Back to Journals » OncoTargets and Therapy » Volume 11
MCL-1 inhibition in cancer treatment
Authors Xiang W, Yang CY ![]() , Bai L
, Bai L ![]()
Received 8 July 2018
Accepted for publication 10 September 2018
Published 23 October 2018 Volume 2018:11 Pages 7301—7314
DOI https://doi.org/10.2147/OTT.S146228
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Weiguo Xiang,1 Chao-Yie Yang,1,2 Longchuan Bai1,2
1Department of Internal Medicine, University of Michigan Medical School, 2Rogel Cancer Center, University of Michigan, Ann Arbor, MI, USA
Abstract: Myeloid cell leukemia-1 (MCL-1), a member of antiapoptotic BCL-2 family proteins, is a key regulator of mitochondrial homeostasis. Frequent overexpression of MCL-1 in human primary and drug-resistant cancer cells makes it an attractive cancer therapeutic target. Significant progress has been made in the development of small-molecule MCL-1 inhibitors in recent years, and three MCL-1 selective inhibitors have advanced to clinical trials. This review briefly discusses recent advances in the development of small molecules targeting MCL-1 for cancer therapy.
Keywords: BCL-2, BAX, BAK, apoptosis, BH3 mimetics
Introduction
MCL-1 is an antiapoptotic member of BCL-2 family proteins, which include the following three groups classified by their functions: antiapoptotic, proapoptotic, and BH3-only proteins.1 Antiapoptotic BCL-2 proteins include MCL-1, BCL-2, BCL-W, BCL-XL, BCL-B, and BFL-1/A1. Proapoptotic proteins include BCL-2 homologous antagonist killer (BAK) and BCL-2 associated X protein (BAX). BH3-only proteins include BCL-like protein 11 (BIM), BCL-2-associated death (BAD), BH3 interacting-domain death agonist (BID), phorbol-12-myristate-13-acetate-induced protein 1 (NOXA), and p53 upregulated modulator of apoptosis (PUMA). The interactions between these three groups of proteins determine the fate of cells.
MCL-1 is widely expressed in human tissues2 and is primarily located in the mitochondria in cells, in which it inserts into the mitochondrial membrane via a hydrophobic tail.3 The antiapoptotic function of MCL-1 is essential to cell survival and homeostasis.4,5
Amplification and overexpression of MCL-1 have been reported in various human tumors, including hematological malignancies and solid tumors (eg, non-small-cell lung cancer, breast cancer, ovarian cancer, prostate cancer, and pancreatic cancer).6–11 An analysis of 3,131 cancer specimens has revealed that 36% of breast cancer and 54% of lung cancer specimens exhibit elevated levels of MCL-1 expression.12 MCL-1 amplification and overexpression are also frequently associated with poor prognosis and resistance to anticancer drugs. As an example, breast cancer, especially triple-negative breast cancer, is characterized with the amplification of MCL-1 gene loci (~20%).13,14 Also, high MCL-1 expression level in breast tumors correlates with high tumor grade and poor prognosis in patients.7,15 Similarly, in esophageal squamous cell carcinoma, the copy number of MCL-1 and its variants is correlated with patient’s poor prognosis.16,17
MCL-1 has been shown to be both an intrinsic and acquired resistance factor that limits the efficacy of various antitumor agents, including taxol, cisplatin, erlotinib, and other standard anticancer drugs.13,18,19 Not surprisingly, downregulation of MCL-1 expression increases cancer cell sensitivity to drug treatment. For instance, MCL-1 knock down in neuroblastoma cell lines increases their sensitivities to etoposide, doxorubicin, and ABT-737 by 2–300-folds.20 Depletion of MCL-1 reverses cisplatin and doxorubicin chemoresistance in osteosarcoma cell lines in vitro and xenograft tumors in vivo.21
MCL-1 and apoptosis
MCL-1 exerts its antiapoptotic function by sequestering proapoptotic proteins BAK/BAX through the BH3 domain containing hydrophobic groove. In the prosurvival mode (Figure 1A), BAK/BAX interacts with antiapoptotic BCL-2 proteins and is unable to execute the apoptotic program, thereby allowing cells to maintain homeostasis. In the apoptotic mode (Figure 1B), BAK/BAX can be activated through intrinsic pathways such as stress and upstream signals that act on the BH3-only proteins.22,23 These BH3-only proteins bind the BCL-2 prosurvival proteins releasing BAK/BAX. Unlike BAK, which attaches to the membrane of mitochondria, BAX is located in the cytosol. After the activation of the BH3-only protein BID, BAX is translocated to the mitochondria to exert its apoptotic function; however, this BAX translocation mechanism remains to be fully understood.24,25 After dissociation from prosurvival proteins, BAK/BAX exposes its BH3 domain through which it forms oligomers. The oligomers are able to breach the outer membrane lipid bilayer of mitochondria that leads to mitochondrial herniation and mitochondrial DNA efflux, followed by the release of cytochrome c and the activation of initiator caspases.26,27
