")
Back to Journals » OncoTargets and Therapy » Volume 12
MDM2 promotes epithelial–mesenchymal transition through activation of Smad2/3 signaling pathway in lung adenocarcinoma
Authors Tang Y , Xuan Y, Qiao G , Ou Z, He Z, Zhu Q, Liao M, Yin G
Received 23 August 2018
Accepted for publication 8 January 2019
Published 27 March 2019 Volume 2019:12 Pages 2247—2258
DOI https://doi.org/10.2147/OTT.S185076
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Yong Tang,1,2,* Yiwen Xuan,2,* Guibin Qiao,3 Zhu’an Ou,2 Zhe He,2 Qihang Zhu,2 Ming Liao,2 Guilin Yin1
1Southern Medical University, Guangzhou, China; 2Department of Thoracic Surgery, General Hospital of Southern Theatre Command of PLA, Guangzhou, China; 3Department of Thoracic Surgery, Guangdong General Hospital, Guangdong Academy of Medical Sciences, Guangzhou, China
*These authors contributed equally to this work
Background: Mouse double minute 2 (MDM2) contributes to cancer metastasis and epithelial–mesenchymal transition (EMT). This study aimed to investigate small mothers against decapentaplegic (Smad) signaling in MDM2-mediated EMT in lung adenocarcinoma (LAC).
Materials and methods: Expression patterns of MDM2 in LAC tissues, adjacent tissues, and cell lines (BEAS-2B, PC9, H1975, and A549) were detected. We then overexpressed MDM2 in PC9 cells and knocked it down in H1975 cells. To explore whether MDM2 activates EMT through the Smad2/3 signaling pathway, Smad2 and Smad3 were also silenced by siRNA in H1975 cells. Male BALB/c nude mice were used in in vivo model to validate the effects of MDM2 on LAC cells.
Results: MDM2 was significantly upregulated in LAC tissues compared with adjacent tissues. The expression of MDM2 was relatively higher in PC9 cells and relatively lower in H1975 cells compared with A549 cells. Overexpression of MDM2 significantly increased cell proliferation, migration, and invasion in LAC cells, while inhibiting apoptosis in PC9 cells. On the contrary, silencing of MDM2 significantly inhibited the expression of EMT-related genes N-cadherin and vimentin, while promoting the expression of E-cadherin and β-catenin. In vivo, MDM2 knockdown inhibited tumor growth. In addition, the expression of Smad2/3 was correlated with MDM2 in H1975 cells transfected with Smad2 and Smad3 siRNAs, which inhibited EMT progress.
Conclusion: MDM2 can activate the Smad2/3 signaling pathway, which promotes the proliferation and EMT progress of LAC cells.
Keywords: lung adenocarcinoma, epithelial–mesenchymal transition, Smad signaling pathway, cell metastasis, MDM2
Introduction
Lung adenocarcinoma (LAC), also known as pulmonary adenocarcinoma, is a type of non-small-cell lung cancer (NSCLC). LAC is a malignancy with a 100% mortality rate within 5 years of diagnosis, and metastasis often occurs within months of diagnosis.1,2 Rates of morbidity in LAC have been increasing among nonsmokers and women.2
The leading causes of cancer death include metastasis and chemotherapy resistance. Tumor metastasis is a complex process and is responsible for over ~90% cancer-related deaths.3 Epithelial–mesenchymal transition (EMT) is a metastasis feature that occurs in drug-resistant tumors in vivo and cancer cells in vitro.4,5 Drug- and chemotherapy-resistant cancers always show increased invasion and metastasis ability, accompanied by loss of E-cadherin (epithelial marker) and the increase in vimentin and N-cadherin (mesenchymal markers).4 The loss of E-cadherin is a key inducer of EMT and promotes metastasis through multiple downstream transcriptional pathways, including Twist and Wnt/β-catenin.6 Several transcription factors are involved in the metastasis or metastatic cells undergoing EMT,5 including Snail, Slug, Twist, and Zeb family members Zeb1 and Zeb2/Smad-interacting protein 1 (Sip1).7–10 In addition, TGF-β plays an important role in activating Snail, which is a key repressor of E-cadherin and an inducer of N-cadherin.5,11 Smads are central mediators of TGF-β signaling pathways.12 TGF-β plays a key role during cancer metastasis, and the TGF-β/Smad signaling pathway is important for maintaining the mesenchymal phenotype in metastatic cancers.13,14 For instance, Chen et al showed that EMT and motility of SKOV3 cells in vitro could be activated by the TGF-β/Smad signaling pathway.15 TGF-β/Smad-mediated Snail/E-cadherin expression plays key roles in cancer EMT.16–18
The E3 ubiquitin ligase mouse double minute 2 (MDM2) is an oncoprotein that promotes the rapid degradation of the tumor suppressor p53.19 The inhibition of p53 transcriptional activity is a key characteristic of higher MDM2 expression in tumors.20 Inhibition of the interaction between MDM2 and p53 may stabilize p53 proteins, thus resulting in suppression of tumor growth and metastasis progression.20,21 Evidence that MDM2 promotes EMT and metastasis of human cancers has led to an emergence of anti-MDM2 therapy.15,22 MDM2 overexpression has been observed and considered a prognostic factor for NSCLC.23–25 In A549 cells, p53 inactivation-mediated DEAD/H box 3 (DDX3) loss may suppress MDM2 expression and elevate Slug-inhibited E-cadherin, resulting in NSCLC metastasis via a possible MDM2/Slug/E-cadherin pathway.26 Also, Lin and Hsu showed that recombinant Ling Zhi-8, a protein analogous to an effective medicinal ingredient in the mushroom Ganoderma lucidum, induced focal adhesion kinase inactivation and Slug degradation by promoting MDM2–Slug interaction and E-cadherin expression, thus reducing EMT and metastasis in CL1–5 cells. In contrast, MDM2-shRNA inhibited Slug degradation, resulting in Slug elevation, cancer cell EMT, and metastasis.27 Chen et al showed that MDM2 promoted EMT and motility of SKOV3 cells in vitro by activation of the TGF-β/Smad signaling pathway and Snail/Slug transcription factors and by inhibition of E-cadherin.15 These results suggested important roles of MDM2 in cell EMT and metastasis mediated by the interaction of MDM2 with various signaling pathways. However, little is known about the interaction between MDM2-induced EMT and Smad2/3 signaling pathway in LAC.
We designed this study to investigate the effect of MDM2 expression on LAC progression and metastasis, as well as the possible association of the Smad signaling pathway in this process. The expression patterns of EMT factors and Smad proteins in cell lines with MDM2 overexpression or interference were also detected. This study demonstrated that MDM2 induced LAC cell EMT and metastasis via activation of Smad2 and Smad3 signaling pathway; these findings provide new insights into MDM2-mediated LAC cell EMT and cancer metastasis.
Materials and methods
Study population
Human LAC and corresponding adjacent tissues were collected from 30 patients during surgery before chemoradiotherapy. All patients were admitted into the General Hospital of Guangzhou Military Command of PLA, between October 2016 and May 2017. Each sample was divided into three sections, which were used for quantitative reverse transcription (qRT)-PCR, Western blot, and immunohistochemical analysis. All experimental protocols were approved by the Ethics Committee of General Hospital of Guangzhou Military Command of PLA. All animal experiments were carried out strictly in accordance with international ethical guidelines and the National Institutes of Health Guide concerning the Care and Use of Laboratory Animals. All donors signed the informed consent in accordance with the Declaration of Helsinki.
