Back to Journals » Journal of Hepatocellular Carcinoma » Volume 13
Mechanistic Research on the Crosstalk Between Macrophage Polarization and Energy Metabolic Reprogramming in Hepatocellular Carcinoma
Authors Zhu R, Na J, Tan X, Chen X, Tian X, Luo J, Chen Y, Zhong L
Received 20 November 2025
Accepted for publication 27 February 2026
Published 5 March 2026 Volume 2026:13 583017
DOI https://doi.org/10.2147/JHC.S583017
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mohamed Shaker
Rui Zhu,1,* Jintong Na,1,* Xinyi Tan,1 Xinyi Chen,1 Xiaorui Tian,1 Jingjie Luo,1 Yongbin Chen,1,2 Liping Zhong1,3
1State Key Laboratory of Targeting Oncology, National Center for International Research of Bio-Targeting Theranostics, Guangxi Key Laboratory of Bio-Targeting Theranostics, Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Talent Highland of Major New Drugs Innovation and Development, Targeting Theranostics Research Center of Guangxi Higher Education, Guangxi Medical University, Nanning, Guangxi, 530021, People’s Republic of China; 2State Key Laboratory of Genetic Evolution & Animal Models, the Key Laboratory of Animal Models & Human Disease Mechanisms of Yunnan Province, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan, 650201, People’s Republic of China; 3Pharmaceutical College, Guangxi Medical University, Nanning, Guangxi, 530021, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yongbin Chen, Email [email protected] Liping Zhong, Email [email protected]
Abstract: Liver cancer is ranked as the sixth most prevalent and the third deadliest cancer worldwide. Macrophages play a crucial role in cancer resistance mechanisms, and the regulation of their polarization state can alter their anti-tumor activity. Additionally, alterations in liver cancer pathways can shift the polarization of macrophages. Inflammatory agents and medications have the potential to alter macrophage polarization, favoring M1 differentiation and enhancing their anti-tumor activity. This review explores metabolic alterations in liver cancer, macrophage polarization processes, and their interconnections in hepatic oncogenesis. It further elaborates on the prospects and challenges of investigating targeted macrophage metabolism for liver cancer treatment. Modulating macrophage metabolic pathways represents a promising therapeutic approach for hepatic carcinoma.
Keywords: liver cancer, macrophage polarization, metabolic reprogramming
Introduction
Liver cancer is ranked as the sixth most prevalent cancer and is responsible for the third highest number of cancer-related fatalities worldwide.1 Approximately 830,000 deaths occur annually due to liver cancer. Hepatocellular carcinoma is characterized by a complex pathogenesis and genetic mutations in proto-oncogenes, often resulting from external factors such as viral infections, excessive alcohol consumption, obesity, and exposure to aflatoxins.2 Hepatocellular carcinoma is characterized by high heterogeneity, which affects tumor progression, invasiveness, and response to anti-tumor drugs.3,4 Treatment for advanced liver cancer primarily employs targeted agents and immunotherapeutics.5,6 Sorafenib has been the only systemic first-line treatment drug approved by the US Food and Drug Administration (FDA) for 10 years.7 Nevertheless, sorafenib exhibits drawbacks when utilized for HCC therapy.8 Consequently, additional research is needed to develop effective therapeutic strategies to improve outcomes for individuals with hepatic malignancies.
In recent years, with the extensive research on tumor immunology and cellular metabolism, the role of macrophage polarization in tumor initiation and progression has become an emerging area of focus. As a crucial component of the immune system, macrophages exhibit significant plasticity and heterogeneity,9 and can polarize into distinct phenotypes with various functions within the tumor microenvironment. Tumor-associated macrophages (TAMs) play a central role in hepatocellular carcinoma, acting as key players within the tumor microenvironment (TME). These critical cells play a pivotal role in tumor formation and progression, influencing angiogenesis, promoting immunosuppression, and facilitating drug resistance and cancer metastasis.10 Among these, M1 macrophages exhibit anti-tumor activity and can eliminate tumor cells through cytokine secretion and immune cell activation, whereas M2 macrophages display pro-tumor effects, promoting neoplastic cell growth, dissemination, infiltration, and immunosuppression.11,12 Increasing evidence suggests that TAMs predominantly exhibit an M2 polarization state, which is strongly associated with the unfavorable prognosis of liver cancer. A comprehensive understanding of the biological characteristics and regulatory mechanisms of TAMs is expected to facilitate the development of more effective treatment strategies for HCC.
Although the M1/M2 dichotomy is a widely used framework for describing macrophage polarization in tumor immunity research, recent studies have increasingly suggested that this framework is overly simplistic and does not fully capture the complex role of macrophages within the tumor microenvironment. Particularly in the immune microenvironment of tumors like liver cancer, macrophages exhibit dynamic states and high spatial heterogeneity, with changes in their phenotypes and functions influenced by the local environment and metabolic conditions.
Meanwhile, energy metabolism reprogramming, a key feature of malignant cells, provides the energy and material foundation necessary for the rapid proliferation and sustained growth of cancer cells.13,14 Hepatocellular carcinoma cells satisfy their aberrant metabolic demands by reprogramming metabolic pathways, including those involved in glucose, lipid, and amino acid metabolism.15 Studies have demonstrated a close association between macrophage polarization and metabolic reprogramming in cancer cells. These factors interact and regulate each other to drive the progression of liver cancer.16 Moreover, several signaling pathways play a crucial role in regulating macrophage polarization and reprogramming energy metabolism in liver cancer, with notable examples including the PI3K/AKT, MAPK, and NF-κB pathways.17–19 Their aberrant regulation is associated with the onset, progression, metastasis, and therapeutic resistance of liver cancer. However, the roles of fatty acid oxidation (FAO) and amino acid metabolism in macrophage polarization have not been fully elucidated, and these metabolic pathways exhibit distinct functions across different tumor microenvironments. Although numerous studies have explored the impact of tumor metabolism on macrophage reprogramming, the causal relationship between metabolic changes and macrophage polarization remains unclear. Metabolic reprogramming may serve as a driving factor in altering the polarization state of macrophages, or it may be a consequence of complex signaling within the tumor microenvironment. Furthermore, the effects of metabolic changes may be contingent upon the specific conditions of the microenvironment, manifesting as context-dependent feedback loops.
This review thoroughly examines the relationship between macrophage metabolic reprogramming and immune regulation within the HCC TME, with particular emphasis on the roles of glucose metabolism, FAO, and amino acid metabolism in macrophage polarization. It is proposed that targeting macrophage metabolism—particularly glucose and lipid metabolism—in conjunction with immunotherapy, may significantly enhance anti-tumor immune responses, offering new therapeutic strategies for liver cancer treatment. Furthermore, clinical trial data are incorporated to highlight the potential of combining metabolic regulation with immune checkpoint inhibitors, which have shown promising results, particularly in the treatment of liver cancer and other cancers, thereby providing novel perspectives and directions for clinical applications.
Basic Characteristics of Macrophages
The Concept and Methods of Macrophage Polarization
Monocytes are a subset of white blood cells derived from the bone marrow, comprising approximately 5%-10% of the total white blood cell count in peripheral blood. Among the sources of tissue macrophages, monocytes serve as the primary precursor cells, with the majority of macrophages originating from monocyte differentiation. In terms of their life cycle, circulating monocytes have a relatively short lifespan, lasting only about two days. However, when influenced by chemotactic signals, they migrate to the tissue microenvironment and differentiate into macrophages, leading to a significant extension of their survival time to several months. This “lifespan extension” enables macrophages to continuously perform vital functions, including phagocytosis, clearance, and immune regulation.20 As a central component of the immune system, macrophages play a crucial role in initiating and executing immune responses, thereby forming a key defense barrier that protects the host from microbial invasion. However, studies have demonstrated that the functions of these cells extend beyond the realm of anti-infection. In pathological conditions where immune homeostasis is disrupted, macrophages are closely implicated in the onset and progression of autoimmune diseases, as well as in the proliferation and invasion of malignant tumors, serving as a critical regulatory link between immune defense and pathological damage.21
Macrophage polarization refers to the activation of macrophages in response to pathogenic microorganisms, inflammatory signals, cytokines, or specific physical and chemical factors, leading to their differentiation into distinct phenotypes based on the state and changes within the microenvironment.22 Macrophage polarization appears to serve as an intermediate process during both the onset and regression of diseases. It is activated by specific signals, leading to the generation of distinct phenotypes, which exert regulatory effects by modulating multiple signal transduction pathways.23
Exogenous polarization is the primary mechanism of macrophage polarization, mediated by cytokines secreted by other cells, such as CD4+ Th1 or Th2 cells. In addition to the classic cytokine-mediated regulatory pathways, macrophage polarization is also influenced by various non-cytokine exogenous factors. Among these, the hypoxic microenvironment and the abnormal accumulation of lactic acid in the tumor microenvironment are two key inducing factors. These factors can specifically drive macrophages to polarize into the M2 phenotype through mechanisms such as the regulation of cellular metabolism and the activation of specific signaling pathways, thereby contributing to the modulation of the local immune microenvironment and disease progression.24 The classically activated M1 phenotype and the alternatively activated M2 phenotype are distinguished by distinct surface receptor expressions, secretion profiles, and functional roles.25 Due to the plasticity of macrophages, their polarization state is dynamic. The two primary phenotypes are the classically activated M1 type and the alternatively activated M2 type.26 However, with advancing research, the academic community has recognized that the polarization state of macrophages is not confined to the dichotomy between M1 and M2 types, but exists along a continuous spectrum, dynamically regulated by factors such as local signals within the tumor microenvironment, metabolic status, and spatial location. Recent advancements in single-cell technology have revealed the spatial heterogeneity of macrophage phenotypes. Macrophages within the same tumor can display distinct functional characteristics across different regions. These findings indicate that the polarization and function of macrophages are not solely defined by the classic M1/M2 model, but result from the combined action of multiple factors, exhibiting both reversibility and diversity, which contribute to the complex regulation of the tumor immune microenvironment. For example, studies have demonstrated that M2-type macrophages are predominant in mouse models; however, their specific roles in human liver cancer may differ,27 potentially influenced by various factors within the tumor microenvironment.
M1 Macrophage
Classically activated macrophages, or M1 macrophages, are the most well-documented subset of these immune cells. Typically stimulated by lipopolysaccharide (LPS) and interferon-γ (IFN-γ), they initiate a robust inflammatory response by releasing a range of pro-inflammatory cytokines, including tumor necrosis factor (TNF-α), interleukins (IL-1, IL-6), and inducible nitric oxide synthase (iNOS), effectively signaling the presence of potential threats.28 In the tumor microenvironment, M1 macrophages initiate Th1 immune responses by releasing cytokines such as IL-1, IL-6, IL-12, TNF, and CXCL9, thereby promoting robust anti-tumor immune reactions.29 In addition, as key effector cells in the body’s anti-tumor and anti-infectious immunity, M1-type macrophages can also directly interact with CD8⁺ T cells (cytotoxic T cells) through specific receptors expressed on their surface, thereby precisely regulating the activation, proliferation, killing function, and other activity states of CD8⁺ T cells. In-depth studies have shown that such intercellular interactions do not occur randomly but are highly dependent on the mediation of co-stimulatory molecules. The molecular interaction between M1 macrophages and CD8⁺ T cells relies on the upregulation of co-stimulatory molecules such as CD40, CD80, and CD86 on the macrophage surface, which subsequently initiate signaling cascades in T cells upon receptor binding. These interactions are essential for the precise modulation of CD8⁺ T cell function.30
IFN-γ was the first macrophage-activating factor identified in 1970. It is a soluble cytokine produced by activated CD4+ helper T cells (Th1), CD8+ cytotoxic T cells, and NK cells. It can induce the transformation of resting macrophages into activated cells and enhances their antigen-presenting ability, increases the synthesis of pro-inflammatory mediators and pathogenic substances, and promotes complement-mediated cellular consumption.31 The activation of early macrophages is referred to as the classical pathway. Stimulated by Th1 cells, also known as cytotoxic T cells, these classically activated macrophages are now termed M1 macrophages. In contrast, M1 macrophages promote Th1 and cytotoxic T-cell activation, inducing the production of IFN-γ, which reinforces M1 polarization through positive feedback regulation.32 M1 macrophages release numerous pro-inflammatory cytokines, including IL-1β, IL-6, IL-12, IL-23, IFN-β, and TNF-α. (Figure 1). These factors simultaneously drive the Th1 response.33
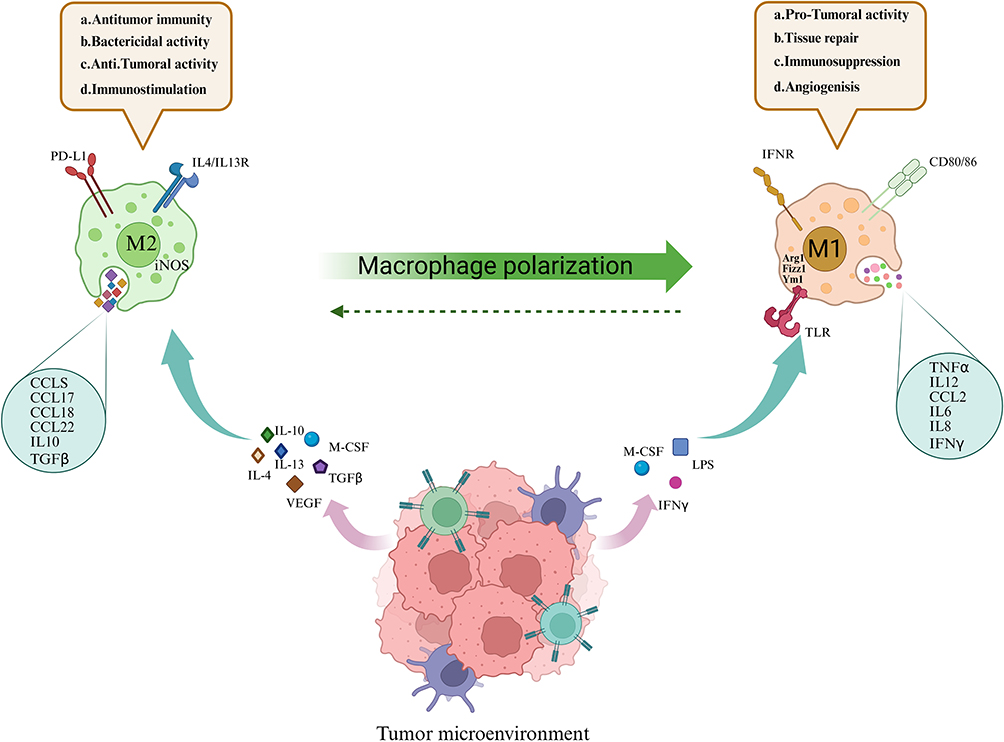 |
Figure 1 M1 and M2 can transform into each other under different stimulating factors and secrete cytokines, producing different effects. M1 macrophages promote anti-tumor immune responses by releasing IL1, IL6, IL12, and TNF-α under the action of interferon and lipopolysaccharide; while M2 macrophages achieve tissue repair and immune tolerance by releasing CCL chemokines under the influence of interleukins and tumor necrosis factors, and have anti-inflammatory and tumor-promoting properties. |
M2 Macrophage
M2 macrophages were first identified in cases of worm infections, where they are distinguished from monocyte progenitor cells that are influenced by IL-4 and IL-13, and exhibit a pronounced Th2 polarization response. Although the recruitment of M2-type macrophages is strongly influenced by Th2-type cytokines, these cells do not solely function as passively recruited effector cells. They can also actively secrete various cytokines to reversely participate in the regulation of immune responses. On the one hand, they can further mediate and amplify Th2-type immune responses, thereby promoting the activation and functional maintenance of Th2 cells. Alternatively, they precisely modulate the severity and extent of regional inflammatory responses by secreting anti-inflammatory cytokines such as IL-10 and TGF-β. This results in the formation of a bidirectional regulatory loop, where Th2 cytokines recruit M2 macrophages, and M2 macrophages regulate Th2 responses and inflammation.34 M2-type activation results from IL-4, IL-10, IL-34, IL-13, vitamin D3, TGF-β, prostaglandin E2 (PGE2), VEGF, EGF, glucocorticoids, and M-CSF.35 M2 macrophages are classified into distinct subsets—IL-4/13-triggered M2a, immune complex-stimulated M2b, IL-10-inhibited M2c, and IL-6/M-CSF-mediated M2d—based on stimulation by various cytokines.36
M2 macrophages primarily contribute to tissue healing and immune tolerance, exhibiting both anti-inflammatory and pro-tumor characteristics, in contrast to the roles of M1 macrophages. In the TME, TAMs in advanced tumors are predominant and serve as key regulators of the TME. Numerous studies have confirmed that, in various cancers, M2-like macrophages constitute a larger proportion of TAMs, and this characteristic is significantly associated with an unfavorable prognosis for patients (Figure 2).
