Back to Journals » Journal of Multidisciplinary Healthcare » Volume 12
Medulloblastoma: optimizing care with a multidisciplinary approach
Received 14 September 2018
Accepted for publication 11 February 2019
Published 30 April 2019 Volume 2019:12 Pages 335—347
DOI https://doi.org/10.2147/JMDH.S167808
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Alice Thomas,1 Georges Noël1,2
1Radiotherapy Department, Centre Paul Strauss, UNICANCER, F-67065 Strasbourg, France; 2Radiobiology Lab, CNRS, IPHC UMR 7178, Centre Paul Strauss, UNICANCER, F-67000 Strasbourg, France
Abstract: Medulloblastoma is a malignant tumor of the cerebellum and the most frequent malignant brain tumor in children. The standard of care consists of maximal resection surgery, followed by craniospinal irradiation and chemotherapy. Such treatment allows long-term survival rates of nearly 70%; however, there are wide disparities among patient outcomes, and in any case, major long-term morbidity is observed with conventional treatment. In the last two decades, the molecular understanding of medulloblastoma has improved drastically, allowing us to revolutionize our understanding of medulloblastoma pathophysiological mechanisms. These advances led to an international consensus in 2010 that defined four prognostic molecular subgroups named after their affected signaling pathways, that is, WNT, SHH, Group 3 and Group 4. The molecular understanding of medulloblastoma is starting to translate through to clinical settings due to the development of targeted therapies. Moreover, recent improvements in radiotherapy modalities and the reconsideration of craniospinal irradiation according to the molecular status hold promise for survival preservation and the reduction of radiation-induced morbidity. This review is an overview of the current knowledge of medulloblastoma through a molecular approach, and therapeutic prospects currently being developed in surgery, radiotherapy and targeted therapies to optimize the treatment of medulloblastoma with a multidisciplinary approach will also be discussed.
Keywords: molecular subgroup, targeted therapies, radiotherapy, proton therapy
Introduction
Medulloblastoma is a highly aggressive malignant tumor of the cerebellum1 and the most common malignant brain tumor in children, accounting for nearly 20% of all central nervous system (CNS) tumors among children1,2 but only 1% of all CNS tumors among adults. In 70–80% of cases, medulloblastoma affects children 16 years or younger.1 The average annual age-adjusted incidence rates range from 0.20 to 0.58 cases per 100,000 persons.3
Currently, the 5-year overall survival (OS) rates reach ~70%, but patients present with very different outcomes. Medulloblastoma is stratified into two main risk levels based on age, presence of metastases, extent of residual disease after surgery and histologic characteristics of the tumor.4 Consequently, the 5-year survival rates for standard and high-risk medulloblastoma are over 80% and ~60%, respectively.5–7
Conventional treatment, for both standard and high-risk patients, involves a combination of maximal resection surgery, craniospinal irradiation (CSI), and cytotoxic chemotherapy (CT). This combination achieves long-term OS in 60–80% of patients but often at the expense of devastating long-term toxicities.8–10 Therefore, a better understanding of the disease appears to be a relevant challenge with the aim of providing more accurate risk-adapted treatment and developing targeted therapies to decrease side effects in low-risk patients and improve efficiency in high-risk patients. In the last two decades, major advances have already been made in understanding the molecular mechanisms underlying medulloblastoma.11,12 These findings have led to an international consensus for defining the prognostic molecular subgroups of medulloblastoma, which have been included in the recently published revised fourth edition of the WHO Classification of Tumors of the CNS.13 A molecular understanding of medulloblastoma is starting to translate through to clinical settings due to the development of subgroup-specific approaches for clinical trials, allowing a more accurate distribution of radiation dosage or CT schedules, and evaluating the efficiency of emerging candidates for targeted therapies.
In this review, we will present the current landscape of the medulloblastoma molecular classification and attempt to correlate this classification with emerging therapeutic strategies to optimize medulloblastoma care through a multidisciplinary approach by finding the right treatments for the right group of patients to increase survival rates and reduce treatment toxicity, hence improving the quality of life (QoL) of survivors.
Clinical classification
The current prognostic classification divides medulloblastomas into “Standard Risk” and “High Risk” based on age, presence of metastases, extent of postsurgical residual disease and histology. Five histological subtypes are individualized: classical, desmoplastic/nodular, with extensive nodularity, anaplastic, and large cell variants. Large cell and anaplastic medulloblastomas overlap in a considerable number of cytological features, and consequently, these types are often considered together and grouped as “large cell/anaplastic histology”,14 which is related to poor prognosis. Anaplastic histology is characterized by marked cytological pleomorphism across most of its area in association with high mitotic and apoptotic counts.15 Large cell medulloblastoma is defined by groups of uniform large round cells with a single nucleolus, which also have higher mitotic and apoptotic indices than in other histologies.16 Medulloblastoma with extensive nodularity, which is closely related to the desmoplastic/nodular variant, has a more favorable outcome.17 Considering the surgical criteria, patients with less than 1.5 cm2 residual disease after surgery present significantly better outcomes.18 Metastatic status is determined by using the Chang criteria, which distinguish four metastatic levels: presence of tumor cells in cerebrospinal fluid (CSF; M1), gross nodular seeding of brain CSF spaces (M2), gross nodular seeding of spinal CSF spaces (M3), and extraneural spread (M4).19 Infants (under 3 years old) have lower survival rates than older children, although this finding could be a result of therapeutic strategies that must eliminate radiotherapy (RT) in this population due to unacceptable long-term morbidity.
Hence, based on this clinical and histological classification, standard-risk patients – 70% of medulloblastomas at the time of diagnosis20 – are children aged >3 years with no evidence of disseminated disease on craniospinal magnetic resonance imaging or CSF cytology, postoperative residual tumor under 1.5 cm2 and non-large cell/anaplastic histology.21 Five-year OS reaches ~85%.18,22 If one or more of these criteria is not available, then the patient is considered to be at high risk, and the 5-year OS declines to ~60%.7
Molecular classification
The existence of distinct molecular subgroups of medulloblastoma was highlighted in 2002 by Pomeroy et al.23 They showed, by studying DNA microarray gene expression data, that PTCH, GLI and MYCN (all three transcriptional targets of SHH) were highly correlated with desmoplastic/nodular medulloblastoma. They also showed molecular markers of the variability of medulloblastoma outcome. On the same topic, several studies started to sub-classify medulloblastoma according to differences in the transcriptome, with largely convergent conclusions. Thompson et al24 in 2006, and Kool et al25 in 2008, concluded the existence of five distinct molecular subtypes named A, B, C, D, and E. Cho et al26 concluded in 2011 the existence of six distinct molecular subtypes named C1 to C6, and Northcott et al27 concluded in 2011 the existence of four distinct molecular subtypes named SHH, WNT, Group C, and Group D. Variations in the number, composition, and nature of the subgroups between studies brought about a consensus conference in Boston in the fall of 2010, where it was agreed there were four main transcriptional subgroups of medulloblastoma named WNT, SHH, Group 3, and Group 4, clearly distinct in terms of demographics, histology, DNA copy-number aberrations, and clinical outcome.28
The WNT subgroup is the rarest (10% of all medulloblastomas) but has the best clinical outcome prediction, with a 5-year OS > 95%.6,29,30 This molecular subgroup is predominantly associated with classical histology. WNT medulloblastomas rarely have a large cell/anaplastic histology, but even with this histology, they present an excellent prognosis.28 WNT medulloblastomas occur in children older than 3 years or teenagers, and the cell of origin derives from the lower rhombic lip.31 The molecular mechanism is defined by the activation of the WNT signaling pathway, which acts via β-catenin expression as a transduction enhancer. In 85–90% of cases, the activation of the WNT signaling pathway results from activating somatic mutations in exon 3 of CTNNB1, leading to the overexpression of β-catenin. Monosomy 6 is also highly recurrent among WNT tumors, presenting in 70–80% of cases.26,31,32 Less frequently, somatic mutations in TP53, SMARCA4, and DDX3X are found.33 WNT medulloblastoma rarely occurs in the context of germline mutations in APC consistent with Turcot syndrome.34 Germline mutations in ALK have also been found in rare cases of WNT medulloblastoma, although the physiopathology is not understood.35
The SHH subgroup represents 30% of all medulloblastomas,36 and the prognosis is intermediate, with a 5-year OS of 70%.26 There is a strong association between desmoplastic/nodular histology and SHH tumors since the vast majority of desmoplastic/nodular cases belong to the SHH subgroup,28,37 but up to 50% of SHH subgroup medulloblastomas are not desmoplastic/nodular.28 SHH medulloblastomas are most frequently found in infants and adults and occur much less frequently in patients aged 4–15 years.29 These tumors derive from the cerebellar granule precursor cells of the external granule layer.38 The molecular mechanism involves the overexpression of the SHH signaling pathway, which, via implication of PTCH1, SMO, GLI, and SUFU, acts as a transduction enhancer.39 The genetic events underlying SHH pathway activation are age-dependent: in infants, germline mutations in PTCH1 (Gorlin syndrome) or SUFU are frequent. Interestingly, in patients with Gorlin syndrome, RT should be avoided because of the major risk of radiation-induced second cancers (mostly meningiomas and basal cell carcinomas).40 Furthermore, infants with SHH medulloblastoma present an excellent prognosis, even with a CT-only regimen. Children between 3 and 16 years mostly present somatic mutations in PTCH1 or germline (or less frequently somatic) mutations in TP53 (Li Fraumeni Syndrome).41 Thus, all pediatric SHH tumors should be referred to the geneticist to diagnose a potential Gorlin syndrome, Li Fraumeni syndrome, or germline SUFU mutation. Somatic TP53 mutations frequently co-occur with GLI2 and MYCN amplifications,41 which induce the activation of the SHH pathway. The TP53 mutation, present in ~30% of SHH medulloblastomas, is related to a very poor prognosis with a 5-year OS of 40%.42 Hence, the fourth edition of the WHO Classification of Tumors of the CNS separates SHH medulloblastoma with or without TP53 mutation. It is well known that cells that express TP53 mutations are less radiosensitive,43 and interestingly, Tchelebi et al44 suggested that RT could even increase tumor growth in medulloblastomas with TP53 mutations.45 In adults, the most frequent mutations are somatic mutations in PTCH1, SMO, and the TERT promoter, or occasionally in IDH1.