Cell lines and culture conditions
The human LAC cell lines A549, PC9, and NCI-H1975 were purchased from ATCC (Manassas, VA, USA). The human bronchial epithelial-derived cells BEAS-2B (ATCC) were used as the “normal” cell line. All cells were maintained in DMEM (Gibco, Carlsbad, CA, USA) supplemented with 10% FBS, 1% penicillin/streptomycin (Sigma, St Louis, MO, USA) at 37°C, with 5% CO2. Each cellular experiment was performed in triplicate.
Plasmids and cell transfection
Based on the expression of MDM2 in LAC cell lines, we selected PC9 cells for gain-of-function studies and the H1975 cells for loss-of-function studies. The lentivirus-packaged MDM2 overexpression vector, MDM2-shRNA, and all negative controls were constructed and verified as previously described.28 These procedures were performed by GenePharma (Shanghai, China). Stably transfected cell lines were screened with 0.6 μg/mL puromycin (Sigma). Knockdown of Smad2 and Smad3 in H1975 cells was performed by transfecting cells with specific siRNAs (Santa Cruz, CA, USA). DNA sequences used in this study are given below. Scrambled negative siRNA sequence: 5′-TTC TCC GAA CGT GTC ACG UTT-3′; Smad2 siRNA: 5′-AUC UAA UCG UCC UGU UUU CUU TT-3′; Smad3: 5′-UCU UCU UGA GUU UCU UGA CCA TT-3′; MDM2 shRNA: 5′-UUG GUA UUG CAC AUU UGC CUG-3′. Lipofectamine 2000 reagents (Invitrogen) were used for cell transfection according to the manufacturer’s instructions.
Cell proliferation analysis
Blank and transfected cells were seeded into 96-well plates (5,000 cells/well) and incubated for 48 hours until cell confluence reached 80%–90%. Then, cell viability was determined using MTT solution (Sigma) according to the manufacturer’s instructions. Optical density at 490 nm (OD490 nm) was recorded for evaluation of cell proliferation rate.
Flow cytometric apoptosis analysis
Blank and transfected cells (2.0×106 cells/well) were placed into 6-well plates for 48 hours until cell confluence reached 80%–90%. Cells were then harvested for apoptosis detection using Annexin V according to the instructions by the manufacturer (BD Biosciences, San Jose, CA, USA). Collected cells were incubated with Annexin V and propidium iodide successively and then analyzed with a BD FACSCalibur™ flow cytometry.
Wound healing assay
Blank and transfected LAC cells were seeded into 24-well plates (2.0×105 cells/well) and incubated for 24 hours for formation of cell monolayers. Then, a straight line was scratched in the middle of adherent cell monolayers using a sterile pipette tip. Subsequently, plates were photographed at 0 and 24 hours after scratching. Migration ability was finally determined by migration index using the formula (W0 h−W24 h) ×100%/W0 h, where W0 h and W24 h indicate the wound width at 0 hour and 24 hours after scratching, respectively.
In vitro invasion assay
The 24-well Transwell (Corning Costar, Cambridge, MA, USA) was first coated with 50 μL Matrigel (BD Biosciences). Subsequently, the upper and lower chambers of Transwell plates were, respectively, filled with serum-free medium and completed DMEM, supplemented with 1% penicillin/streptomycin and 10% FBS. Cells (1.0×105 cells/well) were then placed into the upper chamber, and Transwell plates were maintained at 37°C in 5% CO2 for 48 hours. Cells on the upper surface of the membrane were removed, and those adhered to the lower surface of the membrane (invaded cells) were fixed, stained (crystal violet, 0.1% in 20% ethanol), and counted under an Olympus light microscope (Olympus BX51, Tokyo, Japan, 100× magnification, five random fields).
In vivo animal LAC model
A total of 40 male BALB/c nude mice (4–5 weeks old from the Laboratory Animal Center of Southern Medical University, Guangzhou, China) were kept under standard conditions (22°C–25°C) with unrestricted access to high-pressure sterilized food and water. Five days later, mice were randomly divided into four groups, which were subcutaneously inoculated with PC9 cells with MDM2 overexpression (1×106, PC9 + MDM2 group, n=10), H1975 cells with MDM2 knockdown (1×106, H1975 + MDM2 shRNA group, n=10), and corresponding control PC9 cells (1×106, PC9 + vector group, n=10), or H1975 cells (1×106, H1975 + shRNA-NC, n=10), respectively. Animals were fed with laboratory food, and tumor volume was recorded six times per week. Forty-two days later, all mice were sacrificed, and the tumor tissues were separated. After recording tumor weight, the tumor samples were used for qRT-PCR and Western blot analyses. Animal care and experiments were approved by the Institute Animal Care and Use Committee of the Southern Medical University.
qRT-PCR analysis
Total RNA from cultured cells and tumor tissues (human and mice) was extracted using TRIzol reagent (TaKaRa, Tokyo, Japan). The first-strand cDNA was reverse-transcribed (Bestar qPCR RT Kit, DBI Bioscience, Ludwigshafen, Germany) and used as a DNA template (1 μg) for PCR amplification. Primers were synthesized by Sangon (Shanghai, China) and are listed in Table 1. Amplification was performed using the DBI Bestar® SybrGreen qPCRmasterMix (DBI Bioscience) on an Agilent Stratagene Mx3000P Real-time PCR machine (DBI Bioscience). The following reaction conditions were used: predegeneration at 95°C for 2 minutes, followed by 40 cycles of 94°C for 20 seconds, 58°C for 20 seconds, and 72°C for 20 seconds. Relative expression level was calculated using the 2−ΔΔCt methods with normalization to GAPDH or β-actin.
 | Table 1 Sequence of primers used for qRT-PCR or MDM2 cloning |
Western blot analysis
Protein lysates were isolated using lysis buffer (Beyotime Biotechnology, Shanghai, China) and quantified by the bicinchoninic acid method (Thermo Fisher Scientific, Waltham, MA, USA). Protein lysates (20 μg) were fractionated onto 10% SDS-PAGE (Sangon) and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Whatman, Germany). PVDF membranes were blocked with 5% skim milk-TBS at 4°C overnight, incubated with specific primary antibody solutions at room temperature for 1 hour, and incubated with HRP goat anti-rabbit/mouse IgG (1:20,000) at room temperature for 40 minutes. Enhanced chemiluminescence system (Millipore, Billerica, MA, USA) and Image-Pro Plus 6.0 software analysis system (Media Cybernetics, Inc., Bethesda, MD, USA) were used for protein blot quantitation. Antibodies against E-cadherin (1:1,000), N-cadherin (1:400), MDM2 (1:1,000), vimentin (1:1,000), β-catenin (1:5,000), Snail (1:500), Slug (1:1,000), Smad2/3 (1:2,000), p-Smad2 (1:1,000), p-Smad3 (1:2,000), and secondary antibodies were purchased from Boster Biotechnology (Wuhan, China). GAPDH antibody (1:10,000) was purchased from Yasunari Biological Engineering (Shanghai, China).
