")
Back to Journals » OncoTargets and Therapy » Volume 11
Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma
Authors Kakadia S, Yarlagadda N, Awad R, Kundranda M, Niu J, Naraev B, Mina L, Dragovich T, Gimbel M, Mahmoud F
Received 5 August 2018
Accepted for publication 19 September 2018
Published 17 October 2018 Volume 2018:11 Pages 7095—7107
DOI https://doi.org/10.2147/OTT.S182721
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Sunilkumar Kakadia,1 Naveen Yarlagadda,1 Ramez Awad,2 Madappa Kundranda,3 Jiaxin Niu,3 Boris Naraev,3 Lida Mina,3 Tomislav Dragovich,3 Mark Gimbel,3 Fade Mahmoud3
1Department of Internal Medicine, Division of Hematology and Oncology, University of Arkansas for Medical Sciences, Little Rock, AR, USA; 2Department of Internal Medicine, University of Arkansas for Medical Sciences, Little Rock, AR, USA; 3TW Lewis Melanoma Center of Excellence, Banner MD Anderson Cancer Center, Gilbert, AZ, USA
Abstract: Approximately 50% of melanomas harbor an activating BRAF mutation. Combined BRAF and MEK inhibitors such as dabrafenib and trametinib, vemurafenib and cobimetinib, and encorafenib and binimetinib are US Food and Drug Administration (FDA)-approved to treat patients with BRAFV600-mutated advanced melanoma. Both genetic and epigenetic alterations play a major role in resistance to BRAF inhibitors by reactivation of the MAPK and/or the PI3K–Akt pathways. The role of BRAF inhibitors in modulating the immunomicroenvironment and perhaps enhancing the efficacy of checkpoint inhibitors is gaining interest. This article provides a comprehensive review of mechanisms of resistance to BRAF and MEK inhibitors in melanoma and summarizes landmark trials that led to the FDA approval of BRAF and MEK inhibitors in metastatic melanoma.
Keywords: malignant melanoma, targeted therapy, BRAF inhibitor, MEK inhibitor, resistance
Introduction
Nearly half of patients with metastatic melanomas harbor a valine–glutamine substitution in codon 600 of the serine/threonine kinase BRAF (BRAFV600 mutation).1 These melanomas have all the features of oncogene addiction to the BRAF-mutated gene (Figure 1).2 Vemurafenib, previously known as PLX4032 or RG7204, dabrafenib, known as GSK2118436, and encorafenib are BRAF inhibitors (BRAFi) approved by the US Food and Drug Administration (FDA) in combination with MEK inhibitors to treat patients with BRAFV600E/K-mutated metastatic melanomas.3,4 BRAFi result in high response rates; however, responses are short-lived, with a median time to progression of 5.1–8.8 months.3,5–7 The addition of an MEK inhibitor to a BRAFi extends the median duration of response from 5.6 months to 9.5 months.8,9 Genetic and/or epigenetic changes in melanoma cells (Figure 2) via reactivation of the MAPK pathway and to a lesser extent the PI3K–Akt pathway play a crucial role in acquired resistance to BRAFi10–14 and contribute extensively to tumor heterogeneity.15 There is an intense effort to better understand mechanisms of resistance to BRAFi and develop new agents that target areas of resistance.16–19 Moreover, the role of BRAFi in enhancing immunoresponses and boosting the efficacy of checkpoint inhibitors is an area of extensive research. This article provides a comprehensive review of mechanisms of resistance and summarizes landmark trials that led to the approval of BRAF and MEK inhibitors in metastatic melanoma. We will briefly discuss how BRAFi could modulate the tumor microenvironment and enhance immunoresponses.