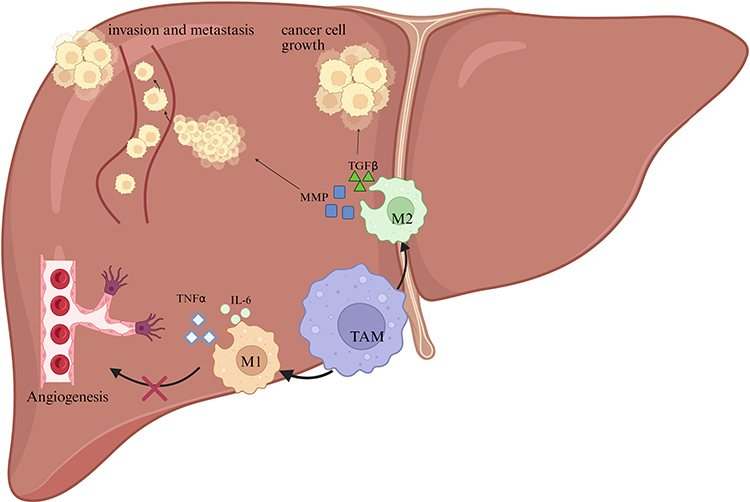 |
Figure 2 The role of TAMs in liver cancer progression. M1 macrophages secrete pro-inflammatory cytokines such as TNFα and interleukin-6 (IL-6), which inhibit angiogenesis and thereby suppress tumor growth. In contrast, M2 macrophages promote cancer cell proliferation and invasion through the secretion of TGFβ and matrix metalloproteinases (MMPs). These factors contribute to the remodeling of the extracellular matrix and facilitate the metastatic spread of cancer cells. |
M2 macrophages act as key drivers of tumor progression through multiple mechanisms, with epidermal growth factor (EGF) released by M2-simulating tumor-associated macrophages directly promoting cancer cell growth and metastasis.37 The transition from M1-like to M2-like macrophage phenotypes reduces NO and iNOS production in TAMs, leading to the loss of the direct tumor cell-killing effect of M1.38 VEGF secreted by M2-like TAMs promotes angiogenesis at the tumor site. M2 macrophages play a crucial role in suppressing NK cell activity through the secretion of inhibitory factors and direct cell-to-cell interactions.39
In summary, M1-like TAMs exhibit anti-cancer properties by initiating inflammatory responses against tumor cells, whereas M2-like TAMs promote tumor growth through anti-inflammatory activities.
Energy Metabolism of Macrophages
Glycometabolism of Macrophages
TME plays a pivotal role in HCC cell proliferation, infiltration, and spread.40 Immune cells are an important component of the TME. Increased infiltration of T cells and NK cells is considered a favorable prognostic indicator, also reflecting the efficacy of immunotherapy in the management of hepatocellular carcinoma.41 Concurrently, accumulating evidence suggests that the metabolic activity of TME cells, including immune cells, plays a crucial role in regulating cancer development.42 In the tumor microenvironment, a competitive struggle often arises between cancerous cells and immune cells, resulting in a metabolic conflict. This intense competition not only promotes tumor progression but also depletes immune cells of essential nutrients, thereby compromising their ability to mount an effective defense against the malignancy.43
Regarding macrophage energy metabolism, M1-type macrophages primarily rely on glycolysis to support their pro-inflammatory functions, whereas M2-type macrophages depend more on fatty acid oxidation and oxidative phosphorylation to sustain their immunosuppressive functions. Although these mechanisms are supported by numerous studies, the specific manifestations and underlying processes of macrophage metabolic reprogramming may differ across tumor types or under varying experimental conditions. Therefore, additional experimental data are required to confirm their generalizability.44
Cellular glucose metabolism primarily relies on three central processes: glycolysis, the pentose phosphate pathway, and the citric acid cycle. Glycolysis, a key metabolic pathway, occurs in the cytoplasm. In the presence of oxygen, glucose is metabolized into pyruvate, which then enters the mitochondria to participate in the tricarboxylic acid (TCA) cycle. However, under conditions of limited oxygen, glucose bypasses the mitochondria and is converted directly into lactic acid, with adenosine triphosphate (ATP) produced as a byproduct.45 Pyruvate, resulting from standard glycolysis, undergoes OXPHOS to produce additional ATP.
Nevertheless, when tumor cells are in oxygen-rich conditions, they bypass the TCA cycle entirely, instead favoring glycolysis. This metabolic shift leads to significant glucose depletion within the tumor microenvironment, while simultaneously producing lactic acid as a byproduct. Consequently, these altered metabolic conditions substantially reshape the function of immune cells within the same microenvironment.46,47 Glycolysis provides energy to macrophages and other cells at a much faster rate than OXPHOS, while simultaneously supplying essential intermediate metabolites needed for the synthesis of biological macromolecules. Under physiological conditions, unactivated naive M0 macrophages primarily rely on OXPHOS, an efficient energy metabolism pathway, to generate energy. Upon polarization, macrophages dynamically select an appropriate metabolic mode to support functional activity, based on the specific conditions of the TME, as well as their energy requirements and metabolic characteristics.48
Metabolic changes in the tumor microenvironment, such as hypoxia and lactic acid accumulation, can subsequently influence the polarization state of macrophages. TAM is the immune cell population with the highest proportion in TME. It consistently exerts an immunosuppressive effect throughout tumor initiation and progression, thereby creating conditions conducive to tumor growth. Conversely, cancer cells exhibit a pronounced reliance on glucose for their energy metabolism. To satisfy the high energy demands required for rapid proliferation, tumor cells competitively consume glucose within the TME and undergo energy conversion via aerobic glycolysis, an efficient energy-producing pathway.49 Therefore, the metabolic characteristics of TAMs undergo adaptive changes, shifting to OXPHOS and FAO as the primary metabolic pathways. Their functions subsequently resemble those of M2 macrophages in a low-glucose TME, ultimately resulting in a pronounced immunosuppressive effect.50 Even slight variations in environmental cues can lead to significant disparities in macrophage profiles and metabolic characteristics. Under identical stimuli, macrophages may display varying reactivity.51 TAMs exhibit increased glucose uptake and enhanced glycolytic activity, similar to M1 macrophages, thereby supporting their cytokine profile and functional efficacy.52 Proteomic profiling research reveals that macrophages activated with breast cancer patient-derived tumor extracts show enhanced glycosylase expression, particularly of hexokinase 2 (HK2), as indicated in S2. Simultaneously, the export of lactic acid produced through glycolysis in malignant cells into the tumor microenvironment enhances HIF-1α expression in tumor-associated macrophages, which subsequently increases glycolysis and drives these immune cells toward an M2-like phenotype.53 Additionally, macrophage polarization remains fluid across physiological and bodily pathological conditions. Macrophages can not only reverse their polarization from the M2 to the M1 phenotype, but also co-express characteristics of both M1 and M2 polarization after the tumor reaches a specific stage, resulting in a “mixed polarization state” that exhibits attributes of both phenotypes.54 Researchers have identified a novel subset of CD19+ tumor-associated macrophages in hepatocellular carcinoma, with evidence suggesting that glycolysis may be an intrinsic feature driving tumor progression. Notably, a glucose-rich environment can induce macrophage polarization, directing them toward an M2-like phenotype.55 Studies have shown that O-GlcNAc modification in TAMs activates the HBP, enhances glucose metabolic flux, and promotes M2 macrophage polarization, thereby accelerating tumor progression and facilitating immune escape.56 Tumor cells are more reliant on glucose than immune cells to support their rapid proliferation. This competition for glucose between these cell populations within the tumor microenvironment may, in fact, inhibit cancer progression.57 In conclusion, a glucose-rich environment promotes the polarization of macrophages toward the M1 subtype and exerts a tumor-suppressive effect through M1 macrophages.
Macrophages in different polarization states exhibit distinct glucose metabolism patterns. Macrophages activated by IFN-γ and Toll-like receptors (TLRs) generate energy through anaerobic glycolysis. During this process, 6-phosphofructo-2-kinase (PFK2) transitions from the liver-type PFK2 (L-PFK2) to the ubiquitous PFK2 (uPFK2), which subsequently increases PFK2 production, thereby promoting the progression of the reaction. However, the glucose metabolism process of macrophages stimulated by IL-4 is the opposite.58 This difference may be attributed to the distinct functions of cells: M1-type macrophages are typically directly involved in combating infections. These cells require rapid bactericidal activity, and the microenvironment at the site of tissue injury is typically hypoxic.59 Therefore, anaerobic glycolysis is the most direct and optimal choice to meet the energy needs of M1-type macrophages. M2 polarization links with the processes of tissue reorganization, restoration, and injury resolution. These activities require a continuous supply of intracellular energy, so aerobic glycolysis is more suitable for meeting the needs of M2 macrophages.60 Macrophages contribute to tumor advancement and treatment resistance by supplying nutrients to cancerous cells.61 Glycolysis in cancerous cells yields lactic acid as an end product. It is essential in signal transduction and facilitates M2-like TAM polarization.62 In HCC, the elevated concentration of lactic acid in the tumor microenvironment can promote the development of a pro-tumor macrophage phenotype through the activation of the AKT signaling pathway.63,64 Conversely, research confirms M1-type macrophages promote glycolysis while suppressing mitochondrial respiration via the AKT/mTOR/HIF-1α pathway.65 The findings emphasize AKT pathway significance in macrophage M1/M2 transition within HCC.
Metabolic pathways are not only driving factors for macrophage polarization but also potential feedback mechanisms for tumor signals. For example, tumor cells affect macrophage metabolism through lactic acid secretion and HIF-1α. The accumulation of lactic acid promotes macrophage polarization toward the M2 phenotype, which in turn enhances tumor cell growth and immune escape. This metabolic polarization of macrophages establishes a context-dependent feedback loop that operates within the metabolism-polarization-tumor progression cycle in the tumor microenvironment.66
Lipid Metabolism of Macrophages
Under physiological conditions, tumor cells display distinct characteristics of metabolic reprogramming, with fatty acid synthesis and associated metabolic pathways exhibiting abnormal activity. Specifically, the expression levels of key regulatory biocatalysts, such as sterol regulatory element-binding protein (SREBP) and fatty acid synthase (FAS), are significantly upregulated, redirecting cellular metabolism toward de novo fatty acid synthesis to support the energy demands and membrane biosynthesis required for rapid tumor proliferation. It is worth noting that fatty acid metabolites synthesized by tumor cells are released into the TME. These metabolites can reshape the metabolic phenotype of immune cells through various pathways, thereby affecting their functional status and contributing to the formation of the tumor immunosuppressive microenvironment.67 Tumor fatty acid metabolites exhibit significant differences in their regulatory effects on macrophages in different polarization states. As M1-polarized macrophages with inflammatory functions, the M1 subtype exhibits a distinct response pattern in this regulatory process. When M1 macrophages uptake excessive unsaturated fatty acids, their intracellular signaling pathways are activated, subsequently stimulating the synthesis and secretion of interleukin-1α (IL-1α). As a key pro-inflammatory cytokine, IL-1α not only directly exacerbates local inflammatory responses but also inversely regulates the metabolic phenotype of M1 macrophages through paracrine or autocrine pathways. This regulation promotes the activation of intracellular fatty acid synthesis pathways, and the enhanced synthesis of fatty acids provides essential support for M1 macrophages to maintain inflammatory functions, such as facilitating inflammatory protein production and preserving cellular membrane integrity, thereby ensuring the effective execution of M1 macrophages’ pro-inflammatory actions.68 Conversely, the fatty acid metabolism pattern of M2 macrophages differs from that of the M1 subtype. M2 macrophages primarily absorb triglycerides via the CD36 fatty acid receptor on their cellular surfaces. Triglycerides entering the cells are initially broken down by lysosomal acid lipase, and their subsequent metabolism depends on a high level of carnitine palmitoyltransferase 1α (CPT1α). CPT1α, as a key enzyme, facilitates the translocation of fatty acids across mitochondrial membranes and regulates the balance between respiratory and oxidative pathways. This metabolic process also reduces the production of inflammatory cytokines while enhancing fatty acid metabolism to support M2 macrophage function.69
Although M1 macrophages predominantly rely on glycolysis for their metabolic processes, intracellular fatty acid levels significantly influence their function. Research has shown that M1 macrophage inflammatory activity requires the involvement of the fatty acid synthesis pathway.70 Upon exposure to inflammatory stimuli like LPS and cytokines, M1 macrophages activate and amplify fatty acid synthesis pathways.71 M2 macrophages specifically mediate immunosuppression through two primary mechanisms: first, by expressing active arginase; and second, by performing S-nitrosylation modifications on the surface proteins of infiltrating T cells.72 M2 macrophages exhibit distinct metabolic profiles compared to M1 macrophages. Among these processes, those involved in triggering the glycolysis switch in M1 macrophages are downregulated in M2 macrophages, enabling M2 macrophages to exhibit higher FAO activity. Additionally, their mitochondrial respiratory rate is significantly elevated.73 Reduction of OXPHOS levels significantly inhibits the polarization of macrophages toward the M2 phenotype, as evidenced by a decrease in the secretion of anti-inflammatory cytokines and a marked reduction in the expression of M2 activation markers.74 CPT1 is anchored to the outer mitochondrial membrane, where it plays a crucial role in facilitating the transport of fatty acids across the membrane. It functions as a critical gatekeeper enzyme in the β-oxidation of fatty acids, with its activity ultimately determining the rate of this metabolic process.75 In macrophage cell lines, the upregulation of CPT1α expression not only effectively promotes the FAO process but also significantly reduces the production of inflammatory cytokines.76,77 This effect is mediated by two key molecules, STAT6 and PGC-1β.78 Studies have shown that CPT1 deficiency does not affect M2 macrophage polarization, although it inhibits FAO.79 In summary, it can be concluded that during M2 macrophage polarization, CPT1, in addition to its well-established role in mediating fatty acid transport, exerts other biological effects through complex mechanisms. Previous studies have confirmed that M2-type macrophages uptake triacylglycerol (TG) substrates via the scavenger receptor CD36, thereby activating the lipolysis process mediated by lysosomal acid lipase (LAL). This research breakthrough has revealed a novel finding: lysosomal lipolysis, occurring within the cell, plays a crucial role in activating M2-type macrophages. This discovery reveals a critical mechanism that activates M2 macrophages, akin to uncovering a hidden lever that triggers a crucial switch. It provides new empirical evidence, offering insights into the complex metabolic processes underlying M2 macrophage activation.80
Amino Acid Metabolism of Macrophages
M2-like tumor-associated macrophages display heightened utilization of glutamine and fatty acids, aligning with glycolytic traits.81 Research indicates GLUL facilitates macrophage M2 polarization through glutamate-to-glutamine conversion.82 Therefore, the inhibitory effect on GLUL facilitates the polarization of M2-like macrophages toward the M1-like phenotype. This process is concurrently characterized by increased glycolytic throughput and enhanced succinate availability.83 This phenomenon suggests a metabolic interaction between glucose and glutamine metabolism in the functional regulation of TAMs.84 Furthermore, limited availability of α-ketoglutarate inhibits M2 polarization by depleting glutamine during epigenetic reprogramming.85
Studies have shown that in environments with high levels of histidine metabolites, the population of macrophages with anti-tumor capabilities—including those associated with heat shock proteins (LA-APO⁺) and those linked to inflammatory responses (Inflam-AREG⁺)—is significantly reduced. In stark contrast, the population of macrophages lacking anti-tumor capabilities or exhibiting immunosuppressive characteristics—such as LA-APO⁺ and tissue-resident RTM-MARCO⁺ macrophages—dramatically increases.86 Enhanced histidine catabolic function increases the transcription of genes associated with various metabolic pathways in macrophages. Simultaneously, the anti-tumor activity of macrophages is inhibited, as evidenced by regulatory changes in functional processes, including cellular signaling, signal propagation, growth, cytokine synthesis, and cytokine receptor-mediated responses. The specific metabolic mechanisms of macrophage capabilities such as (Figure 3).
