to show worse clinical outcomes in kidney transplant patients treated with generics than in patients who received branded ciclosporin.19,20 For tacrolimus, published data are inconsistent: variability in trough concentrations has been observed in transplant patients who switch from the branded to a generic product,21,22 with other reports showing comparable trough data.23,24 Differences in systemic tacrolimus exposure can occur even when trough concentrations are similar between the generic and reference products, as shown in one prospective study in elderly kidney transplant patients.25 Another prospective, randomised study in de novo kidney transplant patients has also reported differences in drug exposure between generic and reference products, with the generic product exhibiting an earlier and higher peak in concentration compared with the reference product.26 In contrast, a different prospective study in stable kidney transplant patients found the pharmacokinetic profiles of generic tacrolimus and the reference product to be similar.27 A systematic review and meta-analysis of clinical efficacy and bioequivalence of generic immunosuppressants in transplant patients sought to clarify this issue.28 Results of the review highlighted the lack of robust bioequivalence data for generic immunosuppressants and their branded counterparts in transplant patients.28
From a practical perspective, performing bioequivalence studies in a more heterogeneous patient population would introduce confounding factors such as variance in age, comorbidities, and concomitant medications. It would be very difficult to control for the effect of these factors on drug levels in plasma. In general, healthy subjects make for a more homogeneous study population, therefore allowing more sensitive detection of differences in bioequivalence caused by differences in formulation between generic and branded products. This is the rationale underlying the current requirements of the EMA and Health Canada, which require bioequivalence studies to be performed in healthy volunteers.16,29
Concerns about bioequivalence are especially pertinent for drugs that have a narrow therapeutic index (NTI), ie, drugs in which there is only a narrow range of drug exposure between lack of efficacy and undesirable toxicity. In the case of tacrolimus, insufficient immunosuppression could lead to acute rejection and graft failure, whereas excess immunosuppression could result in infection, or effects related to extensive calcineurin inhibition such as nephrotoxicity or neurotoxicity.30,31 The need for a stringent demonstration of bioequivalence for approval of generic drugs with an NTI, such as tacrolimus, led to the introduction of tighter bioequivalence limits for these drugs. These were published as part of the EMA Guideline on the Investigation of Bioequivalence in 2010,16 with a similar approach taken by Health Canada in their guidance, last updated in 2012.32 The FDA recommends that bioequivalence of tacrolimus should be evaluated using a scaled average bioequivalence approach.33 These guidelines describe the standards and processes required for a generic drug to be considered bioequivalent to its reference drug. Product-specific guidelines for bioequivalence studies of tacrolimus from the EMA mandate testing in healthy volunteers.34
Despite these stringent regulatory requirements, a number of consensus documents from professional bodies specialising in transplant urge caution in the use of generic immunosuppressants.31,35,36 In one survey of renal transplant patients in the UK, 84% (95% CI: 76–89) of 146 respondents said no, or were uncertain, in response to the question “Do you think that generic medicines are equivalent and have the same quality as branded medicines?”37 Among physicians, there is still some uncertainty about whether generic immunosuppressants can be considered truly bioequivalent to reference products, about the regulatory process that tests this claim, and whether that regulatory process itself is robust enough to ensure therapeutic equivalence. The aim of this article is to illustrate how the EMA and Health Canada regulatory guidance is implemented during the clinical development of generic immunosuppressants. We use a generic, prolonged-release formulation of tacrolimus (Sandoz) that can be taken once daily, which may improve adherence and reduce pill burden, as an example.
Regulatory Requirements and Corresponding Tacrolimus Data
Pharmaceutical Equivalence
Before bioequivalence is considered, it is important to consider pharmaceutical equivalence, as designation of therapeutic equivalence requires that criteria for both pharmaceutical equivalence and bioequivalence are met. According to the EMA, two products are deemed to be pharmaceutically equivalent if they contain the same amount of the same active substance(s), and in the same dosage forms that meet the same, or comparable, standards.16 The once-daily prolonged-release formulation of tacrolimus described in the following sections is indeed the pharmaceutical equivalent of its reference product (Advagraf® 5 mg prolonged-release hard capsules [Astellas Pharma]): it does not differ in its active substance, pharmaceutical form, or route of administration.
