")
Back to Journals » OncoTargets and Therapy » Volume 12
Mechanisms of drug resistance in acute myeloid leukemia
Authors Zhang J , Gu Y, Chen B
Received 20 October 2018
Accepted for publication 18 January 2019
Published 11 March 2019 Volume 2019:12 Pages 1937—1945
DOI https://doi.org/10.2147/OTT.S191621
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Jing Zhang, Yan Gu, Baoan Chen
Department of Hematology and Oncology, Zhongda Hospital, School of Medicine, Southeast University, Nanjing 210009, People’s Republic of China
Abstract: Acute myeloid leukemia (AML) is a kind of malignant hematopoietic system disease characterized by abnormal proliferation, poor cell differentiation, and infiltration of bone marrow, peripheral blood, or other tissues. To date, the first-line treatment of AML is still based on daunorubicin and cytosine arabinoside or idarubicin and cytosine arabinoside regimen. However, the complete remission rate of AML is still not optimistic, especially in elderly patients, and the recurrence rate after complete remission is still high. The resistance of leukemia cells to chemotherapy drugs becomes the main obstacle in the treatment of AML. At present, the research on the mechanisms of drug resistance in AML is very active. This article will elaborate on the main mechanisms of drug resistance currently being studied, including drug resistance-related proteins and enzymes, gene alterations, micro RNAs, and signal pathways.
Keywords: drug resistance, P-glycoprotein, gene alterations, signaling pathway
Introduction
Acute myeloid leukemia (AML) is a kind of malignant clonal disease originating from myeloid progenitors or lymphoid-primed multipotential progenitors.1 With the advancement of chemotherapy, hematopoietic stem cell transplantation, immunotherapy, and molecular targeted therapy, most AML patients can achieve complete remission (CR). The standard regimen, daunorubicin (DA) or idarubicin (IDA) combined with cytosine arabinoside, is still the first-line treatment for AML. The CR rate of first-line treatment is 60%–80% in young adults and 40%–60% in older adults >65 years old.2,3 But nearly 60% of elderly patients failed in inducing chemotherapy due to recurrence, and >85% of patients failed in treatment.4,5 Recently, studies found that drug resistance was the key to treatment failure, which contributed to the short-term survival in AML. Tumor drug resistance is mainly divided into primary drug resistance and acquired drug resistance. Primary drug resistance is the phenomenon that tumor cells, such as cells in the nonproliferative G0 phase, are not sensitive to drugs before the use of antitumor drugs. Acquired resistance refers to the phenomenon that initial tumor cells are sensitive to chemotherapy drugs, but the curative effect of drugs reduces gradually and results in drug resistance after induction therapy.
Residuary drug-resistant cells clone can evolve to predominant clone and make it difficult to be cured.6,7 Although patients can achieve second CR, the relapse-free survival will be worse for patients who did not relapse.8,9 The mechanisms of drug resistance in cancer are still not clear. Many studies have shown that this may be the result of multiple factors. This article reviews the following four common publicly recognized mechanisms of drug resistance in AML and discusses some of the newly discovered specific mechanisms: 1) drug resistance-related protein and enzyme, 2) genetic alterations, 3) miRNAs alterations in drug resistance, and 4) aberrant activation of drug resistance-related signal pathway (Figure 1).
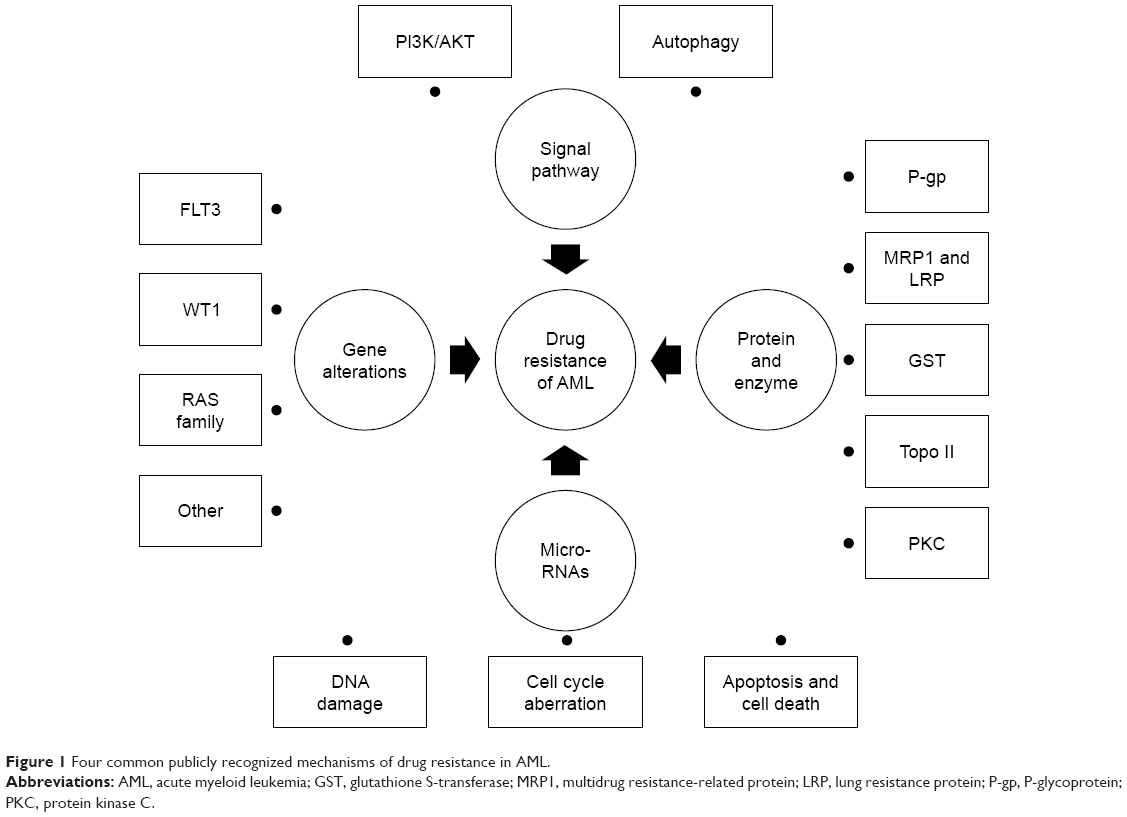 | Figure 1 Four common publicly recognized mechanisms of drug resistance in AML. |
Drug resistance-related protein and enzyme
Overexpression of P-glycoprotein
Multidrug resistance (MDR) gene can make tumor cells obtain drug resistance capability toward a certain antineoplastic agent, and due to the crosslink capacity, the tumor also becomes resistant to other antineoplastic drugs with different structures and functions. Of the many MDR gene products, P-glycoprotein (P-gp), a 170-kDa protein encoded by the MDR1 gene, is an organic positive ion pump with ATP-dependent cross-membrane drug extrusion function. It has two transmembrane domains and two nucleotide-binding domains. It is a kind of efflux pump which can pump out amino acids, organic ions, peptides, drugs, and xenobiotics.10 Overexpression of P-gp is associated with poor outcome in AML, whether newly diagnosed or relapsed AML.2,11 Patients with high-level P-gp were found to have higher white blood cell count, worse chromosomal abnormalities, and shorter overall survival (OS).12 In a cohort of 331 adult AML patients, MDR1 expression was found to be an independent prognostic factor of induction therapy and also of OS in the multivariate analysis.13 Researches found that the drug-resistant variants, SKM-1 and MOLM-13 AML cell lines, had a strong upregulation of P-gp and a downregulation of antiapoptotic protein Bcl-2, but could be reversed by P-gp inhibitor.14–16 P-gp also had a cross-resistance with nestin, which is highly expressed in human solid tumors and related to cell proliferation.17
Nuclear factor kappa B (NF-κB) mediates the expression of multiple genes involved in cell proliferation and antiapoptosis. The activation of NF-κB signal pathway is found to be related to the incidence of leukemia.18 The phosphorylation of PI3K/AKT/mTOR exists in 60%–80% of patients with AML, which has a function in cell apoptosis, cell cycle progression/proliferation, cellular metabolism, and cellular differentiation.19 Studies found that P-gp expression was associated with the activation of NF-κB and PI3K/AKT/mTOR signal pathway. Verapamil, Pantoprazole, Timosaponin A-III, and Balaglitazone were inhibitors of NF-κB and PI3K/AKT/mTOR signal pathways, which can decrease P-gp expression and reverse the resistance in patients with AML.20–23
