Back to Journals » Drug Design, Development and Therapy » Volume 10
Maximizing the encapsulation efficiency and the bioavailability of controlled-release cetirizine microspheres using Draper–Lin small composite design
Authors El-Say KM ![]()
Received 5 December 2015
Accepted for publication 31 December 2015
Published 24 February 2016 Volume 2016:10 Pages 825—839
DOI https://doi.org/10.2147/DDDT.S101900
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Prof. Dr. Wei Duan
Khalid Mohamed El-Say1,2
1Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, King Abdulaziz University, Jeddah, Kingdom of Saudi Arabia; 2Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Al-Azhar University, Cairo, Egypt
Abstract: This study was aimed at developing a controlled-release cetirizine hydrochloride (CTZ)-loaded polymethacrylate microsphere by optimization technique using software-based response surface methodology. The emulsion solvent evaporation method was utilized in the preparation of microspheres. Four process variables were selected, namely, Eudragit RLPO loading percentage in total polymer, the emulsifier hydrophilic lipophilic balance (HLB), the antitacking percentage, and the dispersed phase volume. The desired responses were particle size, angle of repose, production yield, encapsulation efficiency, loading capacity, initial drug release, and the time for 85% of drug release from the microspheres. Optimization was carried out by fitting the experimental data to the software program (Statgraphics Centurion XV). Moreover, 18 batches were subjected to various characterization tests required for the production of dosage form. The pharmacokinetic parameters were evaluated after the oral administration of 10 mg CTZ in both optimized formulation and commercial product on healthy human volunteers using a double-blind, randomized, cross-over design. The optimized formulation showed satisfactory yield (84.43%) and drug encapsulation efficiency (87.1%). Microspheres were of spherical shape, smooth surface, and good flowability with an average size of 142.3 µm. The developed optimized batch of microspheres ensured 28.87% initial release after 2 hours, and the release of CTZ extended for >12 hours. In addition, the relative bioavailability of the optimized formulation was 165.5% with respect to the marketed CTZ tablets indicating a significant enhancement of CTZ bioavailability. Thus, there is an expectation to decrease the administered dose and the frequency of administration, and subsequently minimize the adverse effects that are faced by the patient during the treatment.
Keywords: emulsion solvent evaporation, Eudragit, experimental design, in vivo, optimization, response surface
Introduction
In comparison with the conventional unit dosage form, polymeric multiunit microparticulate dosage forms have received massive scientific and industrial interest as they are distributed more uniformly throughout the gastrointestinal tract, which facilitates a uniform reliable sustained drug release pattern.1 Thus, these dosage forms are mostly suggested by health care professionals and ideal for patient compliance where drug safety is a main concern.2
For instance, emulsion solvent evaporation (ESE) process is a capable technique for the production of microspheres with controlled-release profile using different biocompatible water-insoluble polymers.3,4 Many formulation and processing variables could be involved in the preparation of microspheres by the ESE method, eg, the molecular weight of the polymers and the viscosity of the polymeric solution,5 solvent mixture composition, and stirring speed.6 Also, the drug to polymer ratio affects the properties of the prepared microspheres,7,8 while the concentration of polymer, drug to polymer ratio, percentage of the surfactant, and pore inducing agent affect the release and properties of microcapsules.9 In a different study, the yield, the size of microspheres, and the release rate were increased by the addition of a nonsolvent.10 Hariharan and Price11 reported that the properties of polylactic acid microparticles containing hexamethylamine were affected by the surfactant and cosolvent concentration. Moreover, the effect of drug loading, the concentration of emulsifier and polymer as formulation factors, and stirring speed as a process parameter were investigated on the characteristics of microparticles.12 In another study, the effects of the rate of temperature increase on dextran release profile were evaluated.13
Although the ESE technique is a complex process, it depends mainly on the efficient emulsification and the rate of solvent evaporation. The major challenge remains in the encapsulation of water-soluble drugs within microspheres to extend their release. This challenge arises from the escaping or degradation of drug during the preparation. To achieve this goal, the optimization of the process variables using Draper–Lin small composite experimental design has been made. Draper–Lin small composite design (D-LSCD) is a highly efficient optimization tool that is used to develop mathematical models for the estimation of relationships between the responses and key variables.