Bioequivalence Studies in Drugs with an NTI: Regulatory Requirements
Because tacrolimus is designated an NTI drug,38 less variation in drug exposure between the generic and the reference product will be accepted than would be for non-NTI drugs. As such, limits for the ratio of the area under the curve (AUC) of the test and reference products have been tightened compared with the usual requirements for bioequivalence. The 90% confidence interval of this ratio should be contained within a narrower margin than for other drugs: the EMA’s margin for acceptance is 90.00–111.11%16 and Health Canada have set a margin of 90.00–112.00%.32
Guidelines from the EMA also state that the same acceptance interval (90.00–111.11%) should be applied to the maximum plasma concentration (Cmax), if Cmax is of particular importance for safety, efficacy or monitoring of drug levels.16 In the case of once-daily tacrolimus, the EMA have determined that this requirement does not apply.38 This is because total drug exposure (measured by AUC) is considered to be the major factor affecting the safe and efficacious dose titration of tacrolimus. Cmax is thought to have less impact on the safety and efficacy of dose titration, because the long terminal half-life of tacrolimus means that it accumulates during repeated dosing. This means that any potential difference in Cmax between formulations is likely to be less at steady state, as long as AUC remains the same. As such, the usual bioequivalence limits of 80.00–125.00% apply for the 90% confidence interval of the ratio of Cmax of the test and reference products.38
Bioequivalence Studies of Once-Daily Tacrolimus: Design and Pharmacokinetic Assessments
A number of studies were carried out to evaluate the bioequivalence of a generic formulation of once-daily tacrolimus 5 mg prolonged-release hard capsules (manufactured by Lek Pharmaceuticals d.d., Slovenia, a Sandoz company) and the reference product, Advagraf® 5 mg prolonged-release hard capsules (Astellas Pharma). These studies were implemented in line with the EMA and Health Canada guidance for bioequivalence studies: fasting and fed single-dose studies were carried out. In addition, because the generic product is a prolonged-release formulation, a multiple-dose study was done to comply with EMA requirements39 to compare both products at steady state.
Each of the pharmacokinetic studies, whether a single- or multiple-dose study, had an open-label, randomised, two-period, two-treatment, two-sequence, crossover design. Participants were administered one 5 mg prolonged-release hard capsule of the test or reference product in each of the two study periods, with a 14-day washout in between. Dosing was once daily in the multiple-dose study. In the fasted studies, participants received the study product after an overnight fast of at least 10 hours; in the fed study, participants received the study product 30 minutes after the start of a high-fat, high-calorie breakfast. In each period of single-dose studies, blood samples were collected prior to drug administration and up to 144 hours post-dose. In the multiple-dose study, each dosing period lasted for 10 days; pre-dose samples were collected on the mornings of days 1, 8, 9, and 10, and over a 24-hour period after dosing on day 10. Achievement of steady-state was assessed based on three consecutive pre-dose concentrations, measured on the mornings of days 8, 9, and 10.
Levels of tacrolimus in the blood were measured using a fully validated method that had been developed in compliance with the principles of Good Laboratory Practice and according to applicable guidelines.40 Tacrolimus concentrations were determined by chromatographic tandem mass spectrometry, which was confirmed to be reliable and reproducible by conducting incurred sample reanalysis on approximately 7% of the total number of study samples, where 97.8–99.6% of repeated results were within acceptance criteria. Tacrolimus pharmacokinetic parameters were calculated using a non-compartmental approach.
All studies were conducted in accordance with the Declaration of Helsinki, Good Clinical Practice and local regulatory requirements. The clinical study protocols and informed consent documents were reviewed and approved by the relevant Institutional Review Board prior to the start of associated study procedures.
Study Population: Regulatory Requirements
The EMA guideline states that the population for bioequivalence studies “should be selected with the aim of permitting detection of differences between pharmaceutical products” and
In order to reduce variability not related to differences between products, the studies should normally be performed in healthy volunteers unless the drug carries safety concerns that make this unethical.16
The guideline adds that studies in healthy volunteers are
regarded as adequate in most instances to detect formulation differences and to allow extrapolation of the results to populations for which the reference medicinal product is approved. (the elderly, children, patients with renal or liver impairment, etc.)16
Health Canada guidance is similar, highlighting that in order to minimize variability, comparative bioavailability studies are usually conducted with normal, healthy volunteers.29
Study Population in the Bioequivalence Studies of Once-Daily Tacrolimus
The study population for the fasted, multiple-dose study was healthy male participants, and the other studies included healthy male or female participants. Study participants were aged 18–55 years old and had a body mass index between 18.5 and 30.0 kg/m2, inclusive.