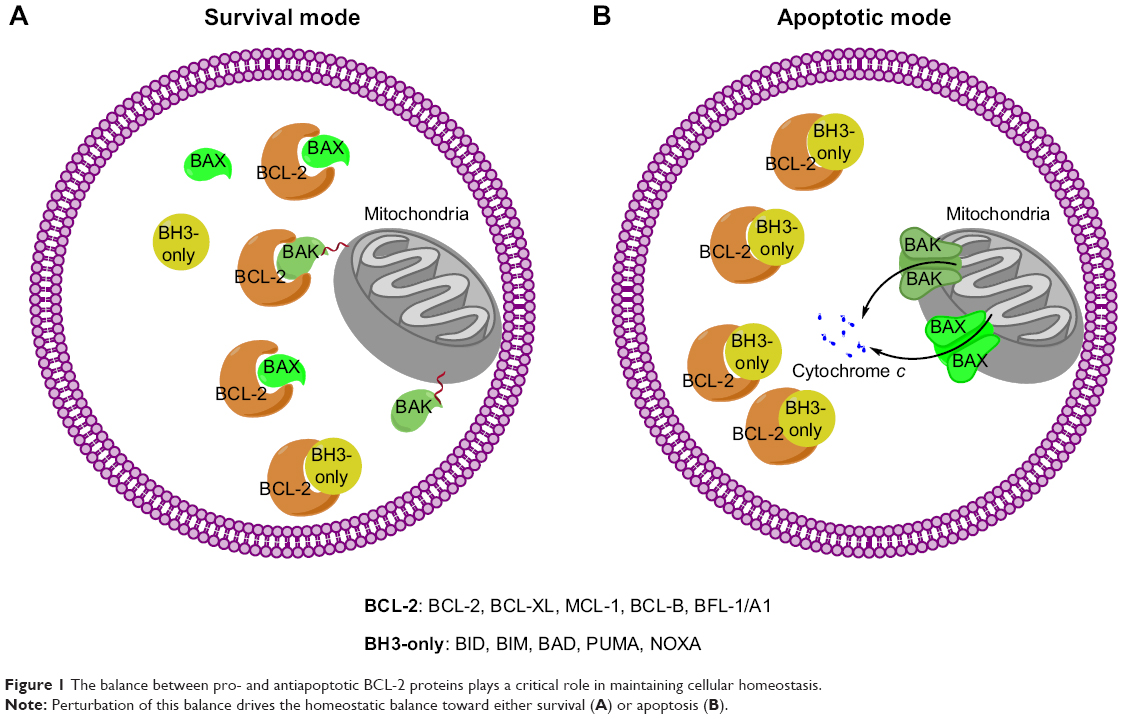 | Figure 1 The balance between pro- and antiapoptotic BCL-2 proteins plays a critical role in maintaining cellular homeostasis. |
BH3-only proteins, BIM, PUMA, BAD, NOXA and BID, restore BAX/BAK activities through interruption of the MCL-1:BAK/BAX complexes.28 There are two proposed BH3-only protein rescuing mechanisms. The first mechanism is the substrate swap model, in which BH3-only proteins bind to MCL-1 to displace BAK/BAX from the MCL-1:BAK/BAX heterodimer.1,29 This model is supported by the evidence that some BH3-only proteins bind tightly to MCL-1 at the site MCL-1 uses to bind with BAK/BAX; thus, some BH3-only proteins retain MCL-1 and prevent it from binding to BAK/BAX. However, this is contested by the reports, which show that when the BH3-only proteins are absent, BAK can still be activated by losing MCL-1 in nontransformed cells.29,30 The second mechanism proposes that BH3-only proteins, competing against MCL-1, instead bind with BAK/BAX and the complexes formed by BH3-only proteins and BAK/BAX activate the apoptotic program. Based on the proposed mechanisms, the BH3-only proteins are also divided into two types, namely sensitizers and activators.31,32 The sensitizer BH3-only proteins maintain cellular homeostasis and can displace BAK/BAX from the MCL-1:BAK/BAX complex by binding to MCL-1. The activators BH3-only proteins not only bind to MCL-1 but also bind to BAK/BAX directly to facilitate BAK/BAX oligomerization by forming macropores embodied on the mitochondrial membrane. This model has been illustrated by monitoring the fluorescence complex of BIM, PUMA, and NOXA with BAK/BAX in living HeLa cells.33
Targeting MCL-1 for cancer therapy
Given the critical roles of BCL-2 family proteins in maintaining cellular homeostasis, perturbation of the complexes between pro- and antiapoptotic BCL-2 proteins or their levels of expression could alter the cellular homeostatic balance and lead to overcoming apoptosis. Such imbalances can lead to the immortalization of cancers. To achieve the effects of controlling cell fate, small molecules have been developed to compensate for the imbalance between pro- and antiapoptotic BCL-2 proteins and restore the apoptotic pathway (Table 1). Several MCL-1 targeting small molecules have advanced into clinical trials (Table 2).
 | Table 1 Approaches of targeting MCL-1 for cancer therapy |
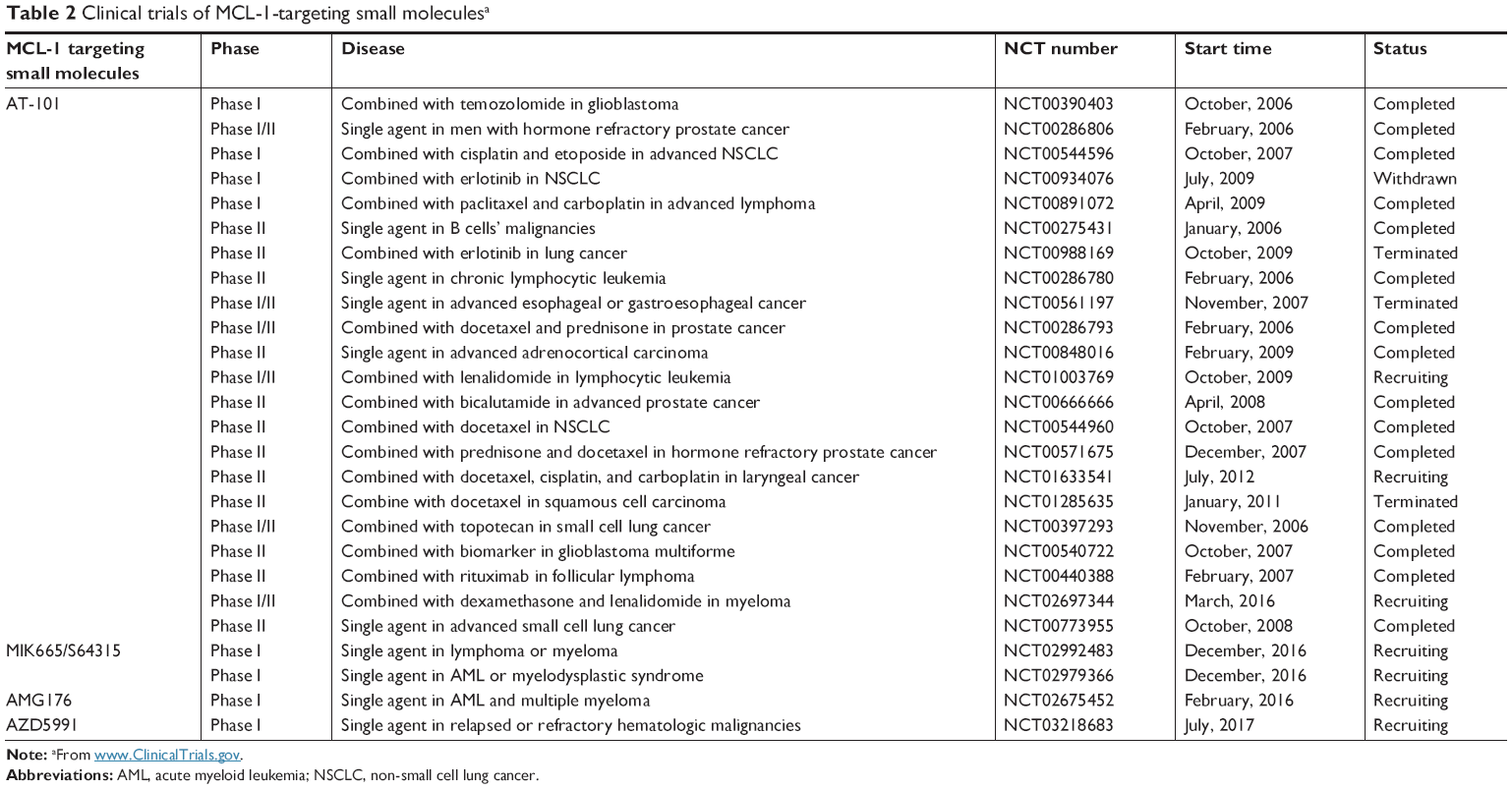 | Table 2 Clinical trials of MCL-1-targeting small moleculesa |
Small molecules targeting MCL-1
MCL-1, and other antiapoptotic BCL-2 proteins, interact with BAK/BAX through the conserved BH3 domain binding groove in MCL-1. BH3-only proteins bind to MCL-1 using the same site to displace BAK/BAX. However, selectivity of BH3-only proteins to different antiapoptotic BCL-2 proteins exists.34,35 For example, BIM and PUMA bind indiscriminately to all antiapoptotic BCL-2 proteins at low nanomolar Kd value. BAD does not bind to MCL-1 but binds tightly to BCL-2, BCL-XL, and BCL-W. BID binds to MCL-1 in the micromolar range but is an activator of BAX for its translocation. NOXA selectively binds to MCL-1 in sub-nanomolar concentration without significant binding to BCL-2 and BCL-XL up to micromolar concentrations. In summary, BIM and PUMA are pan prosurvival BCL-2 binders, BAD is a non-MCL-1 prosurvival BCL-2 protein binder, and NOXA is a MCL-1 selective binder (Figure 2).36
 | Figure 2 Interactions between BCL-2 family proteins. |
BH3 mimetics derived from BH3-only proteins are also able to release BAK/BAX sequestered by prosurvival proteins, therefore leading to apoptosis in cancer cells. Each BH3 mimetic has a similar spectrum of selectivity toward antiapoptotic BCL-2 proteins to its parent BH3-only protein.37 For example, NOXA mimetics demonstrate MCL-1 selectivity over BCL-2, BCL-XL, and BCL-W, whereas BIM and PUMA mimetics are pan-BCL-2 family inhibitors. BH3 mimetics have been extensively investigated and more than two dozen small-molecule BH3 mimetics have shown promising antitumor activities in preclinical studies. Some of these BH3 mimetics have been extensively reviewed elsewhere.38,39 Only those with anti-MCL-1 activity will be discussed here.