Cetirizine hydrochloride (CTZ) is one of the second-generation histamine H1 receptor antagonists, with a rapid onset of action that is used in the treatment of seasonal allergic rhinitis, hay fever, angioedema, and chronic urticaria.14
In this study, we report the influence of Eudragit RLPO loading percentage in total polymer (X1), the emulsifier hydrophilic lipophilic balance (HLB) (X2), the antitacking percentage (X3), and the dispersed phase volume (X4) on the quality attributes of CTZ-loaded polymethacrylate microspheres. These are the mean particle size, angle of repose, production yield percentage, encapsulation efficiency, loading capacity, and release pattern of CTZ as a model of water-soluble drugs from the prepared microspheres. So, the objective of this study was to develop innovative erodible microspheres for the delivery of CTZ and understand the effect of processing factors on the newly developed formula, in an attempt to maximize the encapsulation efficiency and retard the release of CTZ from the microspheres.
Materials and methods
Materials
CTZ was kindly gifted from Deef Pharmaceutical Industries Co. (Al Badayea, KSA). Eudragit RSPO and RLPO were kindly supplied by Evonik Industries (Essen, Germany). Talc powder from Whittaker Clark & Daniels (South Plainfield, NJ, USA); dichloromethane and acetone from Prolabo (Paris, France); n-hexane 95% from Honil Limited (London, UK); acetonitrile high-performance liquid chromatography (HPLC) grade from EMD Millipore (Billerica, MA, USA); potassium dihydrogen orthophosphate from BDH Chemicals Ltd. (England); light liquid paraffin from WINLAB (Leicestershire, UK); and sorbitan monooleate, NF, Span 80 from Sigma Chemicals (Perth, Australia) were purchased. All other materials were of analytical grade and used without any further purification.
Methods
The design of experiment
A full randomized rotatable D-LSCD was adopted to explore the impact of four important process factors on the quality attributes of CTZ-loaded polymethacrylate microspheres and analyze the interaction of each level of factors on the desired responses. The design of experiment was generated within the domain of levels using Statgraphics Centurion XV, version 15.2.05, software (StatPoint, Inc., Warrenton, VA, USA). As listed in Table 1, 18 different batches of CTZ-loaded polymethacrylate microspheres were prepared (eight batches as the cube portion, eight batches as the star points, and two batches as the center points). To make this design rotatable, for each factor, two axial points were chosen to be 1.68 at the lower and higher extreme levels, and the runs were randomized in order to exclude the block effects.15 Besides that, the process factors were Eudragit RLPO loading percentage in total polymer (X1), the emulsifier HLB (X2), the antitacking percentage (X3), and the dispersed phase volume (X4). The selected responses were the mean particle size of the microspheres (Y1), angle of repose for the prepared batches (Y2), production yield percentage (Y3), encapsulation efficiency percentage (Y4), loading capacity (Y5), initial CTZ release from microspheres (Y6), and time for 85% of drug release (Y7). The design matrix including investigated process variables along with their levels is shown in Table 1.