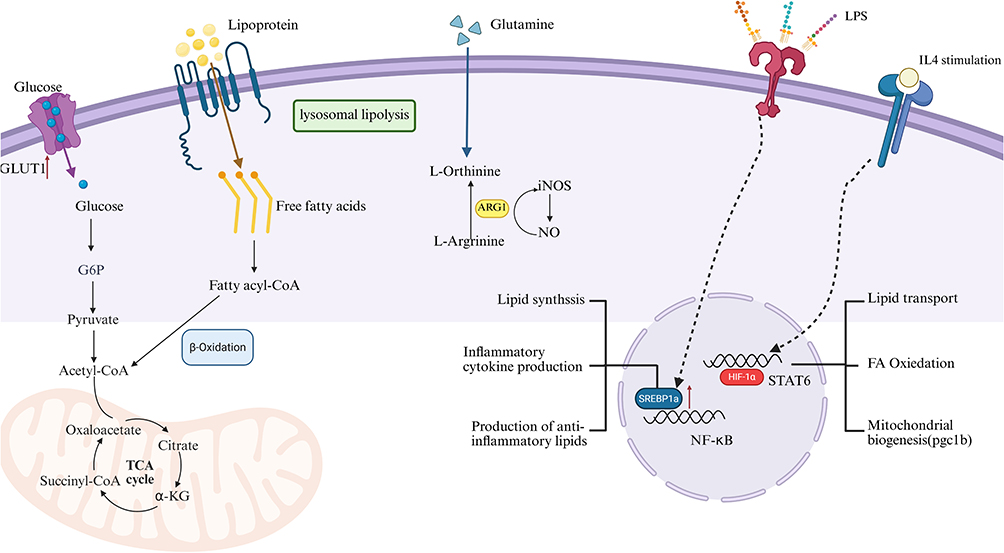 |
Figure 3 This figure illustrates the key pathways of macrophage metabolism and functional regulation, covering two major dimensions: nutrient metabolism and signal pathway activation. The left side shows the process where glucose, after being transported by GLUT1, participates in glycolysis and the TCA cycle. Meanwhile, lipoproteins generate free fatty acids through lysosomal lipolysis, which then participate in β-oxidation. Glutamine metabolism produces L-ornithine and L-arginine; the latter, catalyzed by ARG1 or iNOS, respectively participates in related metabolism or generates NO. The right side shows that under LPS stimulation and IL4 induction, signaling molecules such as SREBP1, NF-κB, STAT6, and PPAR-γ are activated, regulating lipid synthesis, pro-inflammatory cytokine production, anti-inflammatory lipid generation, as well as lipid transport, fatty acid oxidation, mitochondrial biogenesis (mediated by pgc1b, for example) and other processes, ultimately affecting the metabolic phenotype and functional polarization of macrophages. The arrows and symbols in the figure represent different metabolic reactions and signaling pathways. |
Research on targeting macrophage metabolism for cancer treatment is developing rapidly and showing great application prospects and potential. Macrophages play an important role in the tumor microenvironment; they not only participate in immune surveillance but also play a key role in tumor immune escape, metastasis, and the development of drug resistance. In recent years, relevant clinical trials have been conducted (Table 1), which have demonstrated that targeting the metabolic pathways of macrophages can alter their immune response to tumors and enhance the efficacy of tumor immunotherapy.
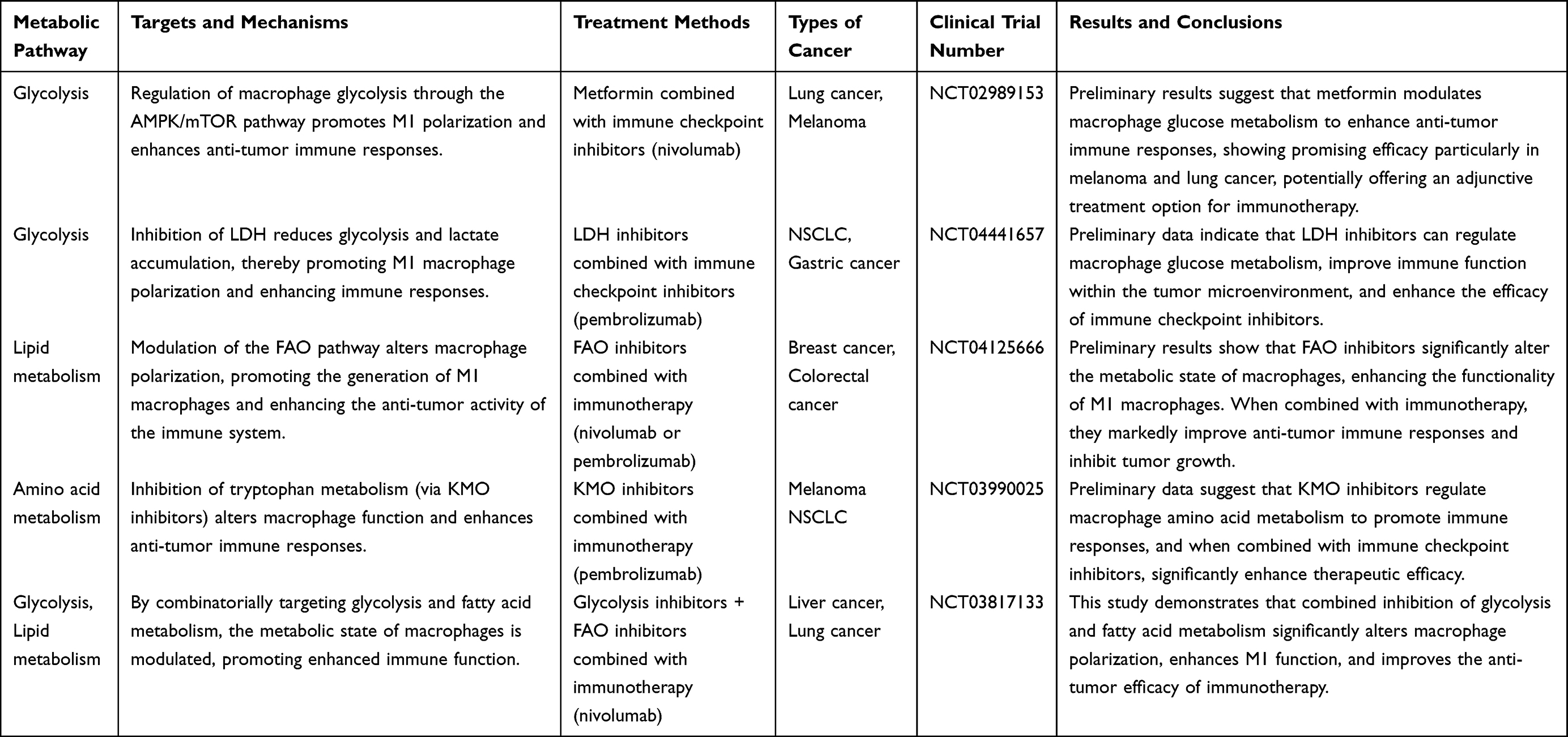 |
Table 1 Clinical Trials Targeting Macrophage Metabolism for Cancer Treatment and Their Targets |
Metabolism of Liver Cancer Cells
Hepatocellular carcinoma cells aggressively spread and grow by relying on their aberrantly remodeled metabolic pathways. Compared to normal hepatocytes, hepatocellular carcinoma cells primarily rely on the “Warburg effect” to favor glycolysis for energy production, utilize glutamine catabolism to maintain redox balance, and increase lipid synthesis to meet the demands of membrane expansion.87 This “metabolic addiction” not only provides a survival advantage but also represents a key target for intervention. In recent years, the inhibition of energy supply through the targeting of metabolic enzymes has emerged as a prominent area of research (Table 2). The shift in focus from “attacking cellular structures” to “disrupting metabolic homeostasis” in intervention strategies targeting the metabolic vulnerabilities of liver cancer provides a more precise direction for treatment. It also fosters the deeper integration of anti-tumor research, from molecular targeting to metabolic regulation.
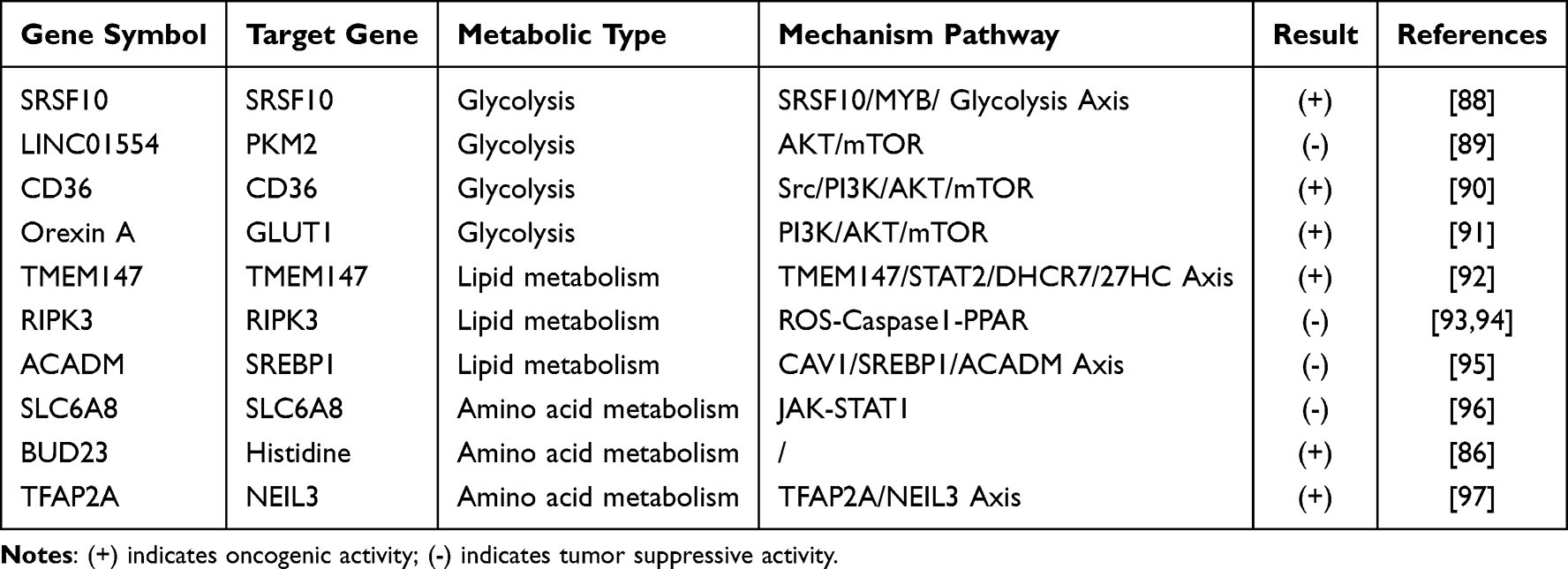 |
Table 2 Overview of Influencing the Progression of Liver Cancer by Targeting Energy Metabolism |
The metabolic characteristics of tumor cells, including lactate accumulation and glucose uptake, not only support their growth but also influence the function of immune cells by modifying the metabolic conditions of the tumor microenvironment. Lactate accumulation specifically regulates the metabolic state of macrophages through the GPR81 receptor and HIF-1α pathway, promoting their polarization toward the M2 phenotype, thereby enhancing the immune escape capacity of tumors. Additionally, competition for metabolic resources occurs between tumor cells and immune cells. Tumor cells preferentially consume glucose and oxygen, resulting in alterations to the metabolism of immune cells and promoting the development of an immunosuppressive microenvironment.42
Glycometabolism of Hepatocellular Carcinoma Cells
During the progression of liver cancer, the typical metabolic functions of hepatocytes, including gluconeogenesis, bile acid (BA) metabolism, detoxification, and urea production (related to ammonia metabolism), gradually diminish. Furthermore, the decline in these functions correlates with the progression of malignant tumors.98,99 The stress adaptation and unlimited proliferation of tumor cells are usually highly dependent on the reprogrammed metabolic state. In contrast to typical cells, which primarily rely on OXPHOS for energy, tumor cells preferentially utilize glycolysis for energy production, even under oxygen-rich conditions. This characteristic is known as the “Warburg effect”.100 Despite the significantly lower energy conversion rate of aerobic glycolysis compared to OXPHOS, its ATP production rate is markedly higher, providing a more efficient energy supply to support the abnormal proliferation of tumor cells.101 Conversely, excess lactic acid produced during aerobic glycolysis can serve as an energy source for surrounding oxygenated cells, thereby fostering a metabolic symbiosis between glycolytic and oxidative metabolism.102 In addition to glucose metabolism, other key metabolic pathways in the TME are frequently reprogrammed, triggering a series of changes, including imbalanced nutrient consumption, accumulation of oncometabolites, and dysregulated signaling pathways.103,104
A hallmark of cancerous metabolism is the tendency towards enhanced aerobic glycolysis. Research indicates that the disruption of the AKT/mTOR signaling cascade leads to a significant reduction in aerobic glycolysis within hepatocellular carcinoma cells, thereby effectively inhibiting their proliferation.105 The development of liver cancer is frequently driven by the activation of the AKT/mTOR signaling pathway, accompanied by increased glycolytic activity during tumor progression. These findings suggest that inhibiting AKT/mTORC1 could be a potential strategy to prevent HCC progression.106 HCC cell lines exhibit an enhanced Warburg effect, which correlates with increased glucose consumption. Notably, miR-873 activates the AKT/mTOR pathway by suppressing NDFIP1, upregulates glycolytic protein expression, and promotes liver cancer progression and metastasis.107 The laminin subunit Lamc1 inhibits the expression of PKM2 and reduces the concentrations of GLUT1 and LDHA, thereby suppressing glucose transport and inhibiting the growth of HCC cells. This confirms that the AKT pathway can promote HCC progression by remodeling glucose metabolism.108 Additionally, as a key downstream molecule of AKT, mTOR activation promotes eIF4E-mediated protein translation by inhibiting eIF4E-binding protein (4EBP1). Consequently, enhancing the integration of GLUT1 into membranes and the function of HK2, thereby reinforcing glucose absorption and metabolic pathways.109 According to researchers Cui et al, pharmacological agents targeting the AKT and mTOR pathways effectively reduce the migratory capabilities of liver cancer cells, while simultaneously decreasing glucose uptake and lactic acid secretion. Moreover, these compounds have been shown to suppress HK2 expression, strongly suggesting that interrupting the AKT/mTOR signaling cascade impedes cellular motility by effectively inhibiting glycolytic processes.110 Reduced AKT activity induces cell cycle arrest, inhibits glycolytic and TCA cycle metabolism, and consequently suppresses tumor progression.111
Glucose metabolism occurs via two concurrent routes: glycolysis and the pentose phosphate pathway (PPP),112 and the metabolic flux of glycolysis can be shunted to the PPP.113 As a metabolic pathway branching from G-6-P, the first step of glycolysis, the PPP plays a critical role in cancer cell proliferation. Its key enzyme, G6PD, has been shown to be involved in tumorigenesis,114 while activation of the PI3K/AKT signaling pathway enhances G6PD activity, increases cellular metabolic activity, and promotes cancer cell growth.115 Studies have shown that overexpression of G6PD can promote the proliferation of normal hepatocytes,116 while targeted inhibition of G6PD can reduce the growth and survival of HCC cells. Cheng et al demonstrated that the PI3K/AKT signaling pathway interacts closely with PPP metabolism. AKT activation stabilizes G6PD by inhibiting the E3 ligase TRIM21, thereby promoting PPP metabolism. Metabolites from the PPP, in turn, reinforce AKT activation by suppressing its inhibitor PHLDA3, thereby facilitating cancer metabolic reprogramming.117 Furthermore, the extracellular matrix protein Versican V1 enhances the Warburg effect in HCC cells via the EGFR-PI3K-AKT pathway, thereby promoting cell proliferation and metastasis.118 The transmembrane glycoprotein CD36 also promotes mTOR-mediated oncogenic glycolysis by activating the PI3K/AKT signaling axis.89
Lipid Metabolism in Hepatocellular Carcinoma Cells
Fatty acids (FAs) serve as key energy sources for cells and also support cell growth and proliferation. In cancer, elevated levels of lipogenesis have emerged as a distinct metabolic characteristic. This is because lipogenesis can work in synergy with lipolysis to jointly promote the growth, proliferation, and survival of tumors.119 Liver cancer is characterized by two significant metabolic features: regulation of the Warburg effect and upregulation of lipid catabolism.120 Alterations in lipid metabolism significantly contribute to the development of liver cancer, with excessive lipid accumulation not only promoting obesity but also increasing the risk of HCC. Acetyl-CoA is a central metabolic compound intersecting numerous biochemical pathways and processes. This substance plays a dual role in cellular processes: it serves as a building block for essential macromolecules while actively participating in both the breakdown and synthesis of biochemical compounds. Specifically, it is involved in the synthesis of fatty acids, steroids, and various non-steroidal compounds essential for proper cellular function.121,122 Acetyl-CoA is derived from multiple sources and can be generated through various metabolic reactions within cells, including those involving glucose, fatty acids, and amino acids. As a key metabolic molecule, its importance is primarily reflected in its diverse functional roles in various cellular processes.123 Firstly, it participates in mitochondrial TCA cycle activity to facilitate energy production; secondly, it acts as a key cellular component in the cytoplasm, contributing to the synthesis of macromolecules such as FA and sterols; third, it functions as an essential nucleocytoplasmic molecule involved in histone acetylation, thereby activating gene expression.124 Acetyl-CoA traverses mitochondrial and cytosolic compartments utilizing carnitine as a carrier.76 The sources of mitochondrial acetyl-CoA (mAc-CoA) include pyruvate, FAO, and acetic acid, while cytosolic acetyl-CoA (cAc-CoA) is generated from acetic acid, citric acid, and ketone bodies.125,126
