")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Mechanistic research holds promise for bacterial vaccines and phage therapies for Pseudomonas aeruginosa
Authors Hoggarth A, Weaver A, Pu Q, Huang T, Schettler J, Chen F, Yuan X, Wu M
Received 5 October 2018
Accepted for publication 26 January 2019
Published 20 March 2019 Volume 2019:13 Pages 909—924
DOI https://doi.org/10.2147/DDDT.S189847
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Austin Hoggarth,1,* Andrew Weaver,1,* Qinqin Pu,1,* Ting Huang,1,2 Jacob Schettler,1 Feng Chen,3 Xiefang Yuan,3 Min Wu1
1Department of Biomedical Sciences, University of North Dakota School of Medicine and Health Sciences, Grand Forks, ND, USA; 2Key Laboratory of Bio-resources and Eco-environment (Ministry of Education), College of Life Sciences, Sichuan University, Chengdu, China; 3Pulmonary and Allergy Institute, Affiliated Hospital of Southwestern Medical University, Luzhou, China
*These authors contributed equally to this work
Abstract: Vaccines for Pseudomonas aeruginosa have been of longstanding interest to immunologists, bacteriologists, and clinicians, due to the widespread prevalence of hospital-acquired infection. As P. aeruginosa becomes increasingly antibiotic resistant, there is a dire need for novel treatments and preventive vaccines. Despite intense efforts, there currently remains no vaccine on the market to combat this dangerous pathogen. This article summarizes current and past vaccines under development that target various constituents of P. aeruginosa. Targeting lipopolysaccharides and O-antigens have shown some promise in preventing infection. Recombinant flagella and pili that target TLR5 have been utilized to combat P. aeruginosa by blocking its motility and adhesion. The type 3 secretion system components, such as needle-like structure PcrV or exotoxin PopB, are also potential vaccine targets. Outer membrane proteins including OprF and OprI are newer representatives of vaccine candidates. Live attenuated vaccines are a focal point in this review, and are also considered for novel vaccines. In addition, phage therapy is revived as an effective option for treating refractory infections after failure with antibiotic treatment. Many of the aforementioned vaccines act on a single target, thus lacking a broad range of protection. Recent studies have shown that mixtures of vaccines and combination approaches may significantly augment immunogenicity, thereby increasing their preventive and therapeutic potential.
Keywords: lipopolysaccharide, flagella, pili, exoenzymes, outer membrane proteins, phage therapy
Introduction to Pseudomonas aeruginosa vaccines
P. aeruginosa is a Gram-negative bacterium that often infects immunocompromised individuals with cystic fibrosis (CF), HIV-1, cancer, and surgical or burn wounds. P. aeruginosa can also cause serious infections in the corneas of patients with extended use of contact lenses and also in the urinary tract.1 P. aeruginosa accounts for 10.1% of nosocomial infections, making it a highly significant bacterium in hospital settings.2 Because of P. aeruginosa’s ubiquitous nature and extremely complex genomic diversity, scientists have put earnest effort into developing effective vaccines over the years, but the results are not so encouraging.3
Thus far, a number of vaccine candidates for P. aeruginosa have been tested and most of them are based on one of the major components of the bacterium, especially lipopolysaccharide (LPS), polysaccharide, polysaccharide conjugates, extracellular protein, outer membrane protein (OMP), flagella, type 3 secretion system (T3SS), pili, and so on (Table 1). Furthermore, whole organisms, such as killed whole-cell and live attenuated P. aeruginosa, are being tested for preventive ability.4 However, the search for a truly applicable vaccine still proves difficult, and no single vaccine has surpassed all preclinical tests and been approved for the clinics. Although the use of various cellular components of P. aeruginosa, such as OMP and LPS, has been shown to be immunogenic and/or beneficial for hosts, the high diversity of P. aeruginosa serotypes has created much difficulties in designing a vaccine to be effective for all serotypes of the pathogen.5 Many such vaccines that have been created show fantastic effects against their homologous strain; but when used against differing serotypes, they prove to be largely inefficient in eliciting an effective immune response. The following sections highlight vaccine research targeting various pathogenic components, analyze the potential difficulties, and discuss the future directions (Figure 1).
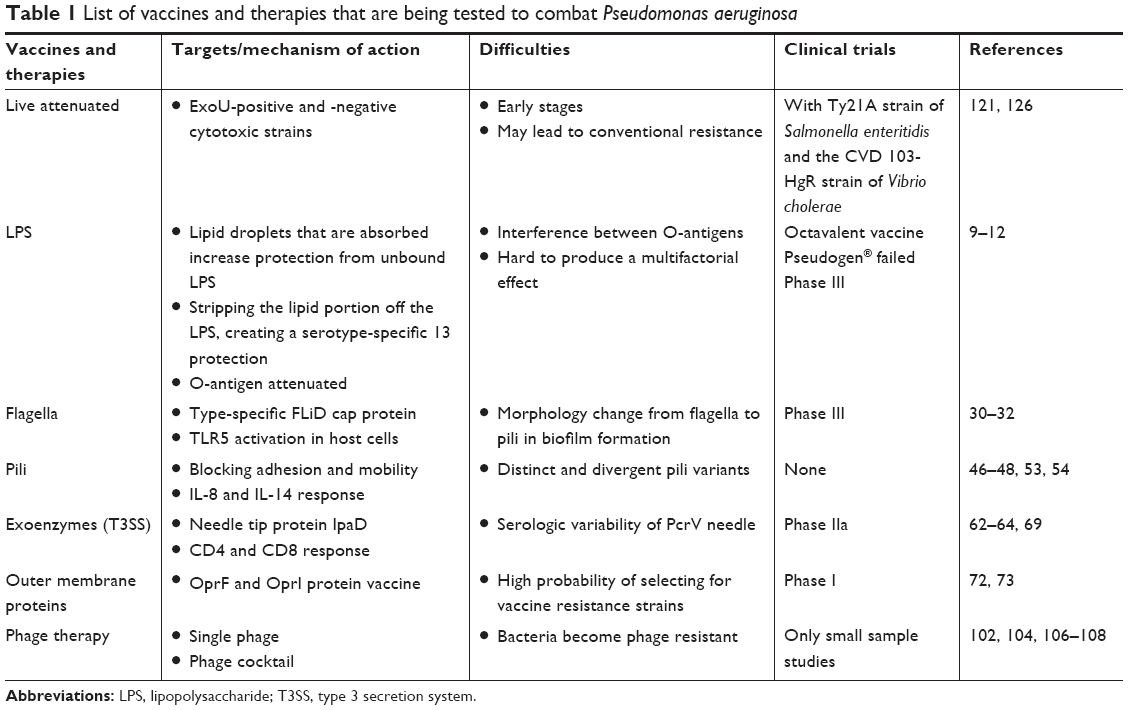 | Table 1 List of vaccines and therapies that are being tested to combat Pseudomonas aeruginosa |
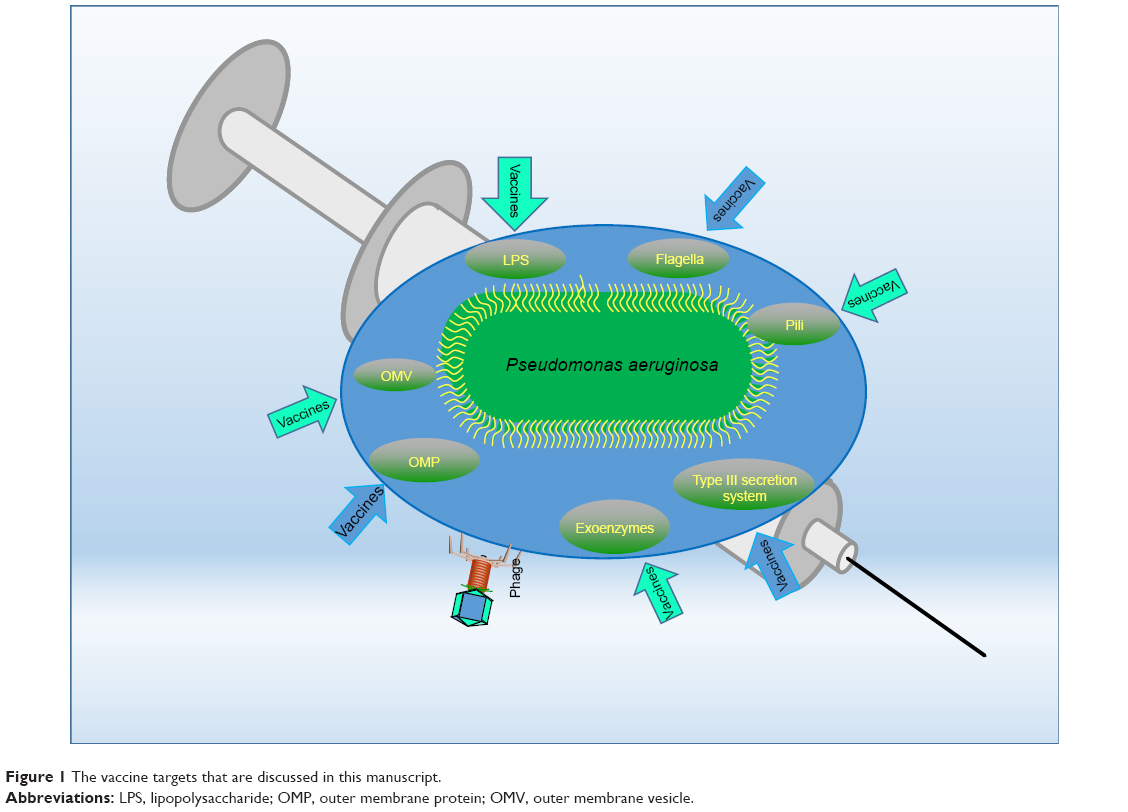 | Figure 1 The vaccine targets that are discussed in this manuscript. |
Lipopolysaccharide
The LPS of P. aeruginosa has been a fascinating candidate for possible vaccines. LPS is a major constituent of the outer leaflet of the Gram-negative cell membranes – a defining feature that distinguishes these bacteria from their Gram-positive counterparts, which lack the outer LPS cell membrane and consist primarily of peptidoglycans and teichoic acids.6 LPS is highly pyogenic and toxic when used in a purified state, primarily due to the lipid component.7 Current research has been directed toward two separate avenues of LPS-based vaccines.8 The first strategy utilizes lipid droplets to carry entire LPS components into the host. These lipid droplets are used to protect the body from the otherwise virulent effects of pure, unbound LPS.9 The second and more widely used strategy is based upon stripping the lipid portion off the LPS, creating serotype-specific protection.10 The serotype or serogroup is determined by varying surface antigens of the pathogen, giving rise to 20 different O-antigens according to the International Antigenic Typing System (IATS).11
The classification proposed by IATS is highly complex and can be broken down further based on O-antigens. O-antigens found on the surface of P. aeruginosa are linked to lipid A, which anchors LPS to the outer portion of the cell membrane.12 As there are 13 different O-serogroups, generating a multivalent vaccine is required for stronger antibacterial effects. Both the lipid A and core protein are considered to be the most conserved portions of LPS, but new data demonstrate their intricate variable regions relating to the number of O-antigen, and increased O-acetylation of the protein core.13 Thus, these regions may be unlikely candidates for vaccination. Little is known about variability of the LPS protein core, although previous research has shown its consistency between laboratory grown P. aeruginosa and CF isolates.14 Although O-polysaccharides have great diversity, there is sequence and structure overlapping among 11 of the 13 O-serogroups.15 Hence, much P. aeruginosa vaccine research has been directed toward the O-antigen in LPS. One of the problems with multivalent LPS O-antigen vaccines composed of antigens from serologically different strains within the same serogroup, though, is that there can be interference between the antigens, sacrificing the immunogenicity of the vaccine.16 Furthermore, when an octavalent vaccine, Aerugen® (Crucell, Berne, Switzerland), was finally created, it failed to reduce the incidence and severity of P. aeruginosa from the airways in a Phase III clinical trial.17 Even though the O-antigen of LPS is believed to mediate immunity in acute P. aeruginosa infections, a vaccine with multifactorial effects has been extremely hard to find.18 The possible reasons for this difficulty are poor immunogenicity of purified O-antigens, serological diversity, and the need to conjugate isolated O-antigens to various protein carriers for maximal effects.19 To help boost the immunogenicity of the polysaccharide vaccines, a study used a detoxified LPS (D-LPS) conjugated with diphtheria toxoid (DT), where the DT was used as a protein carrier. Fortunately, the immune potency was improved compared to D-LPS alone and DT alone.20 Studies like this may show that the key to increasing LPS immunogenicity is to conjugate it with a proper protein carrier.