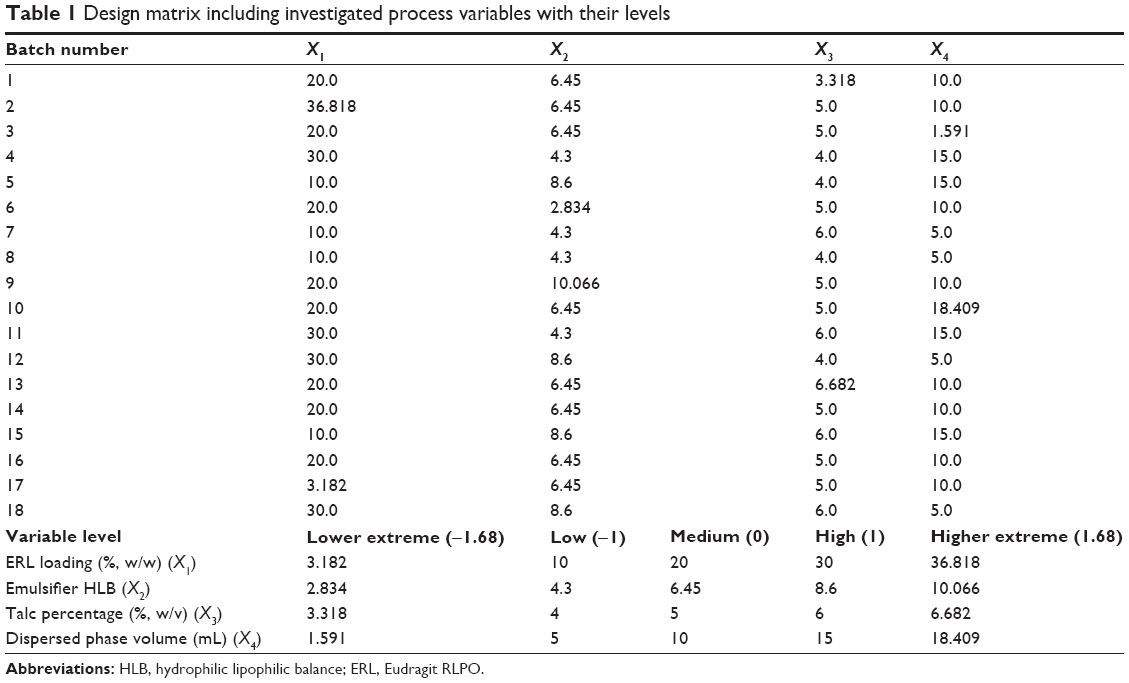 | Table 1 Design matrix including investigated process variables with their levels |
Preparation of the required HLB from the emulsifier blend
To prepare the emulsifier with HLB of 2.83, 58.8% of Span 85 was mixed with 41.2% of Span 80, while 46.07% of Span 80 and 53.93% of Tween 80 would produce an emulsifier with HLB value =10.07. Also, the emulsifier with HLB of 6.45 was prepared by blending 50% of both Span 80 and Span 20.
Preparation of microspheres
Each batch of CTZ-loaded polymethacrylate microspheres was prepared by loading fixed quantity of pure CTZ (500 mg). Furthermore, accurately weighed amount of drug, talc, and the mixture of Eudragit RSPO (ERS) and Eudragit RLPO (ERL) polymers, at drug to polymer ratio of 1:7, were dispersed in the specified volume of mixed solvent system of dichloromethane and acetone (1:1, v/v) forming the dispersed phase. The obtained dispersion was stirred at 1,000 rpm (Steady stir Digital, 855; Thermo Fisher Scientific, Waltham, MA, USA) in mineral oil (light liquid paraffin, 100 mL) to form oil-in-oil emulsion using the specified emulsifiers with different hydrophilic lipophilic balance (2%, w/w). Also, the emulsion was continuously stirred for a period of 6 hours till complete evaporation of solvent from the dispersed phase. The hardened microspheres were filtered off and washed with n-hexane (50 mL, four times) and dried under vacuum for a period of 24 hours and stored in a desiccator for further use.