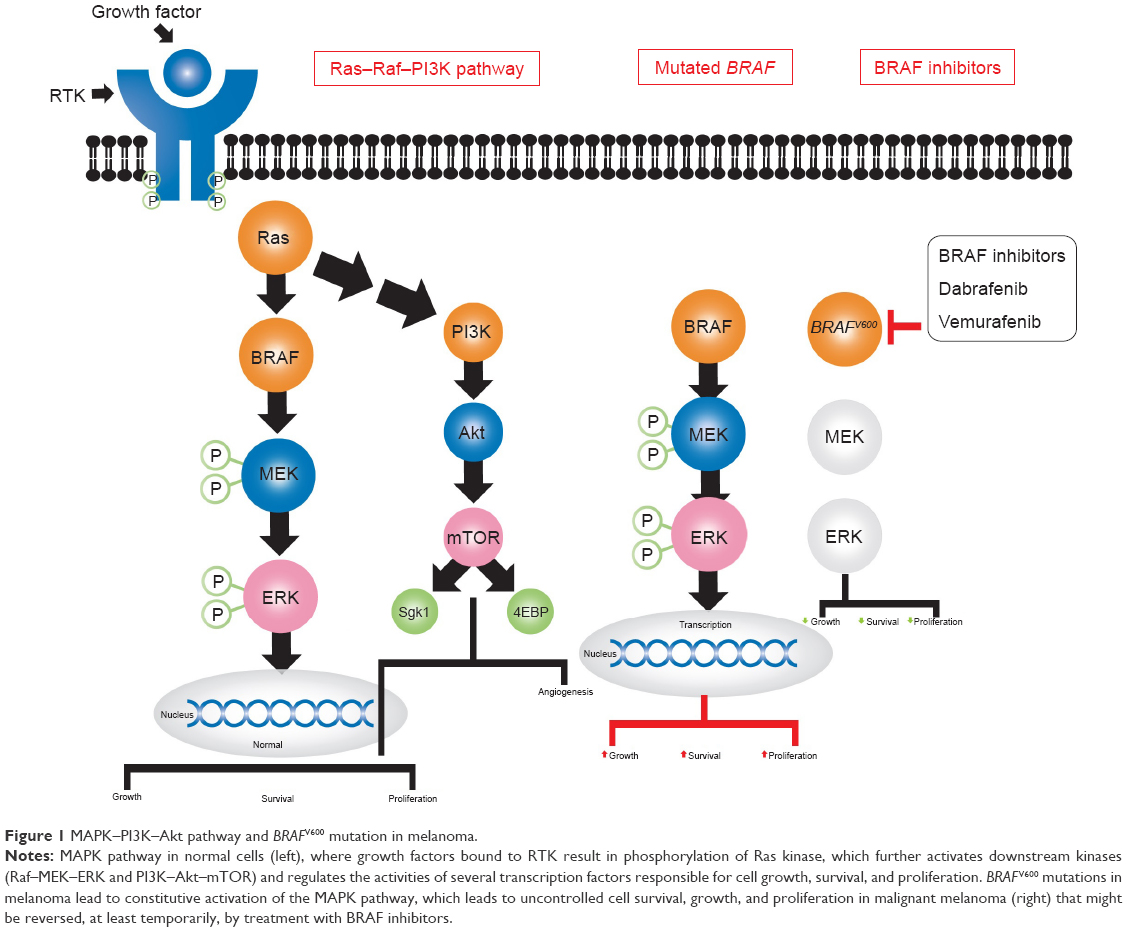 | Figure 1 MAPK–PI3K–Akt pathway and BRAFV600 mutation in melanoma. |
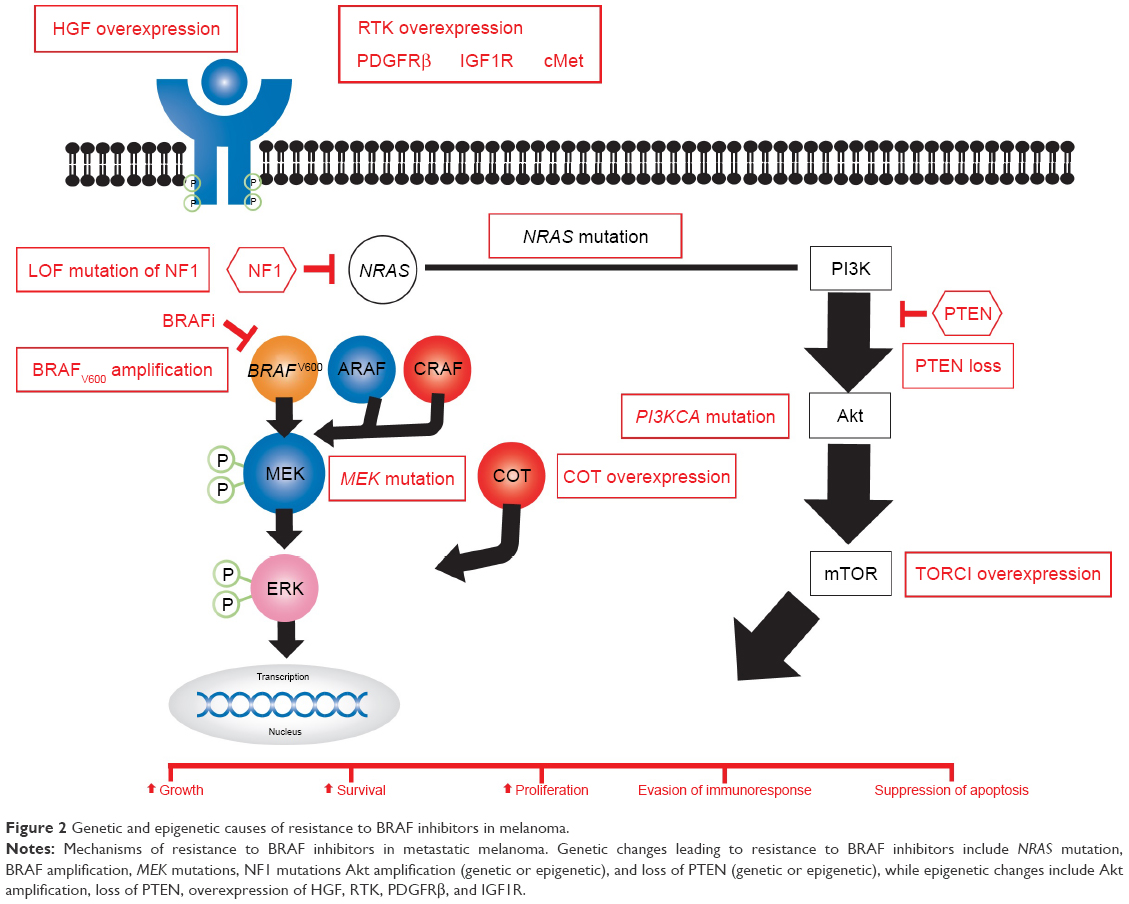 | Figure 2 Genetic and epigenetic causes of resistance to BRAF inhibitors in melanoma. |
Genetic causes of resistance to BRAFi in melanoma
MAPK-pathway activation is a fundamental step in several intracellular processes, including cell growth and differentiation. Physiological upstream negative feedback prevents persistent MAPK-pathway activation in normal cells, but this is lost in melanoma cells that harbor the BRAFV600 mutations, leading to constitutive activation of the MAPK pathway.11
Whole-exome sequencing of serial melanoma biopsies obtained at baseline and upon progression reveal a spectrum of genetic alterations in ~51%–58% of patients with BRAFV600-mutated metastatic melanoma who receive vemurafenib or dabrafenib (Table 1). These genetic alterations (Figure 2) mainly result in reactivation of MAPK and to lesser extent activation of PI3K–Akt pathways.10,11,15,16,20–26
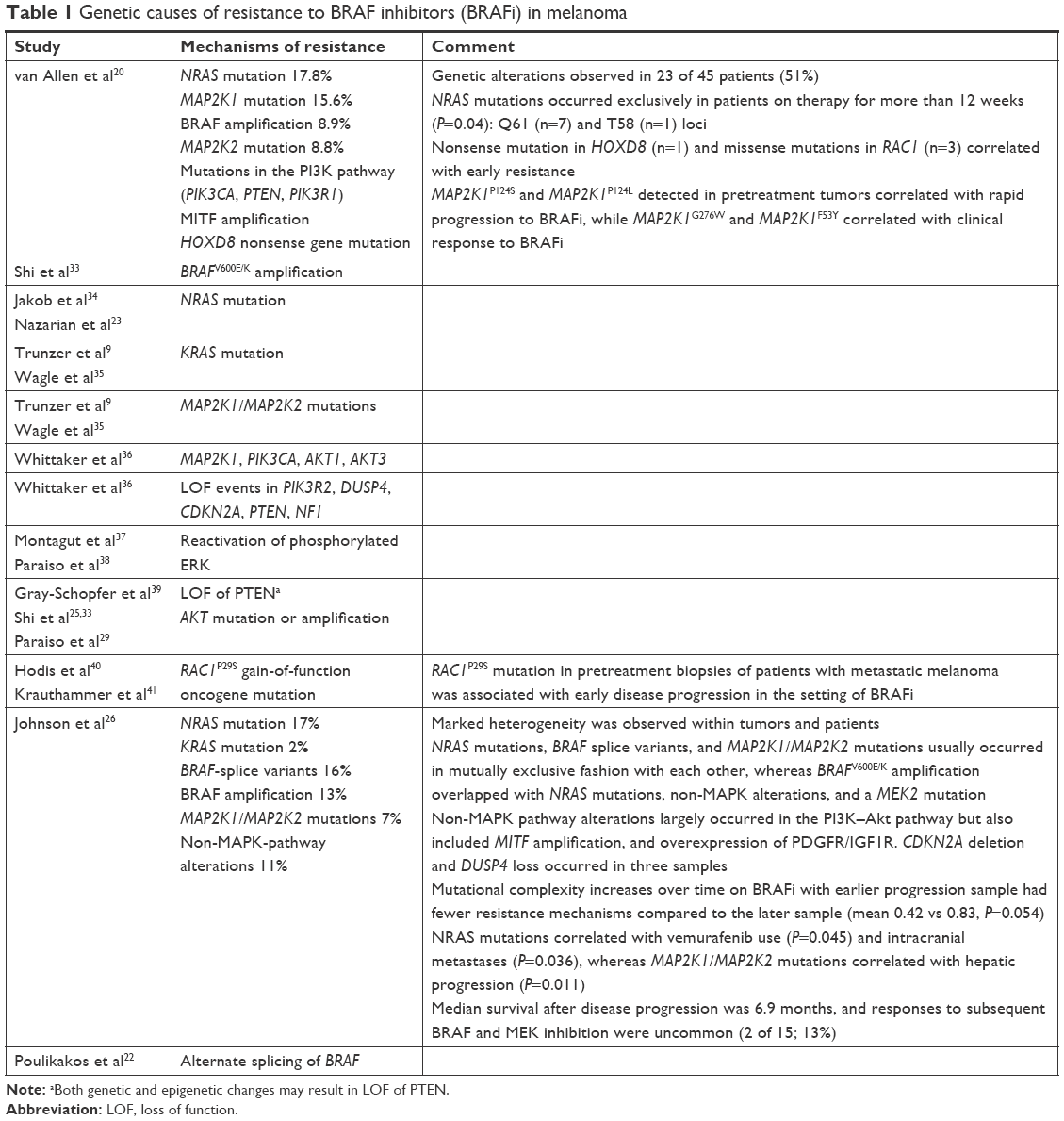 | Table 1 Genetic causes of resistance to BRAF inhibitors (BRAFi) in melanoma |
Secondary mutations in NRAS or MAP2K suggest acquired resistance mechanisms that maintain dependence on the MAPK pathway.27 Mutations in RAS (25%) and BRAFV600 (22%) are mutually exclusive, representing the most frequently detected genetic alterations leading to resistance.15,24,25,28 Resistance to BRAFi or combined BRAF and MEK inhibitors is also associated to a lesser extent with activation of the PI3K–Akt pathways (Table 1).29 Preclinical data have shown that PI3K-pathway activation via loss of PTEN prevents apoptosis of melanoma cells treated with BRAFi or activation of Akt can contribute to BRAFi resistance.29,30 Patients with PTEN loss have shorter progression-free survival (PFS) on dabrafenib.31 The presence of PI3K-pathway alterations does not necessarily preclude clinical response.9,20,32
Epigenetic or transcriptomic changes
Epigenetic or transcriptome-based changes were speculated to be the likely drivers of resistance to BRAFi among 39%–42% of melanomas that progressed on BRAFi and lacked any identifiable genetic abnormality to explain such resistance (Table 2).15,42,43
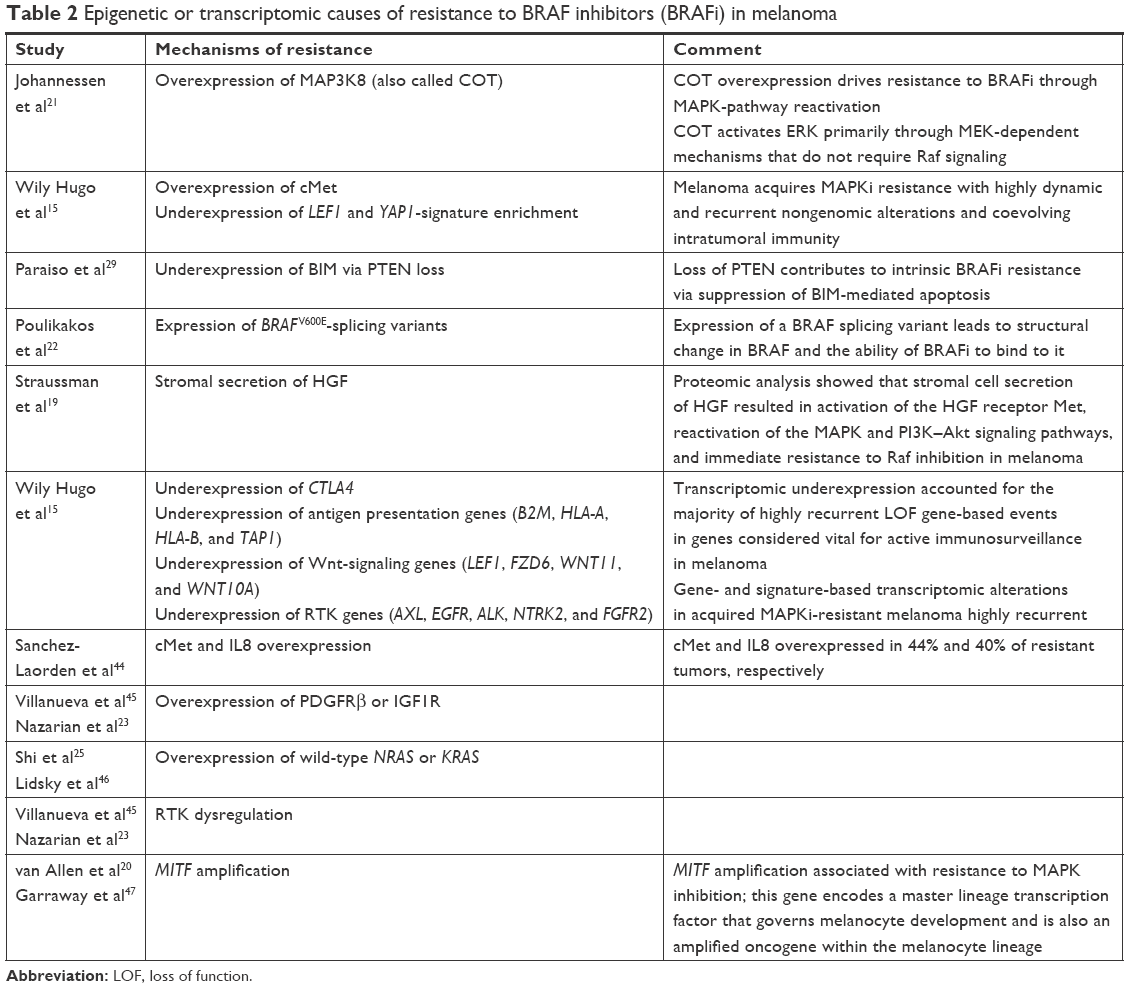 | Table 2 Epigenetic or transcriptomic causes of resistance to BRAF inhibitors (BRAFi) in melanoma |
BRAFi and the immune system
Preclinical data suggest that BRAFV600E mutation contributes to immunoescape and that both BRAFi and MEKi have beneficial effects on antitumor immunity and the tumor microenvironment as a whole, which is mediated by different mechanisms.50 Treatment with BRAFi and MEK inhibitors may modulate the immunomicroenvironment.15,49,50 Increased expression of melanoma antigens (MART, TYRP1, TYRP2, and Gp100), CD8+ T-cell infiltrates, and markers of T-cell cytotoxicity (perforin, granzyme B) and decreased levels of immunosuppressive cytokines occur during BRAFi therapy.51 However, immunoresponses may be limited, due to increased markers of T-cell exhaustion, such