The largest downfall for O-antigen–based vaccines is their single serotype, which is not protective against other serotypes of the pathogen and is essentially unreliable as a protective measure against P. aeruginosa.21 Nevertheless, LPS O-antigens have been at the forefront of P. aeruginosa research for a long time due to their great serotype-specific immunogenicity. It has been suggested by Gerald Pier et al that the O2/O5 serogroup is the most complex in various strains including PAO1, a common laboratory strain that has a fully sequenced genome. Pier’s group used an aroA deletion mutant of P. aeruginosa serogroup O2/O5 strain PAO1, naming it PAO1ΔaroA. This strain achieved high levels of protection against the ExoU-expressing cytotoxic variant of the parental strain PAO1, but failed to protect against serogroup heterologous strains.22 Other studies creating live attenuated vaccines have shown that multivalent vaccination elicits broader in vivo protection than monovalent vaccination. Bivalent vaccines, surprisingly, provided in vivo protection against a non-cytotoxic O-antigen–heterologous strain – something that has never been previously achieved in studies with P. aeruginosa strains, namely, PA14ΔaroA. One of the key findings by Pier’s group, though, is that live attenuated vaccines avoid many of the limitations that prior purified LPS O-antigen vaccines had.23 Among these limitations are competition among related polysaccharide antigens for the limited amount of antibodies on B cells, in vivo antigenic variation of the O-antigen during infection, evolutionary selection for P. aeruginosa strains making O-antigens that elicit antibody primarily to non-protective epitopes, and decreased immunogenicity in the absence of conjugation of polysaccharide O-antigen to a carrier.24 Creating an attenuated vaccine that targets the LPS core, of which there are only four variants as opposed to the 20–30 serotypes in O-antigens, results in stronger immunity response.25 A recent development of vaccine KBPA-101 that targets LPS has been tested against P. aeruginosa serotype O11 pneumonia in a Phase 1/2 trial (NCT00851435).26 Due to the continuing efforts and progress, the vaccines targeting LPS may have real opportunity to become effective preventive products for the clinics.
Flagella
Due to their high immunogenicity, flagellin and flagella have also been considered as potential targets for vaccination against P. aeruginosa.27 Flagellin is the major structural protein component forming the flagellar filament. This protein is formally subdivided into A and B groups.28 Flagella not only serve primarily for motility in P. aeruginosa, but also assist attachment to the respiratory epithelium and activation of TLR5.29 TLR5 is a type of pathogen recognition receptor that recognizes pathogen-associated molecular patterns and initiates innate immunity. Once the bacterium has attached to the respiratory epithelium, the flagella are shed, and with the aid of pili, P. aeruginosa is able to infiltrate the epithelium.30 It has been found that purified flagellin is much more potent in activating TLR5 and also elicits a higher TLR5–neutralizing effect through antibodies than immunization with crude flagella.31 However, flagella, composed of FliC protein, type-specific FliD cap protein, and basal body components, are a better candidate for vaccines against P. aeruginosa than simple monomeric flagellin-based vaccines.32 P. aeruginosa flagellin is required for virulence and targeted by protective antibodies in animal models. A combination vaccine of Klebsiella pneumoniae with P. aeruginosa glycoconjugate (comprising four common types of human infections, O1, O2, O3, and O5) has been developed through chemical linking to various flagellin types of P. aeruginosa.33 By generating proper antibodies without hampering TLR5 activation, flagella have demonstrated better vaccine capabilities than flagellin in other Gram-negative bacteria including Escherichia coli.34 Although there has been some success with flagella-based vaccines – even in Phase III studies – the overall impact including opsonic phagocytosis, antibody response, and inflammasome activation is not as efficient as LPS O-antigens, which explains why O-antigen–based immunity remains the major vaccination strategy. Overall, flagella have become a new option for P. aeruginosa vaccines, but other components of P. aeruginosa display higher probability of producing a vaccine that is both safe and effective in overall protection of the body than flagella.35 Taking this into account, fusion proteins have been created to combat P. aeruginosa. In one particular study, mice were immunized with ExoA and flagellin that were cloned in E. coli as well as a ExoA–flagella recombinant fusion protein. All showed a marked increase in IgG, but the ExoA–flagella showed significant protection against intraperitoneal infection by P. aeruginosa.36 Hence, flagella vaccines can be complementary to other therapies.
Immunotherapy with both recombinant proteins and antibodies is at the forefront of cancer therapy, but the application of biologics in bacterial therapy is much less explored. Behrouz et al developed an effective recombinant type B flagellin (r-b-flagellin) to elicit a response that cross-reacts with different heterologous bacterial strains, consistent with past reports on flagellin being a potent antibody stimulator. Specifically, r-b-flagellin prompts TH1 and TH2 responses involving both B- and T-cells. In addition, r-b-flagellin can induce a Th17 response critical for neutrophil recruitment. TH17-mediated immunity has been demonstrated to play a substantial role in the development of autoimmune diseases, but has also been recently linked to infectious disease. Furthermore, r-b-flagellin achieves a desired protective effect in a burn murine model, suggesting that it is worth further testing for clinical relevance.37
P. aeruginosa strains display heterogeneity, and isolates from chronic CF patients exhibit different properties from individuals with acute infections. After acute infection, bacteria start losing their flagella and pili and begin to downregulate their T3SS.38 Strains derived from chronic CF patients tend to form more biofilms and overexpress exopolysaccharide alginate, becoming mucoid.39 A vaccine based on flagella is only directed toward motile bacteria and not established biofilm-forming strains often seen in chronic infections, so flagella may be a reasonable target for the initial onset of infection. Because of the great number of hospital-acquired acute infections, even targeting acute infection alone can provide substantial relief by preventing pathogenic invasion and spread.
Recent studies have revealed novel important functions for flagella involving the type 6 secretion system (T6SS), especially in coordinating secretion toxins or delivering outer membrane vesicles (OMVs) critical for bacterial pathogenesis.40,41 del Tordello et al exploited the T6SS using sheaths composed of ClpV-interacting protein A and B heterodimers to generate immunogenic multivalent particles for vaccine delivery.42 Despite a long road ahead, flagella may still hold potential for future vaccine development as their molecular function continuously unfolds.