Drug Exposure and Absorption Results from Studies of Once-Daily Tacrolimus
As shown in Figure 1, the concentration–time profiles for test and reference products are very similar. As shown in Table 1, EMA and Health Canada criteria were met in bioequivalence studies under single-dose fasting and single-dose fed studies, and EMA criteria were met in multiple-dose fasting conditions, for once-daily tacrolimus (Sandoz).
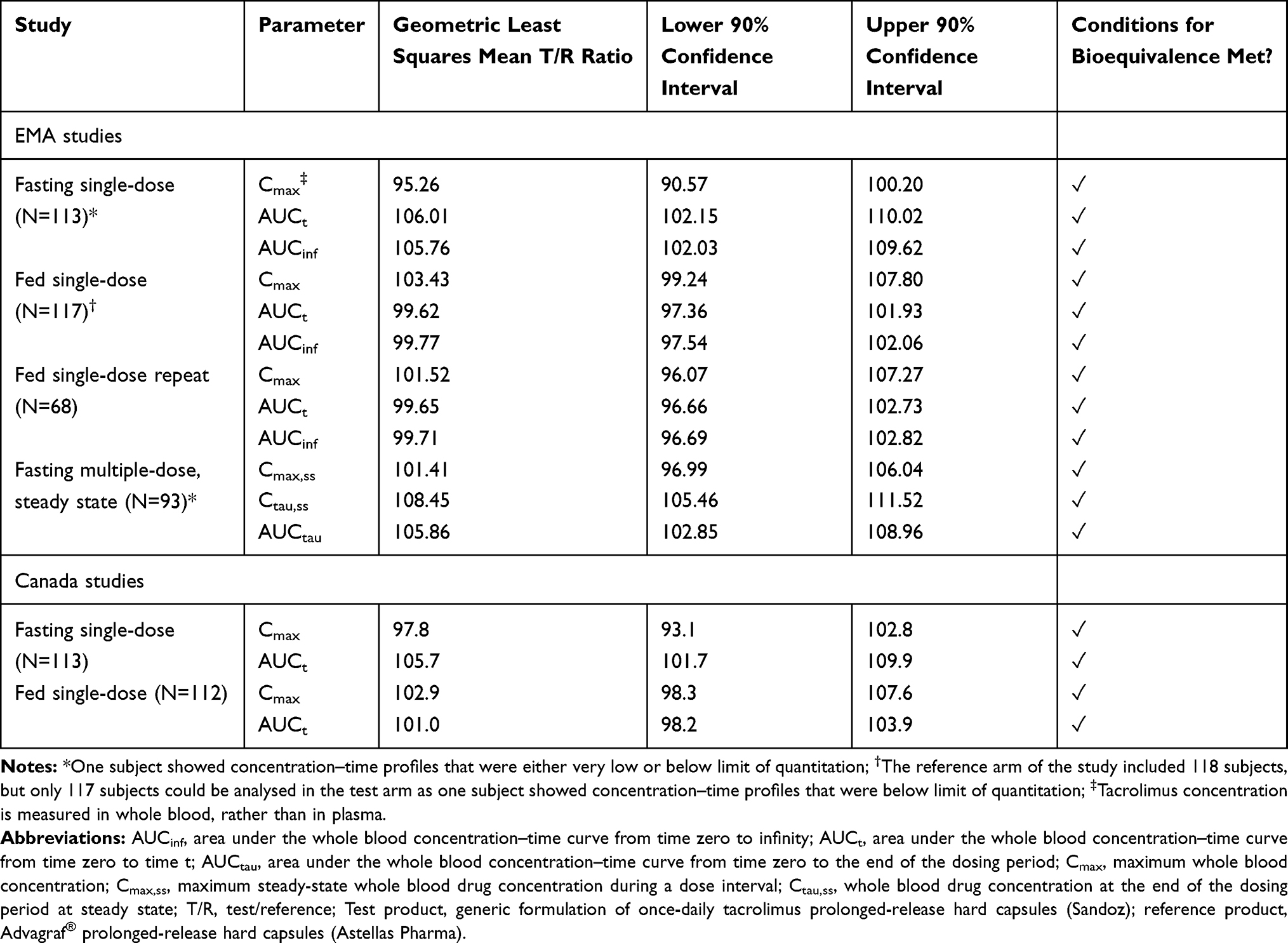 |
Table 1 EU and Canada Regulatory Bioequivalence Study Data for Once-Daily Tacrolimus (Sandoz) |
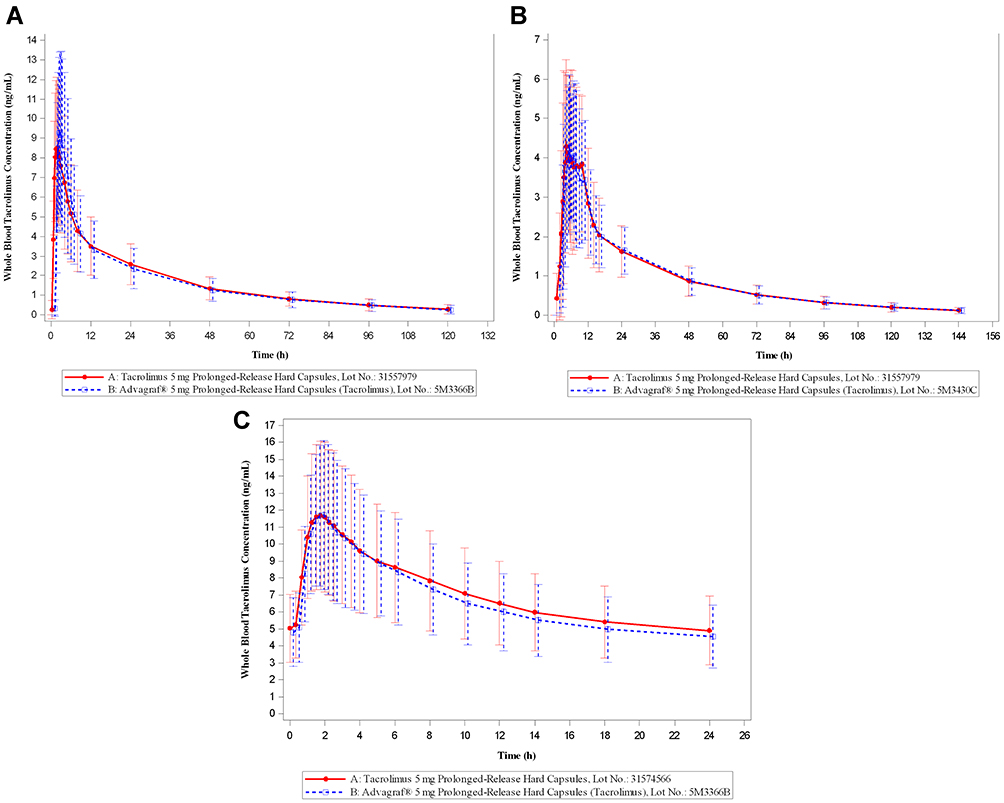 |
Figure 1 Mean whole blood concentration–time profiles for once-daily tacrolimus (Sandoz) versus the reference drug once-daily tacrolimus (Astellas); linear scale. (A) fasting single-dose; (B) fed single-dose (data from repeated study are shown); (C) fasting multiple-dose. Artwork was created using SAS (version 9.4). |
All studies completed for the EMA were sufficiently powered and no adverse events were reported that would have a significant impact on subject safety. However, as noted in Table 1, one subject in each study apart from the repeat fed single-dose study showed concentration–time profiles for the test and/or reference product that were either very low or below limit of quantitation. Further details of these three cases are as follows:
- One subject in the fasting single-dose study was excluded from the pharmacokinetic (PK) analysis in line with EMA guidance, owing to very low whole blood concentrations for the reference product (AUC <5% of reference product geometric mean AUC, calculated without inclusion of data from this outlier).