Group 3 medulloblastoma represents 25% of all medulloblastomas and has a particularly bad prognosis, with a 5-year OS of 58% in children29 and an even poorer OS in nonirradiated infants (5-year OS of 45%).29,41 Tumors present a predominantly classical histology, but this group also has a high ratio of large cell/anaplastic histology (40%), especially in infants.28,46 Group 3 medulloblastomas occur mostly in males (2:1) and in subjects under 16 years of age and derive from cerebellar stem cells.47 Unlike the WNT and SHH subgroups, in which a misfunctioning molecular pathway has clearly been identified, the underlying cause is not well defined in Group 3 medulloblastomas. Recurrent genetic events have been identified: MYC amplification (10% to 20% of Group 3), OTX2 amplification, SMARCA4 mutation, GFI1 enhancer activation,48 isochromosome 17q (42%),29,49,50 gain of 1q (35%),29 gain of chromosome 7 (55%),29 loss of 8p (33%) or gain of 8q (22%),29 loss of 10q (49%),29 gain of 12q (17%),29 loss of 16q (50%),29 and gain of chromosome 18 (26%).29 Isochromosome 17q, as well as MYC amplification, confers a particularly poor prognosis, with a 5-year OS of 20%.26 Group 3 medulloblastoma has a great capacity for metastatic dissemination since 40%–45% have leptomeningeal dissemination at diagnosis and the recurrence pattern is mostly metastatic.51
Group 4 medulloblastoma, although the most frequent (35% of all medulloblastomas), is the least understood of all molecular subgroups.52 This subgroup mostly presents a classical histology and occurs at all ages with a major masculine predominance (3:1).52 The clinical outcome of Group 4 medulloblastoma is intermediate, with a 5-year OS of 75–90%,5,6 but is poor in infants who cannot benefit from RT. Overall, 30–40% of Group 4 cases are metastatic at diagnosis.53 For group 4, no underlying cause has been well defined. Isochromosome 17q is frequent in Group 4 tumors (loss of 17 p 63%, gain of 17q 73%),29 although, unlike in Group 3 tumors, this abnormality does not confer poor prognosis in this subgroup.49 Other recurrent genetic events have been identified: MYCN or CDK6 amplification, SNCAIP duplication, loss of one X chromosome in women, and inactivating the mutation of KDM6A (10% of Group 4),32 chromosome 7 gain (47%),29 8p loss (41%),29 10q loss (15%),29 12q gain (20%),29 chromosome 18 gain (16%),29 and loss of chromosome 11, which is a favorable prognostic marker.41,49
Metastatic medulloblastoma tumor cells harbor molecular alterations that are not present in the primary tumor. Further preclinical work must be performed to identify the molecular mechanisms underlying metastases,54 which remain largely unknown. A recent study involving a deep proteome analysis of metastatic medulloblastomas identified ~1,400 significantly altered proteins between primary and metastatic cell lines, including known factors such as placental growth factor, LIM homeobox 1, prominin 1, and secreted protein acidic and rich in cysteine.55 Additional analysis of clinical medulloblastoma samples implicated yes-associated protein 1 as a potential key factor contributing to metastasis.55
Thus, although molecular subgrouping has revolutionized medulloblastoma classification, there is great heterogeneity within subgroups, and much research is needed to improve the molecular understanding of medulloblastoma.56–58
In addition to the lack of knowledge, an international consensus defining the four molecular subgroups of medulloblastoma has recently been published and included in the revised fourth edition of the WHO Classification of Tumors of the CNS.13
Recent studies have recommended more accurate molecular screening. Better characterization of medulloblastoma subgroups is especially important for Group 3 and Group 4 since, as discussed above, the transcriptomes of Group 3 and Group 4 medulloblastomas are similar, and several cytogenetic features, such as isochromosome 17q (i17q), are found in both groups. However, the outcomes of Group 3 and Group 4 medulloblastoma patients differ, particularly regarding the tendency of metastatic dissemination. In 2017, through the analysis of genome-wide DNA methylation and gene expression data by using the similarity network fusion method, Cavalli et al56 suggested that clinically and biologically relevant subtypes exist for each subgroup. They concluded that the four molecular subgroups could be further split into 12 different subtypes that differ on a molecular, clinical, and prognostic basis (WNT: 2, SHH: 4, Group 3: 3, Group 4: 3). Similarly, in 2017, Northcott et al57 analyzed the somatic landscape across 491 sequenced medulloblastoma samples and the molecular heterogeneity among 1,256 epigenetically analyzed cases and discovered new tumor subtypes enriched for specific genetic and transcriptional signatures, especially those of Group 3 and Group 4. In 2017, Schwalbe et al58 conducted molecular profiling analyses of 428 primary medulloblastoma samples, including a DNA methylation microarray analysis, and identified seven molecular subgroups of childhood medulloblastoma (WNT subgroup remained unchanged, and each remaining consensus subgroup was split in two). These data hold promise that improving disease risk stratification and treating patient subtypes according to their genotype are likely to emerge.
Standard of care
The standard of care for medulloblastomas is currently based on their clinical classification. For all medulloblastomas, treatment is multimodal, and the first step consists of maximal safe resection surgery. Within 30 days after surgery, RT is initiated. This delay is based on two previous trials comparing immediate RT following surgery vs delayed RT, which found significantly lower outcomes when RT was delayed more than 4–6 weeks after surgery.57,58 Conversely, a recent trial60 found decreased 5-year OS when RT was initiated ≤3 weeks after surgery and no adverse impact on OS when RT was initiated after >5 weeks but within 90 days of surgery.