Micromeritics evaluation of microspheres
The mean particle size of the prepared batches was evaluated by sieving method as previously reported,16 whereby the arithmetic mean diameter of the microspheres was calculated using Equation 1. Also, the flowability of the microspheres expressed as the angle of repose was assessed. The standard procedures for these determinations were followed as described earlier.16–19
|
|
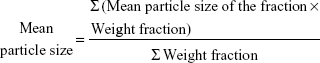
Production yield
On calculation of the weight of the raw materials used in the preparation and the final weight of the produced microspheres, the production yield percentage of the microspheres was calculated (Equation 2).
|
|
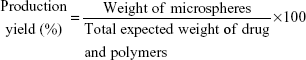
Encapsulation efficiency and loading capacity
A total of 50 mg of each formulation was dissolved in freshly prepared phosphate buffer (50 mL/pH 7.4) and maintained on a mechanical shaker for 24 hours, filtered and suitably diluted, then determined using HPLC (Agilent 1200 series, equipped with an ultraviolet diode array detector and an automatic sampling system, Agilent, CA, USA) at 229 nm (n=3). The encapsulation efficiency and the actual loading capacity expressed in percentage were calculated according to Equations 3 and 4.
|
|

|
|
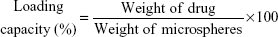
In vitro release study
In this section, United State Pharmacopoeia dissolution tester apparatus II (Erweka GmbH DT 700; Heusenstamm, Germany) was applied for evaluating the in vitro release of CTZ from the prepared microspheres. A hard gelatin capsule of suitable size was filled with the prepared microspheres equivalent to 10 mg of CTZ. Besides that, the experiment was carried out using 900 mL of freshly prepared phosphate buffer (pH 7.4), at 37°C±0.5°C and 50 rpm. Sample aliquots were withdrawn and replenished with fresh media at time intervals of 0.5 hours, 1 hour, 2 hours, 4 hours, 6 hours, 9 hours, 12 hours, 16 hours, 20 hours, and 24 hours. The CTZ content in the filtered samples was analyzed using HPLC (Agilant 1200 series) using reversed-phase column 25 cm ×4.6 mm (id) C18, 5 mm – Inertsil® ODS-3 (GL Sciences Inc., Tokyo, Japan). The mobile phase consisted of a mixture of (28:72) acetonitrile/0.05 M potassium dihydrogen orthophosphate (pH 3.5), and the flow rate was 1 mL/min. In addition, the CTZ content was determined at 229 nm after injecting a volume of 10 L, in which CTZ was detected at a retention time of 3.5 minutes. Assay performance was further evaluated through determination of specificity, recovery, linearity, the limit of quantification, the limit of detection, precision, and accuracy as reported in the International Conference on Harmonisation guidelines. The release experiments were done in triplicate.
Kinetic treatment of CTZ release data
The release data of CTZ were mathematically fitted to different kinetic models: zero-order,20 first-order,21 Higuchi,22 and Korsmeyer–Peppas models that are often utilized to designate the drug release mechanism from the microspheres.23,24
Scanning electron microscopy
The morphology of CTZ-loaded polymethacrylate microspheres, including shape and surface, was examined using scanning electron microscope (Philips XL30; FEI, Hillsboro, OR, USA). At this stage, the samples were mounted onto stubs covered with clean glass and coated with gold palladium film (thickness 2 nm) for 120 seconds at 10 mA, under argon at low pressure and investigated at 4–25 kV. The obtained photomicrographs of the formulation with suitable magnification are given in Figure 1.