Pan-BCL-2 inhibitors with anti-MCL-1 activity
The development of potent and selective MCL-1 inhibitors has proven challenging, in part due to key structural differences between the BH3-binding grooves of MCL-1 and other BCL-2 proteins. Several pan-BCL-2 family protein inhibitors, such as AT-101, TW-37, sabutoclax (BI-97C1), marinopyrrole A, and gambogic acid (GA), have been reported to possess anti-MCL-1 activities (Figure 3).
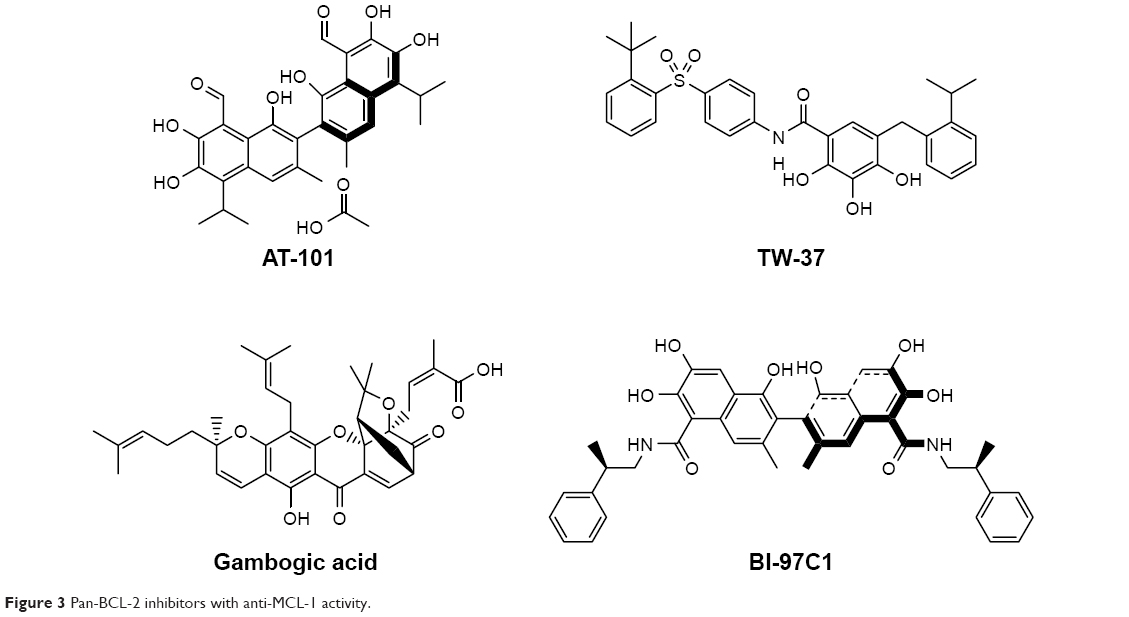 | Figure 3 Pan-BCL-2 inhibitors with anti-MCL-1 activity. |
AT-101
AT-101, the R-(–) enantiomer of gossypol acetic acid, binds with BCL-2, BCL-XL, and MCL-1 with the Ki of 0.32, 0.48, and 0.18 μM, respectively, in cell-free assays.40 In NSCLC and myeloma mouse models, AT-101 demonstrates potent antitumor activities either administered alone or in combination with gefitinib and cisplatin.41,42 In a Phase II clinical trial in CLL, with a single 80 mg dose of AT-101, apoptosis of CLL cells was detected in 18%–45% of cells by Annexin V flow cytometry analysis in four of the six patients.43 As a single agent or in combination with the standard of care chemotherapies, AT-101 has been evaluated in more than 20 Phase I and II clinical trials in different types of cancers (Table 2). AT-101 is the first small molecule with potent anti-MCL-1 activity that has been evaluated in clinical trials.