For standard-risk patients, a CSI of 23.4 Gy in 13 fractions, followed by a tumor bed boost to reach 54–55.8 Gy is administered.61–63 High-risk patients are irradiated with CSI delivering 36–39.6 Gy in 20–22 fractions, followed by a tumor bed boost to 54–55.8 Gy and, when appropriate, 50 Gy CSI is administered to local sites of metastases.64 RT is normofractionated. At 6 weeks after the end of radiotherapy, the patients are treated with four cycles of high-dose chemotherapy, each of which is followed by stem-cell or bone-marrow rescue.64 Each cycle lasts 4 weeks and comprises the following64:
- Day 1: Cisplatin IV 75 mg/m2+ vincristine 1.5 mg/m2 (maximum 2 mg)
- Days 2 and 3: Cyclophosphamide 2 g/m2
- Day 5: Infusion of peripheral blood or bone-marrow progenitor cells
- Day 11: Vincristine 1.5 mg/m2
This treatment scheme, tested in a prospective trial by Gajjar et al64 in 1996, enabled a 5-year OS of 85% (95% CI 75–94) for patients in the average-risk group and 70% (54–84) for those in the high-risk group (P=0·04). The 5-year progression free survival (PFS) was 83% (73–93) and 70% (55–85), respectively (P=0·046).
Notably, many other protocols have been used in clinical trials, for example the “maintenance strategy” from trial HIT’91,65 adapted from the historical “Packer protocol”,66 which consists of vincristine given weekly concomitantly with radiotherapy at 6 weeks after the completion of radiotherapy with eight cycles of lomustine, vincristine, and cisplatin (Table 1).
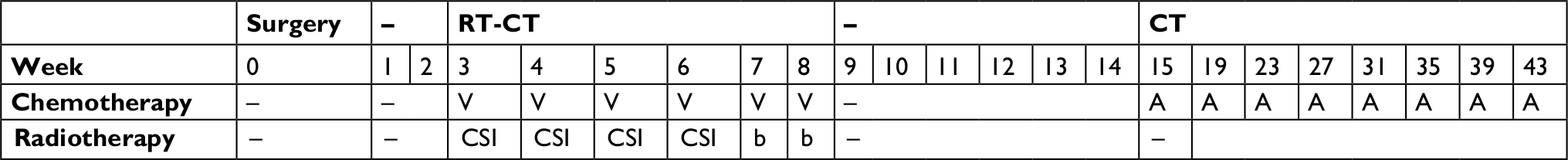 | Table 1 Maintenance strategy from trial HIT’91 Notes: “–” indicates no treatment during this period. Data from von Hoff et al.65 Abbreviations: A, lomustine, vincristine, and cisplatin; b, boost to the posterior fossa to 55.2 Gy (20 Gy given in 10 fractions) and to metastatic sites to 50 Gy; CSI, craniospinal irradiation (35.2 Gy given in 22 fractions); CT, chemotherapy; RT, radiotherapy; V, vincristine. |
In infants, treatment consists exclusively of surgery, followed by high-dose CT due to the particularly devastating radiation-induced morbidity at this young age.67
Standard-risk adults are treated, after maximal safe resection surgery, by a normofractionated CSI of 30–36 Gy followed by a tumor bed boost to 54–55.8 Gy. CT is less well tolerated in adults than in children, and no randomized clinical trial has demonstrated its benefit. Thus, CT is currently only delivered in high-risk adults before RT and in metastatic patients, CT is delivered after RT.68 The molecules used in CT are cyclophosphamide, etoposide, and cisplatin.68 Approximately 30% of medulloblastoma patients are diagnosed with metastasis;69 however, no gold standard treatment has been highlighted for metastatic medulloblastomas. In the trial HIT’91, children with medulloblastoma were randomized to receive either postoperative chemotherapy followed by CSI (“sandwich strategy”) or postoperative CSI followed by chemotherapy (“maintenance strategy”).59,65 The study showed significantly higher OS after maintenance than that after sandwich treatment for M0 and M1 patients, and a moderate trend toward better survival for children with M2/M3 disease who were treated with the “sandwich strategy” compared with the “maintenance strategy”.65 Therefore, a sandwich concept was chosen for the HIT 2000 trial, which was designed to assess an intensified treatment of metastatic medulloblastoma in children and adolescents (4–21 years).70 Compared with the HIT’91 sandwich regimen, the treatment was intensified at the level of neoadjuvant chemotherapy, of radiotherapy – via the introduction of hyperfractionated CSI, to achieve a biologically more effective dose and preserve normal tissue-,71 and the addition of maintenance chemotherapy. As a result, the treatment consisted of two cycles of induction CT, starting 2–4 weeks after surgery, and comprised intravenous cyclophosphamide, vincristine, methotrexate, carboplatin, etoposide, and concomitant intraventricular methotrexate. Radiotherapy, starting at 3–6 weeks after the end of induction chemotherapy, was hyperfractionated with two fractions of 1 Gy per day, and the doses were 40 Gy CSI in combination with 20 Gy on the posterior fossa, 8 Gy on the tumor site bed, 10 Gy on spinal metastases, and 28 Gy on supratentorial metastases. Maintenance CT started at 6 weeks after the end of RT and consisted of four cycles of cisplatin, lomustine, and vincristine.70 OS was superior with this treatment regimen compared with that in the preceding HIT’91 trial, with a 5-year OS of 74% (95% CI, 66–82).70 Independent risk factors were histology (large cell/anaplastic) and nonresponse to the first chemotherapy cycle. Survival rates were different between molecular subgroups: WNT, SHH, Group 4, and Group 3 with or without MYCC/MYCN amplification (P<0.001). Thus, this study showed that molecular subgroup, MYCC/MYCN status, response to induction chemotherapy, and histologic subtype may improve treatment stratification.70 Regarding adult metastatic medulloblastomas, a trial comparing the sandwich strategy from the HIT 2000 protocol (postoperative CT, hyperfractionated CSI, and maintenance CT) with the HIT’91 maintenance strategy (postoperative CSI and maintenance CT) found that after a 4-year follow-up, the patients showed a global PFS and OS of 52% and 91%, respectively, with no significant difference between the two treatment arms.72
Regarding refractory/recurrent medulloblastoma, temozolomide has been shown to be an effective agent, achieving a 6-month PFS and OS of 30% and 42.5%, respectively.73
Side effects
The current treatment of medulloblastoma allows decent survival rates but often at the expense of life-long morbidity. Iatrogenic morbidity occurs in an age-dependent manner; in older patients, the greatest toxicity results from surgery, while in younger patients, CSI confers troublesome morbidity.20
Major postsurgical morbidities are due to critical structures – particularly brainstem – close or adherent to medulloblastoma. Moreover, cerebellar mutism (also called posterior fossa syndrome) occurs in 25% of cases.20 This neurological syndrome develops within 1–4 days following posterior fossa surgery74 and consists of speech reduction, axial hypotonia, ataxia, and emotional lability. Cerebellar mutism takes weeks to months to fade away, although speech troubles can last lifelong.75 The precise etiology of cerebellar mutism is unknown, but compared to other posterior fossa tumors, this condition is overrepresented after medulloblastoma surgery.76 The size and wholeness of resection seem to have an impact on postsurgical morbidities since overall neurological morbidity is 24%, whereas morbidity after gross total resection (GTR) is 44%.77,78 Other studies have shown that the incidence of posterior fossa syndrome is more important than less aggressive resections after GTR.76,79
Considering RT, acute toxicity consists mostly of anorexia and nausea, and particular attention must be paid to the nutritional condition of the patient. Additionally, in most children, the bone marrow of the vertebral bodies actively contributes to hematopoiesis; therefore, cytopenia must be detected, followed by pneumocystis prophylaxis during and in the weeks following CSI.20,80 Considering long-term side effects, the toxicity of RT is more important when the patient is young. Morbidity mostly consists of cognitive decline, inversely proportional to the age of the patient, with a reduction in the IQ score as high as 40 points in the youngest patients.8,10 Attempts have been made to decrease cognitive decline through RT hyperfractionation, but the results were not conclusive.21 The risk of radio-induced malignancy, especially meningioma and glioblastoma, is also high.10,81–83 Other studies have observed endocrine dysfunction, bone growth and development dysfunctions, gynecological, cardiac and pulmonary toxicity, ototoxicity,10 and vascular toxicity with an increased frequency of stroke.84
Regarding CT, a common adverse effect is vincristine-induced peripheral neuropathy (VIPN), which affects sensory, motor, and autonomic nerves. VIPN incidence rates reach 37% during medulloblastoma treatment, and symptoms may not resolve over time.59 Chronic peripheral neuropathy favors physical activity decrease, obesity, type 2 diabetes mellitus, metabolic syndrome, and cardiovascular disease.85 Other toxicities attributed to CT are mainly cisplatin-induced ototoxicity (incidence rates of 34% reaching grade III/IV in 9% of patients) and myelosuppression.59
Optimizing care from molecular classification
The current challenge of the neuro-oncologist is precision medicine. To limit iatrogenic morbidity, we must first avoid the overtreatment of patients with good prognosis who do not need aggressive treatments or, in contrast, patients for whom standard treatments have shown failure to induce good survival rates. Then, new therapeutic approaches must be developed to improve survival rates in treatment failure patients.