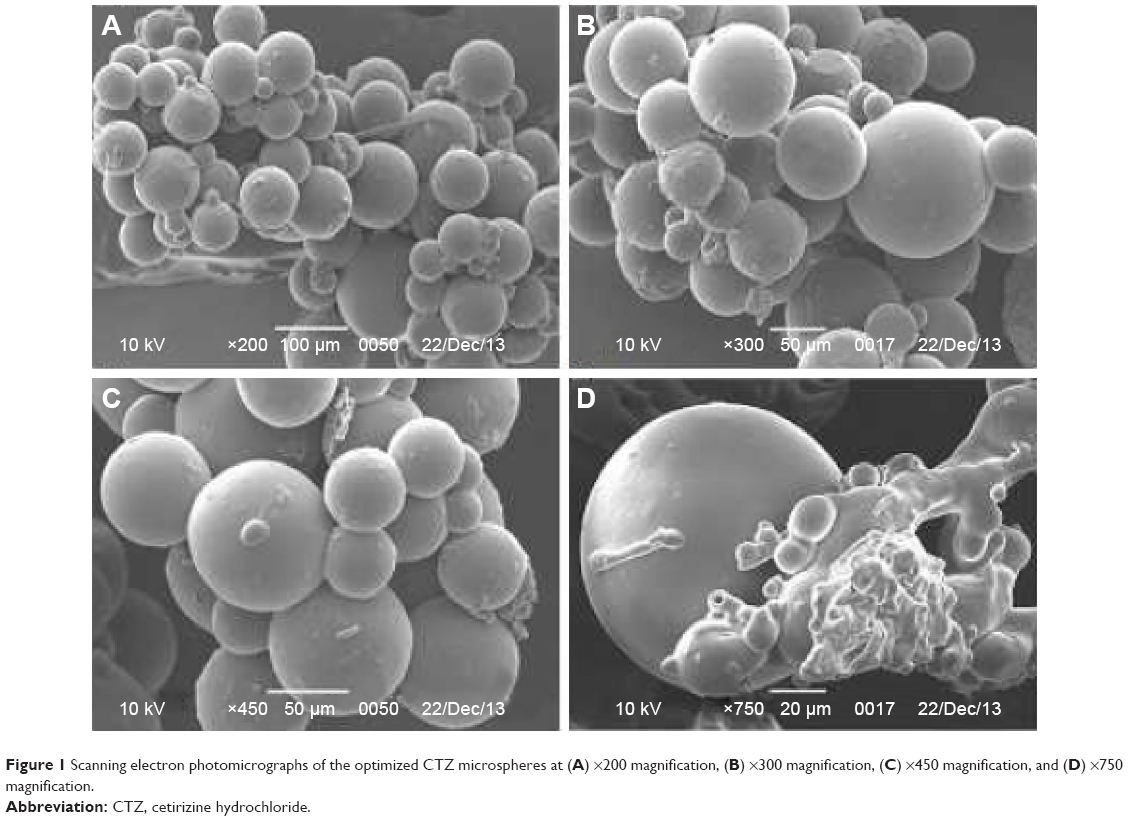 | Figure 1 Scanning electron photomicrographs of the optimized CTZ microspheres at (A) ×200 magnification, (B) ×300 magnification, (C) ×450 magnification, and (D) ×750 magnification. |
In vivo study on human volunteers
The pharmacokinetic parameters of the optimized CTZ microspheres (test) were evaluated in comparison with those of Zyrtec® tablet (reference; GlaxoSmithKline plc, London, UK) after the oral administration of 10 mg single dose to healthy adult, male and nonpregnant female volunteers by means of noncompartmental analysis.
Study design and conduct
The study used single-center, open-label, randomized, single dose with a two-way cross-over design. Two weeks of screening preceded two 36-hour study periods, which were separated by a washout period of at least 1 week. During these study periods, the subjects were given a single oral dose of the optimized CTZ microspheres equivalent to 10 mg (test) or the marketed tablets (reference) with 250 mL of tap water in each period of the study. Also, the subjects were confined in-house for 36 hours prior to and after drug administration, so that regular blood sampling could be done at predetermined times (as described in the “Blood sampling” section).
The study was conducted at the Institutional Review Board of the Egyptian Research and Development Company (ERDC), Cairo, Egypt, that has been endorsed for the ethical conduct of the study and had approved the protocol on its expedited meeting on October 19, 2014. Also, the study was performed in compliance with the Declaration of Helsinki and the International Conference on Harmonisation of Good Clinical Practices and with the approval from the study site’s Independent Ethics Committee.