TW-37
TW-37 is a rationally designed small molecule targeting the BH3-binding groove in BCL-2 where proapoptotic BCL-2 proteins bind and has a higher affinity and selectivity for MCL-1 and BCL-2 over BCL-XL with the Ki values of 0.26, 0.29, and 1.11 μM, respectively.44 TW-37 exerts strong antiproliferative effect in de novo chemoresistant lymphoma cells without a significant effect on normal peripheral blood lymphocytes. Mechanistically, TW-37 disrupts heterodimer complexes between BAX and MCL-1 or BCL-2, resulting in apoptotic cell death. Pre-exposure of lymphoma cells to TW-37 significantly enhances the antitumor activity of cyclophosphamide–doxorubicin–vincristine–prednisone regimen both in vitro and in vivo. A Kaplan–Meier analysis reveals that the combination of TW-37 with AT-101 significantly extends the time to the failure of human oral squamous cell carcinoma OSCC-3 tumors in severe combined immunodeficiency disease mice as compared with monotherapies with TW-37 or cisplatin.45
GA
GA is a caged xanthone derived from Garcinia hanburyi and has a strong cytotoxic activity in human cancer cell lines. Employing BFL-1/A1 as a target for screening of a library of natural products, Reed’s group identified GA as a competitive inhibitor of BFL-1 in a fluorescence polarization (FP) assay.46 Analysis of competition for the BH3 peptides binding revealed that GA competitively inhibits BCL-B, MCL-1, BFL-1/A1, BCL-2, BCL-XL, and BCL-W, with the IC50 of 0.66, 0.79, 1.06, 1.21, 1.47, and 2.02 μM, respectively. However, GA retains a cytotoxic activity against BAX−/−/BAK−/− cells, indicating that additional targets contribute to the cytotoxic activity of GA. GA has a half-life of 1 hour and a relative low Cmax of 12.8 ng/mL in rats after 6.25 mg/kg oral administration.47 GA also exerts moderate efficacy in H22 xenograft tumors with only 45% tumor inhibition after 2 mg/kg ip dosing.48
Sabutoclax (BI-97C1)
Sabutoclax, an apogossypol derivative, binds to BCL-2, BCL-XL, MCL-1, and BFL-1/A1 with the IC50 values of 0.32, 0.31, 0.20, and 0.20 μM, respectively, measured by a BIM FP competitive assay. Sabutoclax also demonstrates low micromolar IC50 values in various breast cancer and prostate cancer cell lines.49 In vivo studies demonstrated that sabutoclax possesses the ability to overcome drug resistance in breast cancer and mda-7/IL-24-mediated prostate cancer. Sabutoclax treatment effectively reduces the tumor sizes in both xenograft tumor models and transgenic mouse models of prostate cancer.50,51
Selective MCL-1 small-molecule inhibitors
The recent few years have witnessed the significant progress in the development of selective MCL-1 inhibitors. A cohort of highly potent and selective small molecule MCL-1 inhibitors has been reported. These include maritoclax,52 UMI-77,53 A-1210477,54 a diverse collection of compounds from Fesik’s group at Vanderbilt University,55,56 AMG176,16 AZD5991,57 and the MIK665/S64315 and S63845 series (Figure 4).42
 | Figure 4 Selective MCL-1 inhibitors. |
Maritoclax
Maritoclax is the renamed natural product of marinopyrrole A. It is reported to selectively bind MCL-1 over BCL-XL by the ELISA and blocks the interaction between MCL-1 and BIM in a biochemical assay.52 Maritoclax selectively kills MCL-1-overexpressed but not BCL-2- or BCL-XL-overexpressed K562 leukemia cells. Moreover, maritoclax induces MCL-1 degradation via the proteasome system, which is associated with the proapoptotic activity of maritoclax.58
UMI-77
UMI-77 was identified by high-throughput screening using an FP-based binding assay.53 It binds to MCL-1, BFL-1/A1, BCL-W, BCL-2, and BCL-XL with the Ki of 0.49, 5.33, 8.19, 23.83, and 32.99 μM, respectively. UMI-77 blocks the heterodimerization of MCL-1/BAX and MCL-1/BAK in cells, thus antagonizing the MCL-1 function. UMI-77 inhibits cell growth and induces intrinsic apoptosis in pancreatic cancer cells. In a pancreatic cancer cell line BxPC-3 xenograft mouse model, UMI-77 (60 mg/kg iv) exhibits tumor growth inhibition activity without overt toxicity. UMI-77 also demonstrated tumor inhibitory activity in triple-negative breast cancer cell line MDA-MB-468 xenograft mouse model.53
A-1210477
A-1210477, developed by the AbbVie team, which also developed highly selective and potent BCL-2-specific and BCL-XL-specific small-molecule inhibitors, is a highly potent and selective MCL-1 inhibitor with the Ki and IC50 values of 0.454 and 26.2 nM, respectively, and gives >100-fold selectivity over other BCL-2 family members.59 A-1210477 potently induces apoptosis and inhibits MCL-1-dependent cell viability in a panel of non-small-cell lung cancer cell lines. Moreover, A-1210477 synergizes with navitoclax (ABT-263) to kill various cancer cell lines. In SKBR3 breast cancer cells, A-1210477 inhibits MCL-1:BIM interaction and induces intrinsic apoptosis.60 A recent study shows that A-1210477 suppresses the formation of carcinogen 4-nitroquinoline oxide-induced esophageal squamous cell carcinoma in mice.61
Fesik’s compounds
Stephen Fesik’s group at Vanderbilt University has discovered a series of potent and selective MCL-1 inhibitors with picomolar to low nanomolar binding affinities utilizing fragment-based drug design and structure-based drug design approaches.55,56,62 Most of these compounds bear a benzo-fused bicyclic and a dimethyl chloro phenyl ring as exemplified in Figure 4. Further structural rigidification has yielded compounds with reduced human serum albumin binding and improved physicochemical properties. Importantly, these compounds bind to the hydrophobic groove of MCL-1 and exhibit cell killing activities in MCL-1-dependent cell lines in vitro.
MIK665/S64315
MIK665/S64315, developed by Servier, Vernalis, and Novartis, belongs to the same series of compounds as S63845.42 Unlike S63845, very little information on S64315 has been disclosed. S63845 initiates BAK/BAX-dependent apoptosis following its binding to the BH3 binding groove of MCL-1. It demonstrates high binding selectivity for human MCL-1 (FP Ki<1.2 nM, and surface plasmon resonance Kd=0.19 nM) over human BCL-2 and BCL-XL (both FP Ki>10,000 nM). It has single-digit to two-digit nanomolar IC50 values in most myeloma, myeloid leukemia, and acute myeloid leukemia (AML) cell lines. However, solid tumor cell lines such as non-small cell lung cancer (NSCLC) cell lines can circumvent S63845 inhibition by overexpressing BCL-XL. In a mouse toxicity study, S63845 is well tolerated at an efficacious dose (25 mg/kg), but significant mice weight loss is observed at a 60 mg/kg dosage. In efficacy studies, S63845 at 25 mg/kg, iv dosing, achieves tumor regression in AMO1, H929, and MV4-11 xenograft tumor models.63 For S64315, two Phase I clinical trials, one in AML and myelodysplastic syndrome (MDS) (NCT02992483) and the other in multiple myeloma (MM) and lymphoma (NCT02979366), have started patient recruitment to determine the tolerated dosage and the toxicity profile.
AMG176
AMG176 is selected from more than a thousand compounds during lead optimization by Amgen.64 AMG176 has Ki<1 nM to MCL-1 in biochemical assays and IC50 <0.1 μM in cellular assays and is a selective MCL-1 inhibitor. A profiling of >200 cell lines treated with AMG176 reveals its robust effects on the viability of hematological cell lines (MM, AML, and non-Hodgkin’s lymphoma) and a subset of solid tumor cell lines (breast cancer and non-small-cell lung cancer cell lines). AMG176 was shown to disrupt the MCL-1:BAK complex, leading to the rapid induction of intrinsic apoptosis and the loss of cell viability. Oral administration of AMG176 to mice bearing MM OPM-2 xenografts results in a dose-dependent increase in activated BAK. Treatment of AMG176 at 20–60 mg/kg oral, once a day, demonstrates robust tumor growth inhibition, whereas complete tumor regression has been achieved at an elevated dose. AMG176 is currently evaluated in Phase I clinical trials in relapsed or refractory MM and myeloid leukemia (NCT02675452).
AZD5991
AZD5991, developed by AstraZeneca, possesses high MCL-1 selectivity with sub-nanomolar affinity for MCL-1 in biochemical assays and induces rapid apoptosis in MCL-1-dependent myeloma cell lines with half concentration of growth inhibition as low as 10 nM.57 Mechanism studies reveal that AZD5991 disrupts the MCL-1:BAK complex to exert its antitumor activity. As such, depletion of BAK by siRNA blocks the activity of AZD5991. AZD5991 achieves excellent antitumor activity in mouse xenograft models of AML, non-Hodgkin’s lymphoma, and MM. Ex vivo studies in primary tumor samples from leukemia patients suggest that leukemia patients would respond favorably to AZD5991 treatment. AZD5991 is now in Phase I clinical trials in patients with relapsed or refractory hematologic cancers (NCT03218683).