Multidrug resistance-related protein and lung resistance protein
Multidrug resistance-related protein (MRP1), also known as ABCC1, is a member of ABC cassette superfamily of transporters, locating in the long arm of chromosome 16.24 The substrate specificity of MRP is glutathione (GSH), which is similar to but more limited than that of P-gp.25 It is a GSH transport pump which can identify and transport the substrate coupling with GSH, including antineoplastic drugs.13,26 Also, it can affect the distribution of drugs in cells, making drugs limited to perinuclear vesicles, preventing drugs from entering the nucleus to play a role of cytotoxic. A study found that MRP1 inhibitor can reverse drug resistance related to MRP1 overexpression by decreasing intracellular ATP.26
Lung resistance protein (LRP) is also a member of major vault protein, which locates in chromosome 16 and is close to the MDR-associated protein gene.27 It has a poor prognosis in AML and is associated with resistance to drugs such as doxorubicin, vincristine, and platinum compounds in drug-resistant cell lines.28 The role of LRP in drug resistance in AML is controversial. Studies found that LRP could independently decrease the effectiveness of induction chemotherapy.29,30 However, research also found that LRP could lead to drug resistance only when it coexisted with P-gp overexpression.31 There are two ways in which LRP can lead to drug resistance. One is blocking nuclear pore and preventing drugs from entering the nucleus. Another is transporting the drugs in nucleus to transport vesicle and making it release out of cellular by exocytosis.32
Glutathione S-transferases
Glutathione S-transferase (GST) is a family of enzymes, including GSTα, GSTμ, and GSTπ. GSTπ is found to be related to drug resistance in leukemia.33 GSTπ plays a wide range of functions in cells, such as maintaining the integrity of cell, resist to oxidation, and protecting from DNA damage.34 However, GSTπ was reported to express highly in AML cells. The GST catalyzes the glutathione-dependent detoxification of reactive electrophiles such as genotoxic chemical carcinogens and cytotoxic therapeutic agents and their oxidative metabolites. The main function of GST is to catalyze the binding of glutathione to chemical drugs (alkylating agents, anthracyclines, and platinum drugs), thereby reducing the cytotoxic effects of chemical drugs.35–37 There are three hypotheses in drug resistance: 1) GST catalyzes the synthesis of anticancer drugs with glutathione to inactivate drug activity directly; 2) intracellular GST inhibits the effect of anticancer drugs on attacking intracellular DNA; 3) GST can catalyze glutathione to bind to metal platinum, making it bind to platinum competitively with DNA and reducing the anticancer effect of platinum preparations.35,38
Topoisomerase II
DNA topoisomerases is a type of ribozymes, which plays an important role in DNA replication, transcription, and chromosome separation. It is found to be related to the cell proliferation, which can result in high expression of AML cells. Topoisomerase II (Topo II) can facilitate changes to DNA topology by allowing one of the double strands to pass through another via an enzyme-bridged DNA double-strand break.39 A series of antitumor drugs such as anthracycline, anthraquinones, and etoposide can act as targets of Topo II. The inhibitors of Topo II can directly bind to it and stabilize Topo II–DNA complex, which prevents the religation of DNA.40 It will trigger tumor cell death pathways after the formation of Topo II–DNA complex induced by Topo II inhibitors.41 Some mutations like K798L and K798P prevent antineoplastic drugs from binding to their targets, which makes AML cells 8- to 12-fold resist to antineoplastic drugs.42 When the number and activity of Topo II decreased, although there was no change in drug accumulation and retention in the cell, the target of anticancer drugs would be reduced or lost, resulting in drug resistance.43,44
Protein kinase C
Protein kinase C (PKC) is a kind of Ca2+/phospholipid-dependent protein kinase, involved in proliferation, antitumor drug resistance, and apoptosis. It can mediate drug transporter regulation and drug disintoxication by transcriptional or translational mechanisms controlling transporter expression, membrane insertion, or internalization processes and phosphorylation status of transporters.45 P-gp was known as a drug resistance pump. It is reported that P-gp could be phosphorylated on serine residues by PKC. So, the pumping activity of P-gp could be enhanced by PKC-mediated phosphorylation.46 Among the isoforms of PKCs, activation of PKCα, PKCe, and PKCq were related to P-gp upregulation, and then resulting in drug resistance in AML.47 According to published reports, the inhibitor of PKC, Bryostatin 1, could reverse the effect of drug efflux, but only in V185 mutant type.48 Moreover, the Midostaurin, inhibitor of Pan-PKC, could directly have effect not only on the leukemic cells but also on the AML neighboring stromal cells in the bone marrow (BM) microenvironment.49
Gene alterations
Molecular targeting drugs play an important role in the treatment of AML. Nevertheless, similar to the classic chemotherapeutic drugs, the drug resistance of molecular targeting drugs will appear in the process of treatment. The change of drug targeting gene is the main reason for the drug resistance of leukemic cells.