This perspective requires a reliable method for the prognostic sorting of medulloblastomas. A consensus conference in Heidelberg in 2015 concluded an updated prognostic classification for children with medulloblastomas based on molecular subgroups,5,6 as shown in Table 2. However, clinical applications face technical difficulties in finding a reliable and easy method for the molecular statement of resected tumors. A recently published study proposes a robust and repeatable molecular classification method based on six epigenetic biomarkers.86 Elsewhere, emerging strategies such as DNA methylation profiling using a comprehensive machine-learning approach enable robust and reproducible classification of central nervous system tumors.87 This method may have a substantial impact on diagnostic precision compared to standard methods, by reducing the substantial inter-observer variability observed in current CNS tumor diagnostics. A uniform implementation of the classification algorithm holds great promise for standardization of tumor diagnostics across centers and across clinical trials.
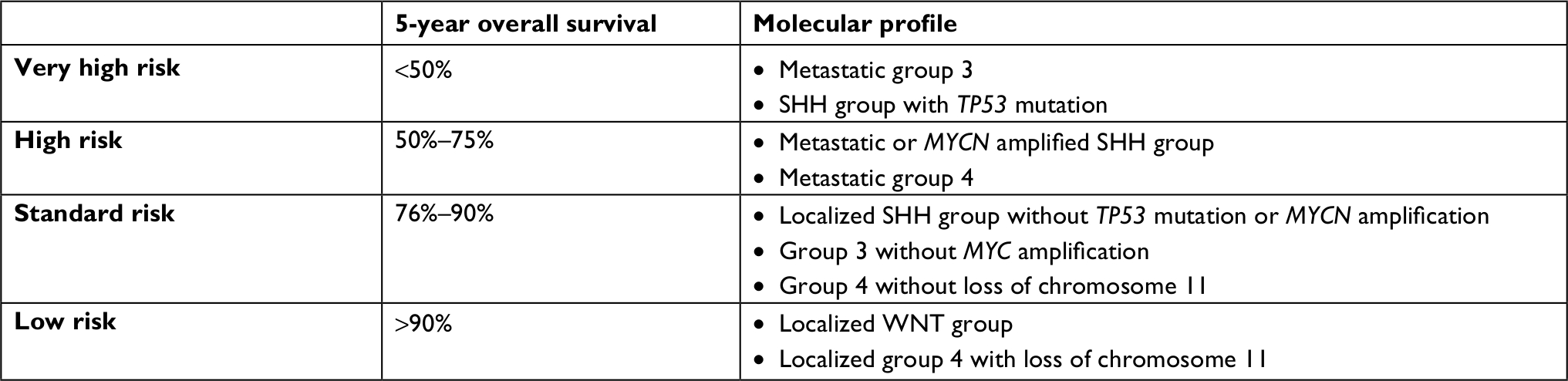 | Table 2 Prognostic classification for pediatric medulloblastomas according to molecular subgroups |
Systemic therapeutic approaches
Overexpression of WNT pathway results in a weaker blood–brain barrier, through vascular dysfunction induced by overexpression of this molecular pathway. A weaker blood–brain barrier enables better penetration of CT molecules into cancer cells. This mechanism could contribute to the good prognosis of this molecular subgroup.88 Hence, targeted therapy inhibiting the WNT pathway could make the tumor less chemosensitive. Furthermore, the WNT pathway participates in many physiological functions, such as bone formation, and inhibiting the WNT pathway would lead to significant toxicity, such as osteoporosis.89 For these reasons, no development of targeted therapy has been undertaken for WNT medulloblastomas, and trials rather focus on decreasing the doses of RT and CT. Several trials are in progress evaluating lower doses of radiation and CT (NCT01878617, NCT02724579) or CT-only approaches (NCT02212574).
Efforts have been made to develop targeted therapies inhibiting the SHH pathway in this subgroup with intermediate prognoses. One therapeutic approach is SMO inhibition with vismodegib90 (currently used for the treatment of locally advanced/metastatic basal cell carcinoma). However, only SHH medulloblastomas with mutations in PTCH1 (upstream SMO) or SMO can benefit from this molecule. A study confirmed that SHH medulloblastoma with SUFU or GLI1 (downstream SMO) mutations do not respond to vismodegib.45 Since 80% of adult SHH medulloblastoma patients have PTCH1 or SMO mutations, vismodegib is likely to be particularly advantageous in this population.91 Moreover, SMO inhibitors could lead to premature bone fusion in children.41 A more recent therapeutic approach refers to epigenetic treatments with bromodomain (BET) inhibitors. BET proteins regulate gene transcription by binding to acetylated histones.92 BET inhibitors have been shown in vitro and in vivo to decrease cell viability and proliferation in SHH medulloblastoma,93,94 but no clinical trial has yet examined BET inhibitors as potential therapeutics for SHH medulloblastoma. Considering metastatic SHH medulloblastoma, for which MET kinase is a marker, the MET inhibitor foretinib has been shown to decrease tumor cell proliferation and induce apoptosis in vitro and in vivo, which confers a strong rationale for its clinical evaluation.95 A clinical trial is ongoing to evaluate doublet therapy comprising the CDK4/6 inhibitor ribociclib with either gemcitabine, trametinib, or sonidegib in adults with refractory or recurrent SHH medulloblastoma (NCT03434262). Clinical trials are also ongoing to evaluate vismodegib in children and adults with refractory or recurrent SHH medulloblastoma (NCT01601184, NCT00939484, NCT01239316).
For Group 3 patients who show poor outcomes when treated with the current standard of care, special expectations rest on the development of targeted therapies. Due to the particularly poor outcome conferred by MYC amplification, efforts have been made concerning MYC inhibition, but no direct MYC inhibitor could successfully be finalized since MYC has no clearly defined ligand-binding domain. A preclinical study designed based on a mouse model of Group 3 medulloblastoma showed the efficacy of palbociclib – a CDK4/6 inhibitor currently used in HR+/Her2– breast tumors, locally advanced or with bone metastases,96 and a clinical trial is in progress for evaluation of palbociclib in pediatric brain tumors (NCT02255461). Another clinical trial showed the efficacy of using HDAC with a PI3K inhibitor in Group 3 medulloblastoma with MYC amplification.97 BET inhibitors also represent a significant therapeutic approach for treating Group 3 medulloblastoma, since BRD4 (a member of BET family) inhibitors have been shown in vitro and in vivo in a mouse model to be an effective therapy against MYC-amplified Group 3 medulloblastoma,98 and the BET inhibitor JQ1 has been demonstrated to reduce tumoral cell viability through the inhibition of MYC transcription.99 However, JQ1 has a very short half-life, which does not allow clinical practice. A phase one clinical trial is ongoing to test other BET inhibitors (CPI-0610, MK-8628) in adults.52 Nevertheless, epigenetic targeting molecules are often found to operate with a cytostatic effect, and this treatment is likely to require a combination of cytostatic and cytotoxic drugs to procure antitumoral efficiency.
Although the most frequent, Group 4 medulloblastoma is the most heterogeneous and least understood medulloblastoma type. No dominant oncogene has been successfully identified, which constitutes a major limitation in the development of targeted therapies. Some studies have suggested a molecular mechanism involving the activation of NFkB.27,100
Another ongoing therapeutic approach is targeting medulloblastoma stem cells, which are a subpopulation of cancer cells largely responsible for medulloblastoma initiation, maintenance, dissemination, and relapse.47 Research efforts are still needed to effectively target medulloblastoma stem cells.