Immunohistochemical analysis
The expression of E-cadherin, N-cadherin, and MDM2 in LAC tissues and adjacent nontumor tissues was detected by immunohistochemistry. In brief, tissues were fixed, transparentized, and embedded in paraffin wax. Sections measuring 4 μm in width were prepared, blocked, and then incubated with specific primary antibodies against E-cadherin (1:500, Boster), N-cadherin (1:200, Boster), and MDM2 (1:800, Boster), followed by secondary antibody incubation following the standard procedure. Protein expression levels were determined by diaminobenzidine staining.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism 6.0 based on data expressed as mean ± SD from at least three biological repeats. The statistical significance of differences between means was analyzed by Student’s t-test (between groups). Significant differences were defined by a P-value of <0.05.
Results
Expression of MDM2 and progression of EMT were enhanced in LAC
In LAC tissues, the relative mRNA expression level of MDM2 was significantly higher than in adjacent tissues (Figure 1C). The expression of epithelial phenotype marker gene E-cadherin was significantly inhibited compared with adjacent tissues (Figure 1A). In contrast, the expression of mesenchymal phenotype marker gene N-cadherin in LAC samples was significantly higher than in adjacent tissues (Figure 1B). Similar trends in protein expression profiles of MDM2, E-cadherin, and N-cadherin were detected by Western blot analysis (Figure 1D and E) and immunohistochemistry (Figure 1F). These results suggested that MDM2 may contribute to the progress of LAC via EMT.
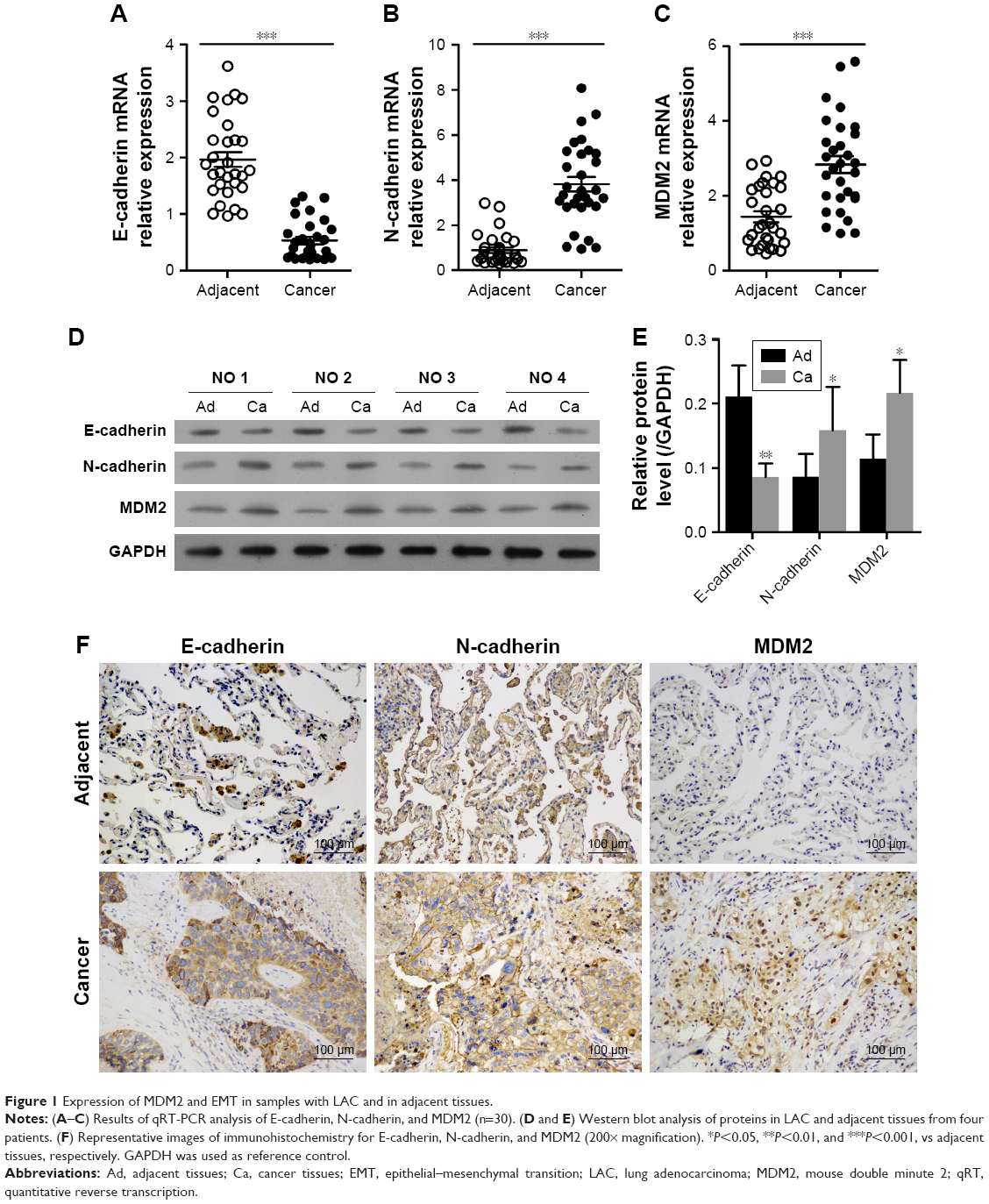 | Figure 1 Expression of MDM2 and EMT in samples with LAC and in adjacent tissues. |
Expression of MDM2 and EMT markers in LAC cell lines
The changes in expression of these proteins were further validated by the expression levels of E-cadherin, β-catenin, N-cadherin, vimentin, and MDM2 observed in several LAC cell lines (A549, PC9, and H1975) in comparison with the human bronchial epithelial-derived cell BEAS-2B (normal control). Among these LAC cell lines, PC9 showed the highest expression of E-cadherin and β-catenin and the lowest expression of MDM2, N-cadherin, and vimentin. H1975 cells exhibited the lowest expression of E-cadherin and β-catenin, but the highest expression of MDM2, N-cadherin, and vimentin (Figure 2A through 2E). Similar results were also noted in Western blot analysis (Figure 2F and G). Considering that MDM2 expression was relatively low in PC9 cells but greatly increased in H1975 cells, these two cell lines were selected for further investigation.