Fms-like tyrosine kinase 3
Fms-like tyrosine kinase 3 (FLT3) is a protooncogene in AML, which is related to cell proliferation, survival, and differentiation. It has two types of mutation, internal tandem duplication (ITD) and tyrosine kinase domain (TKD) mutation. FLT3-ITD, which can be found in one-third of patients with AML, is a molecular marker of poor prognosis, while FLT3-TKD is reported to be a good prognosis marker.50,51 FLT3-ITD mutation is recognized as a relapse-related genetic marker. In paired AML patients detected by next generation sequencing (NGS), diagnosed patients harbored wild-type (WT) FLT3, but 6.25%–16.7% relapsed patients acquired FLT3-ITD mutation.52,53 Patients with FLT3-ITD mutation would relapse in a shorter time than those with FLT3 WT. With routine chemotherapy, FLT3 mutant cells may survive and lead to the next recurrence. Nucleophosmin (NPM1) is associated with favorable prognosis in the newly revised 2016 WHO. It is found that the prognostic impact of the FLT3-ITD mutation depends on the allelic ratio. Patients with a low FLT3-ITD allelic ratio (<0.5) have a better prognosis in the presence of a (NPM1) mutation than those without FLT3-ITD in the presence of NPM1 mutation. However, patients with a high FLT3-ITD allelic ratio (≥0.5) have a poor prognosis without mutations of NPM1.54 A study found that AML cells with FLT3-ITD mutation can constitutively activate the receptor and make cells proliferate uncontrollably,55 which makes AML cells resistant to routine chemotherapeutics. Midostaurin, quizartinib, and gilteritinib are inhibitors of FLT3-ITD, and are considered to be used in the treatment of FLT3-ITD-mutant AML.56–58 However, AML cells with FLT3-ITD point mutations like N676K, F691L, D835V, and Y842C were found to be resistant to FLT3-ITD-specific inhibitors.59 Research found that heat shock protein 90 inhibitors could downregulate FLT3 signal pathway and overcome resistance to FLT3 inhibitors in Ba/F3 transfectants and quizartinib-resistant MV4-11 cells.60
Wilms tumor
Wilms tumor (WT1) is known to be a proto-oncogene that is highly expressed in patients with AML. It can regulate cell proliferation and differentiation by encoding a zinc finger motif. It is an enhancer of silent hematopoietic stem cells and an inducer of cellular differentiation in precursor cells.61 A study found that WT1 was a relapse-related gene that was an independent risk factor in 113 patients cohort analyzed by NGS.62 The higher the expression of WT1, the worse the prognosis of AML. Patients who harbor WT1 mutations will have an increased risk of relapse.63 In a mice transplantation experiment, AML1-ETO could not induce leukemia alone. However, with the transfection of WT1, the mice transplanted with BM cells, which had transfected AML1-ETO, rapidly developed AML.64 In a clinical trial with 842 patients, patients with WTI mutation had a shorter OS and event-free survival, which verified the poor prognosis of WT1.65 A study found that QPRT was the direct target gene of WT1. With the overexpression of WT1, QPRT expression would be upregulated, which conferred partial resistance to the antileukemic drugs.66
RAS family
RAS family includes KRAS, HRAS, and NRAS, which is a type of protooncogene in AML. RAS encodes p21 protein, which locates on the inner surface of cell membrane. It has the activity of GTP enzyme and is involved in the regulation system of cell proliferation signal. KRAS is the most common dominant mutation in cancer. RAS mutations will lead to the combination of GTP-activated protein and RAS protein, making RAS/GTP complex continuously activated and leading to the proliferation and metastasis of AML cells.67 RAS family is also involved in the activation of Raf/MEK/ERK (mitogen-activated protein kinase [MAPK]) and PI3K/Akt/mTOR pathways. A mutation KRAS G12D was reported to relapse even when the rat treated with MEK signal pathway inhibitor.68,69 The efficacy of KRAS alterations including mutations, abnormal expression, and copy number was found to be low in relapsed and refractory human cancers treated with MEK inhibitors.70
Other gene mutations and drug resistance
Studies indicate that patients acquired new mutations of IDH1, TP53, and ASXL1 when comparing newly diagnosed and relapsed AML, which should be relapse-related mutations.7,50,71 Also, patients with DNMT3A, CEBPA, IDH2, PTPN11 mutations in diagnosed AML will lose their mutations when relapse, so they should be drug-sensitive mutation types.7,72–74 Maybe, these gene mutations are drug resistance-related alterations, which make partial tumor cells survive after induce chemotherapy and make it a dominant clone and cause recurrence.
MicroRNAs and drug resistance
miRNAs are not involved in genome transcription or translation, but they play a critical role in AML by modifying or controlling a lot of hallmarks including cell division, self-renewal, invasion, and DNA damage.75 miRNAs are a family of small 18–24 bp noncoding double-stranded RNAs, which can be used to suppress protein expression by degrading and blocking translation of mRNA transcripts. The important role played by miRNA in drug resistance has been proved by more studies.