Surgery optimization
In the current clinical classification, a postsurgical residual disease >1.5 cm2 is a marker of worse prognosis and distinguishes the patient as high risk, hence requiring higher CSI doses. However, considering the important side effects of CSI and surgery, the prognostic implication of the extent of resection (EOR) is worth updating, now that the prognostic classification has been revised as a result of molecular advances. Indeed, experts highlighted this issue at a consensus conference in 2016.5 A recent review analyzed 50 articles about the implication of EOR in clinical outcome,101 showing a nearly equal number of studies with and without a significant association between EOR and survival. Only three of these articles accounted for molecular subgrouping,31,58,102 and no association was found between EOR and survival when molecular subgrouping was considered. To make a reliable determination of the prognostic implication of EOR, it would be worth setting up a prospective trial of patients with residual disease >1.5 cm2 and randomizing these patients into 24 Gy CSI vs 36 Gy CSI to assess whether intensified CSI can improve the disease control of patients with a residual disease >1.5 cm2. Subsequently, molecular stratification would be useful. Thompson et al101 showed that such a study would require 2,890 patients with residual disease >1.5 cm2 to detect a difference with 90% power, unfortunately making stratification technically not feasible, particularly in a subgroup-stratified approach.
Radiotherapy optimization
The survival of medulloblastoma patients improved tremendously in 1950 due to the introduction of CSI, since long-term survival evolved from nearly 0% to 50%.103 The development of a linear accelerator with megavoltage (Linac) and 3D conformational RT (3D-CRT) allowed the establishment of CSI due to better precision and the higher intensity and penetrance of the radiations.104 Since this major advance, many inroads have been made in the RT field, and we will discuss how medulloblastoma irradiation could be optimized.
The randomized multicenter HIT-SIOP PNET 4 trial was initiated to estimate the amelioration of radiation-induced toxicity related to hyperfractionation. The hyperfractionated arm received 36 Gy in 36 fractions of CSI, with an additional 24 Gy in 24 fractions on the posterior fossa and 8 Gy in 8 fractions on the tumor bed, and the standard RT arm received 23.4 Gy in 13 fractions of CSI with an additional 32.4 Gy in 18 fractions on the posterior fossa. The results revealed no significant benefit of hyperfractionation in toxicity or in survival,21 and normofractionation remains the standard of care.
The randomized COG ACNS0331 clinical trial compared survival in the arm with a standard dose of RT (23.4 Gy CSI and 54 Gy posterior fossa RT) vs a decreased dose of RT (18 Gy CSI and a reduction in boost volume). The results showed no significant difference in 5-year OS or PFS for a decrease in radiation boost volume, but concerning CSI, noninferiority of lower-dose CSI to standard dose CSI was not established.105 Thus, CSI seems to be a crucial component of medulloblastoma treatment, and doses do not appear to be reducible without affecting survival. However, in light of molecular understanding, it is legitimate to question whether CSI can be reduced or more importantly removed in some patients. Indeed, WNT patients rarely relapse, and trials are already ongoing to determine the feasibility of irradiation de-escalation (NCT01878617, NCT02724579) or removal (NCT02212574). Considering SHH medulloblastoma, the pattern of relapse has been shown to be predominantly local.51 Consequently, a future clinical trial could focus on establishing whether it is feasible to reduce or even remove CSI and focus RT on the tumor bed in SHH patients. Group 3 and Group 4 medulloblastomas present a significant proportion of metastatic relapse; therefore, CSI is unlikely to be removed in those patients. However, future studies could focus on optimizing the radiation dose distribution to maintain efficiency while lowering toxicity.
Since the 2000s, the development of intensity-modulated irradiation modalities, such as intensity-modulated radiotherapy (IMRT), volumetric-modulated arc therapy, and tomotherapy, have enabled the optimization of radiation dose distribution in CSI. However, those modalities involved more radiation fields and therefore induced a larger volume of normal tissues exposed to low-dose radiation, hence increasing the risk of second neoplasms.106
Several studies suggested an association between the radiation dose delivered to the hippocampus and temporal lobes and neurocognitive decline,107,108 and the feasibility of a hippocampal sparing approach with IMRT modality has been studied. The results revealed a significant amount of perihippocampal relapse among patients with brain metastases at the time of diagnosis. In contrast, no patient without brain metastases at diagnosis developed secondary lesions in the perihippocampal area.109 Hippocampal sparing may thus be considered in high-risk medulloblastoma patients without brain metastases at diagnosis and, by extension, this approach is likely to be safely feasible in standard-risk patients.
In the last decade, the use of proton therapy has rapidly increased as a result of its capacity to better spare organs at risk by eliminating the exit radiation dose due to the characteristic dose distribution of the proton beam modeled by the Bragg peak. This treatment is particularly relevant in childhood malignancies since it offers the promise of decreased late radiation-related morbidities, especially second neoplasms.110 Medulloblastoma, specifically due to a particularly large irradiation field, is an excellent candidate for proton therapy. Indeed, protons eliminate the dose of exit radiation into the chest, abdomen, and pelvis as well as the cochlea, pituitary, and hypothalamus of children after CSI.110 Translation into quality of life (QoL) has been studied in a prospective trial.111 QoL scores were found to improve over time after proton CSI, and after 5 years, children-reported scores were statistically similar to those of healthy children, but the parent-reported scores remained statistically lower than those reported by the parents of healthy children.112 To evaluate the superiority of proton therapy in medulloblastoma treatment with an evidence-based approach, a review recently compared the outcomes of pediatric medulloblastoma patients between proton- and photon-mediated CSI,113 and revealed the advantage of proton therapy in organs at risk sparing, normal organ dysfunction, and secondary malignancy risks compared to various (mostly 3D-CRT) photon techniques. A comparison of target coverage between both radiation modalities showed either similar or better results with proton therapy. However, proton therapy is a modern radiation modality, and the earliest study considered in this review was from 1997. For that reason, data regarding late toxicity after proton therapy are not available. On the other hand, we cannot ignore that second neoplasms after CSI mostly occur in the neuraxis, and this effect cannot be avoided with any irradiation modality as long as CSI is performed. The only way to determine with any certainty whether proton therapy should be developed as a standard of care for CSI would be through a prospective randomized controlled trial comparing both treatment modalities. Such a trial should include cost-effectiveness analysis since proton therapy is undoubtedly associated with higher initial infrastructural costs than those for photon therapy.112 At the present time, proton therapy remains a limited resource, and socioeconomic factors impact access to this treatment.113
Overview and prospects
Table 3 summarizes the current molecular understanding of medulloblastoma.