- For one subject in the fed single-dose study, PK parameters for the test product could not be calculated as the test product concentration was below the limit of quantitation. Instead, the PK analysis was completed without including this subject (data shown in Table 1), and criteria for bioequivalence were met
- A post hoc sensitivity analysis was done in which all values that were below the limit of quantitation were substituted for the lower limit of quantitation (0.05 ng/mL). In this way, the analysis could be performed including all 118 subjects. Results show that the outputs are within bioequivalence limits (Table 2)
- Despite the original study and the sensitivity analysis showing that the test and reference products were bioequivalent, an additional fed single-dose study was carried out at the request of the EMA. The reason for this request was the low concentrations of the test product observed in the original fed, single-dose study. Results of the repeat study (Table 1) confirmed that the test and reference products were indeed bioequivalent, and no outliers were reported
- For one subject in the multiple-dose study, PK parameters for both the reference and test products were below the limit of quantitation. Data from this subject were excluded from the PK analysis in line with EMA guidance; without these data, criteria for bioequivalence were met.
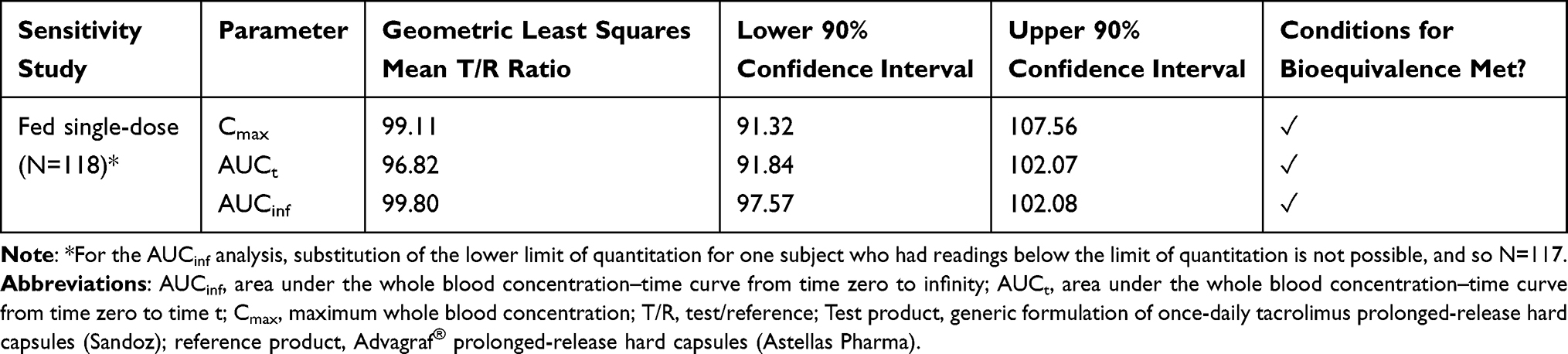 |
Table 2 Post Hoc Sensitivity Analysis of the EU Fed, Single-Dose Study for Once-Daily Tacrolimus (Sandoz) |
Discussion
This paper has described how, in the case of once-daily tacrolimus (Sandoz), the regulatory processes set by the EMA and Health Canada have been followed comprehensively, demonstrating bioequivalence to its reference product. At the moment, there is no regulatory requirement for clinical outcome data to be provided in support of generic drug registration: therapeutic equivalence is assumed once bioequivalence has been demonstrated.41 Based on the EMA and Health Canada guidelines, therefore, clinical efficacy is assumed if bioequivalence is shown. Once-daily tacrolimus can therefore be used in transplant recipients beginning treatment, or as an alternative formulation for patients already undergoing treatment, with appropriate dose modifications if required.42
There have been calls for more controlled, prospective studies that test the validity of the regulatory approach to bioequivalence for NTI immunosuppressants. One such example is a prospective comparison of branded twice-daily tacrolimus with two generic formulations in kidney and liver transplant patients. This study design enabled intra-patient variability in drug exposure to be evaluated over time and across products.43 Future, similar studies would have potential to further address some of the concerns described by physicians and patients about generic immunosuppressants. However, it is important to note that the bioequivalence limits set by the EMA and Health Canada were set based on studies in healthy volunteers for a reason: studies in healthy volunteers provide a more homogeneous population than transplant patients, making these studies more sensitive to differences between formulations.43 This is the most practical way of ensuring that any PK differences detected during bioequivalence studies are caused by differences in the formulations being tested, and are not due to patient, disease or medication-related factors.