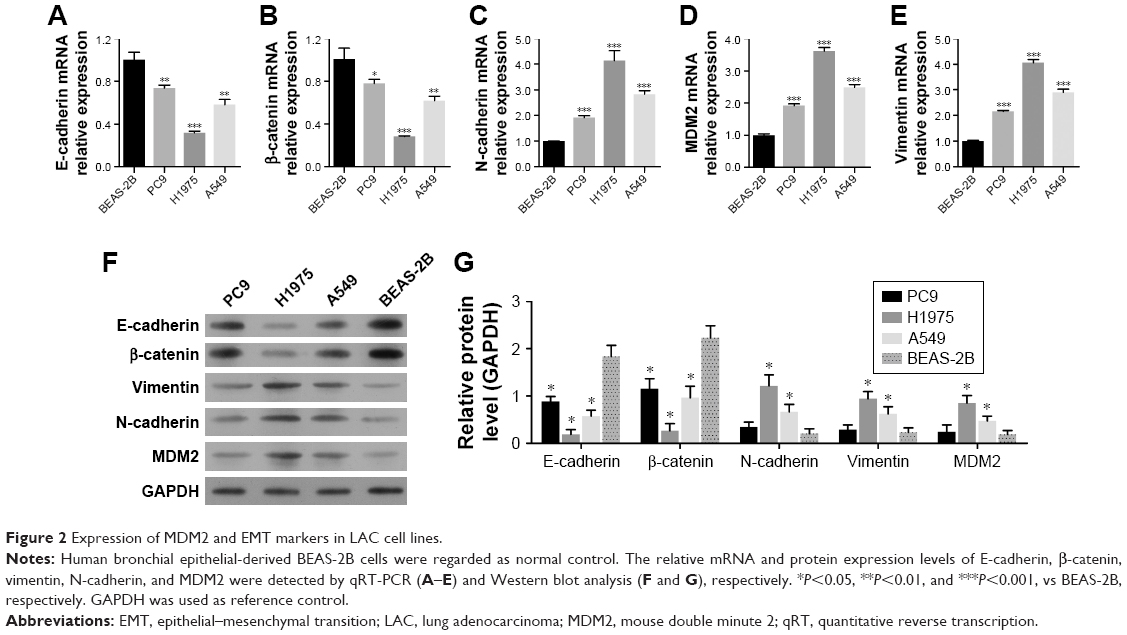 | Figure 2 Expression of MDM2 and EMT markers in LAC cell lines. |
MDM2 expression enhances LAC cell proliferation, migration, and invasion but inhibits cell apoptosis
We transduced PC9 cells with MDM2 overexpressing lentivirus plasmids and established a cell line with stable MDM2 overexpression (Figure 3A). In PC9 cells with MDM2 overexpression, the cell viability was enhanced at 48 hours (Figure 3C), migration index was increased at 24 hours (Figure 3D and E), and the number of invaded cells was upregulated (Figure 3F and G) compared with cells transfected with empty plasmid (PC9+ vector). The percentage of apoptotic cells was decreased after transfection with MDM2-overexpressing plasmid in comparison with the control (PC9+ vector) (Figure 3H and I).
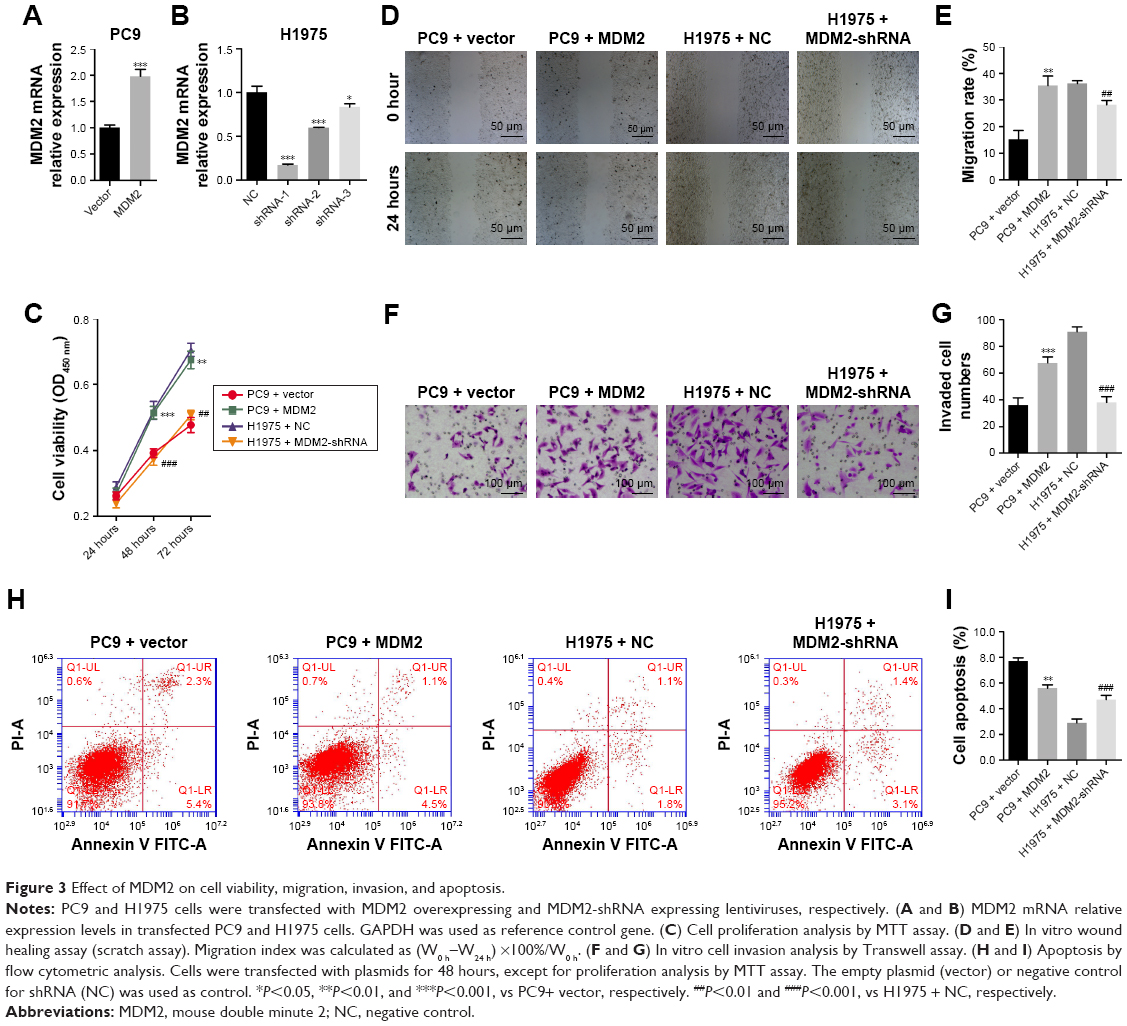 | Figure 3 Effect of MDM2 on cell viability, migration, invasion, and apoptosis. |
Compared with control cells, MDM2-silenced H1975 cells (Figure 3B) had significant reductions in viability, migration, and invasion (Figure 3C through 3G) and upregulation of cell apoptosis (Figure 3H and I). Taken together, these findings demonstrated that MDM2 expression promoted proliferation, migration, and invasion but inhibited apoptosis in LAC cells.
MDM2 expression promotes EMT of LAC cell
We subsequently confirmed that the upregulation of MDM2 in PC9 cells increased mRNA levels of Snail, Slug, N-cadherin, and vimentin but inhibited mRNA levels of E-cadherin and β-catenin in comparison with the control (Figure 4A through 4F). In contrast, knockdown of the MDM2 gene by shRNA plasmid transfection dramatically inhibited the mRNA levels of Snail, Slug, N-cadherin, and vimentin and upregulated the mRNA levels of E-cadherin and β-catenin (Figure 4A through 4F). Similar expression changes were observed in the protein abundances of Snail, Slug, N-cadherin, and vimentin in cancer cells with MDM2 overexpression or knockdown (Figure 4G), suggesting that MDM2 expression promoted EMT of LAC cells. The changes in protein levels of E-cadherin and N-cadherin were further supported by immunofluorescence (Figure 4H).