Targeting MCL-1 expression
MCL-1 is a short-lived protein,65 and its expression is tightly regulated at transcriptional, translational, and post-translational levels. MCL-1 can be upregulated by trophic factor cytokines, including interleukins IL-3,65 IL-6,66 IL-10,67 and IL-15,68 and growth factors, such as EGF,69 vascular endothelial growth factor (VEGF),70 and platelet-derived growth factor (PDGF) (Figure 5).71 STAT3 and STAT5, activated by upstream interleukins (IL-3, -6, -10, and -15) and Janus kinases (JAKs), bind to the promoter region of MCL-1 and potentiate MCL-1 transcription. EGF activates MCL-1 translation through RAS–RAF–MEK–ERK and ELK1 pathway. EGF and VEGF upregulate MCL-1 translation either through PI3K–AKT or through PI3K–mTOR signal transduction pathways. PDGF–β-catenin–HIF1α pathway increases MCL-1 mRNA via functional hypoxia response elements in the promoter region of MCL-1. Activation of GSK-3β phosphorylates MCL-1 and causes proteasomal degradation of MCL-1.72
 | Figure 5 Regulation of MCL-1 transcription, translation, and degradation through different upstream pathways. |
Inhibition of MCL-1 upstream signal pathways can downregulate MCL-1 expression by decreasing transcription and translation. For example, CDK9 inhibitors, roscovitine and CR8, and Na+/K+-ATPase inhibitor cardiac glycosides UNBS1450 effectively inhibit MCL-1 transcription.73–75 Benzyl isothiocyanate inhibits the phosphorylation of eukaryotic initiation factor 4G, resulting in a decreased MCL-1 translation, followed by cell cycle arrest and apoptosis in leukemia cells.53,64 EGFR/VEGFR inhibitor BAY43-9006 downregulates MCL-1 translation cofactor ELK-1 via RAS/RAF–MEK–ERK pathway and inhibits MCL-1 expression.76 Inhibition of PI3K/mTOR pathway by BEZ235/AZD8055 also results in reduced MCL-1 translation.77 Pharmacological promotion of MCL-1 degradation is another approach to reduce cellular MCL-1 protein levels. In this case, activation of GSK-3β phosphorylation by arsenic trioxide and bufalin enhances MCL-1 degradation.78,79
BAK/BAX agonist
During the apoptotic process, BAK/BAX undergoes conformational changes, which is followed by homo-oligomerization to form macropores in the membrane of mitochondria.80 In this regard, direct activation of BAK/BAX to potentiate macropore formation by bypassing MCL-1 inhibition offers another promising approach to target MCL-1-dependent tumors for treatment. The hydrophobic groove formed by the α2-α5 helices in BAK/BAX interacts with BH3-only proteins that are required for BAX/BAK activation during apoptosis. Besides the pocket formed by the α2-α5 helices, multiple sites on BAK/BAX have also been targeted by different approaches. For example, monoclonal antibody Fab 7D10 recognizes the α1 loop of BAK/BAX and its binding with BAK/BAX leads to BAK/BAX oligomerization and apoptosis. The α1-α2 loop of BAK/BAX was shown to be another potential druggable site.81 The α6 helix of BAK is also identified as another site recruited by BH3-only proteins to facilitate BAK homo-oligomerization.82 Unlike the sites on the α1-α2 loop, the α6 site on BAK/BAX requires the BH3-only proteins binding to activate BAK/BAK. Small molecules SMBA1–3 identified by virtual screening bind to the BAX S184 pocket and potentiate BAX function by blocking S184 phosphorylation without affecting the binding of BCL-2 antiapoptotic family protein to BAX.83
While these aforementioned strategies have been explored to antagonize the antiapoptotic functions of MCL-1, studies to directly compare these approaches for their therapeutic effects are lacking. Most of the earlier reported BH3 mimetics are neither very selective nor potent enough to achieve significant tumor growth inhibition in preclinical models and, thus, are mainly used as tool compounds. Most recently discovered BH3 mimetics are highly selective and able to achieve tumor regression in xenograft tumor models. Drugs targeting MCL-1 upstream signaling are highly unlikely to selectively modulate the expression of MCL-1 without affecting other genes. Yet mechanistic studies of these drugs’ actions may shed light on the development of combination therapies. Although BAK/BAX agonists showed promising future, it still needs significant medicinal chemistry efforts to identify drug-like molecules.
Drugs’ combination
Depletion of MCL-1 combined with standard chemotherapies is tolerable in mice, attesting the feasibility of combining MCL-1 inhibitors with other anticancer drugs.84 However, the overlapping toxicities from MCL-1 inhibitors and standard chemotherapies need to be carefully monitored and addressed. Nevertheless, pan-BCL-2 inhibitor AT-101 in combination with a variety of anticancer drugs has been evaluated in multiple Phase I and II clinical trials (Table 2).
Aberrant MCL-1 expression has been linked to anticancer drug resistance in almost every tumor type. Overcoming MCL-1-mediated drug resistance necessitates combination therapies of MCL-1 inhibitors with other anticancer drugs (Table 3). In a study using ABT-199-resistant acute myeloma lymphoma cell lines, MCL-1 selective inhibitor A-1210477 combined with ABT-199 exerts synergistic apoptotic effects and circumvents ABT-199 resistance.85 Another study shows that selective MCL-1 inhibitor maritoclax increases the efficacy of ABT-737 against multiple drug-resistant hematologic cancer cell lines.52
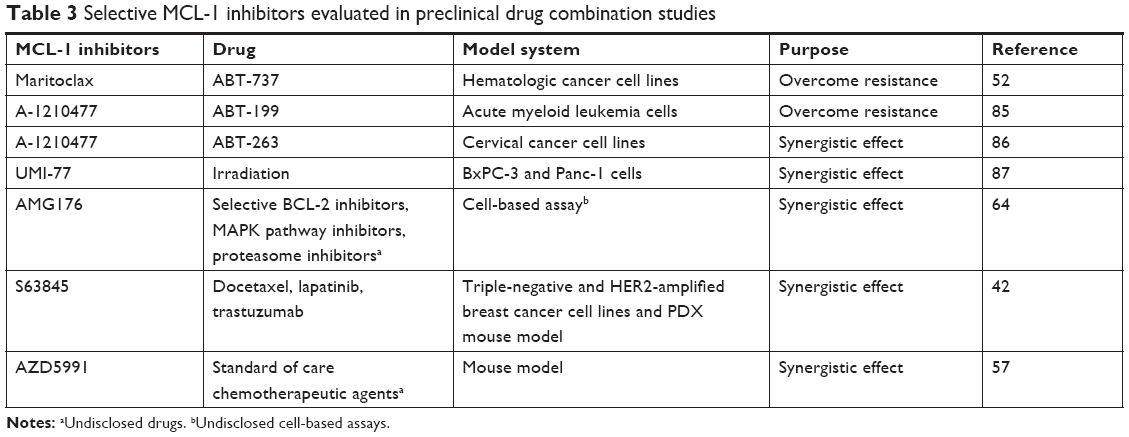 | Table 3 Selective MCL-1 inhibitors evaluated in preclinical drug combination studies |
In addition to overcoming drug resistance, synergistic effect is another benefit of MCL-1 combination therapies (Table 3). A-1210477,86 UMI-77,87 AMG176,64 and S6384542 have been shown to synergize with other anticancer drugs. The synergistic effects between S63845 and docetaxel, lapatinib, and trastuzumab and between AZD5991 and standard anticancer agents were observed in mouse tumor models.42,57
Assessment of small-molecule MCL-1 inhibitors
A specific and bona fide MCL-1 inhibitor should 1) selectively bind to MCL-1 in homogeneous biochemical assays and engage with MCL-1 in cellular context, 2) dissociate the heterodimeric interactions between MCL-1 and proapoptotic BCL-2 family proteins in the cellular context, 3) cause changes in mitochondrial outer membrane permeabilization (MOMP) and the release of cytochrome c in cells, and 4) induce BAX/BAK-dependent apoptosis.