Subjects
Six healthy Egyptian male and nonpregnant female volunteers with a mean age of 27±1.9 years, median weight of 69.5±5.9 kg, and median height of 168.2±4.7 cm participated in the study after understanding and signing an informed written consent about the nature and consequences of the study. The ERDC staff informed the subjects, in nontechnical terms, of the objectives, dates, drugs, diet, potential risks, and general activities during the clinical part of the study. Also, the subjects had been asked to read the consent forms carefully before signing. Upon examination and from results of clinical laboratory tests of all volunteers, the data revealed that all volunteers had normal results and they were advised to stay away from any medications at least 2 weeks prior to and during the study period.
Blood sampling
A total of 5 mL of blood was drawn just before and at 0.5 hours, 1 hour, 1.5 hours, 2 hours, 2.5 hours, 3 hours, 4 hours, 6 hours, 8 hours, 10 hours, 12 hours, 18 hours, 24 hours, and 36 hours after the oral administration of both the test and reference and collected in heparinized tubes. Samples were centrifuged at 3,000 rpm for 5 minutes, and plasma samples were collected and stored at −20°C until analysis.
Chromatographic conditions
Plasma CTZ concentrations were analyzed by liquid chromatography-mass spectrometry (LC-MS)/MS method applying the procedure reported by Tan et al.25 An HPLC method coupled with mass spectrometric detection (LC-MS/MS) was developed, optimized, and validated at ERDC laboratories for the determination of CTZ in human plasma. Moreover, the method was fully validated according to the US Food and Drug Administration Bioanalytical Method Validation Guidelines 2003. HPLC Agilent 6420 LC-MS Technologies (Deutschland GmbH, Waldbronn, Germany) system was used with a mass spectrometer detector, Agilent 6420, Triple Quad with G1311A quaternary pump, G1329 autosampler, and G1322A vacuum degasser, whereby the separation was performed on Inertsil ODS-3 column (4.6 mm ×50 cm, dp =5.0 μm). The mobile phase consisted of methanol:water in a volume ratio of 60:40, containing 0.5% triethylamine, and was adjusted to pH 3 using 0.1 N HCl. The components of the mobile phase were mixed and filtered through 0.45 mm nylon Millipore filter and the flow rate of the mobile phase was 1.2 mL/min. Also, the linearity of the assay method was verified within the concentration range of 0.1–30 ng/mL with a regression coefficient (R2) =0.9997±0.0002 for CTZ. All the results were within the acceptance criteria as stated in the recommended guidelines. The mean recovery of CTZ was 94.97% at 0.3 ng/mL and 103.92% at 30 ng/mL.
Plasma analysis
Plasma sample was mixed with acetonitrile and the internal standard (erythromycin), having the mixture vortexed, and then centrifuged at 3,000 rpm. The upper layer was separated and transferred to another tube, then filtered through 0.45 mm Millipore® filter for analysis with HPLC. In all, 20 mL of each sample was injected to the column for analysis.
Pharmacokinetic analysis
Pharmacokinetic parameters from plasma data following the oral administration of the two formulations were estimated using Kinetica® (version 4; Thermo Electron Corp., Waltham, MA, USA). Two-way analysis of variance (ANOVA) followed by Sidak’s multicomparison test using GraphPad Prism 6 (GraphPad Software, Inc., San Diego, CA, USA) were used to verify the differences in drug bioavailability between the two investigated groups. The level of statistical significance was chosen as P<0.05. Maximum plasma concentration (Cmax) (ng/mL), time to reach maximum plasma concentration (tmax) (hours), area under the time-concentration curve from time zero to infinity (AUC0–∞) (ng h/mL), and mean residence time (hours) were calculated from which the relative bioavailability (AUCtest/AUCstandard ×100) was determined.