TIM3, PD1, and the immunosuppressive ligand PDL1.52 In melanoma cell lines, BRAFi/MEKi increase the rates of PD1+ melanoma cells that may sustain tumor relapse.48 These findings are intriguing, as immunocheckpoint blockade may be critical if combined with BRAF, in enhancing antitumor immunity and augmenting therapeutic responses.48,52 This proimmunotherapy microenvironment is lost upon melanoma progression on BRAFi. Several studies have revealed a decrease in melanoma antigen expression, an increase in T-cell exhaustion, and a decrease in CD8+ T-cell infiltrates in melanoma tumor specimens obtained at time of progression on BRAFi.15,51,52
The number of infiltrating macrophages and levels of macrophage-produced factors (such as growth factors, cytokines, chemokines, extracellular matrix, and proteinases) correlates inversely with patient outcomes in melanoma.53–60 Infiltrating macrophages contribute to cancer resistance to chemotherapy, radiotherapy, and immunotherapy.61–64 BRAFi paradoxically activate the MAPK pathway in macrophages to produce VEGF, which directly activates the MAPK pathway and stimulates cell growth in both macrophages and melanoma cells.10,65–67 The TNFα produced by activated macrophages may also contribute to melanoma resistance to BRAFi.68 BRAFi may transition the macrophage from being a passenger to a driver of melanoma progression, and hence agents that target infiltrating macrophages may overcome resistance to BRAFi.10,59
Potential ways to overcome resistance to BRAF inhibitors
BRAFi cause increased expression of melanocyte differentiation antigens, increased recognition by antigen-specific T cells, increased CD8+ T-cell infiltration in the melanoma microenvironment, decreased expression of immunoinhibitory cytokines, decreased myeloid-derived suppressor cells, and increased T-cell-exhaustion markers (eg, TIM3, PD1, and PDL1). Reactivation of the MAPK pathway causes suppression of melanoma antigens and reemergence of an immunosuppressive tumor microenvironment. Subsequent MAPK-pathway inhibition by an MEK inhibitor has restored melanoma antigen expression and promoted infiltration of CD8+ T cells.48,69,70 Adding MEK inhibitors to BRAFi can overcome resistance to BRAFi and enhance immunosurveillance. In a mouse model of BRAFV600E melanoma, improved antitumor activity, in vivo cytotoxic activity, and intratumoral cytokine secretion have been reported after combining adoptive T-cell transfer with BRAFi.71 As such, BRAF-targeted therapy may be critical in augmenting responses to immunocheckpoint blockade in patients with metastatic melanoma. The optimal treatment sequence remains to be elucidated.76
Preclinical studies have shown a decreased in CD8+ T cells in the tumor microenvironment upon progression to targeted therapy. Indeed, patients with disease progression on targeted therapy have lower response rates to immunocheckpoint blockade in retrospective studies.72–75 Therefore, theoretically we should not treat BRAF-mutated melanoma patients to progression with targeted therapy before starting immunocheckpoint blockade. Instead, we should consider adding it soon after initiation of targeted therapy.51,77 The critical question of therapy sequencing is being tested in a randomized Phase III trial (NCT02224781).