In cellular context, target engagement of MCL-1 small molecules can be conveniently evaluated by a cellular thermal shift assay88,89 or pull down assay using a biotin conjugated MCL-1 ligand. The dissociation of MCL-1 with proapoptotic BCL-2 family proteins can be detected by immunoprecipitation assay. A key feature for the regulation of apoptosis by the BCL-2 family proteins is to control MOMP and release of cytochrome c, which can be evaluated by conventional cellular and biochemical assays. Orthogonal approaches, such as the application of small-interfering RNA-mediated knockdown or clustered regularly interspaced short palindromic repeats-mediated knockout can be used to determine the on-target cytotoxicity of MCL-1 inhibitors.
Mechanisms of resistance to MCL-1 targeted therapies
While several previous studies have revealed that the sensitivity to the inhibition or depletion of MCL-1 is inversely correlated with the expression levels of BCL-XL,14,60,90–92 there are very few studies on the resistant mechanisms to small-molecule MCL-1 inhibitors. With the recent advancement of multiple selective MCL-1 inhibitors to clinical trials, more studies on the resistant mechanisms to small-molecule MCL-1 inhibitors are expected in the near future.
Conclusion
MCL-1 is frequently overexpressed in human cancers and identified as the linchpin of cancer drug resistance in a variety of tumor types, making it an attractive therapeutic target. This stresses the necessity to develop MCL-1 inhibitors that can be used either as a single agent or in combination regimens. Efforts to develop MCL-1 inhibitors have been predominantly focused on BH3-mimetics, and now several small molecules with high MCL-1 affinities and selectivity have been discovered. However, most of the MCL-1 inhibitors are still at the stages of preclinical or early clinical development. Beside the MCL-1 inhibitors, newly identified druggable sites on BAK/BAX may offer an alternative approach to target MCL-1-dependent cancers. Furthermore, the emerging proteolysis targeting chimera technology to pharmacologically induce MCL-1 degradation may provide a novel strategy to target MCL-1.93
Disclosure
The authors report no conflicts of interest in this work.
References
Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5(4):a008714. | ||
Krajewski S, Bodrug S, Krajewska M, et al. Immunohistochemical analysis of Mcl-1 protein in human tissues. Differential regulation of Mcl-1 and Bcl-2 protein production suggests a unique role for Mcl-1 in control of programmed cell death in vivo. Am J Pathol. 1995;146(6):1309–1319. | ||
Perciavalle RM, Opferman JT. Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell Biol. 2013;23(1):22–29. | ||
Peperzak V, Vikström I, Walker J, et al. Mcl-1 is essential for the survival of plasma cells. Nat Immunol. 2013;14(3):290–297. | ||
Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281(5381):1322–1326. | ||
Labelle JL, Katz SG, Bird GH, et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. 2012;122(6):2018–2031. | ||
Campbell KJ, Dhayade S, Ferrari N, et al. MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis. 2018;9(2):19. | ||
Young AI, Law AM, Castillo L, et al. MCL-1 inhibition provides a new way to suppress breast cancer metastasis and increase sensitivity to dasatinib. Breast Cancer Res. 2016;18(1):125. | ||
Zervantonakis IK, Iavarone C, Chen HY, et al. Systems analysis of apoptotic priming in ovarian cancer identifies vulnerabilities and predictors of drug response. Nat Commun. 2017;8(1):365. | ||
Reiner T, de Las Pozas A, Parrondo R, et al. Mcl-1 protects prostate cancer cells from cell death mediated by chemotherapy-induced DNA damage. Oncoscience. 2015;2(8):703–715. | ||
Akgul C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci. 2009;66(8):1326–1336. | ||
Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. | ||
Balko JM, Giltnane JM, Wang K, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4(2):232–245. | ||
Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. | ||
Ding Q, He X, Xia W, et al. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res. 2007;67(10):4564–4571. | ||
Xu C, Liu Y, Huang J, et al. The prognostic significance of MCL1 copy number gain in esophageal squamous cell carcinoma. Oncotarget. 2017;8(50):87699–87709. | ||
Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010;584(14):2981–2989. | ||
Wertz IE, Kusam S, Lam C, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471(7336):110–114. | ||
Wei SH, Dong K, Lin F, et al. Inducing apoptosis and enhancing chemosensitivity to gemcitabine via RNA interference targeting Mcl-1 gene in pancreatic carcinoma cell. Cancer Chemother Pharmacol. 2008;62(6):1055–1064. | ||
Lestini BJ, Goldsmith KC, Fluchel MN, et al. Mcl1 downregulation sensitizes neuroblastoma to cytotoxic chemotherapy and small molecule Bcl2-family antagonists. Cancer Biol Ther. 2009;8(16):1587–1595. | ||
Osaki S, Tazawa H, Hasei J, et al. Ablation of MCL1 expression by virally induced microRNA-29 reverses chemoresistance in human osteosarcomas. Sci Rep. 2016;6:28953. | ||
Glab JA, Doerflinger M, Puthalakath H. BH3-only proteins: the thorny end of the ER stress response. Cell Death Dis. 2017;8(6):e2889. | ||
Shamas-Din A, Brahmbhatt H, Leber B. BH3-only proteins: Orchestrators of apoptosis. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research. 2011;1813:508–520. | ||
Haneef J, Parvathy M, Thankayyan RSK, Sithul H, Sreeharshan S. Bax translocation mediated mitochondrial apoptosis and caspase dependent photosensitizing effect of Ficus religiosa on cancer cells. PLoS One. 2012;7(7):e40055. | ||
D’Alessio M, de Nicola M, Coppola S, et al. Oxidative Bax dimerization promotes its translocation to mitochondria independently of apoptosis. Faseb J. 2005;19(11):1504–1506. | ||
Mcarthur K, Whitehead LW, Heddleston JM, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. 2018;359(6378):eaao6047. | ||