Conclusion
Routine drug monitoring is an established process in transplant patients. This pharmacokinetic monitoring can also be used if an individual patient is switched from branded to generic tacrolimus, to permit dose adjustment as necessary. Nonetheless, many transplant physicians and patients remain reluctant to change tacrolimus formulation. In detailing the stringent regulatory requirements that have been met during the development of once-daily tacrolimus (Sandoz), our aim is to assure physicians and patients that the generic formulation was established to be equivalent to the reference formulation, if used according to the label. In order to ensure that patients benefit from use of generic tacrolimus, we urge that use of a generic as part of a treatment plan is communicated clearly to the transplant recipient. Information should be provided on the process that has been followed to ensure bioequivalence, and on the expected efficacy and safety of the product based on clinical data from the reference product.
Abbreviations
AUC, area under the curve; AUCinf, area under the whole blood concentration–time curve from time zero to infinity; AUCt, area under the whole blood concentration–time curve from time zero to time t; AUCtau, area under the whole blood concentration–time curve from time zero to the end of the dosing period; CI, confidence interval; Cmax, maximum plasma concentration; Cmax,ss, maximum steady-state whole blood drug concentration during a dose interval; Ctau,ss, whole blood drug concentration at the end of the dosing period at steady state; EMA, European Medicines Agency; NTI, narrow therapeutic index; PK, pharmacokinetic; T/R, test/reference; US FDA, United States Food and Drug Administration.
Ethics Approval
All studies were conducted in accordance with the Declaration of Helsinki, Good Clinical Practice and local regulatory requirements. The clinical study protocols and informed consent documents were reviewed and approved by the relevant Institutional Review Board prior to the start of associated study procedures. The relevant Institutional Review Boards were as follows:
- Salus IRB, Austin, Texas, USA approved the multiple-dose study.
- Optimum Clinical Research Inc. Ethics Review Board, Oshawa, Ontario, Canada approved all single-dose studies.
Consent
All study participants provided informed consent prior to participation.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
KG, KŠ and AB are employees of Lek Pharmaceuticals d.d. IM is an employee of Sandoz International GmbH. The authors report no other conflicts of interest in this work.
References
1. Boehm G, Yao L, Han L, Zheng Q. Development of the generic drug industry in the US after the Hatch-Waxman Act of 1984. Acta Pharm Sin B. 2013;3:297–311. doi:10.1016/j.apsb.2013.07.004
2. Administration USFaD Generic Drug Facts. Available from: https://www.fda.gov/drugs/generic-drugs/generic-drug-facts.
3. Wouters OJ, Kanavos PG, Mc KM. Comparing generic drug markets in Europe and the United States: prices, volumes, and spending. Milbank Q. 2017;95(3):554–601. doi:10.1111/1468-0009.12279
4. Kesselheim AS, Gagne JJ, Eddings W, et al. Prevalence and predictors of generic drug skepticism among physicians: results of a national survey. JAMA Intern Med. 2016;176(6):845–847. doi:10.1001/jamainternmed.2016.1688
5. Kesselheim AS, Gagne JJ, Franklin JM, et al. Variations in patients’ perceptions and use of generic drugs: results of a national survey. J Gen Intern Med. 2016;31(6):609–614. doi:10.1007/s11606-016-3612-7
6. Ferrario A, Dedet G, Humbert T, Vogler S, Suleman F, Pedersen HB. Strategies to achieve fairer prices for generic and biosimilar medicines. BMJ. 2020;368:l5444. doi:10.1136/bmj.l5444
7. Sivashanker K, Fanikos J, Kachalia A. Addressing the lack of competition in generic drugs to improve healthcare quality and safety. J Gen Intern Med. 2018;33(11):2005–2007. doi:10.1007/s11606-018-4548-x
8. Evans RW, Applegate WH, Briscoe DM, et al. Cost-related immunosuppressive medication nonadherence among kidney transplant recipients. Clin J Am Soc Nephrol. 2010;5(12):2323–2328. doi:doi:10.2215/CJN.04220510
9. Adoport 0.5 mg capsules: summary of product characteristics. Sandoz Limited. Available from: https://www.medicines.org.uk/emc/product/585/smpc.