Results and discussion
The proficient encapsulation of water-soluble drugs in controlling their release from the microspheres is considered as a major challenge for the researchers in this field. In our previous work, we have succeeded in encapsulating CTZ, a model water-soluble drug, in a reasonable percent; however, the control of drug release did not exceed 8 hours.19 Therefore, the prime features of this study were the use of mixed solvent system as a dispersed phase and light liquid paraffin as an immiscible continuous phase to form an oil-in-oil emulsion.26,27 Moreover, the use of mixed polymethacrylate polymer as a retardant polymer by increasing the drug to polymer ratio to 1:7 and incorporating ERLPO with different percentages in the total polymers were aimed at extending the release of drug from the microspheres as well as to maximize its encapsulation efficiency. Besides that, the highly efficient D-LSCD was utilized to explore the effect of the four process factors on the quality attributes of CTZ-loaded polymethacrylate microspheres.
Draper–Lin small composite design
It is known that, typical response surface methodologies are Box–Behnken design and central composite design.28 According to the data listed in Table 1, the study deals with four factors, namely, central level of Eudragit RLPO (ERL) loading, emulsifier HLB, talc percentage, and dispersed phase volume, which were 20% w/w, 6.45, 5% w/v, and 10 mL, respectively. In this case, central composite design and Box–Behnken design required 30 runs and 27 runs, respectively. To reduce the number of runs and increase the efficiency, D-LSCD was developed and considered as a highly efficient statistical design.29 In our article, for four independent variables, the quadratic equations (Equations 5–11) included 15 coefficients (ie, an independent term, four linear terms, four quadratic terms, and six interaction terms), and the total design runs were only 18 (Table 1).
Emulsification and formation of microspheres
The oil-in-oil (O/O) ESE technique was used to prepare CTZ-loaded polymethacrylate microspheres. The technique is correctly referred to as O/O instead of water-in-oil (W/O) as the organic solvent that contains a polymeric solution is considered as oil in microencapsulation terminology.30 This method was established to be the most common, effective, and robust in the encapsulation of drugs with different properties.12 Furthermore, the insolubility of the drug and polymers in liquid paraffin makes it a good candidate as an external phase in the emulsification process. Different emulsifiers with different HLB were used to prevent aggregation of droplets during emulsification and solvent evaporation by reducing the interfacial tension between the droplets and the external phase.31 Besides that, talc powder was added as an antitacking agent to stabilize the droplet and prevent its sticking behavior. Applying 1,000 rpm stirring speed during the microencapsulation process was intended to decrease the size of microspheres. Finally, the prepared microspheres were washed with n-hexane several times to clean their surfaces from residual liquid paraffin without disturbing the integrity of the microspheres.32
Evaluation of CTZ-loaded polymethacrylate microspheres