There is also growing interest in modifying our current approach of identifying and targeting driver mutations to one that focuses more on targeting elements of resistance in the tumor microenvironment.10 The presence of macrophages in the tumor microenvironment plays a critical role in melanoma resistance and predicts early relapse to targeted therapy, as aforementioned. Targeting macrophages alone in vivo can inhibit melanoma growth and increase the efficacy of BRAFi, which provides a rationale for combining BRAFi with therapies that target macrophages (NCT01826448, NCT03101254).10
Activation of the PI3K–Akt pathways contributes to melanoma resistance to targeted therapy. Therefore, approaches aiming simultaneously to inhibit both the MAPK and PI3K–Akt pathways have been proposed in melanoma.78 Preclinical data have demonstrated the superior antitumor activity of a combination of MAPK and PI3K–Akt–mTOR pathway inhibitors in BRAFV600E-mutant cell lines.29,45,79 Melanoma cells resistant to BRAFi have an MEK-independent survival driver that can be blocked by inhibitors of the PI3K–Akt–mTOR pathway.80 Upon progression, the addition of an Akt or mTOR inhibitor to continued therapy with vemurafenib or switching to a combination of an MEK inhibitor plus an Akt or an mTOR inhibitor may provide additional inhibitory activity.27
NRAS mutations and BRAF amplifications may still prove responsive to subsequent MEK-inhibitor-based regimens, although the existing clinical data suggest that patients who progress following single-agent Raf inhibition are less likely to benefit from MEK inhibitors.81 Complete NRAS extinction is difficult to achieve pharmacologically, due to redundant feedbacks and the likely induction of toxicity in patients, and thus NRAS-mutant melanoma remains without effective therapy.82 Combined MEK and CDK4 inhibition has revealed synergistic antitumor effects in a human NRAS-mutant melanoma xenograft model, providing a rationale for combining a CDK4 inhibitor with MEKi to achieve therapeutic synergy.83 Vemurafenib-resistant cell lines with acquired NRASQ61K mutation exhibit some sensitivity to sequential treatment with an MEK inhibitor and combinations of drugs inhibiting both Akt and MAPK pathways. This may be due to possible cross talk between mutated NRAS and the Akt pathway.27 Therapeutically, these findings imply that multiple pathways may need to be targeted simultaneously if not limited by toxicity or sequentially as part of an intermittent-dosing schedule.84
Update on FDA-approved targeted therapy in metastatic melanoma
Historically, the prognosis of metastatic melanoma has been poor, with 5-year survival ~6% and median overall survival (OS) of 7.5 months.85,86 Dacarbazine was the mainstay of treatment for metastatic melanoma until 2011, with an overall response rate of 7%–12% and median OS of 5.6–7.8 months.96–98 BRAF-targeted therapy and immunotherapy have transformed the landscape of melanoma treatment drastically.99–101 Vemurafenib and dabrafenib have shown significant improvement in response rates, PFS, and OS in BRAFV600E/K-mutated metastatic melanoma.87,88
In the BRIM-3 randomized Phase III trial, patients (n=675) with stage IIIC unresectable or stage IV BRAFV600E-mutated melanoma with no prior therapy were randomized 1:1 to vemurafenib (960 mg orally twice daily) or dacarbazine (1,000 mg/m2 intravenously every 3 weeks; Table 3).87 Median OS and PFS were 13.6 and 5.3 months in the vemurafenib arm compared to 9.7 (HR 0.81) and 1.6 months in the dacarbazine arm, respectively. Vemurafenib reduced the risk of death by 63% (HR 0.37) and risk of progression by 74% (HR 0.26). Normal baseline LDH, Eastern Cooperative Oncology Group performance status score 0, and stage M1a/b melanoma predicted long-term response to vemurafenib. Adverse events seen more frequently in vemurafenib included skin rash, arthralgia, alopecia, fatigue, and photosensitivity reactions. Squamous cell carcinoma of the skin or keratoacanthoma occurred in 18% of patients on vemurafenib. Based on the results of the BRIM-3 trial, the FDA approved vemurafenib on August 17, 2011 for the treatment of patients with unresectable or metastatic BRAFV600E-mutated melanoma.