DNA damage
miRNA alterations can upregulate drug resistance by repairing DNA damage caused by antineoplastic drugs. Ataxia telangiectasia mutated (ATM) is a kind of DNA damage response protein. miRNA-181a was reported to be overexpressed in AML cells, which downregulated ATM expression. Therefore, the DNA damage cannot be repaired by ATM, leading to uninhibited growth of AML cells and drug resistance.76 Rad51 is a key protein that directly mediates DNA damage repair. miRNA-182 was found to be overexpressed by inhibiting HADC and the level of Rad51 protein would decrease, which led to increased levels of residual damage and decreased survival after exposure to double-strand damage-inducing agents.77
Cell cycle aberration
In healthy cells, a series of proteins such as cyclin-dependent kinases (CDK), ATM, and CHK1/2 guarantee the veracity of cell division. If errors are found in cell division, checkpoint protein will inhibit CDK and terminate the process of cell cycling.78 In AML, miRNA-638 was reported to be an inhibitor of CDK and overexpressed in AML, which downregulated CDK, resulting in cell cycle arrest in G1/S phase.79 In addition, miRNA-26a was found to downregulate E2F7, which contributed to cell cycling arrest.80 miRNA-17-92 was related to downregulation of p2181 and miRNA-223 was a regulatory factor of E2F1,82 both causing cycling arrest and drug resistance.
Apoptosis and cell death
Apoptosis is a spontaneous and orderly death of cells controlled by genes in order to maintain a stable internal environment. Apoptosis is a basic biological phenomenon of cells and plays a necessary role in the removal of unwanted or abnormal cells. Genetic aberrations like Bcl-2 family, caspase family, c-myc, and P53 may lead to downregulation of apoptosis, which in turn cause aberrant cancer growth and also drug resistance. Studies found that the low expression of miRNA-181a would downregulate Bcl-2 in AML and suppress apoptosis.83 Low expression of miRNA-149-5p in AML would reduce activation of the extrinsic apoptosis pathway and result in drug resistance.84
Signal pathway and drug resistance
PI3K/AKT signal pathway
PI3K/AKT signal pathway has a great role in promoting cell growth, proliferation, invasion, angiogenesis, and cell apoptosis inhibition, which makes it the new target of antitumor drugs.85 The tumor suppressor gene, PTEN, would arise heterozygous deletion mutation when treated with antitumor drugs. It can increase the level of AKT phosphorylation significantly, generating in the activation of PI3K/AKT pathway and regulating the expression of P-gp downstream, which is the key in drug resistance regulated by P-gp.86 In addition, excessive activation of the PI3K/AKT pathway in tumor cells can also regulate the activity of the JNK-p38 MAPK pathway, leading to the emergence of drug resistance in tumor cells.87 Moreover, AKT itself can phosphorylate a series of substrates directly and induce tumor cells to resist directly to drugs. Studies manifest that the inhibition of PI3K/AKT pathway could decrease the phosphorylation of Akt and mTOR and increase the antiproliferative activity through downregulating P-gp expression via suppressing the PI3K/Akt/mTOR signaling pathway.88,89
Autophagy
Autophagy has become a research hotspot in recent years. Autophagy is a process of phagocytosis of its own cytoplasmic protein or organelles. The inclusion of its package will be transferred into vesicles, and then fuse with lysosomes to synthesize autolysosomes, and eventually degrade the contents of its package. It is the process of cell metabolism and organelles renewal. Many chemotherapeutic drugs can induce autophagy, which is one of the important factors for tumor cells to develop drug resistance. Autophagy is a double-edged sword. At the initial stage of tumor, autophagy can inhibit the formation of tumor and help to improve the therapeutic effect of chemotherapy drugs on tumor. As a result, inhibiting the effect of autophagy may lead to the development of MDR in tumor cells. However, autophagy can also directly result in drug resistance. Tumor cells can reduce drug concentration and prevent apoptosis by protective autophagy. There are three main ways of autophagy in drug resistance.
Heat shock transcription factor 1-mediated autophagy
The heat shock transcription factor 1 is a type of transcription factor that is known to mediate a kind of cytoprotective response that promotes tumor cell survival and drug resistance.90 It will be activated under external stress, which can directly combine with the promotor of ATG7 and upregulate the expression of ATG7, resulting in the activation of cell autophagy and drug resistance in leukemia cells.91
ROS-mediated autophagy
ROS are metabolic by-products of aerobic respiration. Studies found that ROS were related to cancer. It can impact cancer phenotypes, kill cancer cells, impact secondary signaling networks, generate genetic instability that may cause mutations, and has a great role in cancer drug resistance.92 Many tumor cells can produce ROS when treated with the antitumor drugs. The temozolomide can be used as a treatment for neuroglioma. It can activate intracellular ROS/ERK pathway, promote tumor cell protective cell autophagy, and block the occurrence of apoptosis, which in turn induce tumor drug resistance.93
Met-mediated autophagy
Hepatocyte growth factor (HGF) is a critical factor in AML pathogenesis. Met is a receptor of tyrosine kinase, whose secretion can be activated by the expression of HGF. The maintenance of widespread leukemogenic signaling in AML cells depends on autocrine activation of Met.95 Studies found that 3-MA is an inhibitor of cell autophagy. When cell autophagy was inhibited by 3-MA, the drug-resistant papillary thyroid cancer (PTC) cells would be less sensitive to doxorubicin. By contrary, the cytokine autophagy activator, Everolimus, could significantly increase the sensitivity of PTC cell lines to doxorubicin.94,95 The effect of drug resistance reversed by cell autophagy depends on the inactivation of Met. This suggests that in tumor cells that are resistant to apoptosis, activated autophagy may be able to reverse the effect of drug resistance.
Conclusion
Drug resistance is the leading cause of treatment failure. In patients with AML, some types of gene mutations, abnormal expression of drug resistance-related miRNAs, upregulated PI3K/AKT and autophagy signal pathways, overexpression of some kinds of drug resistance-related enzyme will lead to relapse and drug resistance (Table 1). At present, there have been a lot of studies on MDR, and many inhibitors targeting on these drug-resistant mechanisms were reported. Overcoming these adverse factors may reverse drug resistance. For a diagnosed AML patient, it is important to evaluate whether he or she harbors high-risk factors for drug resistance. Therefore, the risk of drug resistance can be predicted through detecting gene mutations by NGS, detecting the level of PI3K/AKT and autophagy signal pathways, and the expressions of protein and enzyme. More resistance mechanisms are expected to be discovered. Regrettably, how to effectively use the above mechanisms to effectively reverse the clinical drug resistance of AML patients to improve the CR rate, long-term survival rate, and cure rate of AML still needs confirmation by clinical studies based on a large number of cases.