Lipid metabolism plays a crucial role in tumorigenesis by serving as an essential energy source, supporting cellular proliferation, and providing metabolic precursors for tumor cell anabolism.127 Lipids and sterols fuel tumor development as membrane building blocks.128 The liver plays a critical role in energy metabolism, lipid biogenesis, and lipid distribution. Abnormal lipid metabolism is observed in liver cancer cells when activated forms of AKT and Nras are introduced into mouse hepatocytes using a transposon system and hydrodynamic injection.129 LncRNA transcribes the micropeptide SMIM30, which is located in the membranes of the endoplasmic reticulum and mitochondria, while LncRNA LINC00998 promotes liver cancer cell growth and motility.130 Following the activation of the AKT/mTOR signaling cascade triggered by the CD147 transmembrane glycoprotein, a series of events is initiated that leads to the transcription of key genes involved in fatty acid production, such as fatty acid synthase and acetyl-CoA carboxylase. This sequence of actions ultimately facilitates an increase in fatty acid synthesis within hepatocellular carcinoma cells.131,132 The AKT/mTOR/SREBP-1 signaling pathway plays a critical role in the regulation of lipid metabolism in hepatocytes.133 In the HepG2 liver cancer cell line, targeted inhibition of adipogenesis can be achieved by inhibiting AKT.134 The promotion of adipogenesis is mediated by the AKT/mTORC1/S6 pathway through both transcriptional and post-transcriptional mechanisms. These mechanisms involve the inhibition of fatty acid synthase ubiquitination by the USP2a deubiquitinase and the disruption of the degradation complexes of SREBP1 and SREBP2.135
In addition, the inhibition of ATP-citrate lyase, acetyl-CoA carboxylase, fatty acid synthase, stearoyl-CoA desaturase 1, or SREBP-1, all of which are involved in adipogenesis, reduces proliferation and survival, as well as AKT-dependent cell proliferation.136 Celecoxib, a steroidal anti-inflammatory, modulates the COX-2/AKT pathway. By targeting this pathway, it inhibits adipogenesis and effectively suppresses the progression of HCC.137 In the oxygen-deprived tumor microenvironment, the AKT/mTOR signaling pathway becomes hyperactivated, leading to abnormal fat production and lipid storage as liver cancer progresses. This process also drives uncontrolled cell proliferation, enhanced cancer cell vitality, and increased angiogenesis.138 In quiescent, non-dividing liver cells, the NRF2 transcription factor initiates the expression of growth factor genes, specifically those encoding the platelet-derived growth factor receptor C and EGFR ligands such as transforming growth factor alpha and amphiregulin. These growth factors enhance AKT phosphorylation through paracrine signaling, thereby modulating liver glycogen and lipid metabolism.139,140
Amino Acid Metabolism in Hepatocellular Carcinoma Cells
Glutamine is one of the most abundant amino acids in the human body. Immune and rapidly proliferating cancer cells have significantly higher glutamine demands.141 To sustain bioenergetic metabolism and anabolic processes, cancer cells enhance the metabolic pathways of glucose and glutamine. Significant external carbon sources are directed toward specific metabolic pathways, facilitating the synthesis of key molecules, including DNA, proteins, and lipids, essential for the proliferation of cancer cells.142 Cells promote tumor growth by utilizing glutamine to activate the PI3K/AKT signaling pathway.131 Glutamine activates mTORC1 through the mTOR signaling pathway, which can occur via either a Rag GTPase-dependent mechanism or an independent mechanism.143 Liver cancer’s reliance on glutamine metabolism presents a viable therapeutic target for its treatment.144
GLS1, a key enzyme in glutamine catabolism, is highly expressed in HCC compared to normal liver tissue. Furthermore, GLS1 correlates with clinical and pathological characteristics as well as poor outcomes in individuals with HCC. The underlying mechanism likely involves the activation of the AKT signaling cascade by GLS1, which promotes HCC cell proliferation.145 In hepatocellular carcinoma tumorigenesis, glutamine metabolism promotes the activation of the positive feedback loop in the mTORC2/AKT/C-Myc axis. As a result, the expression of glutamine synthetase (GS) increases, and mTORC1 signaling is enhanced. This series of changes ultimately leads to the release of the effect of sirtuin 4 (SIRT4) on glutamate dehydrogenase (GDH).146 In tumor cells, the PI3K/AKT pathway mediates the N-Myc downstream-regulated gene (NDRG2)-dependent inhibition of c-Myc, which subsequently suppresses the process of glutaminolysis.147
Saha et al found in their study that tumors presenting elevated basal PI3K/AKT activity would likely respond favorably to amino acid deficiency.148 Under amino acid deprivation conditions, normal cells are induced to enter a quiescent state, a process that enhances the sensitivity of proliferating cancer cells to tumor-targeted therapies. Meanwhile, the utilization of glutamine is crucial; when glutamine concentration is elevated, it may promote the production of other metabolites, including liver growth-promoting amino acids.149 Furthermore, following the knockout of NAD(P)H dehydrogenase 1 (Nqo1), the activated PI3K/AKT pathway not only downregulates the expression of glutaminolysis-related genes but also drives hepatocytes to adapt their metabolic processes. Conversely, Nqo1 upregulation hyperactivates PI3K/AKT signaling, thereby accelerating metabolic adaptation.150 The specific metabolic mechanism of liver cancer cells is shown in the Figure 4.
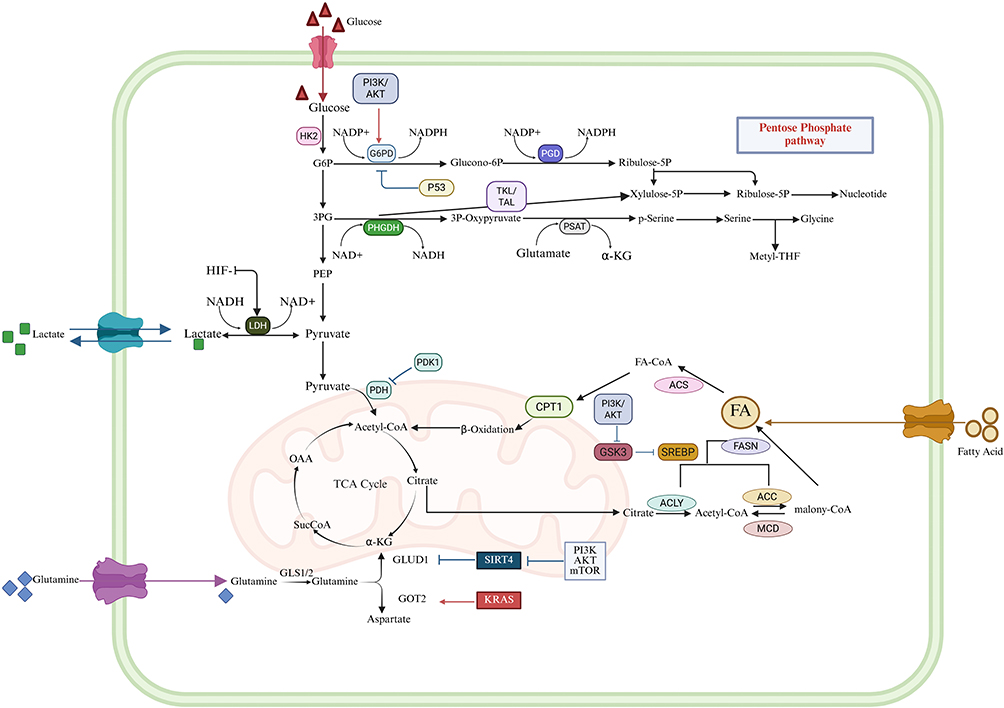 |
Figure 4 Energy metabolism network of hepatocellular carcinoma cells and their regulatory mechanisms: After glucose enters the cell, part of it undergoes glycolysis to generate pyruvate. Under the regulation of HIF-1 and other factors, pyruvate can be converted into lactic acid by the action of LDH and then excreted from the cell; another part enters the pentose phosphate pathway to generate NADPH and other substances, which provide reducing power and raw materials for biosynthesis and also participate in the synthesis of nucleotides and the like. Glutamine, through the action of glutaminase (GLS1/2) and other enzymes, participates in processes related to the tricarboxylic acid cycle, providing intermediates such as α-ketoglutarate (α-KG) for the TCA cycle, and can also generate aspartic acid and the like. In addition, fatty acids can generate Acetyl - CoA through β-oxidation to enter the TCA cycle, and can also undergo anabolic metabolism under the action of a series of enzymes (such as FASN, etc.). Multiple signaling pathways such as PI3K/AKT and mTOR are involved in regulating these metabolic processes. Meanwhile, molecules like p53, SIRT4, and KRAS also play a role in metabolic regulation, collectively maintaining cellular metabolic homeostasis. These metabolic processes are closely related to physiological functions such as cell growth and proliferation, and they also hold significant importance in metabolic reprogramming in diseases like tumors. The arrows in the figure indicate the transformation and flow direction between metabolites, while the symbols represent different enzymes, pathways, and regulatory factors. Red arrows represent pro-tumorigenic pathways, while blue arrows indicate anti-tumorigenic pathways. |
The Role of Macrophage Polarization and Changes in Energy Metabolism in Anti-Hepatocellular Carcinoma
The liver, long thought of as immune-privileged, houses a vast majority of the body’s macrophages 90% to be exact. This diverse group includes liver-specific macrophages, as well as a variety of infiltrating cells, such as the persistent Kupffer cells and transient macrophages that are recruited as needed.151,152 Kupffer cells, typically found lining the sinusoidal blood vessels of the liver, originate from two distinct sources: specialized progenitor cells derived from the yolk sac, which establish themselves in the liver during embryonic development, and monocytes from the bone marrow, which are recruited and transformed to maintain the Kupffer cell population.153,154
In the normal liver, Kupffer cells are the predominant component of liver macrophages, comprising a significant proportion of this population and playing a central role. They primarily maintain liver homeostasis by regulating various physiological processes.155 When external factors invade the liver and induce pathological changes, Kupffer cells are the first to detect these signals and initiate phenotypic differentiation, producing either pro-inflammatory or anti-inflammatory factors. Simultaneously, a wide range of additional macrophages are recruited to the liver. These macrophages exhibit versatility and multifaceted roles similar to those of Kupffer cells, playing a critical role in liver disease pathogenesis.156
The Role of Glucose Metabolism Between Macrophages and Liver Cancer Cells
Metabolic pathways in TAMs serve as a mechanism for modulating macrophage polarization, with M1 utilizing glycolysis and M2 primarily depending on OXPHOS.131,157 Enhancing glucose absorption and redirecting metabolic pathways from oxidative phosphorylation to glycolysis effectively reverses macrophage polarization. Research indicates ACCS modifies immune function via macrophages158 and can also affect biological functions related to metabolism.159
Under normal physiological conditions, macrophages use OXPHOS as their main metabolic pathway to meet energy demands.160 However, when macrophages are stimulated by external pathogens, cytokines (such as LPS, IFN-γ, TNF-α, IL-1, IL-4, IL-10), and tumor metabolites, polarization and metabolic reprogramming occur.161 The metabolic profile of M1 macrophages undergoes a shift from glucose-fueled oxidative phosphorylation to glycolysis. This transition is accompanied by increased lactic acid secretion, reduced oxygen consumption, and enhanced glutaminolysis pathways.70 In pro-inflammatory macrophages, TCA cycle impairment results in succinate and citrate buildup.162 The accumulation of these substances effectively inhibits prolyl hydroxylase, a key enzyme that not only regulates HIF-1α but also promotes IL-1β production. This cascade subsequently accelerates glycolytic metabolism, further amplifying the inflammatory response.163
The metabolic profiles of M2 and M1 macrophages exhibit distinct differences; they have a complete TCA cycle. Activated M2 macrophages not only show a significant increase in oxygen consumption but also primarily rely on oxidative phosphorylation and fatty acid oxidation for metabolic energy production.72 M2 macrophage differentiation similarly relies on glutamine. Glutamine plays a multifaceted role in cellular metabolism, serving not only as an intermediate in the TCA cycle but also as a precursor for lipid biosynthesis. Furthermore, this versatile amino acid serves as a primary nitrogen donor for the synthesis of non-essential amino acids and nucleic acids when required by the cell.164 T Macrophages undergo metabolic reprogramming, with the core trigger being changes in the TME. Additionally, M1 and M2 macrophages can undergo bidirectional conversion through the regulation of lactic acid and specific cytokines. Within the TME, a competition occurs between liver cancer cells and macrophages, both competing for a limited supply of glucose. To avoid the consequences of this intense competition, M2-like macrophages reduce their glucose metabolism, which results in elevated glucose levels within the TME.165 Hence, cancerous cells exponentially increase glucose uptake for enhanced energy and proliferation.