Zhang Y, Qu Y, Gao K, et al. High copy number of mitochondrial DNA (mtDNA) predicts good prognosis in glioma patients. Am J Cancer Res. 2015;5(3):1207–1216. | ||
Wei X, Duan W, Li Y, et al. AT101 exerts a synergetic efficacy in gastric cancer patients with 5-FU based treatment through promoting apoptosis and autophagy. Oncotarget. 2016;7(23):34430–34441. | ||
Senft D, Weber A, Saathoff F. In non-transformed cells Bak activates upon loss of anti-apoptotic Bcl-XL and Mcl-1 but in the absence of active BH3-only proteins. Cell Death Disease. 2015;6(11):e1996. | ||
García Sáez AJ, Villunger A. MOMP in the absence of BH3-only proteins. Genes Dev. 2016;30(8):878–880. | ||
Elkholi R, Floros KV, Chipuk JE. The Role of BH3-Only Proteins in Tumor Cell Development, Signaling, and Treatment. Genes Cancer. 2011;2(5):523–537. | ||
Kodama T, Takehara T, Hikita H, et al. BH3-only activator proteins Bid and Bim are dispensable for Bak/Bax-dependent thrombocyte apoptosis induced by Bcl-xL deficiency: molecular requisites for the mitochondrial pathway to apoptosis in platelets. J Biol Chem. 2011;286(16):13905–13913. | ||
Vela L, Gonzalo O, Naval J, Marzo I. Direct interaction of Bax and Bak proteins with Bcl-2 homology domain 3 (BH3)-only proteins in living cells revealed by fluorescence complementation. J Biol Chem. 2013;288(7):4935–4946. | ||
Zhai D, Jin C, Huang Z, Satterthwait AC, Reed JC. Differential regulation of Bax and Bak by anti-apoptotic Bcl-2 family proteins Bcl-B and Mcl-1. J Biol Chem. 2008;283(15):9580–9586. | ||
Montero J, Letai A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018;25(1):56–64. | ||
Haschka MD, Soratroi C, Kirschnek S, et al. The NOXA-MCL1-BIM axis defines lifespan on extended mitotic arrest. Nat Commun. 2015;6:6891. | ||
Happo L, Strasser A, Cory S. BH3-only proteins in apoptosis at a glance. J Cell Sci. 2012;125(Pt 5):1081–1087. | ||
Chen L, Fletcher S. Mcl-1 inhibitors: a patent review. Expert Opin Ther Pat. 2017;27:163–178. | ||
Nhu D, Lessene G, Huang DCS, Burns CJ. Small molecules targeting Mcl-1: the search for a silver bullet in cancer therapy. Medchemcomm. 2016;7(5):778–787. | ||
Wang G, Nikolovska-Coleska Z, Yang CY, et al. Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. J Med Chem. 2006;49(21):6139–6142. | ||
Ren T, Shan J, Qing Y, et al. Sequential treatment with AT-101 enhances cisplatin chemosensitivity in human non-small cell lung cancer cells through inhibition of apurinic/apyrimidinic endonuclease 1-activated IL-6/STAT3 signaling pathway. Drug Des Devel Ther. 2014;8:2517–2529. | ||
Kotschy A, Szlavik Z, Murray J, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538(7626):477–482. | ||
Castro JE, Loria OJ, Aguillon RA. A Phase II, Open Label Study of AT-101 in Combination with Rituximab in Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia. Evaluation of Two Dose Regimens. Blood. 2007;110:3119. | ||
Mohammad RM, Goustin AS, Aboukameel A, et al. Preclinical studies of TW-37, a new nonpeptidic small-molecule inhibitor of Bcl-2, in diffuse large cell lymphoma xenograft model reveal drug action on both Bcl-2 and Mcl-1. Clin Cancer Res. 2007;13(7):2226–2235. | ||
Ashimori N, Zeitlin BD, Zhang Z, et al. TW-37, a small-molecule inhibitor of Bcl-2, mediates S-phase cell cycle arrest and suppresses head and neck tumor angiogenesis. Mol Cancer Ther. 2009;8(4):893–903. | ||
Zhai D, Jin C, Shiau CW, Kitada S, Satterthwait AC, Reed JC. Gambogic acid is an antagonist of antiapoptotic Bcl-2 family proteins. Mol Cancer Ther. 2008;7(6):1639–1646. | ||
Li X, Liu S, Huang H, et al. Gambogic acid is a tissue-specific proteasome inhibitor in vitro and in vivo. Cell Rep. 2013;3(1):211–222. | ||
Hua X, Liang C, Dong L, Qu X, Zhao T. Simultaneous determination and pharmacokinetic study of gambogic acid and gambogenic acid in rat plasma after oral administration of Garcinia hanburyi extracts by LC-MS/MS. Biomed Chromatogr. 2015;29(4):545–551. | ||
Dash R, Azab B, Quinn BA, et al. Apogossypol derivative BI-97C1 (Sabutoclax) targeting Mcl-1 sensitizes prostate cancer cells to mda-7/IL-24-mediated toxicity. Proc Natl Acad Sci U S A. 2011;108(21):8785–8790. | ||
Jackson RS, Placzek W, Fernandez A, et al. Sabutoclax, a Mcl-1 antagonist, inhibits tumorigenesis in transgenic mouse and human xenograft models of prostate cancer. Neoplasia. 2012;14(7):656–665. | ||
Wei J, Stebbins JL, Kitada S, et al. BI-97C1, an optically pure Apogossypol derivative as pan-active inhibitor of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. J Med Chem. 2010;53(10):4166–4176. | ||
Doi K, Li R, Sung SS, et al. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J Biol Chem. 2012;287(13):10224–10235. | ||
Abulwerdi F, Liao C, Liu M, et al. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther. 2014;13(3):565–575. | ||
Ohmer M, Weber A, Sutter G, Ehrhardt K, Zimmermann A, Häcker G. Anti-apoptotic Bcl-XL but not Mcl-1 contributes to protection against virus-induced apoptosis. Cell Death Dis. 2016;7(8):e2340. | ||
Zhao B, Sensintaffar J, Bian Z, et al. Structure of a Myeloid cell leukemia-1 (Mcl-1) inhibitor bound to drug site 3 of Human Serum Albumin. Bioorg Med Chem. 2017;25(12):3087–3092. | ||
Friberg A, Vigil D, Zhao B, et al. Discovery of Potent Myeloid Cell Leukemia 1 (Mcl-1) Inhibitors Using Fragment-Based Methods and Structure-Based Design. Journal of Medicinal Chemistry. 2013;56:15–30. | ||
Hird AW, Secrist JP, Adam A, et al. Abstract DDT01-02: AZD5991: A potent and selective macrocyclic inhibitor of Mcl-1 for treatment of hematologic cancers. Cancer Research. 2017;77(13):DDT01-02. | ||
Doi K, Gowda K, Liu Q, et al. Pyoluteorin derivatives induce Mcl-1 degradation and apoptosis in hematological cancer cells. Cancer Biol Ther. 2014;15(12):1688–1699. | ||
Leverson JD, Zhang H, Chen J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015;6:e1590. | ||
Xiao Y, Nimmer P, Sheppard GS, et al. MCL-1 Is a Key Determinant of Breast Cancer Cell Survival: Validation of MCL-1 Dependency Utilizing a Highly Selective Small Molecule Inhibitor. Mol Cancer Ther. 2015;14(8):1837–1847. | ||