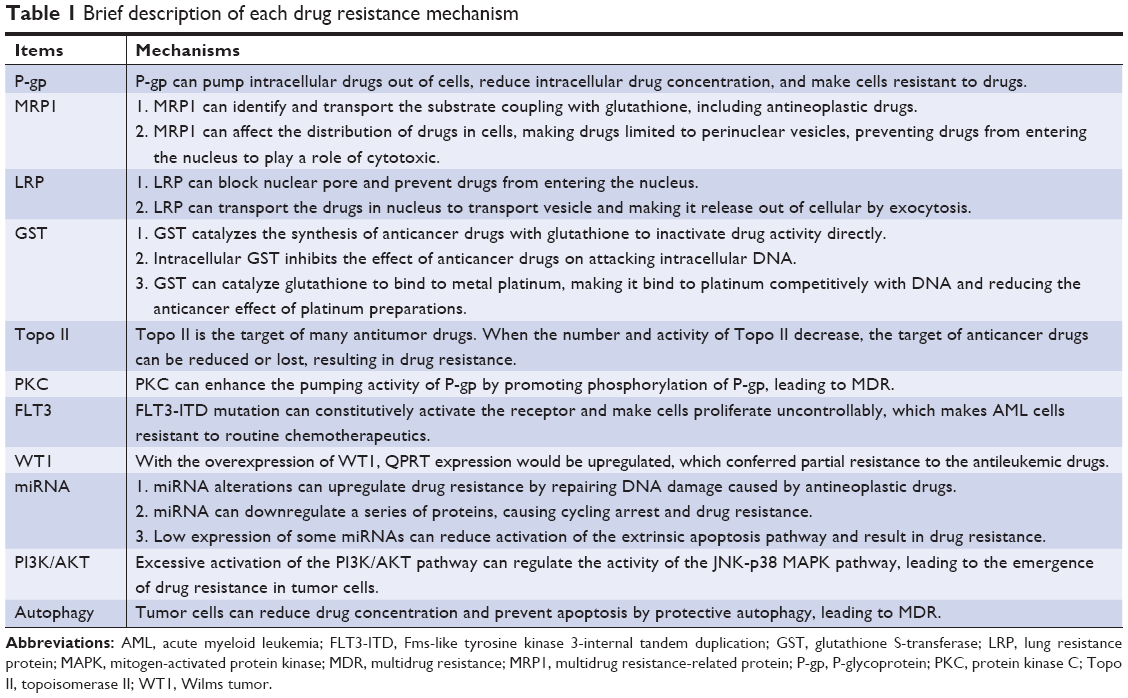 | Table 1 Brief description of each drug resistance mechanism |
Acknowledgments
This work was supported by the Key Medical Projects of Jiangsu Province (BL2014078), the Key Department of Jiangsu Province (ZDXKB201620), the Key Laboratory of Bioelectronics of Zhongda Hospital Affiliated to Southeast University (2018yy-jccx003), Jiangsu Social Development Project (BE2018711), and the Postgraduate Research and Practice Innovation Program of Jiangsu Province (SJCX18_0068).
Disclosure
The authors report no conflicts of interest in this work.
References
Goardon N, Marchi E, Atzberger A, et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 2011;19(1):138–152. | ||
Megías-Vericat JE, Rojas L, Herrero MJ, et al. Influence of ABCB1 polymorphisms upon the effectiveness of standard treatment for acute myeloid leukemia: a systematic review and meta-analysis of observational studies. Pharmacogenomics J. 2015;15(2):109–118. | ||
Luppi M, Fabbiano F, Visani G, Martinelli G, Venditti A. Novel agents for acute myeloid leukemia. Cancers. 2018;10(11):429. | ||
Döhner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European Leukemia Net. Blood. 2010;115(3):453–474. | ||
Bryan JC, Jabbour EJ. Management of relapsed/refractory acute myeloid leukemia in the elderly: current strategies and developments. Drugs Aging. 2015;32(8):623–637. | ||
Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–510. | ||
Krönke J, Bullinger L, Teleanu V, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122(1):100–108. | ||
Saraceni F, Labopin M, Gorin NC, et al. Matched and mismatched unrelated donor compared to autologous stem cell transplantation for acute myeloid leukemia in first complete remission: a retrospective, propensity score-weighted analysis from the ALWP of the EBMT. J Hematol Oncol. 2016;9(1):79. | ||
Forman SJ, Rowe JM. The myth of the second remission of acute leukemia in the adult. Blood. 2013;121(7):1077–1082. | ||
Farawela HM, Khorshied MM, Kassem NM, Kassem HA, Zawam HM. The clinical relevance and prognostic significance of adenosine triphosphate ATP-binding cassette (ABCB5) and multidrug resistance (MDR1) genes expression in acute leukemia: an Egyptian study. J Cancer Res Clin Oncol. 2014;140(8):1323–1330. | ||
Leith CP, Kopecky KJ, Godwin J, et al. Acute myeloid leukemia in the elderly: assessment of multidrug resistance (MDR1) and cytogenetics distinguishes biologic subgroups with remarkably distinct responses to standard chemotherapy. A Southwest Oncology Group study. Blood. 1997;89(9):3323–3329. | ||
Do JH, Oh SH, Song EJ, Chung JS, Kang CD, Lee EY. [Treatment outcome of multidrug resistance related mRNA expression and c-jun-N-terminal kinase activity in patients with acute myeloid leukemia]. Korean J Lab Med. 2007;27(4):229–236. | ||
Schaich M, Soucek S, Thiede C, Ehninger G, Illmer T; SHG AML96 Study Group. MDR1 and MRP1 gene expression are independent predictors for treatment outcome in adult acute myeloid leukaemia. Br J Haematol. 2005;128(3):324–332. | ||
Pavlikova L, Seres M, Imrichova D, et al. The expression of P-gp in leukemia cells is associated with cross-resistance to protein N-glycosylation inhibitor tunicamycin. Gen Physiol Biophys. 2016;35(4):497–510. | ||
Messingerova L, Imrichova D, Kavcova H, Turakova K, Breier A, Sulova Z. Acute myeloid leukemia cells MOLM-13 and SKM-1 established for resistance by azacytidine are crossresistant to P-glycoprotein substrates. Toxicol In vitro. 2015;29(7):1405–1415. | ||
Imrichova D, Messingerova L, Seres M, et al. Selection of resistant acute myeloid leukemia SKM-1 and MOLM-13 cells by vincristine-, mitoxantrone- and lenalidomide-induced upregulation of P-glycoprotein activity and downregulation of CD33 cell surface exposure. Eur J Pharm Sci. 2015;77:29–39. | ||
Imrichova D, Coculova M, Messingerova L, Sulova Z, Breier A. Vincristine-induced expression of P-glycoprotein in MOLM-13 and SKM-1 acute myeloid leukemia cell lines is associated with coexpression of nestin transcript. Gen Physiol Biophys. 2014;33(4):425–431. | ||
Bosman MC, Schuringa JJ, Vellenga E. Constitutive NF-κB activation in AML: causes and treatment strategies. Crit Rev Oncol Hematol. 2016;98:35–44. | ||
Brenner AK, Andersson Tvedt TH, Bruserud Ø. The complexity of targeting PI3K-AKT-mTOR signalling in human acute myeloid leukaemia: the importance of leukemic cell heterogeneity, neighbouring mesenchymal stem cells and immunocompetent cells. Molecules. 2016;21(11):E1512. | ||
Davoudi Z, Akbarzadeh A, Rahmatiyamchi M, et al. Molecular target therapy of Akt and NF-kB signaling pathways and multidrug resistance by specific cell penetrating inhibitor peptides in HL-60 cells. Asian Pac J Cancer Prev. 2014;15(10):4353–4358. | ||