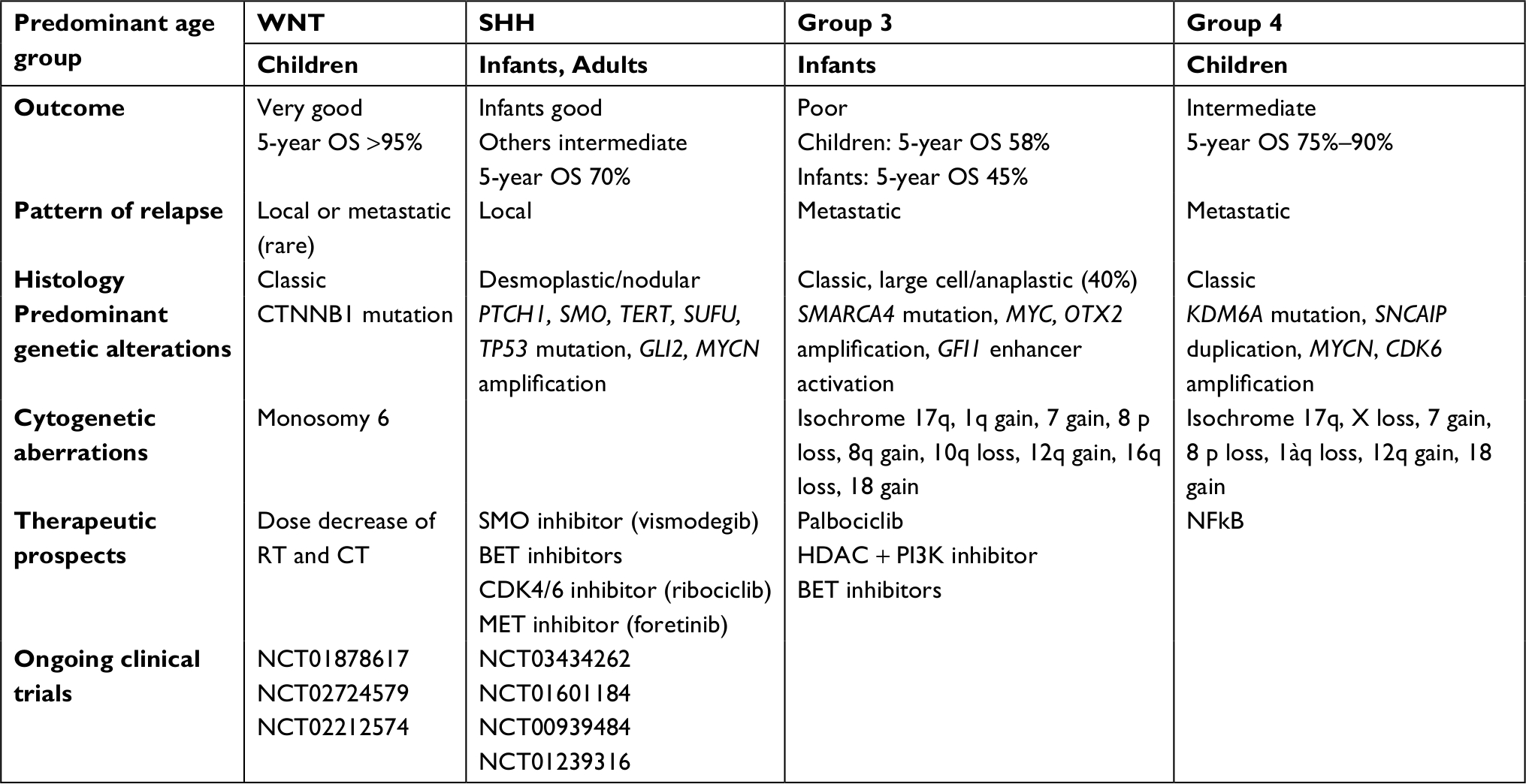 | Table 3 Overview summary of current molecular understanding of medulloblastoma Abbreviations: OS, overall survival; RT, radiotherapy; CT, chemotherapy; BET, bromodomain. |
Although molecular subgrouping for medulloblastoma is important for the prognosis and elaboration of therapeutic agents, this categorization seems insufficient for Group 3 (which undergoes worse prognosis) and Group 4 (which is the most frequent) medulloblastomas. As shown in Table 3, those two subgroups lack targeting agents. Hashimoto et al114 most recently published the results of 36 extensively profiled medulloblastomas. The results revealed the high expression of MRP1, TUBB3, PTEN, TOP2A, thymidylate synthase, RRM1, and TOP1. This finding highlights an all-new therapeutic prospect since targeting agents are available for several of these targets.
Conclusion
Recent advances on the molecular mechanisms of medulloblastoma have allowed the definition of an updated prognostic classification. To optimize medulloblastoma care, efforts must be made to reduce iatrogenic morbidity and improve survival in patients with lower prognoses. Thus, targeted therapies are currently being evaluated in light of a molecular understanding of medulloblastomas. Given the relatively low incidence of medulloblastoma, every oncologist should be particularly aware of the importance of including patients in clinical trials.
RT leads to a significant proportion of the late-onset toxicity observed in medulloblastoma survivors, mainly due to CSI, which is a crucial component of the multimodal treatment. With recent molecular advances, the feasibility of reducing or even removing CSI is currently being evaluated in WNT medulloblastoma, and it would be interesting to study the feasibility of CSI reduction/removal in SHH patients (who present mostly a local pattern of relapse) in a future clinical trial. The necessity of intensifying CSI for all patients with postsurgical residual disease >1.5 cm2 is questionable. In any case where CSI must be maintained (which remains prevalent), the potential solutions to lower radiation-induced morbidity are hippocampal sparing and proton therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Taillandier L, Blonski M, Carrie C, et al. Les médulloblastomes: revue générale [Medulloblastomas: review]. Rev Neurol. 2011;167(5):431–448. | ||
Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014;16(suppl 4): iv1–iv63. | ||
Johnson KJ, Cullen J, Barnholtz-Sloan JS, et al. Childhood brain tumor epidemiology: a Brain Tumor Epidemiology Consortium Review. Canc Epidemiol Biomarkers Prev. 2014;23(12):2716–2736. | ||
Fellay CN, Frappaz D, Sunyach MP, Franceschi E, Brandes AA, Stupp R. Medulloblastomas in adults: prognostic factors and lessons from paediatrics. Curr Opin Neurol. 2011;24(6):626–632. | ||
Ramaswamy V, Remke M, Bouffet E, et al. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol. 2016;131(6):821–831. | ||
Ramaswamy V, Remke M, Adamski J, et al. Medulloblastoma subgroup-specific outcomes in irradiated children: who are the true high-risk patients? Neuro Oncol. 2016;18(2):291–297. | ||
Tarbell NJ, Friedman H, Polkinghorn WR, et al. High-risk medulloblastoma: a pediatric oncology group randomized trial of chemotherapy before or after radiation therapy (POG 9031). J Clin Oncol. 2013;31(23):2936–2941. | ||
Moxon-Emre I, Bouffet E, Taylor MD, et al. Impact of craniospinal dose, boost volume, and neurologic complications on intellectual outcome in patients with medulloblastoma. J Clin Oncol. 2014;32(17):1760–1768. | ||
Moxon-Emre I, Taylor MD, Bouffet E, et al. Intellectual outcome in molecular subgroups of medulloblastoma. J Clin Oncol. 2016;34(34):4161–4170. | ||
Fossati P, Ricardi U, Orecchia R. Pediatric medulloblastoma: toxicity of current treatment and potential role of protontherapy. Cancer Treat Rev. 2009;35(1):79–96. | ||
Ellison DW, Onilude OE, Lindsey JC, et al. Beta-catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol. 2005;23(31):7951–7957. | ||
Herms J, Neidt I, Lüscher B, et al. C-MYC expression in medulloblastoma and its prognostic value. Int. J. Cancer. 2000;89(5):395–402. | ||
Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. | ||
Ellison DW, Dalton J, Kocak M, et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011;121(3):381–396. | ||
Ellison DW. Childhood medulloblastoma: novel approaches to the classification of a heterogeneous disease. Acta Neuropathol. 2010;120(3):305–316. | ||
Giangaspero F, Rigobello L, Badiali M, et al. Large-cell medulloblastomas. A distinct variant with highly aggressive behavior. Am J Surg Pathol. 1992;16(7):687–693. | ||
Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. | ||
Zeltzer PM, Boyett JM, Finlay JL, et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children’s Cancer Group 921 randomized phase III study. J Clin Oncol. 1999;17(3):832–845. | ||
Chang CH, Housepian EM, Herbert C. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology. 1969;93(6):1351–1359. | ||
Martin AM, Raabe E, Eberhart C, Cohen KJ. Management of pediatric and adult patients with medulloblastoma. Curr Treat Options in Oncol. 2014;15(4):581–594. | ||
Lannering B, Rutkowski S, Doz F, et al. Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial. J Clin Oncol. 2012;30(26):3187–3193. | ||
Packer RJ, Gajjar A, Vezina G, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24(25):4202–4208. | ||
Pomeroy SL, Tamayo P, Gaasenbeek M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415(6870):436–442. | ||
Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24(12):1924–1931. | ||
Kool M, Koster J, Bunt J, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3(8):e3088. | ||
Cho Y-J, Tsherniak A, Tamayo P, et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol. 2011;29(11):1424–1430. | ||
Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29(11):1408–1414. | ||
Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathologica. 2012;123(4):465–472. | ||
Kool M, Korshunov A, Remke M, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, group 3, and group 4 medulloblastomas. Acta Neuropathol. 2012;123(4):473–484. | ||
Clifford SC, Lannering B, Schwalbe EC, et al. Biomarker-driven stratification of disease-risk in non-metastatic medulloblastoma: results from the multi-center HIT-SIOP-PNET4 clinical trial. Oncotarget. 2015;6(36):38827–38839. | ||
Pietsch T, Schmidt R, Remke M, et al. Prognostic significance of clinical, histopathological, and molecular characteristics of medulloblastomas in the prospective HIT2000 multicenter clinical trial cohort. Acta Neuropathol. 2014;128(1):137–149. | ||
Northcott PA, Jones DTW, Kool M, et al. Medulloblastomics: the end of the beginning. Nat Rev Cancer. 2012;12(12):818–834. | ||
Robinson G, Parker M, Kranenburg TA, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488(7409):43–48. | ||
Hamilton SR, Liu B, Parsons RE, et al. The molecular basis of Turcot’s syndrome. N Engl J Med. 1995;332(13):839–847. | ||