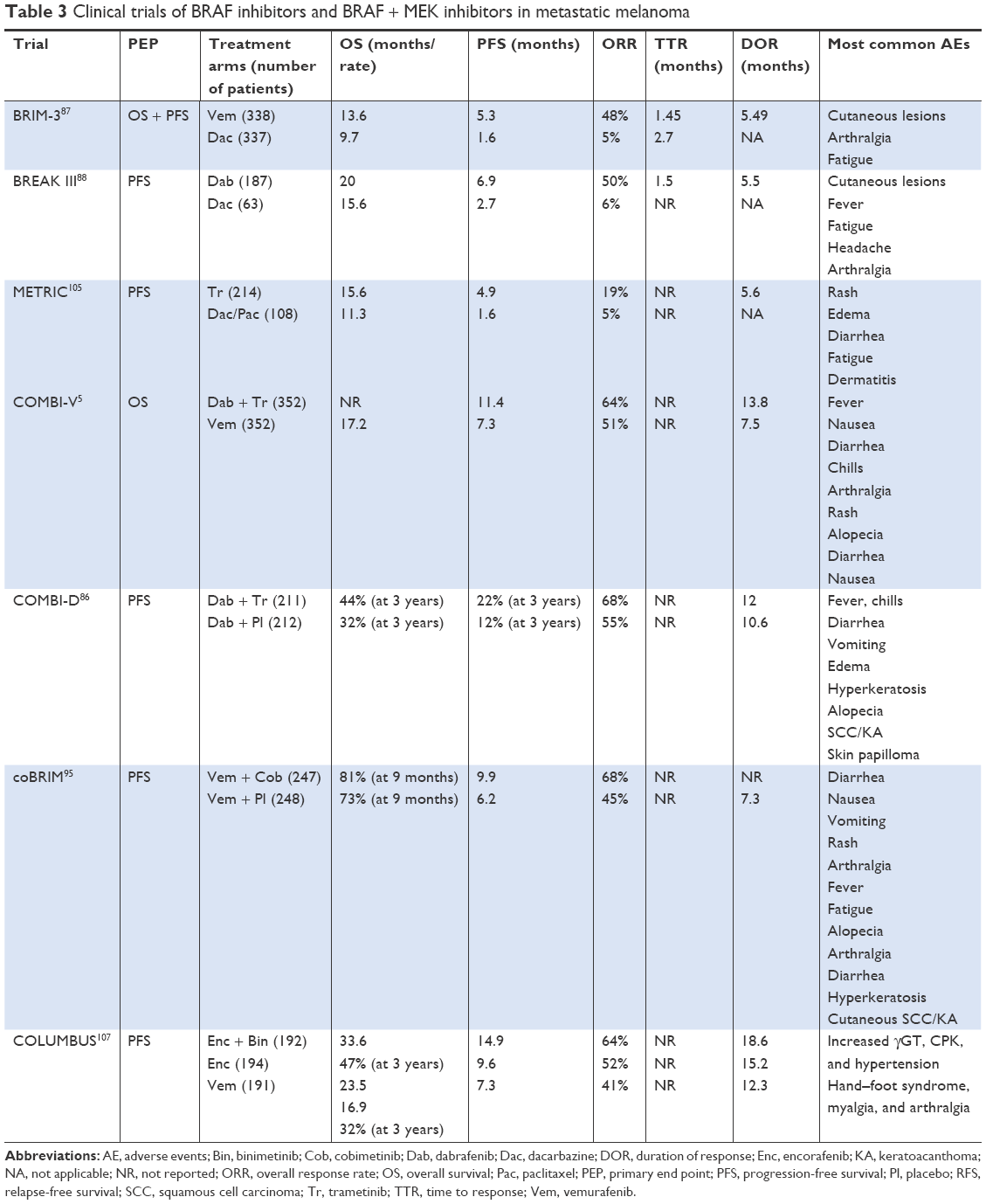 | Table 3 Clinical trials of BRAF inhibitors and BRAF + MEK inhibitors in metastatic melanoma |
BREAK-3 was a multicenter open label Phase III randomized trial where patients (n=250) with unresectable stage III or stage IV BRAFV600E-mutated melanoma were randomized 3:1 to dabrafenib (150 mg orally twice daily) or dacarbazine (1,000 mg/m2 intravenously every 3 weeks; Table 3).88 Median OS and PFS were 20 and 6.9 months for dabrafenib compared with 15.6 (HR 0.61) and 2.7 (HR 0.3) months for dacarbazine, respectively.88 Common adverse events seen with dabrafenib included fever, fatigue, headache, arthralgia, cutaneous squamous cell carcinoma and keratoacanthoma. Based on the results of the BREAK-3 trial, the FDA approved dabrafenib on May 29, 2013 for the treatment of patients with unresectable or metastatic BRAFV600E-mutated melanoma.
Preclinical data have revealed that resistance to BRAFi occurs largely at the level of MEK, and hence increased interest in MEK inhibitors has emerged. Trametinib is a small molecule that selectively inhibits MEK1 and MEK2.102 In Phase I and II trials, trametinib caused tumor regression and stabilization of disease in patients with BRAFV600E/K mutation.103,104 In the METRIC trial, patients (n=322) with stage IIIC or stage IV BRAFV600E/K-mutated melanoma were randomly assigned 2:1 to receive trametinib (2 mg orally daily) or chemotherapy (dacarbazine or paclitaxel; Table 3).105 Median PFS was 4.9 months in the trametinib arm and 1.6 months in the chemotherapy arm (HRprogression 0.45; HRdeath 0.54). OS at 6 months was 81% in the trametinib arm compared to 67% in the chemotherapy arm. Rash, edema, diarrhea, fatigue, and dermatitis acneiform were commonly encountered with trametinib. Cardiomyopathy was observed in 7% of subjects receiving trametinib, with serious cardiac events leading to discontinuation of the drug seen in three patients. Retinal vein occlusion and central serous retinopathy were encountered with trametinib.105 Based on the results of the METRIC trial, the FDA approved trametinib on May 29, 2013 for the treatment of patients with unresectable or metastatic BRAFV600E/K-mutated melanoma.
As discussed earlier, MAPK-pathway activation plays an important role in melanoma resistance to BRAFi, which limit median PFS to 6–8 months after BRAFi therapy alone.18,20,25,87,88 Moreover, paradoxical activation of the MAPK pathway in other normal BRAF wild-type cells, such as keratinocytes, has been associated with development of secondary cutaneous squamous cell carcinoma and keratoacanthomas.90–94 Preclinical and clinical data suggest that inhibition of both MEK and mutant BRAF kinases may result in greater initial tumor response, prevent MAPK-driven acquired resistance, and decrease the incidence and severity of toxicities, such as secondary skin tumors, owing to the paradoxical activation of the MAPK pathway seen in BRAFi monotherapy.18,20,25,89,95
In the COMBI-D double-blinded Phase III randomized control trial, patients (n=423) with unresectable stage IIIC or stage IV BRAFV600E/K-mutated melanoma were randomized 1:1 to receive dabrafenib plus trametinib or dabrafenib plus placebo (Table 3).86 The 3-year landmark analysis of COMBI-D provided evidence that long-term benefit and tolerability are achievable with combination dabrafenib and trametinib in patients with previously untreated BRAFV600E/K-mutant metastatic melanoma. Three-year PFS and OS rates were 22% and 44% with dabrafenib plus trametinib vs 12% (HR 0.71) and 32% (HR 0.75) with monotherapy, respectively.86 These results corroborated the Phase III BRF113220 trial demonstrating OS of 38%.8 The highest 3-year OS of 62% and PFS of 38% was observed in patients with normal baseline LDH level and fewer than three organ-site metastases compared to 45% and 16% in monotherapy.8 Patients with LDH above the upper limit of normal had 3-year OS of 25% vs 14%.86 Common adverse events in the combination vs monotherapy arm included fever (59% vs 33%), chills (32% vs 17%), diarrhea (31% vs 17%), vomiting (26% vs 15%), and peripheral edema (22% vs 9%). Hyperkeratosis, alopecia, cutaneous squamous cell carcinoma, keratoacanthoma, basal cell carcinoma, and skin papilloma were more common in the dabrafenib arm. Extended 5-year follow-up data of the Phase II BRF113220 trial revealed increased OS in patients who received dabrafenib and trametinib with normal baseline LDH (5 years, 45%) and normal LDH with fewer than three organ sites with metastasis (5 years, 51%).89
The COMBI-V open label, multicenter, Phase III trial randomly assigned patients (n=704) with metastatic BRAFV600-mutated melanoma 1:1 to receive either a combination of dabrafenib (150 mg orally twice daily) and trametinib (2 mg orally once daily) or vemurafenib (960 mg orally twice daily) as first-line therapy (Table 3).5 Median PFS and OS were 7.3 months and 17.2 months, respectively, for patients in the vemurafenib group compared with 11.4 months (HR 0.56, 95% CI, 0.46–0.69; P<0.001) and had not been reached for patients in the combination therapy group. The most common reasons for either drug discontinuation or dose modification were pyrexia and decreased left-ventricle ejection fraction in the combination therapy group and arthralgia in the vemurafenib group. Pyrexia was more frequent in the combination therapy group than the vemurafenib group (53% vs 21%). Skin toxicity effects were more frequent in the vemurafenib group, as a result of paradoxical reactivation of the MAPK pathway in BRAF wild-type keratinocytes, compared to the combination group, in particular rash (43% vs 22%), photosensitivity reaction (22% vs 4%), hand–foot syndrome (25% vs 4%), skin papilloma (23% vs 2%), squamous cell carcinoma and keratoacanthoma (18% vs 1%), and hyperkeratosis (25% vs 4%).5
coBRIM was a multinational trial that randomly assigned 495 patients with previously untreated unresectable locally advanced or metastatic BRAFV600-mutation-positive melanoma to vemurafenib and cobimetinib in the combination group vs vemurafenib and placebo in the control group (Table 3).95 Median PFS was 9.9 months in the combination group compared with 6.2 months in the control group (HR 0.51, P<0.001), and 68% of patients in the combination group had an objective response compared with 45% in the control group (P<0.001). The rate of complete response was also significantly higher in the combination group compared with the control group (10% vs 4%). Central serous retinopathy, gastrointestinal events, photosensitivity, and elevated aminotransferase and creatinine levels were seen with high frequency in the combination group. More than 50% of such events were grade 1 or 2. Equivalent rates of grade 3 events were noted in both study groups. CK elevation is a known class effect of MEK blockade and was noted be to the most common grade 4 event (4%) in the combination group. It is notable that the majority of events related to CK elevation were grade 1 or 2. Keratoacanthoma and cutaneous squamous cell carcinoma were less common in the combination group. Six deaths were attributed to adverse events in the combination group compared with three deaths in controls.5