Pili
Pili have also historically been at the forefront of research on vaccines against P. aeruginosa. The polar type 4 secretion system (pili) is located on the outer cell membrane, a characteristic common among all Gram-negative bacteria.43 Pili are hair-like appendages that have the capability of being extended or retracted through the utilization of pilin monomers arranged helically. One of the most important functions of pili is attachment and infiltration of the airway epithelium and also in twitching motility which allows bacterial translocation over moist surfaces.44 The pilus is composed mainly of major pilin subunits, while the less-abundant minor pilins FimU-PilVWXE and the putative adhesion PilY1 prime pilus assembly are proposed to form the pilus tip.45 By pulling the bacterium closer to the epithelium surface, pili allow for the release of substances such as exoenzyme S, enhancing the interaction between P. aeruginosa and the epithelium.46 Twitching motility is also essential for the bacterium’s success in infiltrating the airway by acting as a grapple and hook, allowing P. aeruginosa to essentially glide across the airway surface to its desired location. This movement is accomplished through the expression of various genes and through multiple signal transduction systems. Twitching motility and the overall structure of piliated bacteria, especially P. aeruginosa, allow for easy accumulation, attachment, and movement upon the surface of the respiratory epithelium.47 Users of soft contact lenses, especially extended-use and overnight wearers, are especially prone to corneal P. aeruginosa infection. These infections, similar to infections of the airways, are very difficult to treat.48
All the above characteristics make pili a potential candidate for vaccines. Similar to other vaccines previously discussed, one of the greatest difficulties in creating a pili-based vaccine against P. aeruginosa is that different bacterial strains produce distinct and sometimes very divergent pilin variants. The pili group can be further divided into five distinct phylogenetic groups. Nevertheless, pilin vaccines have demonstrated some success in a preclinical setting utilizing PAO1 as a challenge strain. Loss of specific minor pilins relieves feedback inhibition of FimS-AlgR, increasing transcription of the AlgR regulon and delaying Caenorhabditis elegans killing. Reporter assays demonstrate that FimS-AlgR is required for increased expression of the minor pilin operon upon loss of select minor pilins.45 Only time will tell what success pilin will bring into the clinics as a future P. aeruginosa vaccine.49 Mechanistically, both the pili and flagella of P. aeruginosa induce IL-8 secretion by binding to GalNAcβ1–4 Gal receptors on the respiratory epithelium.50 These receptors are drastically elevated in CF patients.51 Upon binding, the respiratory epithelial cells begin to release IL-8, which is followed by recruitment of polymorphonuclear leukocytes that produce elastase and superoxide to defend against the invading pathogen.52 This further stimulates IL-8, cycling through further inflammation until the pili and flagella are no longer directly opposed to the epithelium due to the formation of microcolonies that eventually form biofilm.53 Once these components are masked by the biofilm, IL-8 peters off, and the body has great difficulty in ridding itself of the pathogen. PilY1 and the minor pilins inhibit their own expression, and loss of these proteins leads to FimS-mediated activation of AlgR that suppresses expression of acute-phase virulence factors and delays killing. This mechanism could contribute to adaptation of P. aeruginosa in chronic lung infections, as mutations in the minor pilin operon result in the loss of piliation and increased expression of AlgR-dependent virulence factors that are characteristic of such infections.45
Another study found that PA14 with an aroA deletion mutation (PA14ΔaroA) immunization exhibited much higher levels of IL-17 in bronchoalveolar lavage fluid than unimmunized or E. coli-immunized mice, leading to protection against lethal pneumonia. When IL-17 was depleted before a challenge or it was lacking altogether, PA14ΔaroA mutant vaccination lost its protective abilities. These results indicate that IL-17 plays an important role in protection against LPS-heterologous strains of P. aeruginosa in the lung.54 Growing research on IL-17 has shown that a lineage of IL-17–secreting T helper cells (Th17) plays an important role in neutrophil recruitment and activation of memory CD4+ T-cells.55 Many other cytokines, chemokines, and other factors along with multiple intracellular signaling cascades may be involved in immunization of the lower respiratory tract.56 These cascades, including Ras, Src, and EEK1/2 MAP kinases, are activated by several bacterial components.57 These components can either be disrupted in parts of the cascades or aberrantly overexpressed: overstimulation of matrilysin by flagella causes lung pathology in CF.58 There are countless other secreted factors and cascades involved with P. aeruginosa infection, a topic that is also intensely studied. A study using a recombinant PilA (r-PilA) showed increased humoral immunity in a murine burn model. This r-PilA also reduced bacterial burden on organs, such as the liver and spleen, in infected mice and increased overall survivability of the P. aeruginosa challenge.59 The same investigators took it one step further by mixing r-PilA and a recombinant full-length type b flagellin (r-b-flagellin). By attacking both virulence factors, the r-PilA + r-b-flagellin immunization induced vigorous opsonophagocytic killing of both the PAO1 strain and a multidrug-resistant strain.60 Vaccines utilizing recombinant PilA are currently under clinical test.59
Exoenzymes and T3SS
Once in close contact with the surface of its host, P. aeruginosa is internalized into multiple types of epithelial cells in vitro and in vivo, although it was often regarded as an exclusively extracellular pathogen that utilizes a T3SS61 to deliver exoenzymes, such as ExoS, ExoT, ExoU, and ExoY, into eukaryotic cells.62 Paradoxically, ExoS, a T3SS effector, has antiphagocytic activities required for intracellular survival of P. aeruginosa and its occupation of bleb niches in epithelial cells.61
This is accomplished through a system of 20 proteins that utilize a needle-like structure to penetrate the plasma membrane of the target cell, injecting effector proteins from the bacterial cytoplasm into the membrane and cytoplasm of the host cell.63 Current research on T3SS vaccines is directed toward utilizing needle tip proteins such as IpaD in Shigella flexneri, LcrV in Yersinia spp., and PcrV in P. aeruginosa as possible candidates. The specific PcrV class of needle tip proteins has been found to localize at the extracellular end of the T3SS apparatus needle, working to regulate flow of proteinaceous contents into the target cell. Like pilin, T3SS vaccines have only been studied in a preclinical setting; thus, more research needs to be accomplished before determining its efficacy in the clinic. It is also true that the protective activity of T3SS is not as good as that of LPS O-antigens, and little is still understood about the genetic, protein, and serologic variability in PcrV among diverse P. aeruginosa isolates.64 MEDI3902 is an antibody vaccine targeting P. aeruginosa virulence factors PcrV and Psl65,66 that enhance neutrophil uptake. Pre-clinical data have shown that MEDI3902 can protect against Pseudomonas pneumonia.67,68
Recently, the use of T3SS-based bacterial live vectors for immunotherapy has been explored, notably because of its potential in triggering antigen-specific cytotoxic CD8+ T-lymphocyte responses, a very important component of the body’s defense against P. aeruginosa infection.69 P. aeruginosa and corresponding effector-null isogenic T3SS mutants, effector-null mutants of cytotoxic bacterium with and without ExoS transformation, antibiotic exclusion assays, and imaging using a T3SS-GFP reporter were internalized into target cells. Intracellular bacteria showed that T3SS activation continued in replicating daughter cells.61
It has been shown that some pathogens (Mycobacterium tuberculosis or Salmonella typhimurium) activate CD4 and CD8 lymphocytes;70 however, P. aeruginosa has been demonstrated to specifically activate a CD8+ T-cell response. One of the limitations of this type of research is the intrinsic toxicity of these vectors; however, lesser virulence has been achieved through deletion of various genes of the T3SS toxin exoenzymes, ExoS, and ExoT, and ExoU.71 Through further attenuating P. aeruginosa T3SS-based vectors, toxicity can be reduced and overall vaccination efficacy can be improved. Wang et al used T3SS to deliver ovalbumin (OVA) antigen into respiratory epithelial cells to induce an immune response against OVA-expressing cells. The OVA antigen was greatly attenuated by deleting various genes, such as quorum-sensing (QS) genes and the aroA gene.72 The aroA mutation causes T3SS to function at a higher level and reduces toxicity greatly, while a QS mutation shows slightly reduced toxicity and a reduced negative effect of QS molecules on the immune system. Overall, research involving the utilization of T3SS P. aeruginosa live vector to deliver antigens to the host’s immune system is a very promising and novel approach for creating attenuated vaccines. Due to their high immunogenicity and low toxicity, the use of Polack et al’s ΔaroA mutants (CHA-OA and CHA-OAL) may have the potential for further development of a vaccine. Quite notably, Polack et al also demonstrated that the T3SS of P. aeruginosa could also be utilized to deliver in vivo tumor antigens, which activate the body’s innate immunity. Certain characteristics of the epitopes’ composition and/or peptide length, such as long peptides with immunodominant, cryptic CD8+ epitopes and strong CD4+ TH epitopes, direct the overall immune response, especially upon the activation of cytotoxic T-lymphocyte–mediated immunity.73
Sawa et al generated a murine monoclonal anti-PcrV antibody, mAB166, which inhibits the T3SS toxin translocation. Phase IIa clinical trials have begun in France to evaluate a recombinant, PEGylated, human Fab’ anti-PcrV fragment (KB001) involving ten intensive care units to see its effect on ventilator-associated pneumonia. They looked at 39 mechanically ventilated patients who had been colonized, but not infected. The KB001 has been shown to be well tolerated by patients and could neutralize PcrV for at least a month and prevented patients from developing pneumonia within that period.74 In addition, clinical trials led by researchers in the USA found a dose-dependent reduction in IL-1β, IL-8, and myeloperoxidase when administering the KB001 vaccine. Sputum samples showed a decrease in neutrophil elastase and neutrophil counts at a 10 mg/kg dose. These findings indicate an auxiliary treatment to reduce inflammation and harm to patients with chronic pneumonia and CF.74
PcrV is not the only promising target for a P. aeruginosa vaccine. PopB/PopD, which helps the translocation of exotoxins across the host’s cell membrane, is a target for Th17 cells. These subsets of helper T-cells elicit a response via production of IL-17 to enhance bacterial clearance. A live attenuated PopB vaccine showed reduced bacterial loads within the lungs and spleen 18 hours post-infection.75 An issue with these findings is that PopB is likely not exposed on the surface of live P. aeruginosa, making it difficult to recognize whole PAO1 cells along with opsonophagocytic killing of invading bacteria.75
Outer membrane proteins
Other important vaccine candidates for P. aeruginosa infections include OMPs, flagellin–OMP fusion proteins, ExoA, alginate, DNA and viral vector vaccines, and passive immunization. Many of these candidates have shown moderate success in preclinical and even in some clinical studies.76 Some of the conserved antigens of P. aeruginosa are not required for full virulence, and vaccines targeting any one of these components may select for the emergence of vaccine-resistant strains instead of producing protection. Conversely, utilizing an aggregation of targets of P. aeruginosa genes may enhance the immune potency and, in turn, achieve better protection. Westritschnig et al attempted to create systemic, nasal, and oral live vaccines against P. aeruginosa. These vaccines were designed to utilize immunogenic epitopes of the OMPs OprF and OprI of P. aeruginosa.77 One vaccine that has recently been developed to target OprF and OprI is the VLA43 vaccine.78 Understanding P. aeruginosa pathogenesis mechanisms could immensely benefit the development of novel prevention and treatment strategies. To identify novel vaccine candidates, researchers comprehensively examined the expression levels of all known OMPs in two P. aeruginosa strains in a murine acute pneumonia model. OprH was one of the most highly expressed proteins during infection. In addition, OprH is known to be highly immunogenic and accessible by host proteins.79
As the OMPs are far more conserved than other virulence factors of P. aeruginosa, OMP-based vaccines may engender total immunogenicity to provide better protection.80 Herein, the OprF–OprI protein showed protective capabilities against systemic P. aeruginosa infection, and was found to be safe with tolerable side effects in a clinical setting. Importantly, attenuated Salmonella was further used to express OprF–OprI as an oral live vaccine81 and demonstrate cross-reactivity with all known serotypes of P. aeruginosa, resulting in vigorous antibody induction in the lower respiratory tract.82 The OMP oral and live vaccines have both resulted in greater levels of IgA antibodies at the bronchial surface, whereas only nasal vaccination induced a strong serum antibody response.83,84 Recent studies indicate that Salmonella invasion requires CF transmembrane conductance regulator protein, a component of the respiratory airways but not the gastrointestinal tract. Oral live vaccination of CF patients using Salmonella strains may be as effective as airway delivery.84,85 A Phase I study was conducted using a recombinant OMP called IC43 that consisted of both OprF and OprI epitopes, given twice intramuscularly. The OprF–OprI titers were markedly different between 7 and 14 days after vaccination. A dose of 50 μg or greater induced a plateau in IgG antibody response.86 A different Phase III study on 483 CF patients produced high and long-lasting serum anti-flagella IgG titers with an overall 34% degree of protection.87 Overall, both nasal and oral OprF–OprI vaccines may represent promising attenuated vaccines for P. aeruginosa.