Trubicka J, Szperl M, Grajkowska W, et al. Identification of a novel inherited ALK variant M1199L in the WNT type of medulloblastoma. Folia Neuropathol. 2016;1(1):23–30. | ||
Northcott PA, Korshunov A, Pfister SM, Taylor MD. The clinical implications of medulloblastoma subgroups. Nat Rev Neurol. 2012;8(6):340–351. | ||
Ellison DW, Kocak M, Dalton J, et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol. 2011;29(11):1400–1407. | ||
Gibson P, Tong Y, Robinson G, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468(7327):1095–1099. | ||
Taylor MD, Liu L, Raffel C, et al. Mutations in Sufu predispose to medulloblastoma. Nat Genet. 2002;31(3):306–310. | ||
O’Malley S, Weitman D, Olding M, Sekhar L. Multiple neoplasms following craniospinal irradiation for medulloblastoma in a patient with nevoid basal cell carcinoma syndrome. Case report. J Neurosurg. 1997;86(2):286–288. | ||
Ramaswamy V, Taylor MD. Medulloblastoma: from myth to molecular. JCO. 2017;35(21):2355–2363. | ||
Zhukova N, Ramaswamy V, Remke M, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol. 2013;31(23):2927–2935. | ||
Williams JR, Zhang Y, Zhou H, et al. A quantitative overview of radiosensitivity of human tumor cells across histological type and TP53 status. Int J Radiat Biol. 2008;84(4):253–264. | ||
Tchelebi L, Ashamalla H, Graves PR. Mutant p53 and the response to chemotherapy and radiation. Subcell Biochem. 2014;85:133–159. | ||
Kool M, Jones DTW, Jäger N, et al. Genome sequencing of Shh medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25(3):393–405. | ||
Coluccia D, Figuereido C, Isik S, Smith C, Rutka JT. Medulloblastoma: tumor biology and relevance to treatment and prognosis paradigm. Curr Neurol Neurosci Rep. 2016;16(5):43. | ||
Huang G-H, Xu Q-F, Cui Y-H, Li N, Bian X-W, Lv S-Q. Medulloblastoma stem cells: promising targets in medulloblastoma therapy. Cancer Sci. 2016;107(5):583–589. | ||
Northcott PA, Lee C, Zichner T, et al. Enhancer hijacking activates Gfi1 family oncogenes in medulloblastoma. Nature. 2014;511(7510):428–434. | ||
Shih DJH, Northcott PA, Remke M, et al. Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol. 2014;32(9):886–896. | ||
Zhou L, Picard D, Ra Y-S, et al. Silencing of thrombospondin-1 is critical for Myc-induced metastatic phenotypes in medulloblastoma. Cancer Res. 2010;70(20):8199–8210. | ||
Ramaswamy V, Remke M, Bouffet E, et al. Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol. 2013;14(12):1200–1207. | ||
Archer TC, Mahoney EL, Pomeroy SL. Medulloblastoma: molecular classification-based personal therapeutics. Neurotherapeutics. 2017;14(2):265–273. | ||
Ramaswamy V, Remke M, Shih D, et al. Duration of the pre-diagnostic interval in medulloblastoma is subgroup dependent. Pediatr Blood Cancer. 2014;61(7):1190–1194. | ||
Miranda Kuzan-Fischer C, Juraschka K, Taylor MD. Medulloblastoma in the molecular era. J Korean Neurosurg Soc. 2018;61(3):292–301. | ||
Gu S, Chen K, Yin M, Wu Z, Wu Y. Proteomic profiling of isogenic primary and metastatic medulloblastoma cell lines reveals differential expression of key metastatic factors. J Proteomics. 2017;160:55–63. | ||
Cavalli FMG, Remke M, Rampasek L, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. 2017;31(6):737–754. | ||
Northcott PA, Buchhalter I, Morrissy AS, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547(7663):311–317. | ||
Schwalbe EC, Lindsey JC, Nakjang S, et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol. 2017;18(7):958–971. | ||
Kortmann R-D, Kühl J, Timmermann B, et al. Postoperative neoadjuvant chemotherapy before radiotherapy as compared to immediate radiotherapy followed by maintenance chemotherapy in the treatment of medulloblastoma in childhood: results of the German prospective randomized trial HIT’91. Int J Radiat Oncol Biol Phys. 2000;46(2):269–279. | ||
Chin AL, Moding EJ, Donaldson SS, et al. Survival impact of postoperative radiotherapy timing in pediatric and adolescent medulloblastoma. Neuro Oncol. 2018;20(8):1133–1141. | ||
Packer RJ, Goldwein J, Nicholson HS, et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: a children’s cancer group study. J Clin Oncol. 1999;17(7):2127–2136. | ||
Merchant TE, Kun LE, Krasin MJ, et al. Multi-institution prospective trial of reduced-dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int J Radiat Oncol Biol Phys. 2008;70(3):782–787. | ||
Wahba HA, Abu-Hegazy M, Wasel Y, Ismail EI, Zidan AS. Adjuvant chemotherapy after reduced craniospinal irradiation dose in children with average-risk medulloblastoma: a 5-year follow-up study. J BUON. 2013;18(2):425–429. | ||
Gajjar A, Chintagumpala M, Ashley D, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (ST Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7(10):813–820. | ||
von Hoff K, Hinkes B, Gerber NU, et al. Long-term outcome and clinical prognostic factors in children with medulloblastoma treated in the prospective randomised multicentre trial HIT’91. Eur J Cancer. 2009;45(7):1209–1217. | ||
Packer RJ, Sutton LN, Goldwein JW, et al. Improved survival with the use of adjuvant chemotherapy in the treatment of medulloblastoma. J Neurosurg. 1991;13(3):433–440. | ||
Lafay-Cousin L, Smith A, Chi SN, et al. Clinical, pathological, and molecular characterization of infant medulloblastomas treated with sequential high-dose chemotherapy. Pediatr Blood Cancer. 2016;63(9):1527–1534. | ||
Brandes AA, Ermani M, Amista P, et al. The treatment of adults with medulloblastoma: a prospective study. Int J Radiat Oncol Biol Phys. 2003;57(3):755–761. | ||
Packer RJ, Rood BR, Macdonald TJ. Medulloblastoma: present concepts of stratification into risk groups. Pediatr Neurosurg. 2003;39(2):60–67. | ||
von Bueren AO, Kortmann R-D, von Hoff K, et al. Treatment of children and adolescents with metastatic medulloblastoma and prognostic relevance of clinical and biologic parameters. J Clin Oncol. 2016;34(34):4151–4160. | ||
Allen J, Donahue B, Mehta M, et al. A phase II study of Preradiotherapy chemotherapy followed by hyperfractionated radiotherapy for newly diagnosed high-risk medulloblastoma/primitive neuroectodermal tumor: a report from the Children’s Oncology Group (CCG 9931). Int J Radiat Oncol Biol Phys. 2009;74(4):1006–1011. | ||
von Bueren AO, Friedrich C, von Hoff K, et al. Metastatic medulloblastoma in adults: outcome of patients treated according to the HIT2000 protocol. Eur J Cancer. 2015;51(16):2434–2443. | ||
Cefalo G, Massimino M, Ruggiero A, et al. Temozolomide is an active agent in children with recurrent medulloblastoma/primitive neuroectodermal tumor: an Italian multi-institutional phase II trial. Neuro-Oncology. 2014;16(5):748–753. | ||
Kirk EA, Howard VC, Scott CA. Description of posterior fossa syndrome in children after posterior fossa brain tumor surgery. J Pediatr Oncol Nurs. 1995;12(4):181–187. | ||
Robertson PL, Muraszko KM, Holmes EJ, et al. Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma: a prospective study by the Children’s Oncology Group. J Neurosurg. 2006;105(6):444–451. | ||