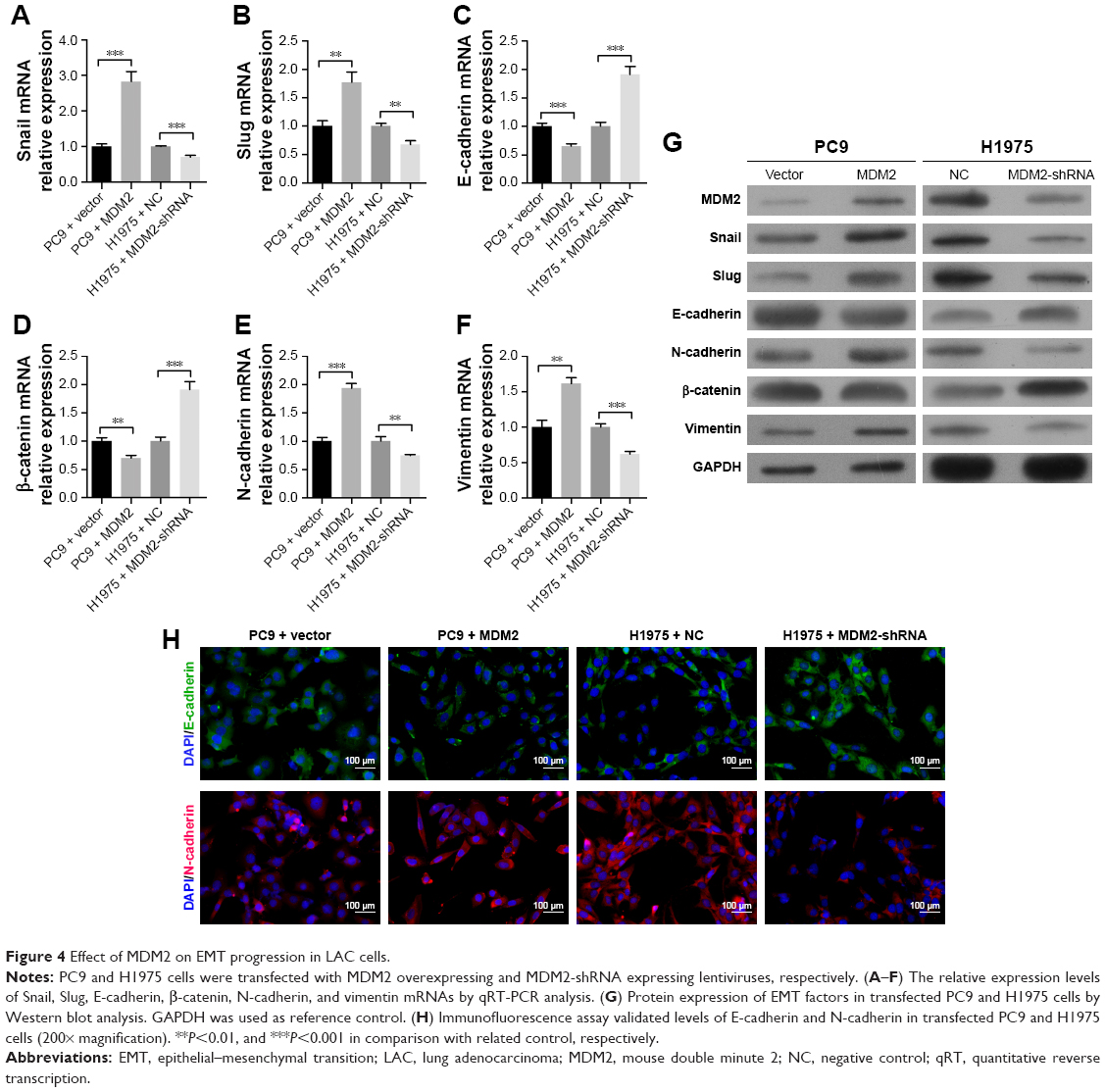 | Figure 4 Effect of MDM2 on EMT progression in LAC cells. |
MDM2 activates Smad2/3 signaling pathway
To investigate whether Smad2/3 was associated with MDM2-mediated EMT in LAC cells, we assessed Smad2/3 activation in response to altered MDM2 expression. We found that the phosphorylation of Smad2/3 was enhanced in MDM2-overexpressing PC9 cells vs PC9 cells transfected with vector (Figure 5A and B) but was inhibited in H1975 cells transfected with MDM2-shRNAs vs H1975 cells transfected with shRNA-NC (Figure 5A and B). These findings suggested consistency in Smad2/3 activation and MDM2 expression. Next, we transfected H1975 cells with Smad2 and Smad3 siRNAs to inhibit Smad2 and Smad3 expression. Following Smad2 and Smad3 inhibition (Figure 5C and D), mRNA and protein levels were significantly increased for E-cadherin and β-catenin and remarkably decreased for N-cadherin and vimentin expression compared with control (Figure 5E through 5J). These results suggested that the Smad2/3 signaling pathway was involved in MDM2-mediated EMT in H1975 cells.