10. Gueta I, Markovits N, Yarden-Bilavsky H, et al. High tacrolimus trough level variability is associated with rejections after heart transplant. Am J Transplant. 2018;18(10):2571–2578. doi:10.1111/ajt.15016
11. Huang CT, Shu KH, Ho HC, Wu MJ. Higher variability of tacrolimus trough level increases risk of acute rejection in kidney transplant recipients. Transplant Proc. 2016;48(6):1978–1980. doi:10.1016/j.transproceed.2016.02.081
12. Rahamimov R, Tifti-Orbach H, Zingerman B, et al. Reduction of exposure to tacrolimus trough level variability is associated with better graft survival after kidney transplantation. Eur J Clin Pharmacol. 2019;75(7):951–958. doi:doi:10.1007/s00228-019-02643-y
13. Susal C, Dohler B. Late intra-patient tacrolimus trough level variability as a major problem in kidney transplantation: a collaborative transplant study report. Am J Transplant. 2019;19(10):2805–2813. doi:10.1111/ajt.15346
14. Vanhove T, Vermeulen T, Annaert P, Lerut E, Kuypers DRJ. High intrapatient variability of tacrolimus concentrations predicts accelerated progression of chronic histologic lesions in renal recipients. Am J Transplant. 2016;16(10):2954–2963. doi:doi:10.1111/ajt.13803
15. Administration TUSFaD. FDA center for drug evaluation and research: approval package for tacrolimus capsules. Sandoz, Inc; 2009. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/anda/2009/065461Orig1s000.pdf.
16. Agency EM. Guideline on the investigation of bioequivalence. European Medicines Agency; 2010. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf.
17. Tsipotis E, Gupta NR, Raman G, Zintzaras E, Jaber BL. Bioavailability, efficacy and safety of generic immunosuppressive drugs for kidney transplantation: a systematic review and meta-analysis. Am J Nephrol. 2016;44(3):206–218. doi:10.1159/000449020
18. Klintmalm GB. Immunosuppression, generic drugs and the FDA. Am J Transplant. 2011;11(9):1765–1766. doi:10.1111/j.1600-6143.2011.03616.x
19. Pollard S, Nashan B, Johnston A, et al; Transplantation CCoSiE. A pharmacokinetic and clinical review of the potential clinical impact of using different formulations of cyclosporin A. Berlin, Germany, November 19, 2001. Clin Ther. 2003;25(6):1654–1669. doi:10.1016/S0149-2918(03)80161-3
20. Taber DJ, Baillie GM, Ashcraft EE, et al. Does bioequivalence between modified cyclosporine formulations translate into equal outcomes? Transplantation. 2005;80(11):1633–1635. doi:10.1097/01.tp.0000188688.15639.03
21. Momper JD, Ridenour TA, Schonder KS, Shapiro R, Humar A, Venkataramanan R. The impact of conversion from prograf to generic tacrolimus in liver and kidney transplant recipients with stable graft function. Am J Transplant. 2011;11(9):1861–1867. doi:10.1111/j.1600-6143.2011.03615.x
22. Rosenborg S, Nordstrom A, Almquist T, Wennberg L, Barany P. Systematic conversion to generic tacrolimus in stable kidney transplant recipients. Clin Kidney J. 2014;7(2):151–155. doi:10.1093/ckj/sfu015
23. McDevitt-Potter LM, Sadaka B, Tichy EM, Rogers CC, Gabardi S. A multicenter experience with generic tacrolimus conversion. Transplantation. 2011;92(6):653–657. doi:10.1097/TP.0b013e31822a79ad
24. Spence MM, Nguyen LM, Hui RL, Chan J. Evaluation of clinical and safety outcomes associated with conversion from brand-name to generic tacrolimus in transplant recipients enrolled in an integrated health care system. Pharmacotherapy. 2012;32(11):981–987. doi:10.1002/phar.1130
25. Robertsen I, Asberg A, Ingero AO, et al. Use of generic tacrolimus in elderly renal transplant recipients: precaution is needed. Transplantation. 2015;99(3):528–532. doi:10.1097/TP.0000000000000384
26. Min SI, Ha J, Kim YS, et al. Therapeutic equivalence and pharmacokinetics of generic tacrolimus formulation in de novo kidney transplant patients. Nephrol Dial Transplant. 2013;28(12):3110–3119. doi:doi:10.1093/ndt/gft300
27. Alloway RR, Sadaka B, Trofe-Clark J, Wiland A, Bloom RD. A randomized pharmacokinetic study of generic tacrolimus versus reference tacrolimus in kidney transplant recipients. Am J Transplant. 2012;12(10):2825–2831. doi:doi:10.1111/j.1600-6143.2012.04174.x
28. Molnar AO, Fergusson D, Tsampalieros AK, et al. Generic immunosuppression in solid organ transplantation: systematic review and meta-analysis. BMJ. 2015;350:h3163. doi:10.1136/bmj.h3163
29. Canada H. Guidance document: conduct and analysis of comparative bioavailability studies. 2018. Available from: https://www.canada.ca/content/dam/hc-sc/documents/services/drugs-health-products/drug-products/applications-submissions/guidance-documents/bioavailability-bioequivalence/conduct-analysis-comparative.pdf.