COMBI-AD enrolled patients (n=870) with stage IIIA/B/C BRAF-mutated melanoma after complete surgical resection.106 Patients were randomized to receive a combination of dabrafenib (150 mg orally twice daily) and trametinib (2 mg orally once daily) or two placebos in a double-blind manner (Table 3). After a median follow-up of 2.8 years, the recurrence rate was 37% in the combination therapy group and 57% in the placebo group. The risk of relapse was 53% lower in the combination therapy group compared with the placebo group (HRrelapse 0.47). The mortality rate was 14% in the combination-therapy group and 22% in the placebo group. OS at 3 years was 86% in the combination therapy group and 77% in the placebo group.106 The most common adverse reactions in the combination therapy group were fever, fatigue, and nausea, and 26% of patients in this group had severe adverse effects leading to permanent discontinuation of the drugs.106
The COLUMBUS trial enrolled patients with stage IIIB, IIIC, or IV unresectable or metastatic BRAFV600E/K-mutated melanoma who were treatment-naïve or had progressed on or after previous first-line immunotherapy.107 Patients were randomized 1:1:1 to receive oral encorafenib (450 mg orally once daily) plus binimetinib (45 mg orally twice daily), oral encorafenib alone (300 mg once daily), or oral vemurafenib alone (960 mg orally twice daily; Table 3). Median PFS was 14.9 months in the encorafenib plus binimetinib group and 7.3 months in the vemurafenib group (HR 0.54, 95% CI 0.41–0.71; two-sided P<0.0001). The most common grade 3–4 adverse events seen in the encorafenib plus binimetinib group were increased γGT (9%), increased CPK (7%), and hypertension (6%). There were no treatment-related deaths, except for one in the combination group, which was considered by the investigator to be possibly related to treatment.107
BRAF inhibitors in management of metastatic melanoma to brain
Up to 60% of patients with metastatic melanoma develop brain metastasis.108 OS for metastatic brain melanoma (MBM) is 4–6 months.108 Despite the recent advances in management of metastatic melanoma with either immunotherapy or BRAFi (among those with BRAFV600-mutated melanoma), MBM remains a therapeutic challenge and an area of unmet need. From the 6,000 patients who were enrolled in the pivotal studies that led to the approval of targeted or immunotherapy in metastatic melanoma, none included patients with previously untreated MBM.87,88,108
Prior use of cytotoxic chemotherapy or temozolomide, a second-generation oral alkylating agent, showed poor activity against MBM with intracranial response of <10%.108,109 A fotemustine Phase III randomized study that included 43 patients with MBM showed a brain response rate of only 5.9% compared with no response in the dacarbazine arm.110 There are a growing number of trials that have shown activity of BRAFi therapy in patients with MBM (Table 4). One involved the use of dabrafenib with stable or tapering doses of steroids in 172 patients with BRAFV600E/K-mutated MBM. Intracranial response rates were 39.2% in treatment-naïve patients and 30.8% in patients with prior central nervous system radiation or surgery. OS >8 months was observed in both groups.108,111 An intracranial response rate of 37% with median OS of 5.3 months was detected in a pilot trial involving the use of vemurafenib in 24 patients with unresectable previously treated symptomatic MBM.112 A larger Phase II trial of vemurafenib showed an intracranial disease control rate and OS of 18% and 8.9 months in previously untreated patients with MBM compared with 20% and 9.6 months in previously treated patients with MBM, respectively.113
 | Table 4 Clinical trials of BRAF inhibitors in metastatic melanoma to brain |
Treatment with BRAFi achieves a rapid response in the majority of patients with BRAFV600-mutated melanoma; however, resistance to BRAFi is almost inevitable, as discussed earlier.108 In a cohort of patients treated with vemurafenib, it was noted that 59% of those who developed brain metastases while receiving vemurafenib had controlled extracranial disease.108,114
To overcome secondary resistance to BRAFi, combinations of BRAFi with other treatment modalities are being studied. Though melanoma is generally considered a radio-resistant tumor, small retrospective case series suggest an increased response by a combination of vemurafenib and radiation therapy, which is consistent with preclinical data suggesting that vemurafenib has a radiosensitizing effect.108,115 Dabrafenib plus trametinib in 125 patients after median follow-up of 8.5 months showed intracranial response in 44 (58%) patients with asymptomatic BRAF-positive brain metastases without previous treatment, nine (56%) with asymptomatic BRAF-positive MBM with previous treatment, seven (44%) with asymptomatic BRAF-positive MBM with or without previous treatment, and 10 (59%) with symptomatic BRAF-positive MBM with or without previous treatment.116
Conclusion
Approximately 50% of melanomas harbor an activating BRAF mutation. Combined BRAF–MEK inhibitor therapy is the standard of care for BRAFV600-mutant advanced melanoma. The three FDA-approved combination (BRAFi + MEKi) therapies in melanoma are vemurafenib plus cobimetinib, dabrafenib plus trametinib, and encorafenib plus binimetinib. Developing grade 3 or 4 adverse events to one combination does not preclude the use of other combinations, as each has its own unique adverse events that might be different than the others. Combination therapy is also effective in MBM; however, responses are short lived.
Common genetic alterations that lead to BRAFi resistance include: NRAS mutation, MAP2K1 and MAP2K2 mutations, BRAF amplification, and mutations in the PI3K pathway. Approaches aiming simultaneously to inhibit both the MAPK and PI3K–Akt pathways have been proposed. The role of epigenetic alterations in the emergence of BRAFi resistance is being recognized. Finally, the relationship between BRAFi and the immune system is of great importance.
BRAFi result in increased expression of melanocyte-differentiation antigens, increased recognition by antigen-specific T cells, increased CD8+ T-cell infiltration in the melanoma microenvironment, decreased expression of immunoinhibitory cytokines, decreased myeloid-derived suppressor cells, and increased T-cell-exhaustion markers (eg, TIM3, PD1, PDL1). This proimmunotherapy microenvironment is lost upon melanoma progression on BRAFi, and hence immunocheckpoint blockade may be critical if combined with BRAFi in enhancing antitumor immunity and augmenting therapeutic responses. The optimal treatment sequence remains to be elucidated in melanoma. Targeting elements of resistance in the melanoma microenvironment, such as macrophages, can inhibit melanoma growth and increase the efficacy of BRAFi.