Outer membrane vesicles
Like other Gram-negative bacteria, P. aeruginosa secretes OMVs that fuse with lipid rafts on its cell membrane as a means to deliver virulence factors to distant locations.88 Due to the growing understanding of how OMVs influence bacterial pathogenesis in the past decade, utilization of this entity for vaccination is a topic that is also heating up. Previous research showed that OMVs elicit heightened innate immune responses by the combined sensing of both LPS and protein components.89–91 Indeed, OMVs may be promising vaccine candidates against several species of bacteria.90–95 Remarkably, vaccines developed based on Neisseria lactamica OMVs rendered effective protection against meningococcal infection and underwent a Phase I clinical trial for safety and immunogenicity studies with adult volunteers.96 Furthermore, OMVs from P. aeruginosa triggered production of cytokines in epithelial cells and macrophages,89 such as IL-1 and IL-6, via the TLR4 signaling pathway requiring LPS.
Unlike whole bacterium vaccines, OMVs have a significant advantage in reducing virulence repertoire, thus drastically quenching safety concerns if applied in clinics. Hence, naturally produced P. aeruginosa OMV vesicles have the potential to protect the immunized host against subsequent pseudomonal infections. Taken together, a well-designed vaccine based on OMVs will offer enhanced safety, improved immunogenicity, and elevated efficacy for prevention of pseudomonal diseases.
Phage therapy
Although practically an ancient topic, phage therapy has been revitalized over the recent years and put back into conversations regarding viable treatment options, especially for patients who are non-responsive to broad spectrum antibiotic treatment. Bacteriophages, which are considered to be among the most ubiquitous and diversified organisms on Earth and, are believed to exist wherever their hosts thrive.97 Although advanced techniques for formulating phage cocktails, training phages, and collecting phage libraries have improved efficacy in vitro, whether personalized or ready-to-use therapeutic approaches or phages and antibiotics combination are effective and safe in vivo, and can reduce P. aeruginosa biofilms, remains to be answered.98
There are two types of phages: lytic and temperate. For therapeutic purposes, only the lytic phages are used, which adsorb on the host cell surface and proceed to inject and replicate their DNA into host cells to induce host cell lysis.99 Subsequently, the progeny phages are released and advance the infection by targeting other bacteria. Phage therapy was proposed >100 years ago by d’Herelle,100 but the notion was subdued after antibiotics proved more impactful and cost-effective. Due to the strong support for antibiotics and arguments against phage therapy experiments, enthusiasm for phage therapy drastically diminished over the years. By the time World War II had ended, phage therapy was nearly abandoned in the western world.101
Due to the growing concerns of rapidly increasing antibiotic resistance or multidrug-resistant superbugs, scientists are beginning to renew the interests in phage therapy as a clinical option. To date, there are 137 known phages that have been completely sequenced on public databases that attack P. aeruginosa, most coming from the Caudovirales order. There have been studies done both in vitro and in vivo, besides many therapeutic cases without academic publications (but searchable on the Internet). One example is a lytic phage that prevents biofilm formation on P. aeruginosa in hydrogel-coated catheters. Fu et al pretreated the catheters with Myoviridae phage M4 for 2 hours prior to bacterial challenge. Results showed a 2.8 log decrease in the number of viable cells compared to the control,102 although at the 24–48-hour period, there was formation of a phage-resistant biofilm. In addition, phages can also affect Pseudomonas spp. by changing their natural environment and influencing the virulence and other pathogenicity. This sudden marked reduction followed by a lagging phage-resistant biofilm seemed to be the trend of the therapy across multiple studies.103 The same phenomenon occurred in another group’s study when phages were administered 2 hours after an onset of infection. The result was a 95% survival rate in immunocompetent mice. There was also a 4-day preventive treatment of bacteriophages that showed a 100% survival rate, which highlights both the curative and preventative aspects that bacteriophages can offer.104 Some researchers use a mixture of bacteriophages and antibiotics to treat P. aeruginosa. This method solves the initial problem, but still involves the disadvantages of antibiotic treatment.105 While most cases have suggested treatment using a single phage, Hall et al utilized a combination of two to four phages – a phage cocktail – which appears to be more effective than use of a single bacteriophage. Therefore, broad host-range phage cocktails potentiate the efficacy of phage therapy.106,107
Human trials of phage therapy have also shown their warrant as a viable therapy. Small studies have shown marked reduction in bacterial colonies with just a single dose of phage therapy.108 Most studies have been done on burn victims where the phages were applied topically on the wounds. Also, very little to no adverse effects are observed with phage therapy, further highlighting its promise.109,110 P. aeruginosa is a ubiquitous pathogen infecting many organs and systems. It would be curious to see phage studies done on individuals with chronic CF or COPD to see its viability as a therapy in that regard to chronic diseases, where the resistance to antibiotics is particularly severe.
There are still some challenges that phage therapy faces. Among the challenges include phage-neutralizing antibodies produced by the body’s immune system and safety concerns regarding endo- and exotoxins in phage preparations. The limited spectrum of activity also limits applications for this therapy. Another disadvantage is that bacteria are rapidly becoming resistant to phages, chiefly due to the existence of clustered regularly interspaced palindromic repeats (CRISPR), a bacterial adaptive immunity system, which can degrade with high specificity invading DNA or RNA via short guide RNAs.111,112 For example, Cady et al found that PA14 CRISPR-Cas systems mediate resistance to engineered phages.113 There is an emergence of research to tackle the problem of CRISPR-Cas system by means of manipulating genes to change the bacterial host defense and pathogenesis. By halting the bacteria’s ability to illicit an immune response, phages are able to gain access and work as intended. The goal then becomes producing reliable pro- or anti-sense proteins to manipulate the CRISPR-Cas system. Maxwell et al found that the anti-CRISPR protein AcrF1 acts a potent inhibitor of CRISPR-Cas systems by locking the Csy complex into a conformation that diminishes its ability to bind to DNA.114 All type I-F CRISPR-Cas systems are vulnerable to inhibition by anti-CRISPR proteins.115 To further expand this field, type I CRISPR-Cas systems may be utilized to invade hosts and evade immune response through the QS regulator LasR, hindering TLR4’s ability to recognize the pathogen.111 This CRISPR-Cas system is a double-edged sword, though. By using the same mechanism that the bacteria use to become resistant, scientists can use it to create synthetic phages with desired characteristics and precise genomic content.116,117 With the decreasing cost of DNA sequencing, scientists have a more precise engineering method to combat this bacterial foe.118 In addition to CRISPR resistance, the bacteria still have many other antiviral mechanisms, such as preventing phage adsorption, inhibiting phage DNA entry, and aborting infection systems.119 These evasive strategies will make it increasingly difficult to apply phage therapy generally; but with advancement of basic scientific research as well as clinical testing, we anticipate a rejuvenated research field for phage therapy of bacterial infections.
Live attenuated vaccines
Attenuated bacteria, a lesser-developed field of research with regard to P. aeruginosa vaccination, can also be used for prevention and therapy. Attenuated vaccines for preventing P. aeruginosa are among the least researched, but have shown to be one of the most successful in the preclinical setting in the last few years.120 Since this topic has partially been covered in previous sections, we will just focus on more mechanistic research in the attenuated bacterial vaccine field. One of the main advantages of attenuated whole bacterial vaccines is their broad coverage of bacterial virulence factors and generation of a more complete and effective response. Gerald B Pier and his colleagues at Harvard Medical School have explored P. aeruginosa live attenuated vaccines for quite some time, reporting that vaccine strains provide protection against acute fatal pneumonia in mice caused by serogroup-homologous strains, but little protection against serogroup-heterologous, highly virulent, ExoU-positive cytotoxic strains.35 The high virulence of these latter strains limits the doses of bacterial challenge during survival experiments, thereby rendering less-potent immune effects. In a more recent study, Pier’s team used a mucosal vaccination route to deliver monovalent to quadrivalent combinations of live attenuated P. aeruginosa strains.121 Interestingly, a multifactorial immunity was induced by the vaccination, one that involves both humoral and cellular components – both antibodies and T-cells. Through this simultaneous induction of immune components, a diverse array of antigens from P. aeruginosa was targeted. This multifactorial immunity was associated with broad in vivo protection against acute lung infection caused by a wide range of P. aeruginosa strains, including both ExoU-negative and -positive strains.
Novel ideas also emerged as Francisella tularensis was genetically modified to express the surface proteins OprFPa, PilAPa, and FliCPa of P. aeruginosa. Out of these three surface proteins, only FliCPa showed immune protection in a murine model.122 Currently, multiple efforts are being made to develop other live attenuated vaccines such as Mycobacterium tuberculosis (Mtb).123 These endeavors demonstrate that live attenuated vaccines may be a more viable means of producing a broader protection. Even though these vaccines are different in genus, there is hope to draw parallels in techniques and procedures in the hope of producing viable vaccines. To add even more utility to attenuated P. aeruginosa, there has been a long-term interest of using live attenuated bacterial vectors for antigen delivery in cancer immunotherapy.124
Live attenuated vaccinations against P. aeruginosa are relatively new; in fact, live attenuated bacterial vaccines in humans have only been tested using the Ty21A strain of Salmonella enteritidis and the CVD 103-HgR strain of Vibrio cholerae. Producing a live attenuated vaccine against P. aeruginosa has the possibility of being quite inexpensive and very effective if a proper means to attenuate virulence is discovered while maintaining immunogenicity.125 Despite some promising results, there has been strong resistance from the well-known use of live attenuated bacterial vaccines due to their intrinsic virulence/toxicity and limited commercial success in the past. Also, with the potential use of attenuated vaccines, developers must be aware of the appropriate recombinant antigens to use. Particularly, in Robinson’s F. tularensis LVS, some recombinant antigens were non-immunogenic.122 Despite huge challenges in this area, live attenuated vaccines are offering some great hope for conquering the bacterium. The Pier’s research group has also pushed toward understanding more about the role of effector CD4 T-cells, which seem to be important for overall protection from P. aeruginosa infection.
As the field matures, different techniques may be used for generating attenuated vaccines and understanding host–pathogen interaction may help in technical development. Over the recent years, autophagy has emerged as an important alternative mechanism for delivering pathogen-deprived antigens to the major histocompatibility complex class I and II presentation pathways in Mtb,123 whereas this may be different for P. aeruginosa.126 In both cases, autophagy was utilized as a defense mechanism against phagocytosis by macrophages.127 P. aeruginosa infection induces assembly of the NLRP3 inflammasome, which later leads to the secretion of caspase 1 and IL-1β. Release of IL-1β leads to increased macrophage autophagy and decreased macrophage-mediated phagocytosis along with subdued NO and ROS production.38,128 One hurdle to cross would be to either find a way for the autophagy to become more beneficial in pathogen presentation or for there not to be a loss of macrophage function. Yet, other studies have revealed that P. aeruginosa triggers the TLR2/Lyn pathway, which initiates recruitment of autophagic components. They found that there is increased survival of host cells when autophagy-related phagocytosis occurs.126 Deeper understanding of autophagy or other related host response may open an avenue to attenuate virulence while maintaining immunogenicity.