30. Ball S. Bioequivalence of twice-daily oral tacrolimus in transplant recipients: more evidence for consensus? PLoS Med. 2017;14(11):e1002429. doi:10.1371/journal.pmed.1002429
31. Harrison JJ, Schiff JR, Coursol CJ, et al. Generic immunosuppression in solid organ transplantation: a Canadian perspective. Transplantation. 2012;93(7):657–665. doi:doi:10.1097/TP.0b013e3182445e9d
32. Canada H. Guidance document: comparative bioavailability standards: formulations used for systemic effects. 2012. Available from: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/applic-demande/guide-ld/bio/gd_standards_ld_normes-eng.pdf.
33. Administration USFaD. Draft guidance on tacrolimus. 2014. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Tacrolimus_cap_50708_RV12-12.pdf.
34. Agency TEM. Tacrolimus granules product-specific bioequivalence guidance. 2016. Available from: https://www.ema.europa.eu/en/tacrolimus-granules-product-specific-bioequivalence-guidance.
35. Uber PA, Ross HJ, Zuckermann AO, et al. Generic drug immunosuppression in thoracic transplantation: an ISHLT educational advisory. J Heart Lung Transplant. 2009;28(7):655–660. doi:10.1016/j.healun.2009.05.001
36. van Gelder T; Substitution EACoG. European society for organ transplantation advisory committee recommendations on generic substitution of immunosuppressive drugs. Transpl Int. 2011;24(12):1135–1141. doi:10.1111/j.1432-2277.2011.01378.x
37. Al Ameri MN, Whittaker C, Tucker A, Yaqoob M, Johnston A. A survey to determine the views of renal transplant patients on generic substitution in the UK. Transpl Int. 2011;24(8):770–779. doi:10.1111/j.1432-2277.2011.01268.x
38. Agency TEM. Questions & answers: positions on specific questions addressed to the pharmacokinetics working party. EMA/618604/2008 Rev. 13. 2015. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/questions-answers-positions-specific-questions-addressed-pharmacokinetics-working-party_en.pdf.
39. Agency TEM. Guideline on the pharmacokinetic and clinical evaluation of modified release dosage forms (EMA/CPMP/EWP/280/96 Corr1). 2014. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-pharmacokinetic-clinical-evaluation-modified-release-dosage-forms_en.pdf.
40. Agency TEM. Guideline on bioanalytical method validation. 2011. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf.
41. van Gelder T, Gabardi S. Methods, strengths, weaknesses, and limitations of bioequivalence tests with special regard to immunosuppressive drugs. Transpl Int. 2013;26(8):771–777. doi:10.1111/tri.12074
42. Staatz CE, Tett SE. Clinical pharmacokinetics of once-daily tacrolimus in solid-organ transplant patients. Clin Pharmacokinet. 2015;54(10):993–1025. doi:10.1007/s40262-015-0282-2
43. Alloway RR, Vinks AA, Fukuda T, et al. Bioequivalence between innovator and generic tacrolimus in liver and kidney transplant recipients: a randomized, crossover clinical trial. PLoS Med. 2017;14(11):e1002428. doi:10.1371/journal.pmed.1002428
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.