Lactic acid, produced by glycolysis, plays a crucial role in signaling within the tumor microenvironment. It induces the polarization of TAMs, promoting the expression of VEGF and driving their differentiation into an M2-like phenotype, thereby influencing cancer progression.166
In studies on the source of VEGF, Folkman’s pioneering work has confirmed the decisive significance of neovascularization for tumor growth. The traditional view generally holds that, in growing tumors, hypoxic cancer cells are the primary source of VEGF. However, research by Oscar et al presents a new perspective: in certain tumor contexts, tumor-associated cells, particularly macrophages, are the primary producers of VEGF.167 In addition, TAM can also participate in the homeostasis of the tumor microenvironment by regulating nitrogen metabolism: on the one hand, the expression levels of all urea cycle-related enzymes in TAM are higher than those in other tumor cells; on the other hand, the expression levels of its transaminase GPT and GLU1 are also significantly higher, and these two types of enzymes together promote the nitrogen metabolism process.168
The Role of Lipid Metabolism Between Macrophages and Hepatocellular Carcinoma Cells
Under oxygen-rich conditions, FAO generates more ATP through the TCA cycle than glucose oxidation, making it a critical energy source for cellular growth and function.169 Cellular lipid accumulation significantly enhances liver cancer cell proliferation, thereby accelerating the progression of HCC. Notably, enzymes involved in the FAO pathway exhibit tumor-suppressive properties in HCC.170 Enhancing β-oxidation reduces free fatty acid levels in HCC cells, offering a novel therapeutic approach for HCC. M2-like macrophages have different metabolic characteristics from M1-like macrophages. Upon activation by M2-like inducers, such as IL-4, IL-10, and IL-13, macrophages exhibit a marked increase in FAO and OXPHOS, accompanied by a reduced glycolysis rate.171 The PGC1β signaling pathway regulates these metabolic alterations.172 In contrast, FAO is significantly reduced in the M1-like macrophage phenotype. Furthermore, nuclear receptors such as PPAR and LXR regulate lipid metabolic pathways,173 thereby influencing macrophage polarization. In general, M2-like macrophages exhibit reduced glycolysis, increased fatty acid oxidation, and enhanced oxidative phosphorylation.
Furthermore, previous studies have shown that SIRT4 is involved in lipid metabolic processes. When SIRT4 is knocked out, the expression of FAO-related genes, including peroxisome proliferator-activated receptor δ (PPARδ), is upregulated in TAMs.174 When SIRT4 is knocked down in TAMs, the expression of p-STAT3 protein is significantly increased, a change that is critical for the polarization of the M2 phenotype.175 PPAR 8 and PPAR 8-STAT3 axis drive M2-like macrophage differentiation. Conversely, inhibition of PPAR 8 reverses the SIRT4 knockdown-induced M2-like polarization.176 Upon SIRT4 knockdown, the apoptosis of M1-type TAMs is enhanced, resulting in an increased M2-to-M1 ratio in TAMs. Consequently, SIRT4 suppression in macrophages drives M2-like phenotype polarization. The FAO/PPAR/STAT3 pathway elevates the M2/M1 ratio, facilitating HCC advancement. The complex metabolic functions of hepatic macrophages play a critical role in HCC progression and immune modulation. Targeting macrophage metabolism to counteract immunosuppression holds significant potential as a therapeutic strategy for HCC.
OXCT1 serves as a pivotal enzyme in the ketolysis pathway, and its expression level in mature hepatocytes is low. OXCT1 plays a pivotal role in hepatocellular carcinoma cells, particularly under nutrient-limited conditions. The upregulation of this expression enhances the survival of cancer cells, enabling them to withstand stress and continue proliferating.177 Studies have demonstrated that the ketogenic gene OXCT1 is highly expressed in tumor-associated macrophages. This elevated expression promotes tumor growth by regulating the reprogramming of these macrophages. For TAMs, both strategic drug interventions and the reduction of OXCT1 gene expression can effectively enhance anti-tumor immunity, inhibit tumor growth, and slow its progression.178
Elevated TMEM147 expression at both mRNA and protein levels in hepatocellular carcinoma correlates with diminished patient survival.91,179 This protein interacts with DHCR7, disrupting cholesterol equilibrium in cells and increasing 27HC concentrations outside HCC cells. Additionally, TMEM147 promotes DHCR7 expression by enhancing the activity of signal transducers and activators of transcription.92 Studies have demonstrated that upregulation of 27HC expression in HCC induces an increase in the expression of glutathione peroxidase 4, thereby conferring resistance to ferroptosis and promoting the proliferation of cancer cells.180 When 27HC produced by liver cancer cells accumulates, it enhances the lipid-processing capabilities of macrophages, activates the peroxisome proliferator-activated receptor γ (PPARγ) signaling pathway, which subsequently drives macrophages toward the M2 phenotype, thereby enhancing the ability of hepatocellular carcinoma cells to invade and metastasize.181 TMEM147 enables cells to acquire resistance to ferroptosis and promotes M2 macrophage polarization. Cancer progression and metastasis largely depend on the body’s ability to maintain cellular cholesterol balance and increase the production of 27HC, thereby creating a favorable environment for tumor development.182
Research has confirmed the critical role of RIPK3 in the necroptotic process. Its expression is significantly reduced in liver cancer-associated macrophages, where this modification not only drives cancer progression but also promotes the accumulation and polarization of M2-type TAMs.183 From a molecular mechanism perspective, the deficiency of RIPK3 in TAMs leads to a reduction in reactive oxygen species (ROS) and significantly inhibits the caspase-1-mediated cleavage of PPARs.94 These outcomes trigger the activation of PPARs, promote fatty acid metabolism, and subsequently induce M2 macrophage polarization within the TME. Additionally, increased RIPK3 expression or inhibited FAO can counteract TAM-mediated immunosuppression and inhibit the progression of HCC. RIPK3 expression is decreased in TAMs from both mouse and human HCC samples. In the absence of RIPK3, the ROS-Caspase-1-PPAR axis enhances FAO in TAMs, promoting their polarization toward the M2 phenotype.184 Co-culturing TAMs with H22 cells induces RIPK3 knockout, leading to increased lipid droplet accumulation in TAMs. RIPK3 initiates the E3 ligase activity within the pyruvate dehydrogenase complex, thereby increasing anaerobic glycolysis,185 indicating that RIPK3 influences glucose processing. As the activation of M1 macrophages primarily depends on aerobic glycolysis, RIPK3 may promote glycolysis in macrophages, thereby facilitating M1 polarization and enhancing inflammation. However, most TAMs are of the M2 phenotype, which experience hypoxia and nutrient deficiency (particularly glucose deficiency) in the TME. As a result, they are compelled to adjust their metabolism by enhancing FAO and glutaminolysis to meet their energy demands.186,187 This suggests that the deficiency of RIPK3 in TAMs simultaneously promotes fatty acid metabolism and the M2-type activation of macrophages through the ROS-caspase1-PPAR pathway. In summary, the deletion of RIPK3 in TAMs reprograms fatty acid metabolism through the ROS-caspase1-PPAR pathway. This signaling pathway plays a crucial role in regulating the accumulation and differentiation of TAMs into M2 macrophages within the TME, thereby promoting the progression of HCC.188
The Role of Amino Acid Metabolism Between Macrophages and Liver Cancer Cells
Histidine metabolism plays a critical role in tumor biology. In liver cancer, dysregulated histidine metabolism likely contributes to oncogenesis and facilitates immune evasion.85 Histidine metabolism primarily occurs through two pathways: in one, histidine is decarboxylated by histidine decarboxylase to produce histamine, which is subsequently metabolized into imidazole acetic acid; in the other, histidine undergoes transamination to form glutamic acid, which plays a role in energy metabolism.189
Histidine metabolites, especially histamine, play a critical role in modulating immune responses and the tumor microenvironment. Liver cancer is known to induce alterations in the immune microenvironment. Histamine influences immune cell activities through its various receptors, including H1, H2, H3, and H4. This modulation affects key immune players, such as T cells, B cells, macrophages, and dendritic cells, ultimately shaping the tumor’s immune landscape.190–192 Furthermore, when histidine is metabolized into byproducts such as uric acid, glutamic acid, and cis-uric acid, these compounds interfere with the immune system, suppressing anti-tumor immunity and allowing cancer cells to proliferate unchecked.193 Histidine metabolism also regulates oxidative stress through metabolites that affect redox conditions, thereby maintaining cellular antioxidant balance.194–196 Hepatocellular carcinoma cells often experience high levels of oxidative stress, a condition that promotes tumor cell viability and growth.197–200 Thus, targeting histidine metabolism or oxidative stress pathways may provide a promising therapeutic strategy for liver cancer management. Histidine metabolism is closely linked to the energy demands of hepatic cancer cells. By producing glutamic acid, it supports the TCA cycle, thereby regulating hepatic cellular energy metabolism.201,202 In liver cancer, metabolic reprogramming increases cellular energy demand,203,204 and histidine metabolic processes influence cancer cell growth and lifespan by modulating energy metabolic pathways.
Liu et al demonstrated that in a high histidine metabolome, the number of hepatocytes is significantly increased, whereas the number of immune cells and stromal cells remains relatively low. Additionally, histidine metabolism-related genes are highly expressed in hepatocytes, moderately expressed in macrophages, and minimally expressed in lymphocytes and stromal cells. These findings suggest that the elevation in histidine metabolism is not only associated with the expansion of liver cancer cells but also correlates with a reduced infiltration of immune cells and stromal cells within the TME.85
In recent years, single-cell RNA sequencing technology (scRNA-seq) has been widely used to depict the heterogeneity of macrophages in the tumor microenvironment. scRNA-seq analysis has identified macrophage subsets with different functional and metabolic states in non-small cell lung cancer (NSCLC), showing the high diversity and regional differences of TAMs in tumor tissues.205 Similarly, in gastric cancer, researchers characterized tens of thousands of single cells and identified multiple macrophage subsets with different functional characteristics, further confirming the phenotypic diversity of macrophages in different locations within tumors.206 In addition, studies have found that the heterogeneity of TAMs is closely related to their specific localization in the microenvironment, suggesting that spatial factors play an important role in shaping their phenotypes.207
Conclusion
Based on the analysis of macrophage polarization and energy metabolism reprogramming in liver cancer, macrophages play a key role in the progression of liver cancer by altering energy metabolism and activating signaling pathways. Specifically, M1-type macrophages are associated with inhibiting the metabolism and signaling pathways of cancer cells, thereby exerting an anti-tumor effect, while M2-type macrophages promote tumor growth by facilitating energy metabolism pathways. Several potential therapeutic targets mentioned in the article include specific markers on the surface of M2 macrophages, as well as key signaling pathway molecules. For example, the polarization of M2-type macrophages is closely related to the FAO pathway. Nuclear receptors such as PPAR-γ and STAT3 regulate lipid metabolism pathways, and these receptors can serve as targets for intervention. Furthermore, the study also pointed out that interventions targeting the SIRT4, RIPK3, and AKT/mTOR signaling pathways can effectively regulate the polarization state of macrophages, improve the immunosuppressive environment of liver cancer, and inhibit tumor progression. Despite notable advances in this research area, several challenges persist. For instance, interspecies variations in immune system function may compromise the efficacy of targeted therapies. The distinct metabolic profiles and functional characteristics of macrophages across species pose significant limitations to the translational applicability of experimental models. Current metabolic detection techniques, such as mass spectrometry and nuclear magnetic resonance (NMR), are hindered by issues related to sensitivity and quantitative precision, making it difficult to capture the subtle metabolic alterations of macrophages within the complex TME. Furthermore, the metabolic state of macrophages is profoundly influenced by the TME, with significant variability across tumor types and individual patients. This variability introduces considerable challenges for personalized therapeutic strategies, particularly when addressing the intricacies and dynamic nature of metabolic pathways within the TME. Clinical translation is further complicated by issues such as drug targeting and the sustainability of therapeutic interventions. The highly dynamic nature of macrophage metabolism necessitates that therapeutic approaches be both highly adaptable and precisely controllable to achieve optimal efficacy. Future research should combine single-cell technology to explore the functional heterogeneity of macrophages in different tumor regions, investigate the dynamic relationship between metabolic and polarization states, provide new directions for immunotherapy of liver cancer, achieve significant breakthroughs in liver cancer treatment, and bring new hope to the majority of liver cancer patients.
Data Sharing Statement
All data analyzed in this study are included in this paper. All data are available from the corresponding author upon reasonable request.