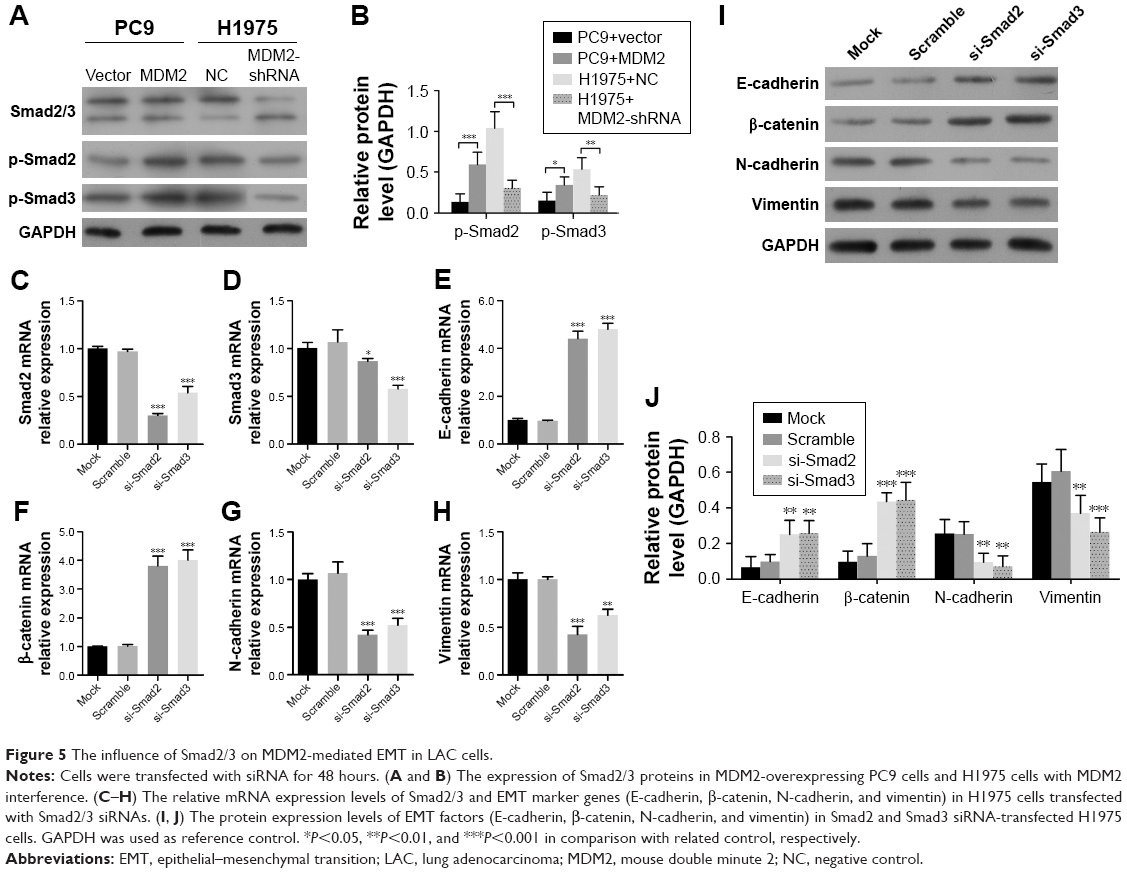 | Figure 5 The influence of Smad2/3 on MDM2-mediated EMT in LAC cells. |
MDM2 accelerates tumor growth in vivo in the mouse LAC model
For in vivo validation, we established the LAC model by subcutaneously inoculating LAC cells into male BALB/c nude mice. The injection of MDM2-overexpressing PC9 cells slightly increased tumor volume and weight compared with control (Figure 6A through 6C). In addition, mice injected with H1975 cells in which MDM2 was downregulated showed significantly decreased tumor volume and weight in comparison with mice injected with control cells (Figure 6A through 6B). Compared with tumor tissues from control PC9 cells, tumor tissues derived from MDM2-overexpressing PC9 cells showed upregulation of transcription factors Snail and Slug as well as MDM2 in Figure 6D through 6F. Also, the expression of EMT factors E-cadherin and β-catenin was downregulated (Figure 6G and H) while N-cadherin and vimentin were upregulated (Figure 6I and J) in tumor tissues derived from MDM2-overexpressing PC9 cells compared with control cells (PC9+ vector). In contrast, the downregulation of MDM2 inhibited tumor growth and expression of Snail, Slug, N-cadherin, and vimentin while enhancing the expression of E-cadherin and β-catenin in tumor tissues in comparison with tumors derived from H1975 cells. These data suggested that MDM2 expression promotes LAC tumor growth and metastasis. Moreover, we found that the expression levels of Smad2 and Smad3 in tumors generated from MDM2 overexpressing PC9 cells were relatively higher than in tumors generated from PC9 control cells (Figure 6K and L). Furthermore, the expression levels of Smad2 and Smad3 were higher in tumors derived from H1975 cells than in tumors generated from H1975 cells with MDM2 silencing. These data also supported that MDM2 promotes the Smad2/3 signaling pathway and that inhibition of MDM2 produced the opposite effect.
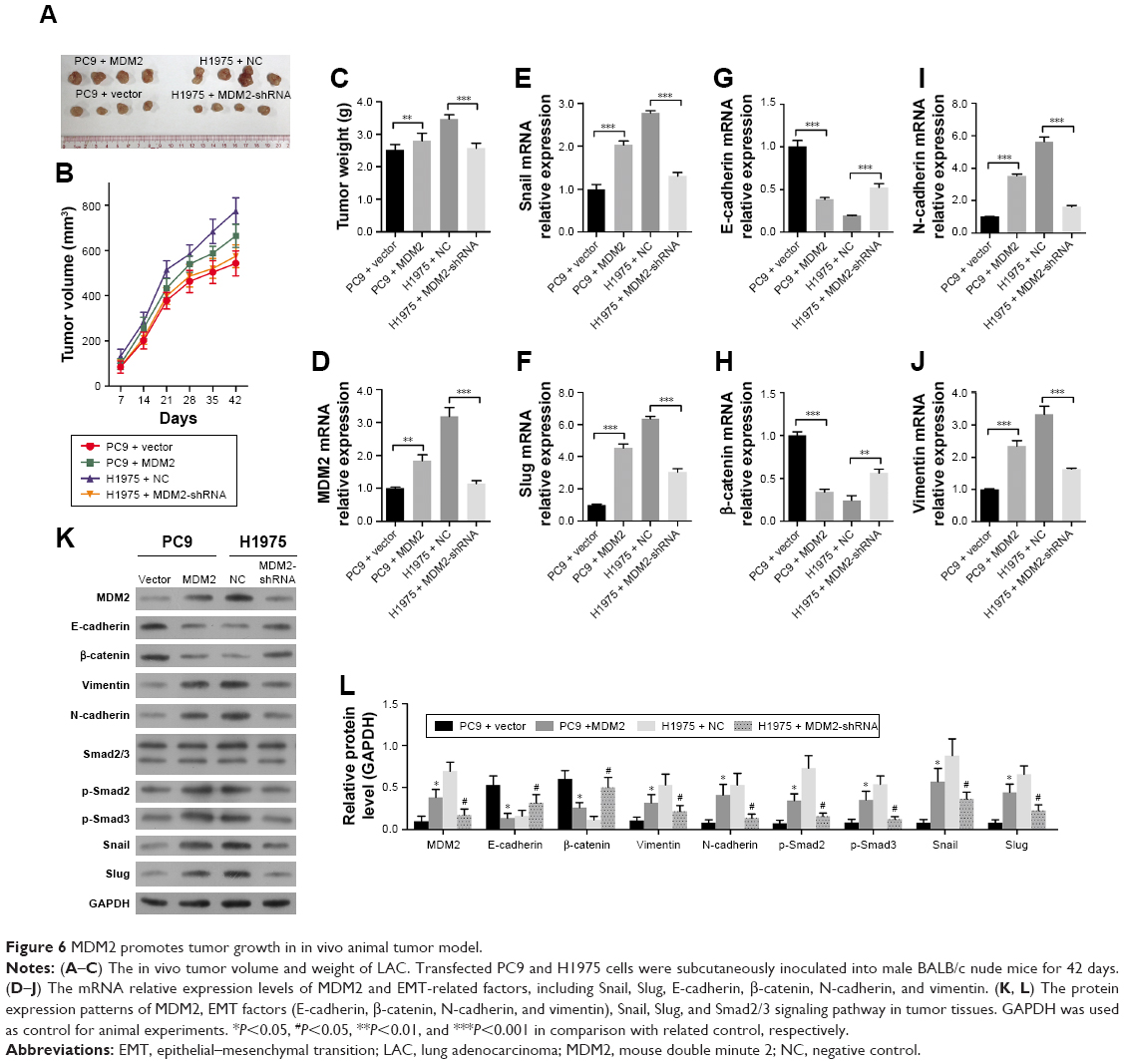 | Figure 6 MDM2 promotes tumor growth in in vivo animal tumor model. |
Discussion
MDM2 is an oncoprotein that binds to and promotes the rapid degradation of p53 tumor suppressor.19 Inhibition of MDM2–p53 interaction has been suggested to suppress tumor growth and induce tumor cell apoptosis in NSCLC and several other human cancers.20,29,30 We found that MDM2 expression was upregulated in LAC tumor tissues compared with adjacent tissues. The expression of MDM2 has been correlated with EMT and metastasis in some human cancers.15,31–34 EMT involves the Twist transcriptional pathway during migration and metastasis of cancer cells. Zhang and Hill showed that hypoxia treatment in KHT cells could upregulate MDM2 expression, increase cell resistance to apoptosis, and increase metastatic efficiency.35 Khor et al showed that higher expression level of MDM2 was related to distant metastasis in prostate cancer.33 There is increasing evidence of MDM2 overexpression in metastasis and in tumor patients with higher failure rates.20,33,36 In our investigation, upregulation of MDM2 in LAC tumor tissues compared with adjacent tissues was in agreement with studies of other cancers. In addition, we also determined that the expression of MDM2 in H1975 cells (gefitinib-resistant cells) was significantly higher than that in PC9 cells (gefitinib-sensitive cells). Taken together, these results suggested that MDM2 may be associated with LAC pathogenesis and treatment efficacy.