New mechanistic insight into vaccine research
As mentioned above and shown in Figure 2, the main problem with vaccine research is the lack of full protection with various components targeted at P. aeruginosa. Another is to strike a balance between attenuation and immunogenicity. To achieve full protection against various P. aeruginosa clinical isolates, vaccine-based defense should cover multiple bacterial antigenic components with diverse immunologic effects generated by the host. However, there is currently only a partial understanding of the range of effectors involved in acquired immunity contributing to protection against P. aeruginosa infections in humans. A limited array of effectors may be insufficient to protect against all types of clinically relevant strains encountered by humans.121 Current data from preclinical experiments show almost all aspects of the humoral and cellular immune systems as mediators of adaptive immunity against P. aeruginosa infection.129 Mucosal induced tolerance is a major direction of research. This tolerance involves the induction of serum antibodies – specifically IgG and to a lesser extent IgA – and upregulation of the secretion of various cytokines and chemokines, such as IL-8, IL-17, IL-4, IL-10, TGF-β, and interferon gamma.130 Currently, there is debate over the Th1/Th2 balance of the immune response as to which components of the cellular immune system will provide the greatest results. While systemic vaccination, especially with vaccines that are alive, tends to induce a Th2-directed response, nasal and oral vaccination with Salmonella induces a mixed, but more so Th1-biased type of response.131 There are examples, however, such as r-b-flagellin, that elicit both Th1 and Th2 responses.37 To add more to the debate, Th17 and its IL-17 cytokine bear great promise in light of the recent discovery of their origin and function.55 But when it comes to vaccine design with Th17 in mind, there seems to be a sharp contrast with individuals suffering from chronic forms of pneumonia such as CF to more acute forms such as pneumonia. With chronic forms, IL-17 levels are elevated and may be linked to inflammation causing damage in the lungs, although with acute forms, the elevated IL-17 is needed for neutrophil recruitment and ultimately for bacterial removal.132,133
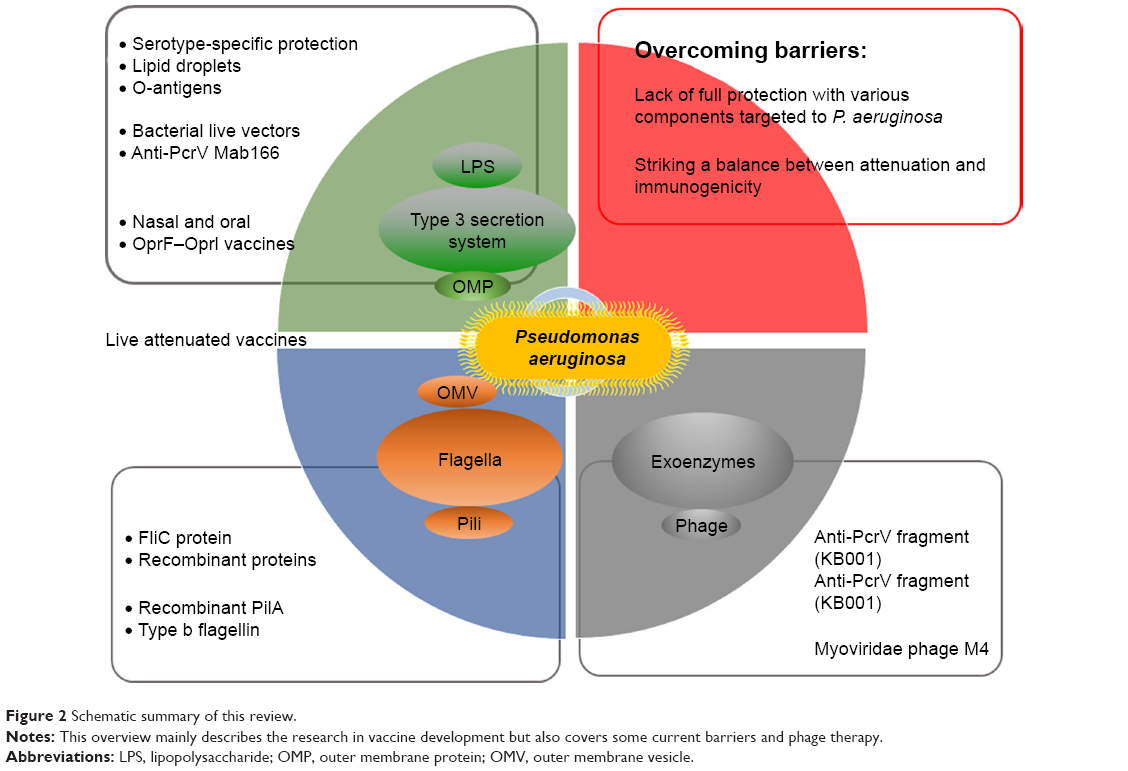 | Figure 2 Schematic summary of this review. |
A proposition can be made in this review on providing a broad range of protection. There may not be a single factor-targeting vaccine, but a combination of different vaccines to provide full or nearly full protection. The examples above were based on the r-PilA + r-b-flagellin or the phage cocktail. This could be the most viable option of closing the gap of immunity. The bottleneck is to develop a technology for easily targeting multiple components, and fortunately, the Cas gene editing tools provide a fresh air in the vaccine field.134 CRISPR/Cas has enabled the possibility of quick diagnosis of multiple pathogens.135 We may soon see more effective vaccines or preventive weapons being developed with improved CRISPR-Cas technology.
Besides working to produce a live attenuated vaccine against P. aeruginosa, researchers have also studied other aspects of the pathology involved in lower respiratory tract infection. The discovery of QS systems in Gram-negative bacteria left researchers perplexed for quite some time, because little was known about the capability that the bacteria have to communicate with each other.136 QS serves as a special form of communication in P. aeruginosa, helping in the control of biofilm production, cell aggregation, and exopolysaccharide production. QS allows P. aeruginosa to grow quietly until the population is high enough to attack host cells.137 Precise coordination by QS gives P. aeruginosa an advantage in successfully growing within the host, especially in conditions where patients have ventilator-associated pneumonia and other previous conditions.138 This is proving to be a major hurdle in finding a valid vaccine for P. aeruginosa. Many of P. aeruginosa’s virulence factors have been shown to be regulated by QS, including phospholipase C, pyocyanin, lecithinase, elastase, and rhamnolipids. It is not yet known whether live attenuated vaccines can combat this cell-to-cell communication. This may be one of the keys to unlocking a reliable prevention strategy, which should be scrutinized in vaccine research.
QS also activates three key facets of the prokaryotic adaptive immune system, also called the CRISPR-Cas system. The QS-mediated activation of CRISPR-Cas shows downregulation of phage receptors, leading to reduced ability to combat phage invasion. When a phage enters the cytoplasm of a bacterium, an immune response is activated through the CRISPR-Cas system. Modifying the CRISPR-Cas system may make bacteria more susceptible to phage therapies,139 which may point out new avenues of therapies as well as fresh challenges.
Studying other older therapies with new modern solutions has also emerged, for instance, the study of antimicrobial peptides (AMPs). The creation of synthetic analogs that mimic natural AMPs can produce a broad spectrum of protection from microbial challenges.140,141 Also, photodynamic therapy (PDT) has shown some promise in studies. This technique was discovered over 100 years ago and has been used in localized treatments of cancers and macular degeneration.142–144 With the use of protease-stable polycationic photosensitizers, PDT has been highly effective against Gram-positive bacteria. New studies have shown ways of high selectivity of Gram-negative bacteria with incubation rates as short as 1 minute.145 This shows great potential for the rapid eradication of localized infections.
Concluding remarks
Scientists have searched for P. aeruginosa vaccines for over half a century. While many vaccines, such as antitoxin antibodies, opsonophagocytic killing antibodies, and other antibodies, are under clinical trials or preclinical studies now, there is currently no product available for clinical application.78 Further understanding of the molecular pathogenesis of P. aeruginosa may hold the key to development of novel vaccines and therapies against this pathogen. There will always be new development for improved vaccine candidates. A critical recent vaccine candidate, targeting a heme-containing catalase KatA (bacterial defense against osmotic stress and H2O2 attack by phagocytes), effectively eradicates bacterial growth in cells and in mice with better outcomes than the leading vaccine candidate OprF–OprI.146 Another development is the vaccine created for serogroup B Neisseria meningitidis (MenB). MenB is a serious and frequent cause of meningitis and septicemia. Vaccines directed against MenB have been used for years by targeting the OMV,147 and new recombinant protein-based MenB vaccines, MenB-4C and MenB-FHbp,148,149 can induce a long-lasting immune response. This is encouraging, representing many ongoing efforts to find better vaccines to combat Gram-negative bacteria. These and other transformative research will advance into clinical application. Technologically, rapid and continued development of core genomics, proteomics, pan-genomics, high-throughput genomic sequencing, and reverse vaccinology will be beneficial for inventing clinically effective vaccine candidates because only the vaccine that covers multiple bacterial antigenic components with the most diverse immunologic effects generated by the host will have the potential of broad-spectrum targeting.150,151 In addition, pipelines, such as VacSol,152 VaxiJen,153 and Vaxign,154 for automating the high-throughput in silico vaccine candidates are highly promising. Peptide-based vaccines hold strong potential to develop multi-epitope-based constructs/formulation along with suitable adjuvants and linkers to target various antigenic and immunogenic epitopes.155 It is clear that we should not put all of our eggs in one basket, and with fruitful results continuing to be published, the future may look bright on finding a potential prevention as well as treatment (Figure 1).
Disclosure
The authors report no conflicts of interest in this work.