Acknowledgments
We would like to thank BioRender (https://app.biorender.com/) for their help in creating the charts. During the preparation of this work the authors used Doubao in order to improve language. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work is supported by the following funding: Guangxi Key Technologies R&D Program (Guike AB23026058). Guangxi Science and Technology Major Program (No. AA24263028). Guangxi Science and Technology Major Program (Guike AA24011004). The Scientific and Technological Innovation Major Base of Guangxi (No. 2022–36-Z05).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–24. doi:10.3322/caac.21834
2. Rumgay H, Ferlay J, de Martel C, et al. Global, regional and national burden of primary liver cancer by subtype. Eur J Cancer. 2022;161:108–118. doi:10.1016/j.ejca.2021.11.023
3. Yin Y, Feng W, Chen J, et al. Immunosuppressive tumor microenvironment in the progression, metastasis, and therapy of hepatocellular carcinoma: from bench to bedside. Exp Hematol Oncol. 2024;13(1):72. doi:10.1186/s40164-024-00539-x
4. Liu D, Song T. Advances in neoadjuvant therapy for hepatocellular carcinoma. Holistic Integr Oncol. 2025;4(1):37. doi:10.1007/s44178-025-00173-5
5. Yu SJ. Immunotherapy for hepatocellular carcinoma: recent advances and future targets. Pharmacol Ther. 2023;244:108387. doi:10.1016/j.pharmthera.2023.108387
6. Feng X, Li X, Zhang X. Combining immunogenic cell death and cuproptosis to construct a prognostic signature and predict the immune status and treatment efficacy for hepatocellular carcinoma. Holistic Integr Oncol. 2024;3(1):65. doi:10.1007/s44178-024-00143-3
7. Chen S, Du Y, Xu B, et al. Vaccinia-related kinase 2 blunts sorafenib’s efficacy against hepatocellular carcinoma by disturbing the apoptosis-autophagy balance. Oncogene. 2021;40(19):3378–3393. doi:10.1038/s41388-021-01780-y
8. Zhang W, Hong X, Xiao Y, Wang H, Zeng X. Sorafenib resistance and therapeutic strategies in hepatocellular carcinoma. Biochim Biophys Acta Rev Cancer. 2025;1880(3):189310. doi:10.1016/j.bbcan.2025.189310
9. Wu S, Zhao S, Hai L, et al. Macrophage polarization regulates the pathogenesis and progression of autoimmune diseases. Autoimmun Rev. 2025;24(7):103820. doi:10.1016/j.autrev.2025.103820
10. Zheng H, Peng X, Yang S, et al. Targeting tumor-associated macrophages in hepatocellular carcinoma: biology, strategy, and immunotherapy. Cell Death Discov. 2023;9(1):65. doi:10.1038/s41420-023-01356-7
11. Tong T, Gao W, Jian H, et al. The role and potential mechanisms of exosomes in the progression of hepatocellular carcinoma. Holistic Integr Oncol. 2025;4(1). doi:10.1007/s44178-025-00171-7
12. Li D, Zhang T, Guo Y, Bi C, Liu M, Wang G. Biological impact and therapeutic implication of tumor-associated macrophages in hepatocellular carcinoma. Cell Death Dis. 2024;15(7):498. doi:10.1038/s41419-024-06888-z
13. Nong S, Han X, Xiang Y, et al. Metabolic reprogramming in cancer: mechanisms and therapeutics. MedComm. 2023;4(2):e218. doi:10.1002/mco2.218
14. Song Q, Wang H, Bao J, et al. Systems biology approach to studying proliferation-dependent prognostic subnetworks in breast cancer. Sci Rep. 2015;5(1):12981. doi:10.1038/srep12981
15. Hu Y, Liu W, Fang W, Dong Y, Zhang H, Luo Q. Tumor energy metabolism: implications for therapeutic targets. Mol Biomed. 2024;5(1):63. doi:10.1186/s43556-024-00229-4
16. Zeng W, Li F, Jin S, Ho PC, Liu PS, Xie X. Functional polarization of tumor-associated macrophages dictated by metabolic reprogramming. J Exp Clin Cancer Res. 2023;42(1):245. doi:10.1186/s13046-023-02832-9
17. Glaviano A, Foo A, Lam HY, et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. 2023;22(1):138. doi:10.1186/s12943-023-01827-6
18. Lioulia E, Mokos P, Panteris E, Dafou D. UBE2T promotes β-catenin nuclear translocation in hepatocellular carcinoma through MAPK/ERK-dependent activation. Mol Oncol. 2022;16(8):1694–1713. doi:10.1002/1878-0261.13111
19. Zheng W, Wang Y, Sun H, Bao S, Ge S, Quan C. The role of Fusobacterium nucleatum in macrophage M2 polarization and NF-κB pathway activation in colorectal cancer. Front Immunol. 2025;16:1549564. doi:10.3389/fimmu.2025.1549564
20. Cox N, Pokrovskii M, Vicario R, Geissmann F. Origins, biology, and diseases of tissue macrophages. Annu Rev Immunol. 2021;39(1):313–344. doi:10.1146/annurev-immunol-093019-111748
21. Chi Y, Jiang H, Yin Y, et al. Macrophage signaling pathways in health and disease: from bench to bedside applications. MedComm. 2025;6(7):e70256. doi:10.1002/mco2.70256
22. Zhang Y, Liu H, Niu M, et al. Roles of long noncoding RNAs in human inflammatory diseases. Cell Death Discov. 2024;10(1):235. doi:10.1038/s41420-024-02002-6
23. Bai X, Guo YR, Zhao ZM, et al. Macrophage polarization in cancer and beyond: from inflammatory signaling pathways to potential therapeutic strategies. Cancer Lett. 2025;625:217772. doi:10.1016/j.canlet.2025.217772
24. Chaterjee O, Sur D. Artificially induced in situ macrophage polarization: an emerging cellular therapy for immuno-inflammatory diseases. Eur J Pharmacol. 2023;957:176006. doi:10.1016/j.ejphar.2023.176006
25. Cutolo M, Soldano S, Smith V, Gotelli E, Hysa E. Dynamic macrophage phenotypes in autoimmune and inflammatory rheumatic diseases. Nat Rev Rheumatol. 2025;21(9):546–565. doi:10.1038/s41584-025-01279-w
26. Cutolo M, Campitiello R, Gotelli E, Soldano S. The role of M1/M2 macrophage polarization in rheumatoid arthritis synovitis. Front Immunol. 2022;13:867260. doi:10.3389/fimmu.2022.867260
27. Monnier M, Paolini L, Vinatier E, Mantovani A, Delneste Y, Jeannin P. Antitumor strategies targeting macrophages: the importance of considering the differences in differentiation/polarization processes between human and mouse macrophages. J Immunother Cancer. 2022;10(10):e005560. doi:10.1136/jitc-2022-005560
28. Cheng Z, Zhou YZ, Wu Y, et al. Diverse roles of macrophage polarization in aortic aneurysm: destruction and repair. J Transl Med. 2018;16(1):354. doi:10.1186/s12967-018-1731-0
29. Guo Q, Zhu X, Wei R, et al. miR-130b-3p regulates M1 macrophage polarization via targeting IRF1. J Cell Physiol. 2021;236(3):2008–2022. doi:10.1002/jcp.29987
30. Meier P, Legrand AJ, Adam D, Silke J. Immunogenic cell death in cancer: targeting necroptosis to induce antitumour immunity. Nat Rev Cancer. 2024;24(5):299–315. doi:10.1038/s41568-024-00674-x
31. Han J, Wu M, Liu Z. Dysregulation in IFN-γ signaling and response: the barricade to tumor immunotherapy. Front Immunol. 2023;14:1190333. doi:10.3389/fimmu.2023.1190333
32. Chen S, Saeed A, Liu Q, et al. Macrophages in immunoregulation and therapeutics. Signal Transduct Target Ther. 2023;8(1):207. doi:10.1038/s41392-023-01452-1
33. Wang T, Wang S, Jia X, et al. Baicalein alleviates cardiomyocyte death in EAM mice by inhibiting the JAK-STAT1/4 signalling pathway. Phytomedicine. 2024;128:155558. doi:10.1016/j.phymed.2024.155558
34. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. doi:10.1016/j.immuni.2010.05.007
35. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. 2020;877:173090. doi:10.1016/j.ejphar.2020.173090
36. Hallett RM, Bonfill-Teixidor E, Iurlaro R, et al. Therapeutic targeting of LIF overcomes macrophage-mediated immunosuppression of the local tumor microenvironment. Clin Cancer Res. 2023;29(4):791–804. doi:10.1158/1078-0432.CCR-21-1888
37. Beylerli O, Shi H, Begliarzade S, Shumadalova A, Ilyasova T, Sufianov A. MiRNAs as new potential biomarkers and therapeutic targets in brain metastasis. Noncoding RNA Res. 2024;9(3):678–686. doi:10.1016/j.ncrna.2024.02.014
38. Kashfi K, Kannikal J, Nath N. Macrophage reprogramming and cancer therapeutics: role of iNOS-derived NO. Cells. 2021;10(11):3194. doi:10.3390/cells10113194
39. Chaib M, Tanveer UA, Makowski L. Myeloid cells in the era of cancer immunotherapy: top 3 unanswered questions. Pharmacol Ther. 2023;244:108370. doi:10.1016/j.pharmthera.2023.108370
40. Wu Q, Zhou L, Lv D, Zhu X, Tang H. Exosome-mediated communication in the tumor microenvironment contributes to hepatocellular carcinoma development and progression. J Hematol Oncol. 2019;12(1):53. doi:10.1186/s13045-019-0739-0
41. Polak R, Zhang ET, Kuo CJ. Cancer organoids 2.0: modelling the complexity of the tumour immune microenvironment. Nat Rev Cancer. 2024;24(8):523–539. doi:10.1038/s41568-024-00706-6
42. Arner EN, Rathmell JC. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell. 2023;41(3):421–433. doi:10.1016/j.ccell.2023.01.009
43. Liu Y, Zhao Y, Song H, et al. Metabolic reprogramming in tumor immune microenvironment: impact on immune cell function and therapeutic implications. Cancer Lett. 2024;597:217076. doi:10.1016/j.canlet.2024.217076
44. Wang L, Wang D, Zhang T, Ma Y, Tong X, Fan H. The role of immunometabolism in macrophage polarization and its impact on acute lung injury/acute respiratory distress syndrome. Front Immunol. 2023;14:1117548. doi:10.3389/fimmu.2023.1117548
45. Hao X, Ren Y, Feng M, Wang Q, Wang Y. Metabolic reprogramming due to hypoxia in pancreatic cancer: implications for tumor formation, immunity, and more. Biomed Pharmacother. 2021;141:111798. doi:10.1016/j.biopha.2021.111798
46. Kohl A, Kao KC, Ho PC. Can tumor cells take it all away. Cell Metab. 2021;33(6):1071–1072. doi:10.1016/j.cmet.2021.05.010
47. Bouch RJ, Zhang J, Miller BC, et al. Distinct inflammatory Th17 subsets emerge in autoimmunity and infection. J Exp Med. 2023;220(10). doi:10.1084/jem.20221911
48. Wang R, Zhuang J, Zhang Q, et al. Decoding the metabolic dialogue in the tumor microenvironment: from immune suppression to precision cancer therapies. Exp Hematol Oncol. 2025;14(1):99. doi:10.1186/s40164-025-00689-6
49. Wang J, Mi S, Ding M, Li X, Yuan S. Metabolism and polarization regulation of macrophages in the tumor microenvironment. Cancer Lett. 2022;543:215766. doi:10.1016/j.canlet.2022.215766
50. de Goede KE, Driessen A, Van den Bossche J. Metabolic cancer-macrophage crosstalk in the tumor microenvironment. Biology. 2020;9(11). doi:10.3390/biology9110380
51. Thibaut R, Orliaguet L, Ejlalmanesh T, Venteclef N, Alzaid F. Perspective on direction of control: cellular metabolism and macrophage polarization. Front Immunol. 2022;13:918747. doi:10.3389/fimmu.2022.918747
52. Shi Q, Shen Q, Liu Y, et al. Increased glucose metabolism in TAMs fuels O-GlcNAcylation of lysosomal Cathepsin B to promote cancer metastasis and chemoresistance. Cancer Cell. 2022;40(10):1207–1222.e10. doi:10.1016/j.ccell.2022.08.012
53. Kooshan Z, Cárdenas-Piedra L, Clements J, Batra J. Glycolysis, the sweet appetite of the tumor microenvironment. Cancer Lett. 2024;600:217156. doi:10.1016/j.canlet.2024.217156
54. Van den Bossche J, Baardman J, Otto NA, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 2016;17(3):684–696. doi:10.1016/j.celrep.2016.09.008
55. Liu J, Cao X. Glucose metabolism of TAMs in tumor chemoresistance and metastasis. Trends Cell Biol. 2023;33(11):967–978. doi:10.1016/j.tcb.2023.03.008
56. Rodrigues Mantuano N, Stanczak MA, Oliveira IA, et al. Hyperglycemia enhances cancer immune evasion by inducing alternative macrophage polarization through increased O-GlcNAcylation. Cancer Immunol Res. 2020;8(10):1262–1272. doi:10.1158/2326-6066.CIR-19-0904
57. Park JH, Pyun WY, Park HW. Cancer metabolism: phenotype, signaling and therapeutic targets. Cells. 2020;9(10):2308. doi:10.3390/cells9102308
58. Li M, Yang Y, Xiong L, Jiang P, Wang J, Li C. Metabolism, metabolites, and macrophages in cancer. J Hematol Oncol. 2023;16(1):80. doi:10.1186/s13045-023-01478-6
59. Shi Z, Hu C, Zheng X, Sun C, Li Q. Feedback loop between hypoxia and energy metabolic reprogramming aggravates the radioresistance of cancer cells. Exp Hematol Oncol. 2024;13(1):55. doi:10.1186/s40164-024-00519-1
60. Du Q, Dickinson A, Nakuleswaran P, et al. Targeting macrophage polarization for reinstating homeostasis following tissue damage. Int J Mol Sci. 2024;25(13):7278. doi:10.3390/ijms25137278
61. Zhang X, Ji L, Li MO. Control of tumor-associated macrophage responses by nutrient acquisition and metabolism. Immunity. 2023;56(1):14–31. doi:10.1016/j.immuni.2022.12.003
62. Fang X, Zhao P, Gao S, et al. Lactate induces tumor-associated macrophage polarization independent of mitochondrial pyruvate carrier-mediated metabolism. Int J Biol Macromol. 2023;237:123810. doi:10.1016/j.ijbiomac.2023.123810
63. Ajam-Hosseini M, Heydari R, Rasouli M, et al. Lactic acid in macrophage polarization: a factor in carcinogenesis and a promising target for cancer therapy. Biochem Pharmacol. 2024;222:116098. doi:10.1016/j.bcp.2024.116098
64. Zhang C, Cheng W, Yang T, Fang H, Zhang R. Lactate secreted by esophageal cancer cells induces M2 macrophage polarization via the AKT/ERK pathway. Thorac Cancer. 2023;14(22):2139–2148. doi:10.1111/1759-7714.14998
65. Mortezaee K, Majidpoor J. Dysregulated metabolism: a friend-to-foe skewer of macrophages. Int Rev Immunol. 2023;42(4):287–303. doi:10.1080/08830185.2022.2095374
66. Keeney JN, Winters AD, Sitcheran R, West AP. NF-κB–inducing kinase governs the mitochondrial respiratory capacity, differentiation, and inflammatory status of innate immune cells. J Immunol. 2023;210(8):1123–1133. doi:10.4049/jimmunol.2200596
67. Li Y, Pan Y, Zhao X, et al. Peroxisome proliferator-activated receptors: a key link between lipid metabolism and cancer progression. Clin Nutr. 2024;43(2):332–345. doi:10.1016/j.clnu.2023.12.005
68. Sun X, Gao S, Chang R, et al. Fatty acids promote M1 polarization of monocyte-derived macrophages in healthy or ketotic dairy cows and a bovine macrophage cell line by impairing mTOR-mediated autophagy. J Dairy Sci. 2024;107(9):7423–7434. doi:10.3168/jds.2023-24357