The EMT process is controlled by several key regulators, including suppressors of E-cadherin and β-catenin, promoters of N-cadherin and vimentin, and transcription factors including Snail, Slug, and the Zeb family. Both Snail and Slug transcription factors are zinc finger transcriptional repressors; they are key regulators of TGF-β-induced EMT and the expression of tumor suppressors such as E-cadherin.37,38 The suppression of E-cadherin and mesenchymal phenotype maintenance are initiation phenotype of EMT and metastasis. In this study, we demonstrated that MDM2 overexpression in PC9 cells promoted cell proliferation, migration, and invasion but inhibited apoptosis. In in vitro and in vivo experiments, MDM2 upregulation was followed by upregulated expression of Snail, Slug, N-cadherin, and vimentin and inhibition of E-cadherin and β-catenin. In contrast, both in vitro and in vivo experiments using MDM2-shRNA transfection into H1975 cells showed the opposite. Thus, our finding suggested that MDM2-induced invasion and metastasis in LAC cells were mediated by EMT.
Chen et al showed that MDM2 promoted EMT and motility of SKOV3 cells in vitro by activating TGF-β-Smad pathways and Snail/Slug transcription factors and inhibiting E-cadherin.15 TGF-β plays a dual role during the progression and metastasis of cancer. In early stages, TGF-β inhibits tumor growth; in later stages, it promotes cell invasion and metastasis.37 In later stages, TGF-β induced EMT by Snail in a Smad-dependent manner.14,39 Some regard E-cadherin repressors, especially the overexpression of Snail and TGF-β, as novel parameters of aggressiveness in metastatic cancers.40–44 TGF-β is a key inducer of EMT in many cancer types. In metastatic cancers, Smad-dependent TGF-β signaling pathways are potent inducers of EMT and transcription of Snail, Slug, and Zeb family to maintain the mesenchymal phenotype.13,14 Reports have shown that Smad2 and Smad3, in TGF-β/Smad signaling pathways, are associated with Zeb proteins, Zeb1 and Zeb2/Sip1, which mediates the switch from E-cadherin to N-cadherin.7–9 Another study reported that Sip1 overexpression inhibited E-cadherin and promoted clonogenicity of glioma cells.45 In our study, we demonstrated that expression of Smad2 and Smad3 was correlated with MDM2 expression. The inhibition of MDM2 suppressed Smad2 and Smad3 expression, and further inhibition of Smad2 and Smad3 by siRNA significantly suppressed EMT progression in H1975 cells by upregulating EMT suppressors E-cadherin and β-catenin and inhibiting EMT promoters N-cadherin and vimentin. These findings suggested that MDM2-induced migration and EMT in LAC cells and cancer metastasis were mediated by the Smad2/3 signaling pathways. The specific mechanisms for this process deserve further investigation. For instance, MDM2 promotes p53 degradation,19 and p53 also regulates Smad protein activities,46,47 suggesting a role for p53 as a link between MDM2 and Smad signaling in LAC. Whether this modulation is regulated by TGF-β requires additional study.
Conclusion
In summary, we confirmed that MDM2 overexpression in LAC cells was correlated with cell migration, invasion, and metastasis in in vitro and in vivo studies. MDM2-induced tumor metastasis was associated with the Smad2/3 signaling pathway. The inhibition of MDM2 suppressed tumor growth, cell proliferation, migration, and invasion but promoted cell apoptosis. Therefore, MDM2 may be a potential therapeutic target for LAC treatment. Finally, the role of TGF-β in Smad2/3 pathway-associated metastasis in LAC merits further investigation.
Acknowledgment
This work is supported by the Science and Technology Program of Guangzhou, China (No 201607010117).
Disclosure
The authors report no conflicts of interest in this work.
References
Nguyen DX, Chiang AC, Zhang XH, et al. Wnt/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell. 2009;138(1):51–62. | ||
Schuller HM, Al-Wadei HA, Majidi M. Gamma-aminobutyric acid, a potential tumor suppressor for small airway-derived lung adenocarcinoma. Carcinogenesis. 2008;29(10):1979–1985. | ||
Song Q, Xu Y, Yang C, et al. miR-483-5p promotes invasion and metastasis of lung adenocarcinoma by targeting RhoGDI1 and ALCAM. Cancer Res. 2014;74(11):3031–3042. | ||
Li J, Wang Y, Song Y, Fu Z, Yu W. miR-27a regulates cisplatin resistance and metastasis by targeting RKIP in human lung adenocarcinoma cells. Mol Cancer. 2014;13(1):193. | ||
Heerboth S, Housman G, Leary M, et al. EMT and tumor metastasis. Clin Transl Med. 2015;4(1):6. | ||
Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68(10):3645–3654. | ||
Cong N, Du P, Zhang A, et al. Downregulated microRNA-200a promotes EMT and tumor growth through the Wnt/β-catenin pathway by targeting the E-cadherin repressors ZEB1/ZEB2 in gastric adenocarcinoma. Oncol Rep. 2013;29(4):1579–1587. | ||
Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–4751. | ||
Rogers CD, Saxena A, Bronner ME. SIP1 mediates an E-cadherin-to-N-cadherin switch during cranial neural crest EMT. J Cell Biol. 2013;203(5):835–847. | ||
Roberts AB, Tian F, Byfield SD, et al. Smad3 is key to TGF-beta-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev. 2006;17(1–2):19–27. | ||
Yu H, Shen Y, Hong J, Xia Q, Zhou F, Liu X. The contribution of TGF-β in epithelial-mesenchymal transition (EMT): down-regulation of E-cadherin via snail. Neoplasma. 2015;62(1):1–15. | ||
Katsuno Y, Hanyu A, Kanda H, et al. Bone morphogenetic protein signaling enhances invasion and bone metastasis of breast cancer cells through Smad pathway. Oncogene. 2008;27(49):6322–6333. | ||
Brandl M, Seidler B, Haller F, et al. IKK(α) controls canonical TGF(β)-SMAD signaling to regulate genes expressing SNAIL and SLUG during EMT in Panc1 cells. J Cell Sci. 2010;123(Pt 24):4231–4239. | ||
Wang Y, Shi J, Chai K, Ying X, Zhou B. The role of Snail in EMT and tumorigenesis. Curr Cancer Drug Targets. 2013;13(9):963–972. | ||