Chen JR, Jia XH, Wang H. Timosaponin A-III reverses multi-drug resistance in human chronic myelogenous leukemia K562/ADM cells via downregulation of MDR1 and MRP1 expression by inhibiting PI3K/Akt signaling pathway. Int J Oncol. 2016;48(5):2063–2070. | ||
Liu M, Tang R, Jiang Y. Pantoprazole induces apoptosis of leukemic cells by inhibiting expression of P-glycoprotein/multidrug resistance-associated protein-1 through PI3K/Akt/mTOR signaling. Indian J Hematol Blood Transfus. 2017;33(4):500–508. | ||
Yousefi B, Azimi A, Majidinia M, et al. Balaglitazone reverses P-glycoprotein-mediated multidrug resistance via upregulation of PTEN in a PPARγ-dependent manner in leukemia cells. Tumour Biol. 2017;39(10):101042831771650. | ||
Kruh GD, Chan A, Myers K, Gaughan K, Miki T, Aaronson SA. Expression complementary DNA library transfer establishes MRP as a multidrug resistance gene. Cancer Res. 1994;54(7):1649–1652. | ||
Huh HJ, Park CJ, Jang S, et al. Prognostic significance of multidrug resistance gene 1 (MDR1), multidrug resistance-related protein (MRP) and lung resistance protein (LRP) mRNA expression in acute leukemia. J Korean Med Sci. 2006;21(2):253–258. | ||
Ji Q, Qiu L. Mechanism study of PEGylated polyester and β-cyclodextrin integrated micelles on drug resistance reversal in MRP1-overexpressed HL60/ADR cells. Colloids Surf B Biointerfaces. 2016;144:203–213. | ||
Damiani D, Tiribelli M, Raspadori D, et al. The role of MDR-related proteins in the prognosis of adult acute myeloid leukaemia (AML) with normal karyotype. Hematol Oncol. 2007;25(1):38–43. | ||
Izquierdo MA, Shoemaker RH, Flens MJ, et al. Overlapping phenotypes of multidrug resistance among panels of human cancer-cell lines. Int J Cancer. 1996;65(2):230–237. | ||
Tsuji K, Wang YH, Takanashi M, et al. Overexpression of lung resistance-related protein and P-glycoprotein and response to induction chemotherapy in acute myelogenous leukemia. Hematol Rep. 2012;4(3):e18. | ||
Scheper RJ, Broxterman HJ, Scheffer GL, et al. Overexpression of a M(r) 110,000 vesicular protein in non-P-glycoprotein-mediated multidrug resistance. Cancer Res. 1993;53(7):1475–1479. | ||
Tsuji K, Motoji T, Sugawara I, et al. Significance of lung resistance-related protein in the clinical outcome of acute leukaemic patients with reference to P-glycoprotein. Br J Haematol. 2000;110(2):370–378. | ||
Hart SM, Ganeshaguru K, Scheper RJ, Prentice HG, Hoffbrand AV, Mehta AB. Expression of the human major vault protein LRP in acute myeloid leukemia. Exp Hematol. 1997;25(12):1227–1232. | ||
Nasr AS, Sami RM, Ibrahim NY, Darwish DO, Sami RM. Glutathione S transferase (GSTp 1, GSTM 1, and GSTT 1) gene polymorphisms in Egyptian patients with acute myeloid leukemia. Indian J Cancer. 2015;52(4):490–495. | ||
Chatterjee A, Gupta S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. 2018;433:33–42. | ||
Hatem E, El Banna N, Huang ME. Multifaceted roles of glutathione and glutathione-based systems in carcinogenesis and anticancer drug resistance. Antioxid Redox Signal. 2017;27(15):1217–1234. | ||
Mossallam GI, Abdel Hamid TM, Samra MA. Glutathione S-transferase GSTM1 and GSTT1 polymorphisms in adult acute myeloid leukemia; its impact on toxicity and response to chemotherapy. J Egypt Natl Canc Inst. 2006;18(3):264–273. | ||
Weich N, Nuñez MC, Galimberti G, et al. Polymorphic variants of GSTM1, GSTT1, and GSTP1 genes in childhood acute leukemias: a preliminary study in Argentina. Hematology. 2015;20(9):511–516. | ||
Li S, Li C, Jin S, et al. Overcoming resistance to cisplatin by inhibition of glutathione S-transferases (GSTs) with ethacraplatin micelles in vitro and in vivo. Biomaterials. 2017;144:119–129. | ||
Cowell IG, Austin CA. Mechanism of generation of therapy related leukemia in response to anti-topoisomerase II agents. Int J Environ Res Public Health. 2012;9(6):2075–2091. | ||
de Isabella P, Palumbo M, Sissi C, et al. Topoisomerase II DNA cleavage stimulation, DNA binding activity, cytotoxicity, and physico-chemical properties of 2-aza- and 2-aza-oxide-anthracenedione derivatives. Mol Pharmacol. 1995;48(1):30–38. | ||
Zhou R, Frostvik Stolt M, Kronenwett U, Gruber A, Liliemark J, Liliemark E. Real-time RT-PCR for the determination of topoisomerase II mRNA level in leukaemic cells. Leuk Res. 2002;26(5):487–494. | ||
Okada Y, Tosaka A, Nimura Y, Kikuchi A, Yoshida S, Suzuki M. Atypical multidrug resistance may be associated with catalytically active mutants of human DNA topoisomerase II alpha. Gene. 2001;272(1–2):141–148. | ||
Fukushima T, Inoue H, Takemura H, et al. Idarubicin and idarubicinol are less affected by topoisomerase II-related multidrug resistance than is daunorubicin. Leuk Res. 1998;22(7):625–629. | ||