References
Kaufman MR, Jia J, Zeng L, Ha U, Chow M, Jin S. Pseudomonas aeruginosa mediated apoptosis requires the ADP-ribosylating activity of exoS. Microbiology. 2000;146(Pt 10):2531–2541. | ||
Jones RN. Microbial etiologies of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia. Clin Infect Dis. 2010;51(Suppl 1):S81–S87. | ||
Cooke GS, Hill AV. Genetics of susceptibility to human infectious disease. Nat Rev Genet. 2001;2(12):967–977. | ||
Kamei A, Coutinho-Sledge YS, Goldberg JB, Priebe GP, Pier GB. Mucosal vaccination with a multivalent, live-attenuated vaccine induces multifactorial immunity against Pseudomonas aeruginosa acute lung infection. Infect Immun. 2011;79(3):1289–1299. | ||
Chuanchuen R, Murata T, Gotoh N, Schweizer HP. Substrate-dependent utilization of OprM or OpmH by the Pseudomonas aeruginosa MexJK efflux pump. Antimicrob Agents Chemother. 2005;49(5):2133–2136. | ||
Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harb Perspect Biol. 2010;2(5):a000414. | ||
Chen Q, Ma Y, Ross AC. Opposing cytokine-specific effects of all trans-retinoic acid on the activation and expression of signal transducer and activator of transcription (STAT)-1 in THP-1 cells. Immunology. 2002;107(2):199–208. | ||
Schmiel DH, Moran EE, Keiser PB, Brandt BL, Zollinger WD. Importance of antibodies to lipopolysaccharide in natural and vaccine-induced serum bactericidal activity against Neisseria meningitidis group B. Infect Immun. 2011;79(10):4146–4156. | ||
de Mattos KA, Sarno EN, Pessolani MC, Bozza PT. Deciphering the contribution of lipid droplets in leprosy: multifunctional organelles with roles in Mycobacterium leprae pathogenesis. Mem Inst Oswaldo Cruz. 2012;107(Suppl 1):156–166. | ||
Hamilton VT, Stone DM, Cantor GH. Translocation of the B cell receptor to lipid rafts is inhibited in B cells from BLV-infected, persistent lymphocytosis cattle. Virology. 2003;315(1):135–147. | ||
Knirel YA, Bystrova OV, Kocharova NA, Zähringer U, Pier GB. Conserved and variable structural features in the lipopolysaccharide of Pseudomonas aeruginosa. J Endotoxin Res. 2006;12(6):324–336. | ||
Trent MS. Biosynthesis, transport, and modification of lipid A. Biochem Cell Biol. 2004;82(1):71–86. | ||
Gazzano-Santoro H, Parent JB, Conlon PJ, et al. Characterization of the structural elements in lipid a required for binding of a recombinant fragment of bactericidal/permeability-increasing protein rBPI23. Infect Immun. 1995;63(6):2201–2205. | ||
Qin X, Zerr DM, Mcnutt MA, Berry JE, Burns JL, Kapur RP. Pseudomonas aeruginosa syntrophy in chronically colonized airways of cystic fibrosis patients. Antimicrob Agents Chemother. 2012;56(11):5971–5981. | ||
Arbatsky NP, Drzewiecka D, Palusiak A, et al. Structure of a Kdo-containing O polysaccharide representing Proteus O79, a newly described serogroup for some clinical Proteus genomospecies isolates from Poland. Carbohydr Res. 2013;379:100–105. | ||
Sharma A, Krause A, Worgall S. Recent developments for Pseudomonas vaccines. Hum Vaccin. 2011;7(10):999–1011. | ||
Vincent JL. Vaccine development and passive immunization for Pseudomonas aeruginosa in critically ill patients: a clinical update. Future Microbiol. 2014;9(4):457–463. | ||
Taniguchi Y, Nishizawa T, Honda T, et al. Development and potential use of a monoclonal antibody to the lipopolysaccharide of Pantoea agglomerans (IP-PA1). Anticancer Res. 2007;27(6A):3701–3706. | ||
Digiandomenico A, Rao J, Harcher K, et al. Intranasal immunization with heterologously expressed polysaccharide protects against multiple Pseudomonas aeruginosa infections. Proc Natl Acad Sci U S A. 2007;104(11):4624–4629. | ||
Najafzadeh F, Shapouri R, Rahnema M, Rokhsartalab Azar S, Kianmehr A. Pseudomonas aeruginosa PAO-1 lipopolysaccharide–diphtheria toxoid conjugate vaccine: preparation, characterization and immunogenicity. Jundishapur J Microbiol. 2015;8(6):e17712. | ||
Hemachandra S, Kamboj K, Copfer J, Pier G, Green LL, Schreiber JR. Human monoclonal antibodies against Pseudomonas aeruginosa lipopolysaccharide derived from transgenic mice containing megabase human immunoglobulin loci are opsonic and protective against fatal Pseudomonas sepsis. Infect Immun. 2001;69(4):2223–2229. | ||
Allewelt M, Coleman FT, Grout M, Priebe GP, Pier GB. Acquisition of expression of the Pseudomonas aeruginosa ExoU cytotoxin leads to increased bacterial virulence in a murine model of acute pneumonia and systemic spread. Infect Immun. 2000;68(7):3998–4004. | ||
Shirley DA, Mcarthur MA. The utility of human challenge studies in vaccine development: lessons learned from cholera. Vaccine. 2011;2011(1):3–13. | ||
Dagan R, Givon-Lavi N, Greenberg D, Fritzell B, Siegrist CA. Nasopharyngeal carriage of Streptococcus pneumoniae shortly before vaccination with a pneumococcal conjugate vaccine causes serotype-specific hyporesponsiveness in early infancy. J Infect Dis. 2010;201(10):1570–1579. | ||
Nagy G, Palkovics T, Otto A, et al. “Gently rough”: the vaccine potential of a Salmonella enterica regulatory lipopolysaccharide mutant. J Infect Dis. 2008;198(11):1699–1706. | ||
Que YA, Lazar H, Wolff M, et al. Assessment of panobacumab as adjunctive immunotherapy for the treatment of nosocomial Pseudomonas aeruginosa pneumonia. Eur J Clin Microbiol Infect Dis. 2014;33(10):1861–1867. | ||
Dennehy R, Mcclean S. Immunoproteomics: the key to discovery of new vaccine antigens against bacterial respiratory infections. Curr Protein Pept Sci. 2012;13(8):807–815. | ||
Hales BA, Morgan JA, Hart CA, Winstanley C. Variation in flagellin genes and proteins of Burkholderia cepacia. J Bacteriol. 1998;180(5):1110–1118. | ||
Harms A, Dehio C. Intruders below the radar: molecular pathogenesis of Bartonella spp. Clin Microbiol Rev. 2012;25(1):42–78. | ||
Burrows LL. Pseudomonas aeruginosa twitching motility: type IV pili in action. Annu Rev Microbiol. 2012;66:493–520. | ||
Zhang J, Xu K, Ambati B, Yu FS. Toll-like receptor 5-mediated corneal epithelial inflammatory responses to Pseudomonas aeruginosa flagellin. Invest Ophthalmol Vis Sci. 2003;44(10):4247–4254. | ||
de Zoete MR, van Putten JP, Wagenaar JA. Vaccination of chickens against Campylobacter. Vaccine. 2007;25(30):5548–5557. | ||
Hegerle N, Choi M, Sinclair J, et al. Development of a broad spectrum glycoconjugate vaccine to prevent wound and disseminated infections with Klebsiella pneumoniae and Pseudomonas aeruginosa. PLoS One. 2018;13(9):e0203143. | ||
Yang J, Zhang E, Liu F, et al. Flagellins of Salmonella typhi and nonpathogenic Escherichia coli are differentially recognized through the NLRC4 pathway in macrophages. J Innate Immun. 2014;6(1):47–57. | ||
Campodónico VL, Llosa NJ, Bentancor LV, Maira-Litran T, Pier GB. Efficacy of a conjugate vaccine containing polymannuronic acid and flagellin against experimental Pseudomonas aeruginosa lung infection in mice. Infect Immun. 2011;79(8):3455–3464. | ||
Farajnia S, Peerayeh SN, Tanomand A, et al. Protective efficacy of recombinant exotoxin A–flagellin fusion protein against Pseudomonas aeruginosa infection. Can J Microbiol. 2015;61(1):60–64. | ||
Behrouz B, Mahdavi M, Amirmozafari N, et al. Immunogenicity of Pseudomonas aeruginosa recombinant B-type fagellin as a vaccine candidate: protective efficacy in a murine burn wound sepsis model. Burns. Epub 2016 May 2. | ||
Wu Y, Li D, Wang Y, et al. Pseudomonas aeruginosa promotes autophagy to suppress macrophage-mediated bacterial eradication. Int Immunopharmacol. 2016;38:214–222. | ||
Kipnis E, Sawa T, Wiener-Kronish J. Targeting mechanisms of Pseudomonas aeruginosa pathogenesis. Med Mal Infect. 2006;36(2):78–91. | ||
Tashiro Y, Yawata Y, Toyofuku M, Uchiyama H, Nomura N. Interspecies interaction between Pseudomonas aeruginosa and other microorganisms. Microbes Environ. 2013;28(1):13–24. | ||
Silverman JM, Agnello DM, Zheng H, et al. Haemolysin coregulated protein is an exported receptor and chaperone of type VI secretion substrates. Mol Cell. 2013;51(5):584–593. | ||
del Tordello E, Danilchanka O, Mccluskey AJ, Mekalanos JJ. Type VI secretion system sheaths as nanoparticles for antigen display. Proc Natl Acad Sci U S A. 2016;113(11):3042–3047. | ||
Fernandes PJ, Guo Q, Waag DM, Donnenberg MS. The type IV pilin of Burkholderia mallei is highly immunogenic but fails to protect against lethal aerosol challenge in a murine model. Infect Immun. 2007;75(6):3027–3032. | ||
Kostakioti M, Hadjifrangiskou M, Hultgren SJ. Bacterial biofilms: development, dispersal, and therapeutic strategies in the dawn of the postantibiotic era. Cold Spring Harb Perspect Med. 2013;3(4):a010306. | ||
Marko VA, Kilmury SLN, Macneil LT, Burrows LL. Pseudomonas aeruginosa type IV minor pilins and PilY1 regulate virulence by modulating FimS-AlgR activity. PLoS Pathog. 2018;14(5):e1007074. | ||
Silverblatt FJ. Host–parasite interaction in the rat renal pelvis: a possible role for pili in the pathogenesis of pyelonephritis. J Exp Med. 1974;140(6):1696–1711. | ||
Darling KE, Evans TJ. Effects of nitric oxide on Pseudomonas aeruginosa infection of epithelial cells from a human respiratory cell line derived from a patient with cystic fibrosis. Infect Immun. 2003;71(5):2341–2349. | ||
Al-Mujaini A, Al-Kharusi N, Thakral A, Wali UK. Bacterial keratitis: perspective on epidemiology, clinico-pathogenesis, diagnosis and treatment. Sultan Qaboos Univ Med J. 2009;9(2):184–195. | ||