Chen Y, Wang DD, Wu YP, et al. MDM2 promotes epithelial-mesenchymal transition and metastasis of ovarian cancer SKOV3 cells. Br J Cancer. 2017;117(8):1192–1201. | ||
Ji Q, Han Z, Zhou L. Resveratrol inhibits epithelial-to-mesenchymal transition and metastasis in colorectal cancer through regulating Snail/E-cadherin expression by TGFβ1/Smads signaling pathway. AACR. 2016;76(14 Suppl): Abstract nr 1689. | ||
Wei MG, Sun W, He WM, Ni L, Yang YY. Ferulic acid attenuates TGF-β1-induced renal cellular fibrosis in NRK-52E cells by inhibiting Smad/ILK/Snail pathway. Evid Based Complement Alternat Med. 2015;2015(4):1–7. | ||
Liu X, Ji Q, Deng W, et al. JianPi JieDu recipe inhibits epithelial-to-mesenchymal transition in colorectal cancer through TGF-β/Smad mediated Snail/E-cadherin expression. Biomed Res Int. 2017;2017:Article ID 2613198. | ||
Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362(6423):857–860. | ||
Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13(2):83–96. | ||
Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. | ||
Karni-Schmidt O, Lokshin M, Prives C. The roles of MDM2 and MDMX in cancer. Annu Rev Pathol. 2016;11(1):617–644. | ||
Ko JL, Cheng YW, Chang SL, Su JM, Chen CY, Lee H. MDM2 mRNA expression is a favorable prognostic factor in non-small-cell lung cancer. Int J Cancer. 2000;89(3):265–270. | ||
Heist RS, Zhou W, Chirieac LR, et al. MDM2 polymorphism, survival, and histology in early-stage non-small-cell lung cancer. J Clin Oncol. 2007;25(16):2243–2247. | ||
Dworakowska D, Jassem E, Jassem J, et al. MDM2 gene amplification: a new independent factor of adverse prognosis in non-small cell lung cancer (NSCLC). Lung Cancer. 2004;43(3):285–295. | ||
Wu DW, Lee MC, Wang J, Chen CY, Cheng YW, Lee H. DDX3 loss by p53 inactivation promotes tumor malignancy via the MDM2/Slug/E-cadherin pathway and poor patient outcome in non-small-cell lung cancer. Oncogene. 2014;33(12):1515–1526. | ||
Lin TY, Hsu HY. Ling Zhi-8 reduces lung cancer mobility and metastasis through disruption of focal adhesion and induction of MDM2-mediated slug degradation. Cancer Lett. 2016;375(2):340–348. | ||
Zhang Y, Zhang Q, Zhang M, et al. DC – SIGNR by influencing the lncRNA HNRNPKP2 upregulates the expression of CXCR4 in gastric cancer liver metastasis. Mol Cancer. 2017;16(1):78. | ||
Deben C, Wouters A, Op de Beeck K, et al. The MDM2-inhibitor nutlin-3 synergizes with cisplatin to induce p53 dependent tumor cell apoptosis in non-small cell lung cancer. Oncotarget. 2015;6(26):22666–22679. | ||
Hai J, Sakashita S, Allo G, et al. Inhibiting MDM2-p53 interaction suppresses tumor growth in patient-derived non-small cell lung cancer xenograft models. J Thorac Oncol. 2015;10(8):1172–1180. | ||
Araki S, Eitel JA, Batuello CN, et al. TGF-beta1-induced expression of human MDM2 correlates with late-stage metastatic breast cancer. J Clin Invest. 2010;120(1):290–302. | ||
Wang W, Zhang X, Qin JJ, et al. Natural product ginsenoside 25-OCH3-PPD inhibits breast cancer growth and metastasis through down-regulating MDM2. PLoS One. 2012;7(7):e41586. | ||
Khor LY, Bae K, Paulus R, et al. MDM2 and Ki-67 predict for distant metastasis and mortality in men treated with radiotherapy and androgen deprivation for prostate cancer: RTOG 92-02. J Clin Oncol. 2009;27(19):3177–3184. | ||
Ma C, Nong K, Zhu H, et al. H19 promotes pancreatic cancer metastasis by derepressing let-7’s suppression on its target HMGA2-mediated EMT. Tumour Biol. 2014;35(9):9163–9169. | ||
Zhang L, Hill RP. Hypoxia enhances metastatic efficiency by up-regulating MDM2 in KHT cells and increasing resistance to apoptosis. Cancer Res. 2004;64(12):4180–4189. | ||
Ladanyi M, Cha C, Lewis R, Jhanwar SC, Huvos AG, Healey JH. MDM2 gene amplification in metastatic osteosarcoma. Cancer Res. 1993;53(1):16–18. | ||
Naber HP, Drabsch Y, Snaar-Jagalska BE, Ten Dijke P, van Laar T. Snail and Slug, key regulators of TGF-β-induced EMT, are sufficient for the induction of single-cell invasion. Biochem Biophys Res Commun. 2013;435(1):58–63. | ||
Bolós V, Peinado H, Pérez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with snail and E47 repressors. J Cell Sci. 2003;116(Pt 3):499–511. | ||
Peinado H, Olmeda D, Cano A. Snail, ZEB and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7(6):415–428. | ||
Elloul S, Elstrand MB, Nesland JM, et al. Snail, slug, and Smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer. 2005;103(8):1631–1643. | ||
Yoshida J, Horiuchi A, Kikuchi N, et al. Changes in the expression of E-cadherin repressors, Snail, slug, SIP1, and twist, in the development and progression of ovarian carcinoma: the important role of Snail in ovarian tumorigenesis and progression. Med Mol Morphol. 2009;42(2):82–91. | ||
Zheng M, Jiang Y-P, Chen W, et al. Snail and Slug collaborate on EMT and tumor metastasis through miR-101-mediated EZH2 axis in oral tongue squamous cell carcinoma. Oncotarget. 2015;6(9):6797–6810. | ||
Kurioka K, Wato M, Iseki T, Tanaka A, Morita S. Differential expression of the epithelial mesenchymal transition factors snail, slug, Twist, TGF-β, and E-cadherin in ameloblastoma. Med Mol Morphol. 2017;50(2):68–75. | ||
Siar CH, Ng KH. Differential expression of transcription factors snail, slug, SIP1, and twist in ameloblastoma. J Oral Pathol Med. 2014;43(1):45–52. | ||
Xia M, Hu M, Wang J, et al. Identification of the role of Smad interacting protein 1 (SIP1) in glioma. J Neurooncol. 2010;97(2):225–232. | ||
Liu H, Jia D, Li A, et al. P53 regulates neural stem cell proliferation and differentiation via BMP-Smad1 signaling and Id1. Stem Cells Dev. 2013;22(6):913–927. | ||
Wu B, Li W, Qian C, Zhou Z, Xu W, Wu J. Down-regulated p53 by siRNA increases Smad4’s activity in promoting cell apoptosis in MCF-7 cells. Eur Rev Med Pharmacol Sci. 2012;16(9):1243–1248. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.