Gaur S, Chen L, Yang L, Wu X, Un F, Yen Y. Inhibitors of mTOR overcome drug resistance from topoisomerase II inhibitors in solid tumors. Cancer Lett. 2011;311(1):20–28. | ||
Mayati A, Moreau A, Le Vée M, et al. Protein kinases C-mediated regulations of drug transporter activity, localization and expression. Int J Mol Sci. 2017;18(4):pii: E764. | ||
Hofmann J. Protein kinase C isozymes as potential targets for anticancer therapy. Curr Cancer Drug Targets. 2004;4(2):125–146. | ||
Shtil AA, Ktitorova OV, Kakpakova ES, Holian O. Differential effects of the MDR1 (multidrug resistance) gene-activating agents on protein kinase C: evidence for redundancy of mechanisms of acquired MDR in leukemia cells. Leuk Lymphoma. 2000;40(1–2):191–195. | ||
Spitaler M, Utz I, Hilbe W, Hofmann J, Grunicke HH. PKC-independent modulation of multidrug resistance in cells with mutant (V185) but not wild-type (G185) P-glycoprotein by bryostatin 1. Biochem Pharmacol. 1998;56(7):861–869. | ||
Tvedt TH, Nepstad I, Bruserud Ø. Antileukemic effects of midostaurin in acute myeloid leukemia – the possible importance of multikinase inhibition in leukemic as well as nonleukemic stromal cells. Expert Opin Investig Drugs. 2017;26(3):343–355. | ||
Wakita S, Yamaguchi H, Omori I, et al. Mutations of the epigenetics-modifying gene (DNMT3a, TET2, IDH1/2) at diagnosis may induce FLT3-ITD at relapse in de novo acute myeloid leukemia. Leukemia. 2013;27(5):1044–1052. | ||
Boddu P, Kantarjian H, Borthakur G, et al. Co-occurrence of FLT3-TKD and NPM1 mutations defines a highly favorable prognostic AML group. Blood Adv. 2017;1(19):1546–1550. | ||
Kottaridis PD, Gale RE, Langabeer SE, Frew ME, Bowen DT, Linch DC. Studies of FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood. 2002;100(7):2393–2398. | ||
Cloos J, Goemans BF, Hess CJ, et al. Stability and prognostic influence of FLT3 mutations in paired initial and relapsed AML samples. Leukemia. 2006;20(7):1217–1220. | ||
Schlenk RF, Kayser S, Bullinger L, et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood. 2014;124(23):3441–3449. | ||
El FR, Rasheed W, Hawsawi Y. Targeting FLT3 mutations in acute myeloid leukemia. Cells. 2018;7(1). | ||
Lee LY, Hernandez D, Rajkhowa T, et al. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood. 2017;129(2):257–260. | ||
Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 2017;18(8):1061–1075. | ||
Larrosa-Garcia M, Baer MR. FLT3 inhibitors in acute myeloid leukemia: current status and future directions. Mol Cancer Ther. 2017;16(6):991–1001. | ||
Smith CC, Wang Q, Chin CS, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260–263. | ||
Katayama K, Noguchi K, Sugimoto Y. Heat shock protein 90 inhibitors overcome the resistance to Fms-like tyrosine kinase 3 inhibitors in acute myeloid leukemia. Oncotarget. 2018;9(76):34240–34258. | ||
Hollink IH, van den Heuvel-Eibrink MM, Zimmermann M, et al. Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood. 2009;113(23):5951–5960. | ||
Quek L, Ferguson P, Metzner M, et al. Mutational analysis of disease relapse in patients allografted for acute myeloid leukemia. Blood Adv. 2016;1(3):193–204. | ||
Sugiyama H. Wilms’ tumor gene WT1: its oncogenic function and clinical application. Int J Hematol. 2001;73(2):177–187. | ||
Nishida S, Hosen N, Shirakata T, et al. AML1-ETO rapidly induces acute myeloblastic leukemia in cooperation with the Wilms tumor gene, WT1. Blood. 2006;107(8):3303–3312. | ||
Ho PA, Zeng R, Alonzo TA, et al. Prevalence and prognostic implications of WT1 mutations in pediatric acute myeloid leukemia (AML): a report from the Children’s Oncology Group. Blood. 2010;116(5):702–710. | ||
Ullmark T, Montano G, Jarvstrat L, et al. Anti-apoptotic quinolinate phosphoribosyltransferase (QPRT) is a target gene of Wilms’ tumor gene 1 (WT1) protein in leukemic cells. Biochem Biophys Res Commun. 2017;482(4):802–807. | ||
Stephen AG, Esposito D, Bagni RK. Dragging Ras back in the ring. Cancer Cell. 2014;25(3):272–281. | ||
Brown AP, Carlson TCG, Loi CM, Graziano MJ. Pharmacodynamic and toxicokinetic evaluation of the novel MEK inhibitor, PD0325901, in the rat following oral and intravenous administration. Cancer Chemother Pharmacol. 2007;59(5):671–679. | ||
Burgess MR, Hwang E, Mroue R, et al. KRAS allelic imbalance enhances fitness and modulates MAP Kinase dependence in cancer. Cell. 2017;168(5):817–829. | ||
Infante JR, Fecher LA, Falchook GS, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):773–781. | ||
Nakano Y, Kiyoi H, Miyawaki S, et al. Molecular evolution of acute myeloid leukaemia in relapse: unstable N-ras and Flt3 genes compared with p53 gene. Br J Haematol. 1999;104(4):659–664. | ||