69. Batista-Gonzalez A, Vidal R, Criollo A, Carreño LJ. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front Immunol. 2019;10:2993. doi:10.3389/fimmu.2019.02993
70. Xiao Y, Yang Y, Xiong H, Dong G. The implications of FASN in immune cell biology and related diseases. Cell Death Dis. 2024;15(1):88. doi:10.1038/s41419-024-06463-6
71. Meng T, He D, Han Z, et al. Nanomaterial-based repurposing of macrophage metabolism and its applications. Nanomicro Lett. 2024;16(1):246. doi:10.1007/s40820-024-01455-9
72. Lunj S, Smith T, Reeves KJ, et al. Immune effects of α and β radionuclides in metastatic prostate cancer. Nat Rev Urol. 2024;21(11):651–661. doi:10.1038/s41585-024-00924-5
73. Zaid A, Ariel A. Harnessing anti-inflammatory pathways and macrophage nano delivery to treat inflammatory and fibrotic disorders. Adv Drug Deliv Rev. 2024;207:115204. doi:10.1016/j.addr.2024.115204
74. Yang S, Li Y, Zheng X, et al. Effects of folate-chicory acid liposome on macrophage polarization and TLR4/NF-κB signaling pathway in ulcerative colitis mouse. Phytomedicine. 2024;128:155415. doi:10.1016/j.phymed.2024.155415
75. Liao J, Xie X, Wang N, et al. Formononetin promotes fatty acid β-oxidation to treat non-alcoholic steatohepatitis through SIRT1/PGC-1α/PPARα pathway. Phytomedicine. 2024;124:155285. doi:10.1016/j.phymed.2023.155285
76. Kruglov V, Jang IH, Camell CD. Inflammaging and fatty acid oxidation in monocytes and macrophages. Immunometabolism. 2024;6(1):e00038. doi:10.1097/IN9.0000000000000038
77. Zhang S, Lv K, Liu Z, Zhao R, Li F. Fatty acid metabolism of immune cells: a new target of tumour immunotherapy. Cell Death Discov. 2024;10(1):39. doi:10.1038/s41420-024-01807-9
78. Vats D, Mukundan L, Odegaard JI, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4(1):13–24. doi:10.1016/j.cmet.2006.05.011
79. Nomura M, Liu J, Rovira II, et al. Fatty acid oxidation in macrophage polarization. Nat Immunol. 2016;17(3):216–217. doi:10.1038/ni.3366
80. Huang SC, Everts B, Ivanova Y, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15(9):846–855. doi:10.1038/ni.2956
81. Zhu Y, Chen X, Lu Y, et al. Glutamine mitigates murine burn sepsis by supporting macrophage M2 polarization through repressing the SIRT5-mediated desuccinylation of pyruvate dehydrogenase. Burns Trauma. 2022;10:tkac041. doi:10.1093/burnst/tkac041
82. Chen S, Zeng J, Li R, et al. Traditional Chinese medicine in regulating macrophage polarization in immune response of inflammatory diseases. J Ethnopharmacol. 2024;325:117838. doi:10.1016/j.jep.2024.117838
83. Palmieri EM, Menga A, Martín-Pérez R, et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep. 2017;20(7):1654–1666. doi:10.1016/j.celrep.2017.07.054
84. Xu Y, Yu Z, Fu H, Guo Y, Hu P, Shi J. Dual inhibitions on glucose/glutamine metabolisms for nontoxic pancreatic cancer therapy. ACS Appl Mater Interfaces. 2022;14(19):21836–21847. doi:10.1021/acsami.2c00111
85. Liu S, Yang J, Wu Z. The regulatory role of α-ketoglutarate metabolism in macrophages. Mediators Inflamm. 2021;2021(1):5577577. doi:10.1155/2021/5577577
86. Liu P, Huang F, Lin P, et al. Histidine metabolism drives liver cancer progression via immune microenvironment modulation through metabolic reprogramming. J Transl Med. 2025;23(1):262. doi:10.1186/s12967-025-06267-y
87. Schiliro C, Firestein BL. Mechanisms of metabolic reprogramming in cancer cells supporting enhanced growth and proliferation. Cells. 2021;10(5):1056. doi:10.3390/cells10051056
88. Cai J, Song L, Zhang F, et al. Targeting SRSF10 might inhibit M2 macrophage polarization and potentiate anti-PD-1 therapy in hepatocellular carcinoma. Cancer Commun. 2024;44(11):1231–1260. doi:10.1002/cac2.12607
89. Zheng YL, Li L, Jia YX, et al. LINC01554-mediated glucose metabolism reprogramming suppresses tumorigenicity in hepatocellular carcinoma via downregulating PKM2 expression and inhibiting Akt/mTOR signaling pathway. Theranostics. 2019;9(3):796–810. doi:10.7150/thno.28992
90. Luo X, Zheng E, Wei L, et al. The fatty acid receptor CD36 promotes HCC progression through activating Src/PI3K/AKT axis-dependent aerobic glycolysis. Cell Death Dis. 2021;12(4):328. doi:10.1038/s41419-021-03596-w
91. Liu Y, Zhao Y, Guo L. Effects of orexin A on glucose metabolism in human hepatocellular carcinoma in vitro via PI3K/Akt/mTOR-dependent and -independent mechanism. Mol Cell Endocrinol. 2016;420:208–216. doi:10.1016/j.mce.2015.11.002
92. Cheng S, Li J, Xu M, et al. TMEM147 correlates with immune infiltration and serve as a potential prognostic biomarker in hepatocellular carcinoma. Anal Cell Pathol. 2023;2023(1):4413049. doi:10.1155/2023/4413049
93. Wu L, Zhang X, Zheng L, et al. RIPK3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol Res. 2020;8(5):710–721. doi:10.1158/2326-6066.CIR-19-0261
94. Afonso MB, Islam T, Magusto J, et al. RIPK3 dampens mitochondrial bioenergetics and lipid droplet dynamics in metabolic liver disease. Hepatology. 2023;77(4):1319–1334. doi:10.1002/hep.32756
95. Ma A, Yeung C, Tey SK, et al. Suppression of ACADM-mediated fatty acid oxidation promotes hepatocellular carcinoma via aberrant CAV1/SREBP1 signaling. Cancer Res. 2021;81(13):3679–3692. doi:10.1158/0008-5472.CAN-20-3944
96. Ji L, Zhao X, Zhang B, et al. Slc6a8-mediated creatine uptake and accumulation reprogram macrophage polarization via regulating cytokine responses. Immunity. 2019;51(2):272–284.e7. doi:10.1016/j.immuni.2019.06.007
97. Zhang F, Wang B, Zhang W, Xu Y, Zhang C, Xue X. NEIL3 upregulated by TFAP2A promotes M2 polarization of macrophages in liver cancer via the mediation of glutamine metabolism. Digestion. 2025;106(1):30–44. doi:10.1159/000540804
98. Park S, Hall MN. Metabolic reprogramming in hepatocellular carcinoma: mechanisms and therapeutic implications. Exp Mol Med. 2025;57(3):515–523. doi:10.1038/s12276-025-01415-2
99. Wang X, Klaassen CD, Chen X, Zhang Y. Pathological and therapeutic roles of bile acid metabolism and signaling in hepatocellular carcinoma: insights from human and mouse studies. Pharmacol Rev. 2025;77(5):100073. doi:10.1016/j.pharmr.2025.100073
100. Mitaishvili E, Feinsod H, David Z, et al. The molecular mechanisms behind advanced breast cancer metabolism: warburg effect, OXPHOS, and calcium. Front Biosci. 2024;29(3):99. doi:10.31083/j.fbl2903099
101. Xia L, Oyang L, Lin J, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. 2021;20(1):28. doi:10.1186/s12943-021-01316-8
102. Li X, Yang Y, Zhang B, et al. Lactate metabolism in human health and disease. Signal Transduct Target Ther. 2022;7(1):305.
103. Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17(4):351–359.
104. Chen L, Huang M. Oncometabolites in cancer: from cancer cells to the tumor microenvironment. Holistic Integr Oncol. 2024;3(1):26.
105. Li L, Wang C, Qiu Z, et al. Triptolide inhibits intrahepatic cholangiocarcinoma growth by suppressing glycolysis via the AKT/mTOR pathway. Phytomedicine. 2023;109:154575.
106. Bang J, Jun M, Lee S, Moon H, Ro SW. Targeting EGFR/PI3K/AKT/mTOR signaling in hepatocellular carcinoma. Pharmaceutics. 2023;15(8):2130. doi:10.3390/pharmaceutics15082130
107. Zhang Y, Zhang C, Zhao Q, et al. The miR-873/NDFIP1 axis promotes hepatocellular carcinoma growth and metastasis through the AKT/mTOR-mediated Warburg effect. Am J Cancer Res. 2019;9(5):927–944.
108. Ye G, Qin Y, Wang S, et al. Lamc1 promotes the Warburg effect in hepatocellular carcinoma cells by regulating PKM2 expression through AKT pathway. Cancer Biol Ther. 2019;20(5):711–719. doi:10.1080/15384047.2018.1564558
109. Zheng ZQ, Zhong CR, Wei CZ, et al. Hyperactivation of mTOR/eIF4E signaling pathway promotes the production of tryptophan-to-phenylalanine substitutants in EBV-positive gastric cancer. Adv Sci. 2024;11(35):e2402284.
110. Cui H, Gao Q, Zhang L, Han F, Wang L. Knockdown of FOXK1 suppresses liver cancer cell viability by inhibiting glycolysis. Life Sci. 2018;213:66–73. doi:10.1016/j.lfs.2018.10.018
111. He Y, Sun MM, Zhang GG, et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct Target Ther. 2021;6(1):425.
112. TeSlaa T, Ralser M, Fan J, Rabinowitz JD. The pentose phosphate pathway in health and disease. Nat Metab. 2023;5(8):1275–1289.
113. DeMichele E, Buret AG, Taylor CT. Hypoxia-inducible factor-driven glycolytic adaptations in host-microbe interactions. Pflugers Arch. 2024;476(9):1353–1368. doi:10.1007/s00424-024-02953-w
114. Liu B, Fu X, Du Y, et al. Pan-cancer analysis of G6PD carcinogenesis in human tumors. Carcinogenesis. 2023;44(6):525–534. doi:10.1093/carcin/bgad043
115. Paskeh M, Ghadyani F, Hashemi M, et al. Biological impact and therapeutic perspective of targeting PI3K/Akt signaling in hepatocellular carcinoma: promises and challenges. Pharmacol Res. 2023;187:106553. doi:10.1016/j.phrs.2022.106553
116. Zhang X, Gao F, Ai H, et al. TSP50 promotes hepatocyte proliferation and tumour formation by activating glucose-6-phosphate dehydrogenase (G6PD). Cell Prolif. 2021;54(4):e13015.
117. Cheng J, Huang Y, Zhang X, et al. TRIM21 and PHLDA3 negatively regulate the crosstalk between the PI3K/AKT pathway and PPP metabolism. Nat Commun. 2020;11(1):1880. doi:10.1038/s41467-020-15819-3
118. Zhangyuan G, Wang F, Zhang H, et al. VersicanV1 promotes proliferation and metastasis of hepatocellular carcinoma through the activation of EGFR-PI3K-AKT pathway. Oncogene. 2020;39(6):1213–1230. doi:10.1038/s41388-019-1052-7
119. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. 2020;122(1):4–22.
120. Zhang J, Zhang Z, Wu Z, Wang Y, Zhang Z, Xia L. The switch triggering the invasion process: lipid metabolism in the metastasis of hepatocellular carcinoma. Chin Med J. 2024;137(11):1271–1284. doi:10.1097/CM9.0000000000003144
121. Guertin DA, Wellen KE. Acetyl-CoA metabolism in cancer. Nat Rev Cancer. 2023;23(3):156–172.
122. Chen G, Bao B, Cheng Y, et al. Acetyl-CoA metabolism as a therapeutic target for cancer. Biomed Pharmacother. 2023;168:115741. doi:10.1016/j.biopha.2023.115741
123. Houston R, Sekine S, Calderon MJ, et al. Acetylation-mediated remodeling of the nucleolus regulates cellular acetyl-CoA responses. PLoS Biol. 2020;18(11):e3000981. doi:10.1371/journal.pbio.3000981
124. Liu H, Wang S, Wang J, et al. Energy metabolism in health and diseases. Signal Transduct Target Ther. 2025;10(1):69.
125. Lei C, Guo X, Zhang M, et al. Regulating the metabolic flux of pyruvate dehydrogenase bypass to enhance lipid production in Saccharomyces cerevisiae. Commun Biol. 2024;7(1):1399. doi:10.1038/s42003-024-07103-7
126. Puchalska P, Crawford PA. Metabolic and signaling roles of ketone bodies in health and disease. Annu Rev Nutr. 2021;41(1):49–77. doi:10.1146/annurev-nutr-111120-111518
127. Alannan M, Fayyad-Kazan H, Trézéguet V, Merched A. Targeting lipid metabolism in liver cancer. Biochemistry. 2020;59(41):3951–3964. doi:10.1021/acs.biochem.0c00477
128. Feng T, Zhang H, Zhou Y, et al. Roles of posttranslational modifications in lipid metabolism and cancer progression. Biomark Res. 2024;12(1):141. doi:10.1186/s40364-024-00681-y
129. Cheng YH, Ko YC, Ku HJ, et al. Novel paired cell lines for the study of lipid metabolism and cancer stemness of hepatocellular carcinoma. Front Cell Dev Biol. 2022;10:821224. doi:10.3389/fcell.2022.821224
130. Ge G, Zhao Z, Song Y. The relationship between lncRNA-encoded micropeptides and tumors. Holistic Integr Oncol. 2025;4(1):47. doi:10.1007/s44178-025-00185-1
131. Su WY, Tian LY, Guo LP, Huang LQ, Gao WY. PI3K signaling-regulated metabolic reprogramming: from mechanism to application. Biochim Biophys Acta Rev Cancer. 2023;1878(5):188952. doi:10.1016/j.bbcan.2023.188952
132. Li K, Fan T, Shi Z, Jiang H. CDCA2 promotes HCC cells development via AKT-mTOR pathway. Anal Cell Pathol. 2022;2022:9912254. doi:10.1155/2022/9912254
133. Li Y, Wu S, Zhao X, et al. Key events in cancer: dysregulation of SREBPs. Front Pharmacol. 2023;14:1130747. doi:10.3389/fphar.2023.1130747
134. Williams SG, Wolin SL. The autoantigen repertoire and the microbial RNP world. Trends Mol Med. 2021;27(5):422–435. doi:10.1016/j.molmed.2021.02.003
135. Shi YH, Liu ZD, Ma MJ, et al. Platelet-derived growth factor C facilitates malignant behavior of pancreatic ductal adenocarcinoma by regulating SREBP1 mediated lipid metabolism. Adv Sci. 2024;11(40):e2407069. doi:10.1002/advs.202407069
136. Morrow MR, Batchuluun B, Wu J, et al. Inhibition of ATP-citrate lyase improves NASH, liver fibrosis, and dyslipidemia. Cell Metab. 2022;34(6):919–936.e8. doi:10.1016/j.cmet.2022.05.004
137. Chen H, Qian Z, Zhang S, et al. Silencing COX-2 blocks PDK1/TRAF4-induced AKT activation to inhibit fibrogenesis during skeletal muscle atrophy. Redox Biol. 2021;38:101774. doi:10.1016/j.redox.2020.101774
138. Tian LY, Smit DJ, Jücker M. The role of PI3K/AKT/mTOR signaling in hepatocellular carcinoma metabolism. Int J Mol Sci. 2023;24(3). doi:10.3390/ijms24032652
139. Kineman RD, Del Rio-Moreno M, Waxman DJ. Liver-specific actions of GH and IGF1 that protect against MASLD. Nat Rev Endocrinol. 2025;21(2):105–117. doi:10.1038/s41574-024-01037-0
140. Xia X, Zhang Q, Fang X, et al. Nuclear factor erythroid 2-related factor 2 ameliorates disordered glucose and lipid metabolism in liver: involvement of gasdermin D in regulating pyroptosis. Clin Transl Med. 2025;15(3):e70233. doi:10.1002/ctm2.70233
141. Wang B, Pei J, Xu S, Liu J, Yu J. A glutamine tug-of-war between cancer and immune cells: recent advances in unraveling the ongoing battle. J Exp Clin Cancer Res. 2024;43(1):74.