Disclosure
The authors report no conflicts of interest in this work.
References
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. | ||
Weinstein IB, Joe AK. Mechanisms of disease: oncogene addiction – a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol. 2006;3(8):448–457. | ||
Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–819. | ||
Kefford R, Arkenau H, Brown MP, et al. Phase I/II study of GSK2118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumors. J Clin Oncol. 2010;28(15 Suppl):8503. | ||
Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–39. | ||
Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386(9992):444–451. | ||
Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. | ||
Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. | ||
Trunzer K, Pavlick AC, Schuchter L, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31(14):1767–1774. | ||
Wang T, Xiao M, Ge Y, et al. BRAF inhibition stimulates melanoma-associated macrophages to drive tumor growth. Clin Cancer Res. 2015;21(7):1652–1664. | ||
Amaral T, Sinnberg T, Meier F, et al. The mitogen-activated protein kinase pathway in melanoma part I – activation and primary resistance mechanisms to BRAF inhibition. Eur J Cancer. 2017;73:85–92. Epub 2017 Feb 3. | ||
Flaherty KT, Hodi FS, Fisher DE. From genes to drugs: targeted strategies for melanoma. Nat Rev Cancer. 2012;12(5):349–361. | ||
Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat Rev Clin Oncol. 2011;8(7):426–433. | ||
Tsao H, Chin L, Garraway LA, Fisher DE. Melanoma: from mutations to medicine. Genes Dev. 2012;26(11):1131–1155. | ||
Hugo W, Shi H, Sun L, et al. Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell. 2015;162(6):1271–1285. | ||
Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106(48):20411–20416. | ||
Puzanov I, Burnett P, Flaherty KT. Biological challenges of BRAF inhibitor therapy. Mol Oncol. 2011;5(2):116–123. | ||
Solit DB, Rosen N. Resistance to BRAF inhibition in melanomas. N Engl J Med. 2011;364(8):772–774. | ||
Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–504. | ||
van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4(1):94–109. | ||
Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–972. | ||
Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480(7377):387–390. | ||
Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. | ||
Moriceau G, Hugo W, Hong A, et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell. 2015;27(2):240–256. | ||
Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4(1):80–93. | ||
Johnson DB, Menzies AM, Zimmer L, et al. Acquired BRAF inhibitor resistance: a multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer. 2015;51(18):2792–2799. | ||
Atefi M, von Euw E, Attar N, et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the Akt/mTOR pathway. PLoS One. 2011;6(12):e28973. | ||
Goel VK, Lazar AJ, Warneke CL, Redston MS, Haluska FG. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol. 2006;126(1):154–160. | ||
Paraiso KH, Xiang Y, Rebecca VW, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71(7):2750–2760. | ||
Xing F, Persaud Y, Pratilas CA, et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene. 2012;31(4):446–457. | ||
Nathanson KL, Martin AM, Wubbenhorst B, et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin Cancer Res. 2013;19(17):4868–4878. | ||
Shi H, Moriceau G, Kong X, et al. Preexisting MEK1 exon 3 mutations in V600E/KBRAFmelanomas do not confer resistance to BRAF inhibitors. Cancer Discov. 2012;2(5):414–424. | ||
Shi H, Moriceau G, Kong X, et al. Melanoma whole-exome sequencing identifies (V600E) B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3(1):724. | ||
Jakob JA, Bassett RL, Ng CS, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118(16):4014–4023. | ||
Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29(22):3085–3096. | ||
Whittaker SR, Theurillat J-P, van Allen E, et al. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013;3(3):350–362. | ||
Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68(12):4853–4861. | ||
Paraiso KH, Fedorenko IV, Cantini LP, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102(12):1724–1730. | ||
Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445(7130):851–857. | ||
Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251–263. | ||
Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44(9):1006–1014. | ||
Ascierto PA, Simeone E, Giannarelli D, et al. Sequencing of BRAF inhibitors and ipilimumab in patients with metastatic melanoma: a possible algorithm for clinical use. J Transl Med. 2012;10(1):107. | ||
Ackerman A, McDermott DF, Lawrence DP, et al. Outcomes of patients with malignant melanoma treated with immunotherapy prior to or after vemurafenib. J Clin Oncol. 2012;30(15 Suppl):8569. | ||
Sanchez-Laorden B, Viros A, Girotti MR, et al. BRAF inhibitors induce metastasis in RAS mutant or inhibitor-resistant melanoma cells by reactivating MEK and ERK signaling. Sci Signal. 2014;7(318):ra30. | ||
Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–695. | ||
Lidsky M, Antoun G, Speicher P, et al. Mitogen-activated protein kinase (MAPK) hyperactivation and enhanced NRAS expression drive acquired vemurafenib resistance in V600E BRAF melanoma cells. J Biol Chem. 2014;289(40):27714–27726. | ||
Garraway LA, Widlund HR, Rubin MA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436(7047):117–122. | ||
Sanlorenzo M, Vujic I, Floris A, et al. BRAF and MEK Inhibitors Increase PD-1-positive melanoma cells leading to a potential lymphocyte-independent synergism with anti-PD-1 antibody. Clin Cancer Res. 2018;24(14):3377–3385. | ||
Ferrari de Andrade L, Ngiow SF, Stannard K, et al. Natural killer cells are essential for the ability of BRAF inhibitors to control BRAFV600E-mutant metastatic melanoma. Cancer Res. 2014;74(24):7298–7308. | ||
Kuske M, Westphal D, Wehner R, et al. Immunomodulatory effects of BRAF and MEK Inhibitors: implications for melanoma therapy. Pharmacol Res. 2018;6618(18):S1043. | ||
Cooper ZA, Reuben A, Spencer CN, et al. Distinct clinical patterns and immune infiltrates are observed at time of progression on targeted therapy versus immune checkpoint blockade for melanoma. Oncoimmunology. 2016;5(3):e1136044. | ||
Frederick DT, Piris A, Cogdill AP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19(5):1225–1231. | ||
Gazzaniga S, Bravo AI, Guglielmotti A, et al. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J Invest Dermatol. 2007;127(8):2031–2041. | ||
Jensen TO, Schmidt H, Møller HJ, et al. Macrophage markers in serum and tumor have prognostic impact in American Joint Committee on Cancer stage I/II melanoma. J Clin Oncol. 2009;27(20):3330–3337. | ||
Torisu H, Ono M, Kiryu H, et al. Macrophage infiltration correlates with tumor stage and angiogenesis in human malignant melanoma: possible involvement of TNFalpha and IL-1alpha. Int J Cancer. 2000;85(2):182–188. | ||
Kang K, Hammerberg C, Meunier L, Cooper KD. CD11b+ macrophages that infiltrate human epidermis after in vivo ultraviolet exposure potently produce IL-10 and represent the major secretory source of epidermal IL-10 protein. J Immunol. 1994;153(11):5256–5264. | ||
Payne AS, Cornelius LA. The role of chemokines in melanoma tumor growth and metastasis. J Invest Dermatol. 2002;118(6):915–922. | ||
Varney ML, Olsen KJ, Mosley RL, Bucana CD, Talmadge JE, Singh RK. Monocyte/macrophage recruitment, activation and differentiation modulate interleukin-8 production: a paracrine role of tumor-associated macrophages in tumor angiogenesis. In Vivo. 2002;16(6):471–477. | ||
Wang T, Ge Y, Xiao M, et al. Melanoma-derived conditioned media efficiently induce the differentiation of monocytes to macrophages that display a highly invasive gene signature. Pigment Cell Melanoma Res. 2012;25(4):493–505. | ||