Horzempa J, Held TK, Cross AS, et al. Immunization with a Pseudomonas aeruginosa 1244 pilin provides O-antigen-specific protection. Clin Vaccine Immunol. 2008;15(4):590–597. | ||
Cigana C, Lorè NI, Bernardini ML, Bragonzi A. Dampening host sensing and avoiding recognition in Pseudomonas aeruginosa pneumonia. J Biomed Biotechnol. 2011;2011:1–10. | ||
Kube DM, Fletcher D, Davis PB. Relation of exaggerated cytokine responses of CF airway epithelial cells to PAO1 adherence. Respir Res. 2005;6:69. | ||
Kumar V, Sharma A. Neutrophils: Cinderella of innate immune system. Int Immunopharmacol. 2010;10(11):1325–1334. | ||
Kung VL, Ozer EA, Hauser AR. The accessory genome of Pseudomonas aeruginosa. Microbiol Mol Biol Rev. 2010;74(4):621–641. | ||
Priebe GP, Walsh RL, Cederroth TA, et al. IL-17 is a critical component of vaccine-induced protection against lung infection by lipopolysaccharide-heterologous strains of Pseudomonas aeruginosa. J Immunol. 2008;181(7):4965–4975. | ||
Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361(9):888–898. | ||
Sadikot RT, Blackwell TS, Christman JW, Prince AS. Pathogen–host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med. 2005;171(11):1209–1223. | ||
Roach SK, Schorey JS. Differential regulation of the mitogen-activated protein kinases by pathogenic and nonpathogenic mycobacteria. Infect Immun. 2002;70(6):3040–3052. | ||
Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. 2002;15(2):194–222. | ||
Korpi F, Irajian G, Mahadavi M, et al. Active immunization with recombinant Pila protein protects against Pseudomonas aeruginosa infection in a mouse burn wound model. J Microbiol Biotechnol. Epub 2015 Sep 18. | ||
Korpi F, Hashemi FB, Irajian G, Fatemi MJ, Laghaei P, Behrouz B. Flagellin and pilin immunization against multi-drug resistant Pseudomonas aeruginosa protects mice in the burn wound sepsis model. Immunol Lett. 2016;176:8–17. | ||
Kroken AR, Chen CK, Evans DJ, Yahr TL, Fleiszig SMJ. The impact of ExoS on Pseudomonas aeruginosa internalization by epithelial cells is independent of fleQ and correlates with bistability of type three secretion system gene expression. MBio. 2018;9(3):e00668. | ||
Lee VT, Smith RS, Tümmler B, Lory S. Activities of Pseudomonas aeruginosa effectors secreted by the type III secretion system in vitro and during infection. Infect Immun. 2005;73(3):1695–1705. | ||
Pukatzki S, Ma AT, Revel AT, Sturtevant D, Mekalanos JJ. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc Natl Acad Sci U S A. 2007;104(39):15508–15513. | ||
Hauser AR, Jain M, Bar-Meir M, Mccolley SA. Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin Microbiol Rev. 2011;24(1):29–70. | ||
Sato H, Frank DW. Multi-functional characteristics of the Pseudomonas aeruginosa type III needle-tip protein, PcrV; comparison to orthologs in other gram-negative bacteria. Front Microbiol. 2011;2:142. | ||
Jones CJ, Wozniak DJ. Psl produced by mucoid Pseudomonas aeruginosa contributes to the establishment of biofilms and immune evasion. MBio. 2017;8(3):e00864-17. | ||
Digiandomenico A, Keller AE, Gao C, et al. A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci Transl Med. 2014;6(262):262ra155. | ||
Le HN, Quetz JS, Tran VG, et al. MEDI3902 correlates of protection against severe Pseudomonas aeruginosa pneumonia in a rabbit acute pneumonia model. Antimicrobial Agents Chemother. 2018;62(5):e02565-17. | ||
Epaulard O, Derouazi M, Margerit C, et al. Optimization of a type III secretion system-based Pseudomonas aeruginosa live vector for antigen delivery. Clin Vaccine Immunol. 2008;15(2):308–313. | ||
O’Donnell H, Mcsorley SJ. Salmonella as a model for non-cognate Th1 cell stimulation. Front Immunol. 2014;5:621. | ||
Jones MA, Hulme SD, Barrow PA, Wigley P. The Salmonella pathogenicity island 1 and Salmonella pathogenicity island 2 type III secretion systems play a major role in pathogenesis of systemic disease and gastrointestinal tract colonization of Salmonella enterica serovar Typhimurium in the chicken. Avian Pathol. 2007;36(3):199–203. | ||
Wang Y, Gouellec AL, Chaker H, Asrih H, Polack B, Toussaint B. Optimization of antitumor immunotherapy mediated by type III secretion system-based live attenuated bacterial vectors. J Immunother. 2012;35(3):223–234. | ||
Derouazi M, Wang Y, Marlu R, et al. Optimal epitope composition after antigen screening using a live bacterial delivery vector. Bioeng Bugs. 2010;1(1):51–60. | ||
Sawa T, Ito E, Nguyen VH, Haight M. Anti-PcrV antibody strategies against virulent Pseudomonas aeruginosa. Hum Vaccin Immunother. 2014;10(10):2843–2852. | ||
Wu W, Huang J, Duan B, et al. Th17-stimulating protein vaccines confer protection against Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med. 2012;186(5):420–427. | ||
Thomas LD, Kyd JM, Bastin DA, Dunkley ML, Cripps AW. Immunisation with non-integral OMPs promotes pulmonary clearance of Pseudomonas aeruginosa. FEMS Immunol Med Microbiol. 2003;37(2–3):155–160. | ||
Westritschnig K, Hochreiter R, Wallner G, Firbas C, Schwameis M, Jilma B. A randomized, placebo-controlled Phase I study assessing the safety and immunogenicity of a Pseudomonas aeruginosa hybrid outer membrane protein OprF/I vaccine (IC43) in healthy volunteers. Hum Vaccin Immunother. 2014;10(1):170–183. | ||
Merakou C, Schaefers MM, Priebe GP. Progress toward the elusive Pseudomonas aeruginosa vaccine. Surg Infect (Larchmt). Epub 2018 Nov 15. | ||
Liu C, Pan X, Xia B, et al. Construction of a protective vaccine against lipopolysaccharide-heterologous Pseudomonas aeruginosa strains based on expression profiling of outer membrane proteins during infection. Front Immunol. 2018;9:1737. | ||
Stover CK, Pham XQ, Erwin AL, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406(6799):959–964. | ||
Arnold H, Bumann D, Felies M, et al. Enhanced immunogenicity in the murine airway mucosa with an attenuated Salmonella live vaccine expressing OprF–OprI from Pseudomonas aeruginosa. Infect Immun. 2004;72(11):6546–6553. | ||
Göcke K, Baumann U, Hagemann H, et al. Mucosal vaccination with a recombinant OprF-I vaccine of Pseudomonas aeruginosa in healthy volunteers: comparison of a systemic vs a mucosal booster schedule. FEMS Immunol Med Microbiol. 2003;37(2–3):167–171. | ||
Rudin A, Riise GC, Holmgren J. Antibody responses in the lower respiratory tract and male urogenital tract in humans after nasal and oral vaccination with cholera toxin B subunit. Infect Immun. 1999;67(6):2884–2890. | ||
Bumann D, Behre C, Behre K, et al. Systemic, nasal and oral live vaccines against Pseudomonas aeruginosa: a clinical trial of immunogenicity in lower airways of human volunteers. Vaccine. 2010;28(3):707–713. | ||
Lyczak JB. Commensal bacteria increase invasion of intestinal epithelium by Salmonella enterica serovar Typhi. Infect Immun. 2003;71(11):6610–6614. | ||
Westritschnig K, Hochreiter R, Wallner G, Firbas C, Schwameis M, Jilma B. A randomized, placebo-controlled Phase I study assessing the safety and immunogenicity of a Pseudomonas aeruginosa hybrid outer membrane protein OprF/I vaccine (IC43) in healthy volunteers. Hum Vaccin Immunother. 2014;10(1):170–183. | ||
Döring G, Meisner C, Stern M; Flagella Vaccine Trial Study Group. A double-blind randomized placebo-controlled Phase III study of a Pseudomonas aeruginosa flagella vaccine in cystic fibrosis patients. Proc Natl Acad Sci U S A. 2007;104(26):11020–11025. | ||
Bomberger JM, Maceachran DP, Coutermarsh BA, Ye S, O’Toole GA, Stanton BA. Long-distance delivery of bacterial virulence factors by Pseudomonas aeruginosa outer membrane vesicles. PLoS Pathog. 2009;5(4):e1000382. | ||
Ellis TN, Leiman SA, Kuehn MJ. Naturally produced outer membrane vesicles from Pseudomonas aeruginosa elicit a potent innate immune response via combined sensing of both lipopolysaccharide and protein components. Infect Immun. 2010;78(9):3822–3831. | ||
Lee DH, Kim SH, Kang W, et al. Adjuvant effect of bacterial outer membrane vesicles with penta-acylated lipopolysaccharide on antigen-specific T cell priming. Vaccine. 2011;29(46):8293–8301. | ||
Zughaier S, Steeghs L, van der Ley P, Stephens DS. TLR4-dependent adjuvant activity of Neisseria meningitidis lipid A. Vaccine. 2007;25(22):4401–4409. | ||
Gorringe A, Halliwell D, Matheson M, Reddin K, Finney M, Hudson M. The development of a meningococcal disease vaccine based on Neisseria lactamica outer membrane vesicles. Vaccine. 2005;23(17–18):2210–2213. | ||
Fransen F, Stenger RM, Poelen MC, et al. Differential effect of TLR2 and TLR4 on the immune response after immunization with a vaccine against Neisseria meningitidis or Bordetella pertussis. PLoS One. 2010;5(12):e15692. | ||
Morley SL, Pollard AJ. Vaccine prevention of meningococcal disease, coming soon? Vaccine. 2001;20(5–6):666–687. | ||
Oliver KJ, Reddin KM, Bracegirdle P, et al. Neisseria lactamica protects against experimental meningococcal infection. Infect Immun. 2002;70(7):3621–3626. | ||
Gorringe AR, Taylor S, Brookes C, et al. Phase I safety and immunogenicity study of a candidate meningococcal disease vaccine based on Neisseria lactamica outer membrane vesicles. Clin Vaccine Immunol. 2009;16(8):1113–1120. | ||
Clokie MR, Millard AD, Letarov AV, Heaphy S. Phages in nature. Bacteriophage. 2011;1(1):31–45. | ||
Forti F, Roach DR, Cafora M, et al. Design of a broad-range bacteriophage cocktail that reduces Pseudomonas aeruginosa biofilms and treats acute infections in two animal models. Antimicrob Agents Chemother. 2018;62(6): pii:e02573-17. | ||