142. Julià-Sapé M, Candiota AP, Arús C. Cancer metabolism in a snapshot: MRS(I). NMR Biomed. 2019;32(10):e4054. doi:10.1002/nbm.4054
143. Gollwitzer P, Grützmacher N, Wilhelm S, Kümmel D, Demetriades C. A Rag GTPase dimer code defines the regulation of mTORC1 by amino acids. Nat Cell Biol. 2022;24(9):1394–1406. doi:10.1038/s41556-022-00976-y
144. Ziki RA, Colnot S. Glutamine metabolism, a double agent combating or fuelling hepatocellular carcinoma. JHEP Rep. 2024;6(5):101077.
145. Sun Y, Shen Y, Yan Y, Luo W, Liu C, Tang J. GLS1 promotes lipid metabolism in hepatocellular carcinoma by regulating the PI3K/AKT/mTORC1 signaling pathway through SREBP-1. Am J Transl Res. 2025;17(4):2527–2540. doi:10.62347/ZTGP5030
146. Tong Y, Kai J, Wang S, et al. VHL regulates the sensitivity of clear cell renal cell carcinoma to SIRT4-mediated metabolic stress via HIF-1α/HO-1 pathway. Cell Death Dis. 2021;12(7):621. doi:10.1038/s41419-021-03901-7
147. Xue C, Li G, Lu J, Li L. Crosstalk between circRNAs and the PI3K/AKT signaling pathway in cancer progression. Signal Transduct Target Ther. 2021;6(1):400. doi:10.1038/s41392-021-00788-w
148. Saha A, Connelly S, Jiang J, et al. Akt phosphorylation and regulation of transketolase is a nodal point for amino acid control of purine synthesis. Mol Cell. 2014;55(2):264–276.
149. Kumar MA, Baba SK, Khan IR, et al. Glutamine metabolism: molecular regulation, biological functions, and diseases. MedComm. 2025;6(7):e70120.
150. Dimri M, Humphries A, Laknaur A, et al. NAD(P)H quinone dehydrogenase 1 ablation inhibits activation of the phosphoinositide 3-Kinase/Akt Serine/threonine kinase and mitogen-activated protein kinase/extracellular signal-regulated kinase pathways and blocks metabolic adaptation in hepatocellular carcinoma. Hepatology. 2020;71(2):549–568.
151. Nusse Y, Kubes P. Liver macrophages: development, dynamics, and functions. Cell Mol Immunol. 2025;22(10):1178–1189. doi:10.1038/s41423-025-01298-3
152. Guillot A, Tacke F. Spatial dimension of macrophage heterogeneity in liver diseases. eGastroenterology. 2023;1(1):e000003. doi:10.1136/egastro-2023-000003
153. Huang H, Balzer NR, Seep L, et al. Kupffer cell programming by maternal obesity triggers fatty liver disease. Nature. 2025;644(8077):790–798. doi:10.1038/s41586-025-09190-w
154. Fan X, Lu P, Cui XH, et al. Repopulating Kupffer cells originate directly from hematopoietic stem cells. Stem Cell Res Ther. 2023;14(1):351. doi:10.1186/s13287-023-03569-0
155. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol. 2021;18(1):45–56. doi:10.1038/s41423-020-00558-8
156. Wang Y, Rodrigues RM, Chen C, Feng D, Maccioni L, Gao B. Macrophages in necrotic liver lesion repair: opportunities for therapeutical applications. Am J Physiol Cell Physiol. 2024;326(5):C1556–C1562. doi:10.1152/ajpcell.00053.2024
157. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019;30(1):36–50. doi:10.1016/j.cmet.2019.06.001
158. Jiang T, Huang JB, Xu CY, et al. Arsenic trioxide cooperate cryptotanshinone exerts antitumor effect by medicating macrophage polarization through glycolysis. J Immunol Res. 2022;2022:2619781. doi:10.1155/2022/2619781
159. Wong A, Sun Q, Latif II, Karwi QG. Macrophage energy metabolism in cardiometabolic disease. Mol Cell Biochem. 2025;480(3):1763–1783. doi:10.1007/s11010-024-05099-6
160. Wculek SK, Heras-Murillo I, Mastrangelo A, et al. Oxidative phosphorylation selectively orchestrates tissue macrophage homeostasis. Immunity. 2023;56(3):516–530.e9. doi:10.1016/j.immuni.2023.01.011
161. Xie L, Deng X, Li X, et al. CircMETTL3-156aa reshapes the glycolytic metabolism of macrophages to promote M1 polarization and induce cytokine storms in sHLH. Cell Death Discov. 2024;10(1):431. doi:10.1038/s41420-024-02202-0
162. Kumar M, Sharma S, Kumar J, Barik S, Mazumder S. Mitochondrial electron transport chain in macrophage reprogramming: potential role in antibacterial immune response. Curr Res Immunol. 2024;5:100077. doi:10.1016/j.crimmu.2024.100077
163. Palmieri EM, Gonzalez-Cotto M, Baseler WA, et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat Commun. 2020;11(1):698. doi:10.1038/s41467-020-14433-7
164. Wang Z, Wu X, Chen HN, Wang K. Amino acid metabolic reprogramming in tumor metastatic colonization. Front Oncol. 2023;13:1123192. doi:10.3389/fonc.2023.1123192
165. Wu Y, Pu X, Wang X, Xu M. Reprogramming of lipid metabolism in the tumor microenvironment: a strategy for tumor immunotherapy. Lipids Health Dis. 2024;23(1):35. doi:10.1186/s12944-024-02024-0
166. Goswami KK, Banerjee S, Bose A, Baral R. Lactic acid in alternative polarization and function of macrophages in tumor microenvironment. Hum Immunol. 2022;83(5):409–417. doi:10.1016/j.humimm.2022.02.007
167. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi:10.1016/j.cell.2010.03.014
168. Saha P, Ettel P, Weichhart T. Leveraging macrophage metabolism for anticancer therapy: opportunities and pitfalls. Trends Pharmacol Sci. 2024;45(4):335–349. doi:10.1016/j.tips.2024.02.005
169. Huang Y, Wang F, Lin X, et al. Nuclear VCP drives colorectal cancer progression by promoting fatty acid oxidation. Proc Natl Acad Sci U S A. 2023;120(41):e2221653120. doi:10.1073/pnas.2221653120
170. Cheng Y, He J, Zuo B, He Y. Role of lipid metabolism in hepatocellular carcinoma. Discov Oncol. 2024;15(1):206. doi:10.1007/s12672-024-01069-y
171. Lundahl M, Mitermite M, Ryan DG, et al. Macrophage innate training induced by IL-4 and IL-13 activation enhances OXPHOS driven anti-mycobacterial responses. Elife. 2022;11. doi:10.7554/eLife.74690
172. Gupta S, Sarangi PP. Inflammation driven metabolic regulation and adaptation in macrophages. Clin Immunol. 2023;246:109216.
173. Qiu Y, Gan M, Wang X, et al. The global perspective on peroxisome proliferator-activated receptor γ (PPARγ) in ectopic fat deposition: a review. Int J Biol Macromol. 2023;253(Pt 5):127042.
174. Feng X, Zhang J, Yu B, An W, Jin X. Lipid metabolic reprogramming in tumor-associated macrophages: a key driver of functional polarization and tumor immunomodulation. Crit Rev Oncol Hematol. 2025;215:104881. doi:10.1016/j.critrevonc.2025.104881
175. Xiao C, Tan L, Liu X, et al. OSMR induces M2 polarization of glioblastoma associated macrophages through JAK/STAT3 signaling pathway. Front Oncol. 2025;15:1538649. doi:10.3389/fonc.2025.1538649
176. Sezginer O, Unver N. Dissection of pro-tumoral macrophage subtypes and immunosuppressive cells participating in M2 polarization. Inflamm Res. 2024;73(9):1411–1423. doi:10.1007/s00011-024-01907-3
177. Ma W, Sun Y, Yan R, et al. OXCT1 functions as a succinyltransferase, contributing to hepatocellular carcinoma via succinylating LACTB. Mol Cell. 2024;84(3):538–551.e7. doi:10.1016/j.molcel.2023.11.042
178. Sun JL, Zhang NP, Xu RC, et al. Tumor cell-imposed iron restriction drives immunosuppressive polarization of tumor-associated macrophages. J Transl Med. 2021;19(1):347. doi:10.1186/s12967-021-03034-7
179. Ma W, Zhang X, Lu J, et al. Transmembrane protein 147, as a potential Sorafenib target, could expedite cell cycle process and confer adverse prognosis in hepatocellular carcinoma. Mol Carcinog. 2023;62(9):1295–1311. doi:10.1002/mc.23564
180. Jin M, Yang Y, Dai Y, et al. 27-Hydroxycholesterol is a specific factor in the neoplastic microenvironment of HCC that causes MDR via GRP75 regulation of the redox balance and metabolic reprogramming. Cell Biol Toxicol. 2022;38(2):311–324. doi:10.1007/s10565-021-09607-y
181. Wu X, Cao J, Wan X, Du S. Programmed cell death in hepatocellular carcinoma: mechanisms and therapeutic prospects. Cell Death Discov. 2024;10(1):356. doi:10.1038/s41420-024-02116-x
182. Huang J, Pan H, Sun J, et al. TMEM147 aggravates the progression of HCC by modulating cholesterol homeostasis, suppressing ferroptosis, and promoting the M2 polarization of tumor-associated macrophages. J Exp Clin Cancer Res. 2023;42(1):286. doi:10.1186/s13046-023-02865-0
183. Rucker AJ, Park CS, Li QJ, Moseman EA, Chan FK. Necroptosis stimulates interferon-mediated protective anti-tumor immunity. Cell Death Dis. 2024;15(6):403. doi:10.1038/s41419-024-06801-8
184. Teng Y, Xu L, Li W, Liu P, Tian L, Liu M. Targeting reactive oxygen species and fat acid oxidation for the modulation of tumor-associated macrophages: a narrative review. Front Immunol. 2023;14:1224443. doi:10.3389/fimmu.2023.1224443
185. Yang Z, Wang Y, Zhang Y, et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat Cell Biol. 2018;20(2):186–197. doi:10.1038/s41556-017-0022-y
186. Netea-Maier RT, Smit J, Netea MG. Metabolic changes in tumor cells and tumor-associated macrophages: a mutual relationship. Cancer Lett. 2018;413:102–109. doi:10.1016/j.canlet.2017.10.037
187. Zhang Y, Kurupati R, Liu L, et al. Enhancing CD8+ T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell. 2017;32(3):377–391.e9. doi:10.1016/j.ccell.2017.08.004
188. Afonso MB, Rodrigues PM, Mateus-Pinheiro M, et al. RIPK3 acts as a lipid metabolism regulator contributing to inflammation and carcinogenesis in non-alcoholic fatty liver disease. Gut. 2021;70(12):2359–2372. doi:10.1136/gutjnl-2020-321767
189. Zhao M, Jiang Z, Yu M, Liu Z, Zheng Y. Identification of histidine decarboxylase and its multi-enzyme cascade system for cost-efficient biosynthesis of carcinine. 3 Biotech. 2025;15(9):309. doi:10.1007/s13205-025-04467-3
190. Li H, Xiao Y, Li Q, et al. The allergy mediator histamine confers resistance to immunotherapy in cancer patients via activation of the macrophage histamine receptor H1. Cancer Cell. 2022;40(1):36–52.e9. doi:10.1016/j.ccell.2021.11.002
191. Chen J, Liu G, Wang X, et al. Glioblastoma stem cell-specific histamine secretion drives pro-angiogenic tumor microenvironment remodeling. Cell Stem Cell. 2022;29(11):1531–1546.e7. doi:10.1016/j.stem.2022.09.009
192. Chen S, Luster AD. Antihistamines for cancer immunotherapy: more than just treating allergies. Cancer Cell. 2022;40(1):9–11. doi:10.1016/j.ccell.2021.11.007
193. Bernard JJ, Gallo RL, Krutmann J. Photoimmunology: how ultraviolet radiation affects the immune system. Nat Rev Immunol. 2019;19(11):688–701. doi:10.1038/s41577-019-0185-9
194. Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. 2016;165(6):1332–1345. doi:10.1016/j.cell.2016.05.041
195. Li YC, Li CL, Qi JY, et al. Relationships of dietary histidine and obesity in Northern Chinese adults, an internet-based cross-sectional study. Nutrients. 2016;8(7):420. doi:10.3390/nu8070420
196. Feng RN, Niu YC, Sun XW, et al. Histidine supplementation improves insulin resistance through suppressed inflammation in obese women with the metabolic syndrome: a randomised controlled trial. Diabetologia. 2013;56(5):985–994. doi:10.1007/s00125-013-2839-7
197. Rebouissou S, Nault JC. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J Hepatol. 2020;72(2):215–229. doi:10.1016/j.jhep.2019.08.017
198. Leone V, Ali A, Weber A, Tschaharganeh DF, Heikenwalder M. Liver inflammation and hepatobiliary cancers. Trends Cancer. 2021;7(7):606–623. doi:10.1016/j.trecan.2021.01.012
199. Brahma MK, Gilglioni EH, Zhou L, Trépo E, Chen P, Gurzov EN. Oxidative stress in obesity-associated hepatocellular carcinoma: sources, signaling and therapeutic challenges. Oncogene. 2021;40(33):5155–5167. doi:10.1038/s41388-021-01950-y
200. Huang Z, Zhou L, Duan J, et al. Oxidative stress promotes liver cancer metastasis via RNF25-mediated E-cadherin protein degradation. Adv Sci. 2024;11(13):e2306929. doi:10.1002/advs.202306929
201. Holeček M. Histidine in health and disease: metabolism, physiological importance, and use as a supplement. Nutrients. 2020;12(3):848. doi:10.3390/nu12030848
202. Brosnan ME, Brosnan JT. Histidine metabolism and function. J Nutr. 2020;150(Suppl 1):2570S–2575S. doi:10.1093/jn/nxaa079
203. Horn P, Tacke F. Metabolic reprogramming in liver fibrosis. Cell Metab. 2024;36(7):1439–1455. doi:10.1016/j.cmet.2024.05.003
204. Yang F, Hilakivi-Clarke L, Shaha A, et al. Metabolic reprogramming and its clinical implication for liver cancer. Hepatology. 2023;78(5):1602–1624. doi:10.1097/HEP.0000000000000005
205. Yang Q, Zhang H, Wei T, et al. Single-cell RNA sequencing reveals the heterogeneity of tumor-associated macrophage in non-small cell lung cancer and differences between sexes. Front Immunol. 2021;12:756722. doi:10.3389/fimmu.2021.756722
206. Liu W, Wang C, Liang L, et al. Single-cell RNA sequencing analysis revealed the immunosuppressive remodeling of tumor-associated macrophages mediated by the MIF-CD74 axis in gastric cancer. Sci Rep. 2025;15(1):26883. doi:10.1038/s41598-025-10301-w
207. Laviron M, Petit M, Weber-Delacroix E, et al. Tumor-associated macrophage heterogeneity is driven by tissue territories in breast cancer. Cell Rep. 2022;39(8):110865. doi:10.1016/j.celrep.2022.110865
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.