Ce C-B, van Dongen HR, Mulder PG. Paz y Geuze D, Paquier PF, Lequin MH. Tumour type and size are high risk factors for the syndrome of “cerebellar” mutism and subsequent dysarthria. J Neurol Neurosurg Psychiatry. 1999;67(6):755–757. | ||
Albright AL, Sposto R, Holmes E, et al. Correlation of neurosurgical subspecialization with outcomes in children with malignant brain tumors. Neurosurgery. 2000;47(4):879–887; discussion 885–887. | ||
Douglas Cochrane D, Gustavsson B, Poskitt KP, Steinbok P, Kestle JRW. The surgical and natural morbidity of aggressive resection for posterior fossa tumors in childhood. Pediatr Neurosurg. 1994;20(1):19–29. | ||
Gelabert-González M, Fernández-Villa J. Mutism after posterior fossa surgery. Review of the literature. Clin Neurol Neurosurg. 2001;103(2):111–114. | ||
Groll AH, Ritter J, Müller FM. [Guidelines for prevention of Pneumocystis carinii pneumonitis in children and adolescents with cancer]. Klin Padiatr. 2001;213(Suppl 1):A38–A49. | ||
Neglia JP, Robison LL, Stovall M, et al. New primary neoplasms of the central nervous system in survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2006;98(21):1528–1537. | ||
Packer RJ, Zhou T, Holmes E, Vezina G, Gajjar A. Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: results of children’s Oncology Group trial A9961. Neuro-oncology. 2013;15(1):97–103. | ||
Kotecha RS, Pascoe EM, Rushing EJ, et al. Meningiomas in children and adolescents: a meta-analysis of individual patient data. Lancet Oncol. 2011;12(13):1229–1239. | ||
Bansal LR, Belair J, Cummings D, Zuccoli G. Late-onset radiation-induced vasculopathy and stroke in a child with medulloblastoma. J Child Neurol. 2015;30(6):800–802. | ||
Lavoie Smith EM, Li L, Chiang C, et al. Patterns and severity of vincristine-induced peripheral neuropathy in children with acute lymphoblastic leukemia. J Peripher Nerv Syst. 2015;20(1):37–46. | ||
Gómez S, Garrido-Garcia A, Garcia-Gerique L, et al. A novel method for rapid molecular subgrouping of medulloblastoma. Clin Cancer Res. 2018;24(6):1355–1363. | ||
Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. | ||
Phoenix TN, Patmore DM, Boop S, et al. Medulloblastoma genotype dictates blood brain barrier phenotype. Cancer Cell. 2016;29(4):508–522. | ||
Manolagas SC. WNT signaling and osteoporosis. Maturitas. 2014; 78(3):233–237. | ||
Robinson GW, Orr BA, Wu G, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog–subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J Clin Oncol. 2015;33(24):2646–2654. | ||
Northcott PA, Hielscher T, Dubuc A, et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011;122(2):231–240. | ||
Wu S-Y, Chiang C-M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. 2007;282(18):13141–13145. | ||
Tang Y, Gholamin S, Schubert S, et al. Epigenetic targeting of hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med. 2014;20(7):732–740. | ||
Long J, Li B, Rodriguez-Blanco J, et al. The BET bromodomain inhibitor I-BET151 acts downstream of smoothened protein to abrogate the growth of hedgehog protein-driven cancers. J Biol Chem. 2014;289(51):35494–35502. | ||
Faria CC, Golbourn BJ, Dubuc AM, et al. Foretinib is effective therapy for metastatic sonic hedgehog medulloblastoma. Cancer Res. 2015;75(1):134–146. | ||
Hanaford AR, Archer TC, Price A, et al. DiSCoVERing innovative therapies for rare tumors: combining genetically accurate disease models with in silico analysis to identify novel therapeutic targets. Clin Cancer Res. 2016;22(15):3903–3914. | ||
Pei Y, Liu K-W, Wang J, et al. HDAC and PI3K antagonists cooperate to inhibit growth of MYC-driven medulloblastoma. Cancer Cell. 2016;29(3):311–323. | ||
Venkataraman S, Alimova I, Balakrishnan I, et al. Inhibition of BRD4 attenuates tumor cell self-renewal and suppresses stem cell signaling in MYC driven medulloblastoma. Oncotarget. 2014;5(9):2355–2371. | ||
Bandopadhayay P, Bergthold G, Nguyen B, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res. 2014;20(4):912–925. | ||
Northcott PA, Shih DJH, Peacock J, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488(7409):49–56. | ||
Thompson EM, Bramall A, Herndon JE, Taylor MD, Ramaswamy V. The clinical importance of medulloblastoma extent of resection: a systematic review. J Neurooncol. 2018;139(3):523–539. | ||
Thompson EM, Hielscher T, Bouffet E, et al. Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: a retrospective integrated clinical and molecular analysis. Lancet Oncol. 2016;17(4):484–495. | ||
Paterson E, Farr RF. Cerebellar medulloblastoma: treatment by irradiation of the whole central nervous system. Acta radiol. 1953;39(4):323–336. | ||
Thariat J, Hannoun-Levi J-M, Sun Myint A, Vuong T, Gérard J-P, Past GJ-P. Past, present, and future of radiotherapy for the benefit of patients. Nat Rev Clin Oncol. 2013;10(1):52–60. | ||
Michalski JM, Janss A, Vezina G, et al. Results of cog ACNS0331: a phase III trial of involved-field radiotherapy (IFRT) and low dose craniospinal irradiation (LD-CSI) with chemotherapy in average-risk medulloblastoma: a report from the Children’s Oncology Group. Int J Radiation Oncol Biol Physics. 2016;96(5):937–938. | ||
Hall EJ, Wuu C-S. Radiation-induced second cancers: the impact of 3D-CRT and IMRT. Int J Radiation Oncol Biol Physics. 2003;56(1):83–88. | ||
Armstrong GT, Jain N, Liu W, et al. Region-specific radiotherapy and neuropsychological outcomes in adult survivors of childhood CNS malignancies. Neuro-oncology. 2010;12(11):1173–1186. | ||
Jalali R, Mallick I, Dutta D, et al. Factors influencing neurocognitive outcomes in young patients with benign and low-grade brain tumors treated with stereotactic conformal radiotherapy. Int J Radiat Oncol Biol Phys. 2010;77(4):974–979. | ||
Padovani L, Chapon F, André N, et al. Hippocampal sparing during craniospinal irradiation: what did we learn about the incidence of perihippocampus metastases? Int J Radiation Oncol Biol Physics. 2018;100(4):980–986. | ||
Ladra MM, MacDonald SM, Terezakis SA. Proton therapy for central nervous system tumors in children. Pediatr Blood Cancer. 2018;65(7):e27046. | ||
Kamran SC, Goldberg SI, Kuhlthau KA, et al. Quality of life in patients with proton-treated pediatric medulloblastoma: results of a prospective assessment with 5-year follow-up. Cancer. 2018;124(16):3390–3400. | ||
Ho ESQ, Barrett SA, Mullaney LM. A review of dosimetric and toxicity modeling of proton versus photon craniospinal irradiation for pediatrics medulloblastoma. Acta Oncol. 2017;56(8):1031–1042. | ||
Shen CJ, Hu C, Ladra MM, Narang AK, Ce P, Terezakis SA. Socioeconomic factors affect the selection of proton radiation therapy for children: proton therapy and socioeconomic factors. Cancer. 2017;123(20):4048–4056. | ||
Hashimoto Y, Penas-Prado M, Zhou S, et al. Rethinking medulloblastoma from a targeted therapeutics perspective. J Neurooncol. 2018;139(3):713–720. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.