Guttman BRR, Kutter E. Basic phage biology. Boca Raton, FL: CRC Press; 2005. | ||
d’Herelle F. Sur un microbe invisible antagoniste des bacilles dys-entériques. C R Acad Sci Hebd Seances Acad Sci D. 1917(165):373–375. | ||
Sulakvelidze A, Alavidze Z, Morris JG. Bacteriophage therapy. Antimicrob Agents Chemother. 2001;45(3):649–659. | ||
Fu W, Forster T, Mayer O, Curtin JJ, Lehman SM, Donlan RM. Bacteriophage cocktail for the prevention of biofilm formation by Pseudomonas aeruginosa on catheters in an in vitro model system. Antimicrob Agents Chemother. 2010;54(1):397–404. | ||
Pires D, Sillankorva S, Faustino A, Azeredo J. Use of newly isolated phages for control of Pseudomonas aeruginosa PAO1 and ATCC 10145 biofilms. Res Microbiol. 2011;162(8):798–806. | ||
Morello E, Saussereau E, Maura D, Huerre M, Touqui L, Debarbieux L. Pulmonary bacteriophage therapy on Pseudomonas aeruginosa cystic fibrosis strains: first steps towards treatment and prevention. PLoS One. 2011;6(2):e16963. | ||
Torres-Barceló C, Arias-Sánchez FI, Vasse M, Ramsayer J, Kaltz O, Hochberg ME. A window of opportunity to control the bacterial pathogen Pseudomonas aeruginosa combining antibiotics and phages. PLoS One. 2014;9(9):e106628. | ||
Chan BK, Abedon ST, Loc-Carrillo C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013;8(6):769–783. | ||
Hall AR, De Vos D, Friman VP, Pirnay JP, Buckling A. Effects of sequential and simultaneous applications of bacteriophages on populations of Pseudomonas aeruginosa in vitro and in wax moth larvae. Appl Environ Microbiol. 2012;78(16):5646–5652. | ||
Wright A, Hawkins CH, Anggård EE, Harper DR. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; a preliminary report of efficacy. Clin Otolaryngol. 2009;34(4):349–357. | ||
Marza JA, Soothill JS, Boydell P, Collyns TA. Multiplication of therapeutically administered bacteriophages in Pseudomonas aeruginosa infected patients. Burns. 2006;32(5):644–646. | ||
Merabishvili M, Pirnay JP, Verbeken G, et al. Quality-controlled small-scale production of a well-defined bacteriophage cocktail for use in human clinical trials. PLoS One. 2009;4(3):e4944. | ||
Li R, Fang L, Tan S, et al. Type I CRISPR-Cas targets endogenous genes and regulates virulence to evade mammalian host immunity. Cell Res. 2016;26(12):1273–1287. | ||
Bondy-Denomy J, Garcia B, Strum S, et al. Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature. 2015;526(7571):136–139. | ||
Cady KC, Bondy-Denomy J, Heussler GE, Davidson AR, O’Toole GA. The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J Bacteriol. 2012;194(21):5728–5738. | ||
Maxwell KL, Garcia B, Bondy-Denomy J, Bona D, Hidalgo-Reyes Y, Davidson AR. The solution structure of an anti-CRISPR protein. Nat Commun. 2016;7(1):13134. | ||
Pawluk A, Staals RH, Taylor C, et al. Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat Microbiol. 2016;1(8):16085. | ||
Bikard D, Euler CW, Jiang W, et al. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat Biotechnol. 2014;32(11):1146–1150. | ||
Citorik RJ, Mimee M, Lu TK. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat Biotechnol. 2014;32(11):1141–1145. | ||
Barbu EM, Cady KC, Hubby B. Phage therapy in the era of synthetic biology. Cold Spring Harb Perspect Biol. 2016;8(10):a023879. | ||
Labrie SJ, Samson JE, Moineau S. Bacteriophage resistance mechanisms. Nat Rev Microbiol. 2010;8(5):317–327. | ||
Zaidi TS, Priebe GP, Pier GB. A live-attenuated Pseudomonas aeruginosa vaccine elicits outer membrane protein-specific active and passive protection against corneal infection. Infect Immun. 2006;74(2):975–983. | ||
Priebe GP, Meluleni GJ, Coleman FT, Goldberg JB, Pier GB. Protection against fatal Pseudomonas aeruginosa pneumonia in mice after nasal immunization with a live, attenuated aroA deletion mutant. Infect Immun. 2003;71(3):1453–1461. | ||
Robinson CM, Kobe BN, Schmitt DM, et al. Genetic engineering of Francisella tularensis LVS for use as a novel live vaccine platform against Pseudomonas aeruginosa infections. Bioengineered. 2015;6(2):82–88. | ||
Ng TW, Saavedra-Ávila NA, Kennedy SC, Carreño LJ, Porcelli SA. Current efforts and future prospects in the development of live mycobacteria as vaccines. Expert Rev Vaccines. 2015;14(11):1493–1507. | ||
Chauchet X, Hannani D, Djebali S, et al. Poly-functional and long-lasting anticancer immune response elicited by a safe attenuated Pseudomonas aeruginosa vector for antigens delivery. Mol Ther Oncolytics. 2016;3:16033. | ||
Shippy DC, Fadl AA. Immunological characterization of a gidA mutant strain of Salmonella for potential use in a live-attenuated vaccine. BMC Microbiol. 2012;12(1):286. | ||
Li X, He S, Zhou X, et al. Lyn delivers bacteria to lysosomes for eradication through TLR2-initiated autophagy related phagocytosis. PLoS Pathog. 2016;12(1):e1005363. | ||
Yuan K, Huang C, Fox J, et al. Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J Cell Sci. 2012;125(Pt 2):507–515. | ||
Deng Q, Wang Y, Zhang Y, et al. Pseudomonas aeruginosa triggers macrophage autophagy to escape intracellular killing by activation of the NLRP3 inflammasome. Infect Immun. 2016;84(1):56–66. | ||
König W. Role of the immune system in recovery from infection. Respiration. 1993;60(1):16–24. | ||
Winkler P, Ghadimi D, Schrezenmeir J, Kraehenbuhl JP. Molecular and cellular basis of microflora–host interactions. J Nutr. 2007;137(3 Suppl 2):756S–772S. | ||
Capozzo AV, Cuberos L, Levine MM, Pasetti MF. Mucosally delivered Salmonella live vector vaccines elicit potent immune responses against a foreign antigen in neonatal mice born to naive and immune mothers. Infect Immun. 2004;72(8):4637–4646. | ||
Dubin PJ, Kolls JK. IL-23 mediates inflammatory responses to mucoid Pseudomonas aeruginosa lung infection in mice. Am J Physiol Lung Cell Mol Physiol. 2007;292(2):L519–L528. | ||
Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol. 2007;179(6):4135–4141. | ||
Fan S, Xu X, Liao Y, et al. Attenuated phenotype and immunogenic characteristics of a mutated herpes simplex virus 1 strain in the rhesus macaque. Viruses. 2018;10(5):234. | ||
Gootenberg JS, Abudayyeh OO, Kellner MJ, Joung J, Collins JJ, Zhang F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. 2018;360(6387):439–444. | ||
de Kievit TR, Iglewski BH. Bacterial quorum sensing in pathogenic relationships. Infect Immun. 2000;68(9):4839–4849. | ||
Jimenez PN, Koch G, Thompson JA, Xavier KB, Cool RH, Quax WJ. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol Mol Biol Rev. 2012;76(1):46–65. | ||
Abu-Salah T, Dhand R. Inhaled antibiotic therapy for ventilator-associated tracheobronchitis and ventilator-associated pneumonia: an update. Adv Ther. 2011;28(9):728–747. | ||
Høyland-Kroghsbo NM, Paczkowski J, Mukherjee S, et al. Quorum sensing controls the Pseudomonas aeruginosa CRISPR-Cas adaptive immune system. Proc Natl Acad Sci U S A. 2017;114(1):131–135. | ||
Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24(12):1551–1557. | ||
Giuliani A, Pirri G, Bozzi A, di Giulio A, Aschi M, Rinaldi AC. Antimicrobial peptides: natural templates for synthetic membrane-active compounds. Cell Mol Life Sci. 2008;65(16):2450–2460. | ||
Moan J, Peng Q. An outline of the hundred-year history of PDT. Anticancer Res. 2003;23(5):3591–3600. | ||
Dolmans DE, Fukumura D, Jain RK. Photodynamic therapy for cancer. Nat Rev Cancer. 2003;3(5):380–387. | ||
Brown SB, Mellish KJ. Verteporfin: a milestone in ophthalmology and photodynamic therapy. Expert Opin Pharmacother. 2001;2(2):351–361. | ||
Hamblin MR, O’Donnell DA, Murthy N, et al. Polycationic photosensitizer conjugates: effects of chain length and gram classification on the photodynamic inactivation of bacteria. J Antimicrob Chemother. 2002;49(6):941–951. | ||
Grimwood K, Kyd JM, Owen SJ, Massa HM, Cripps AW. Vaccination against respiratory Pseudomonas aeruginosa infection. Hum Vaccin Immunother. 2015;11(1):14–20. | ||
Balmer P, York LJ. Optimal use of meningococcal serogroup B vaccines: moving beyond outbreak control. Ther Adv Vaccines Immunother. 2018;6(3):49–60. | ||
Green LR, Lucidarme J, Dave N, et al. Phase variation of NadA in invasive Neisseria meningitidis isolates impacts on coverage estimates for 4C-MenB, a MenB vaccine. J Clin Microbiol. 2018;56(9):e00204-18. | ||
Harris SL, Tan C, Andrew L, et al. The bivalent factor H binding protein meningococcal serogroup B vaccine elicits bactericidal antibodies against representative non-serogroup B meningococci. Vaccine. 2018;36(45):6867–6874. | ||
Hassan A, Naz A, Obaid A, et al. Pangenome and immuno-proteomics analysis of Acinetobacter baumannii strains revealed the core peptide vaccine targets. BMC Genomics. 2016;17(1):732. | ||
Soares SC, Silva A, Trost E, et al. The pan-genome of the animal pathogen Corynebacterium pseudotuberculosis reveals differences in genome plasticity between the biovar ovis and equi strains. PLoS One. 2013;8(1):e53818. | ||
Rizwan M, Naz A, Ahmad J, et al. VacSol: a high throughput in silico pipeline to predict potential therapeutic targets in prokaryotic pathogens using subtractive reverse vaccinology. BMC Bioinformatics. 2017;18(1):106. | ||
Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007;8(1):4. | ||
He Y, Xiang Z, Mobley HL. Vaxign: the first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J Biomed Biotechnol. 2010;2010(3–4):1–15. | ||
Skwarczynski M, Toth I. Peptide-based synthetic vaccines. Chem Sci. 2016;7(2):842–854. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.