Hou HA, Kuo YY, Liu CY, et al. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 2012;119(2):559–568. | ||
Shih LY, Liang DC, Huang CF. AML patients with CEBPalpha mutations mostly retain identical mutant patterns but frequently change in allelic distribution at relapse: a comparative analysis on paired diagnosis and relapse samples. Leukemia. 2006;20(4):604–609. | ||
Bachas C, Schuurhuis GJ, Zwaan CM. Gene expression profiles associated with pediatric relapsed AML. PLoS One. 2015;10(4):e121730. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Liu X, Liao W, Peng H, et al. miR-181a promotes G1/S transition and cell proliferation in pediatric acute myeloid leukemia by targeting ATM. J Cancer Res Clin Oncol. 2016;142(1):77–87. | ||
Lai TH, Ewald B, Zecevic A, et al. HDAC inhibition induces MicroRNA-182, which targets Rad51 and impairs HR repair to sensitize cells to sapacitabine in acute myelogenous leukemia. Clin Cancer Res. 2016;22(14):3537–3549. | ||
Beaumont KA, Hill DS, Daignault SM, et al. Cell cycle phase-specific drug resistance as an escape mechanism of melanoma cells. J Invest Dermatol. 2016;136(7):1479–1489. | ||
Lin Y, Li D, Liang Q, et al. miR-638 regulates differentiation and proliferation in leukemic cells by targeting cyclin-dependent kinase 2. J Biol Chem. 2015;290(3):1818–1828. | ||
Salvatori B, Iosue I, Mangiavacchi A, et al. The microRNA-26a target E2F7 sustains cell proliferation and inhibits monocytic differentiation of acute myeloid leukemia cells. Cell Death Dis. 2012;3:e413. | ||
Wong P, Iwasaki M, Somervaille TC, et al. The miR-17-92 microRNA polycistron regulates MLL leukemia stem cell potential by modulating p21 expression. Cancer Res. 2010;70(9):3833–3842. | ||
Pulikkan JA, Dengler V, Peramangalam PS, et al. Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood. 2010;115(9):1768–1778. | ||
Li H, Hui L, Xu W. miR-181a sensitizes a multidrug-resistant leukemia cell line K562/A02 to daunorubicin by targeting Bcl-2. Acta Biochim Biophys Sin. 2012;44(3):269–277. | ||
Tian P, Yan L. Inhibition of MicroRNA-149-5p induces apoptosis of acute myeloid leukemia cell line THP-1 by targeting Fas ligand (FASLG). Med Sci Monit. 2016;22:5116–5123. | ||
Tazzari PL, Cappellini A, Ricci F, et al. Multidrug resistance-associated protein 1 expression is under the control of the phosphoinositide 3 kinase/Akt signal transduction network in human acute myelogenous leukemia blasts. Leukemia. 2007;21(3):427–438. | ||
Chen P, Jin Q, Fu Q, et al. Induction of multidrug resistance of acute myeloid leukemia cells by cocultured stromal cells via upregulation of the PI3K/Akt signaling pathway. Oncol Res. 2016;24(4):215–223. | ||
Roszak J, Smok-Pieniążek A, Stępnik M, Stępnik M. Transcriptomic analysis of the PI3K/Akt signaling pathway reveals the dual role of the c-Jun oncogene in cytotoxicity and the development of resistance in HL-60 leukemia cells in response to arsenic trioxide. Adv Clin Exp Med. 2017;26(9):1335–1342. | ||
Wang L, Wang C, Jia Y, Liu Z, Shu X, Liu K. Resveratrol increases anti-proliferative activity of bestatin through downregulating P-glycoprotein expression via inhibiting PI3K/Akt/mTOR pathway in K562/ADR cells. J Cell Biochem. 2016;117(5):1233–1239. | ||
Chen Y, Wang T, Du J, et al. The critical role of PTEN/PI3K/AKT signaling pathway in shikonin-induced apoptosis and proliferation inhibition of chronic myeloid leukemia. Cell Physiol Biochem. 2018;47(3):981–993. | ||
Wales CTK, Taylor FR, Higa AT, Mcallister HA, Jacobs AT. ERK-dependent phosphorylation of HSF1 mediates chemotherapeutic resistance to benzimidazole carbamates in colorectal cancer cells. Anti-Cancer Drugs. 2015;26(6):1–666. | ||
Vydra N, Toma A, Glowala-Kosinska M, Gogler-Piglowska A, Widlak W. Overexpression of heat shock transcription factor 1 enhances the resistance of melanoma cells to doxorubicin and paclitaxel. BMC Cancer. 2013;13:504. | ||
Okon IS, Zou MH. Mitochondrial ROS and cancer drug resistance: implications for therapy. Pharmacol Res. 2015;100:170–174. | ||
Lin CJ, Lee CC, Shih YL, et al. Resveratrol enhances the therapeutic effect of temozolomide against malignant glioma in vitro and in vivo by inhibiting autophagy. Free Radic Biol Med. 2012;52(2):377–391. | ||
Kentsis A, Reed C, Rice KL, et al. Autocrine activation of the Met receptor tyrosine kinase in acute myeloid leukemia. Nat Med. 2012;18(7):1118–1122. | ||
Lin CI, Whang EE, Donner DB, et al. Autophagy induction with RAD001 enhances chemosensitivity and radiosensitivity through MET inhibition in papillary thyroid cancer. Mol Cancer Res. 2010;8(9):1217–1226. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.