Zaidi MR, Davis S, Noonan FP, et al. Interferon-γ links ultraviolet radiation to melanomagenesis in mice. Nature. 2011;469(7331):548–553. | ||
Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339(6117):286–291. | ||
de Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23(3):277–286. | ||
Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–1616. | ||
Weiskopf K, Ring AM, Ho CC, et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341(6141):88–91. | ||
Liu B, Earl HM, Baban D, et al. Melanoma cell lines express VEGF receptor KDR and respond to exogenously added VEGF. Biochem Biophys Res Commun. 1995;217(3):721–727. | ||
Masood R, Cai J, Zheng T, Smith DL, Hinton DR, Gill PS. Vascular endothelial growth factor (VEGF) is an autocrine growth factor for VEGF receptor-positive human tumors. Blood. 2001;98(6):1904–1913. | ||
Graells J, Vinyals A, Figueras A, et al. Overproduction of VEGF concomitantly expressed with its receptors promotes growth and survival of melanoma cells through MAPK and PI3K signaling. J Invest Dermatol. 2004;123(6):1151–1161. | ||
Smith MP, Sanchez-Laorden B, O’Brien K, et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFα. Cancer Discov. 2014;4(10):1214–1229. | ||
Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18(5):1386–1394. | ||
Wargo JA, Cogdill A, Dang P, et al. Abstract 958: Treatment with a selective inhibitor of BRAFV600E increases melanocyte antigen expression and CD8 T cell infiltrate in tumors of patients with metastatic melanoma. Cancer Res. 2011;71(8 Suppl):958. | ||
Koya RC, Mok S, Otte N, et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res. 2012;72(16):3928–3937. | ||
Ackerman A, Klein O, Mcdermott DF, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120(11):1695–1701. | ||
Ascierto PA, Simeone E, Sileni VC, et al. Sequential treatment with ipilimumab and BRAF inhibitors in patients with metastatic melanoma: data from the Italian cohort of the ipilimumab expanded access program. Cancer Invest. 2014;32(4):144–149. | ||
Ascierto PA, Margolin K. Ipilimumab before BRAF inhibitor treatment may be more beneficial than vice versa for the majority of patients with advanced melanoma. Cancer. 2014;120(11):1617–1619. | ||
Hooijkaas A, Gadiot J, Morrow M, Stewart R, Schumacher T, Blank CU. Selective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma. Oncoimmunology. 2012;1(5):609–617. | ||
Wargo JA, Cooper ZA, Flaherty KT. Universes collide: combining immunotherapy with targeted therapy for cancer. Cancer Discov. 2014;4(12):1377–1386. | ||
Cooper ZA, Reuben A, Amaria RN, Wargo JA. Evidence of synergy with combined BRAF-targeted therapy and immune checkpoint blockade for metastatic melanoma. Oncoimmunology. 2014;3(9):e954956. | ||
Smalley KS, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006;5(5):1136–1144. | ||
Gopal YN, Deng W, Woodman SE, et al. Basal and treatment-induced activation of Akt mediates resistance to cell death by AZD6244 (ARRY-142886) in BRAF-mutant human cutaneous melanoma cells. Cancer Res. 2010;70(21):8736–8747. | ||
Jiang CC, Lai F, Thorne RF, et al. MEK-independent survival of B-RAFV600E melanoma cells selected for resistance to apoptosis induced by the RAF inhibitor PLX4720. Clin Cancer Res. 2011;17(4):721–730. | ||
Kefford R. Preliminary results from a phase Ib/II, open-label, dose-escalation study of the oral BRAF inhibitor LGX818 in combination with the oral MEK1/2 inhibitor MEK162 in BRAF V600-dependent advanced solid tumors. J Clin Oncol. 2013;31(15 Suppl):9029. | ||
Kwong LN, Costello JC, Liu H, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18(10):1503–1510. | ||
Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26(13):2139–2146. | ||
das Thakur M, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494(7436):251–255. | ||
Barth A, Wanek LA, Morton DL. Prognostic factors in 1,521 melanoma patients with distant metastases. J Am Coll Surg. 1995;181(3):193–201. | ||
Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28(7):1631–1639. | ||
Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. | ||
Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. | ||
Long GV, Eroglu Z, Infante J, et al. Long-term outcomes in patients with BRAF V600-mutant metastatic melanoma who received dabrafenib combined with trametinib. J Clin Oncol. 2018;36(7):667–673. | ||
Robert C, Arnault JP, Mateus C. RAF inhibition and induction of cutaneous squamous cell carcinoma. Curr Opin Oncol. 2011;23(2):177–182. | ||
Carnahan J, Beltran PJ, Babij C, et al. Selective and potent Raf inhibitors paradoxically stimulate normal cell proliferation and tumor growth. Mol Cancer Ther. 2010;9(8):2399–2410. | ||
Oberholzer PA, Kee D, Dziunycz P, et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J Clin Oncol. 2012;30(3):316–321. | ||
Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24(6):1691–1697. | ||
Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366(3):207–215. | ||
Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–1876. | ||
Chapman PB, Einhorn LH, Meyers ML, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol. 1999;17(9):2745–2751. | ||
Middleton MR, Grob JJ, Aaronson N, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol. 2000;18(1):158–166. | ||
Bedikian AY, Millward M, Pehamberger H, et al. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: the Oblimersen Melanoma Study Group. J Clin Oncol. 2006;24(29):4738–4745. | ||
Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596–599. | ||
Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105(8):3041–3046. | ||
Joseph EW, Pratilas CA, Poulikakos PI, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci U S A. 2010;107(33):14903–14908. | ||
Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17(5):989–1000. | ||
Infante JR, Fecher LA, Nallapareddy S, et al. Safety and efficacy results from the first-in-human study of the oral MEK 1/2 inhibitor GSK1120212. J Clin Oncol. 2010;28(15 Suppl):2503. | ||
Kim KB, Kefford R, Pavlick AC, et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31(4):482–489. | ||
Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–114. | ||
Long GV, Hauschild A, Santinami M, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med. 2017;377(19):1813–1823. | ||
Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19(5):603–615. Epub 2018 Mar 21. | ||
Glitza Oliva I, Tawbi H, Davies MA. Melanoma brain metastases: current areas of investigation and future directions. Cancer J. 2017;23(1):68–74. | ||
Agarwala SS, Kirkwood JM, Gore M, et al. Temozolomide for the treatment of brain metastases associated with metastatic melanoma: a phase II study. J Clin Oncol. 2004;22(11):2101–2107. | ||
Lyle M, Long GV. The role of systemic therapies in the management of melanoma brain metastases. Curr Opin Oncol. 2014;26(2):222–229. | ||
Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087–1095. | ||
Dummer R, Goldinger SM, Turtschi CP, et al. Vemurafenib in patients with BRAF(V600) mutation-positive melanoma with symptomatic brain metastases: final results of an open-label pilot study. Eur J Cancer. 2014;50(3):611–621. | ||
Mcarthur GA, Maio M, Arance A, et al. Vemurafenib in metastatic melanoma patients with brain metastases: an open-label, single-arm, phase 2, multicentre study. Ann Oncol. 2017;28(3):634–641. | ||
Peuvrel L, Saint-Jean M, Quéreux G, et al. Incidence and characteristics of melanoma brain metastases developing during treatment with vemurafenib. J Neurooncol. 2014;120(1):147–154. | ||
Narayana A, Mathew M, Tam M, et al. Vemurafenib and radiation therapy in melanoma brain metastases. J Neurooncol. 2013;113(3):411–416. | ||
Davies MA, Saiag P, Robert C, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017;18(7):863–873. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.