Lin J, Fu D, Dai Y, Lin J, Xu T. Mcl-1 inhibitor suppresses tumor growth of esophageal squamous cell carcinoma in a mouse model. Oncotarget. 2017;8(70):114457–114462. | ||
Pelz NF, Bian Z, Zhao B, et al. Discovery of 2-Indole-acylsulfonamide Myeloid Cell Leukemia 1 (Mcl-1) Inhibitors Using Fragment-Based Methods. J Med Chem. 2016;59(5):2054–2066. | ||
Merino D, Whittle JR, Vaillant F, et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci Transl Med. 2017;9(401):eaam7049. | ||
Caenepeel SR, Belmontes B, Sun J, Coxon A, Moody G, Hughes PE. Abstract 2027: Preclinical evaluation of AMG 176, a novel, potent and selective Mcl-1 inhibitor with robust anti-tumor activity in Mcl-1 dependent cancer models. Cancer Research. 2017;77(13):2027. | ||
Nijhawan D, Fang M, Traer E, et al. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17(12):1475–1486. | ||
Jourdan M, Veyrune JL, de Vos J, Redal N, Couderc G, Klein B. A major role for Mcl-1 antiapoptotic protein in the IL-6-induced survival of human myeloma cells. Oncogene. 2003;22(19):2950–2959. | ||
Dash R, Richards JE, Su ZZ, et al. Mechanism by which Mcl-1 regulates cancer-specific apoptosis triggered by mda-7/IL-24, an IL-10-related cytokine. Cancer Res. 2010;70(12):5034–5045. | ||
Shenoy AR, Kirschnek S, Häcker G. IL-15 regulates Bcl-2 family members Bim and Mcl-1 through JAK/STAT and PI3K/AKT pathways in T cells. Eur J Immunol. 2014;44(8):2500–2507. | ||
Fu NY, Rios AC, Pal B, et al. EGF-mediated induction of Mcl-1 at the switch to lactation is essential for alveolar cell survival. Nat Cell Biol. 2015;17(4):365. | ||
Véronèse L, Tournilhac O, Verrelle P, et al. Strong correlation between VEGF and MCL-1 mRNA expression levels in B-cell chronic lymphocytic leukemia. Leuk Res. 2009;33(12):1623–1626. | ||
Iqbal S, Zhang S, Driss A, et al. PDGF upregulates Mcl-1 through activation of β-catenin and HIF-1α-dependent signaling in human prostate cancer cells. PLoS One. 2012;7(1):e30764. | ||
Wakatsuki S, Tokunaga S, Shibata M, Araki T. GSK3B-mediated phosphorylation of MCL1 regulates axonal autophagy to promote Wallerian degeneration. J Cell Biol. 2017;216(2):477–493. | ||
Bettayeb K, Baunbæk D, Delehouze C, et al. CDK Inhibitors Roscovitine and CR8 Trigger Mcl-1 Down-Regulation and Apoptotic Cell Death in Neuroblastoma Cells. Genes Cancer. 2010;1(4):369–380. | ||
Delehouzé C, Godl K, Loaëc N, et al. CDK/CK1 inhibitors roscovitine and CR8 downregulate amplified MYCN in neuroblastoma cells. Oncogene. 2014;33(50):5675–5687. | ||
Cerella C, Muller F, Gaigneaux A, et al. Early downregulation of Mcl-1 regulates apoptosis triggered by cardiac glycoside UNBS1450. Cell Death Dis. 2015;6(6):e1782. | ||
Rahmani M, Davis EM, Bauer C, Dent P, Grant S. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. J Biol Chem. 2005;280(42):35217–35227. | ||
Lee JS, Tang SS, Ortiz V, Vo TT, Fruman DA. MCL-1-independent mechanisms of synergy between dual PI3K/mTOR and BCL-2 inhibition in diffuse large B cell lymphoma. Oncotarget. 2015;6(34):35202–35217. | ||
Wang R, Xia L, Gabrilove J, Waxman S, Jing Y. Downregulation of Mcl-1 through GSK-3β activation contributes to arsenic trioxide-induced apoptosis in acute myeloid leukemia cells. Leukemia. 2013;27(2):315–324. | ||
Kang XH, Zhang JH, Zhang QQ, et al. Degradation of Mcl-1 through GSK-3β Activation Regulates Apoptosis Induced by Bufalin in Non-Small Cell Lung Cancer H1975 Cells. Cell Physiol Biochem. 2017;41(5):2067–2076. | ||
Pang YP, Dai H, Smith A, Meng XW, Schneider PA, Kaufmann SH. Bak Conformational Changes Induced by Ligand Binding: Insight into BH3 Domain Binding and Bak Homo-Oligomerization. Sci Rep. 2012;2:257. | ||
Iyer S, Anwari K, Alsop AE, et al. Identification of an activation site in Bak and mitochondrial Bax triggered by antibodies. Nat Commun. 2016;7:11734. | ||
Li MX, Tan IKL, Ma SB, et al. BAK α6 permits activation by BH3-only proteins and homooligomerization via the canonical hydrophobic groove. Proc Natl Acad Sci U S A. 2017;114(29):7629–7634. | ||
Xin M, Li R, Xie M, et al. Small-molecule Bax agonists for cancer therapy. Nat Commun. 2014;5:4935. | ||
Brinkmann K, Grabow S, Hyland CD, et al. The combination of reduced MCL-1 and standard chemotherapeutics is tolerable in mice. Cell Death Differ. 2017;24(12):2032–2043. | ||
Luedtke DA, Niu X, Pan Y, et al. Inhibition of Mcl-1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Signal Transduct Target Ther. 2017;2:17012. | ||
Lian BSX, Yek AEH, Shuvas H, Abdul Rahman SF, Muniandy K, Mohana-Kumaran N. Synergistic anti-proliferative effects of combination of ABT-263 and MCL-1 selective inhibitor A-1210477 on cervical cancer cell lines. BMC Res Notes. 2018;11(1):197. | ||
Wei D, Zhang Q, Schreiber JS, et al. Targeting mcl-1 for radiosensitization of pancreatic cancers. Transl Oncol. 2015;8(1):47–54. | ||
Martinez Molina D, Jafari R, Ignatushchenko M, et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. | ||
Bai L, Chen J, Mceachern D, et al. BM-1197: a novel and specific Bcl-2/Bcl-xL inhibitor inducing complete and long-lasting tumor regression in vivo. PLoS One. 2014;9:e99404. | ||
Zhang H, Guttikonda S, Roberts L, et al. Mcl-1 is critical for survival in a subgroup of non-small-cell lung cancer cell lines. Oncogene. 2011;30:1963–1968. | ||
Wei G, Margolin AA, Haery L, et al. Chemical genomics identifies small-molecule MCL1 repressors and BCL-xL as a predictor of MCL1 dependency. Cancer Cell. 2012;21:547–562. | ||
Kotschy A, Szlavik Z, Murray J, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477. | ||
Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by PROTACs. Pharmacol Ther. 2017;174:138–144. | ||
Dey J, Deckwerth TL, Kerwin WS, et al. Voruciclib, a clinical stage oral CDK9 inhibitor, represses MCL-1 and sensitizes high-risk Diffuse Large B-cell Lymphoma to BCL2 inhibition. Sci Rep. 2017;7:18007. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.