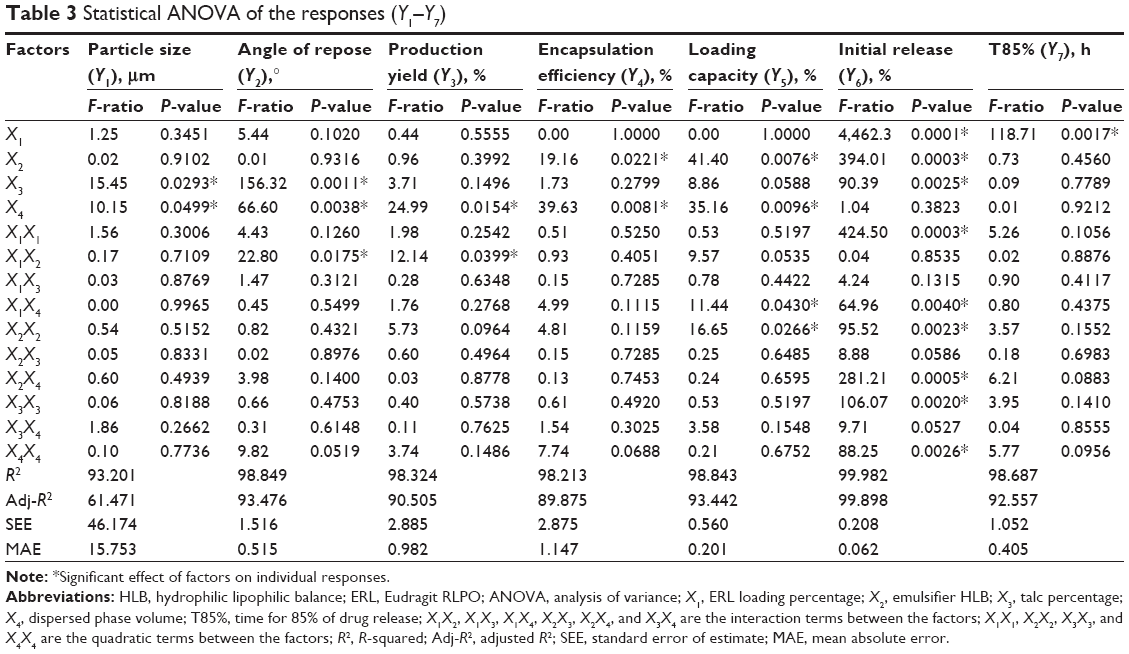
Table 2 summarizes the observed values of the response parameters studied (Y1–Y7) from the 18 formulations. As listed in Table 2, the ESE technique employed in this study produced spherical microspheres with smooth surfaces with different sizes ranging from 87 μm to 330 μm. This variation in particle size changed the angle of repose from 18.5° to 34.5° as an indication of the flow behavior of the microspheres. The size and flowability of the prepared microspheres depended mainly on the percentage of antitacking agent and the volume of the dispersed phase. The particle size of the microspheres can be decreased by the increase in dispersed phase volume, which could be attributed to the increase in volume that lowers the polymer concentration, which produces less viscous dispersion and maximizes the shearing action leading to smaller droplets and subsequently smaller microspheres.33 We can also infer that lower polymer concentration reduces the tendency of collision between the droplets producing smaller microspheres.3 In addition, the dispersed phase volume and the emulsifier HLB significantly (P<0.05) improved the production yield, encapsulation efficiency, and the loading capacity to 97.5%, 86.7%, and 13.9%, respectively. This finding may be due to the increase of the viscosity of the dispersed phase by decreasing its volume, which permits faster solidification and reduces the leaching of CTZ to the continuous phase. As a consequence, the increase in the production yield, encapsulation efficiency, and loading capacity were in agreement with the work reported by Pandit et al.34 However, the release of CTZ from the prepared microspheres was significantly (P<0.05) affected by the ERL loading percentage and HLB of the emulsifier. With respect to different combinations of factors and factor levels, a substantial variation among drug release patterns was obtained. It is obvious from Table 2 and Figure 2A–C that the initial release of CTZ after 2 hours increased from 15.57% in batch 17 to 35.19% in batch 2, while the time required for 85% of drug release decreased in the same batches from 25.35 hours to 9.14 hours reflecting the impact of ERL loading% on the release pattern. In general, the USP monographs for extended drug release dosage forms identify the percent of drug released after more than one time point (USP XXIII). An optimal extended-release dosage form must give a minimal initial release, whereas most of the drugs were released in a specific time period. As expected, when ERL was used in high percentage, CTZ was released at a faster rate as compared with the batch containing lower percentage of ERL. This is due to the fact that ERL is rich in the amount of quaternary ammonium groups, which renders it more permeable and hastens the liberation of drug from the microspheres.35,36 Figures 3 and 4 depict that increasing HLB of the emulsifier increased the hydrophilicity and subsequently increased the drug release. Also, increasing the amount of talc decreased the particle size of the microspheres, which influenced CTZ release owing to the large surface area exposed to the dissolution medium.37 Moreover, the correlations of the factors and the obtained responses are presented in Figure 5A–D.
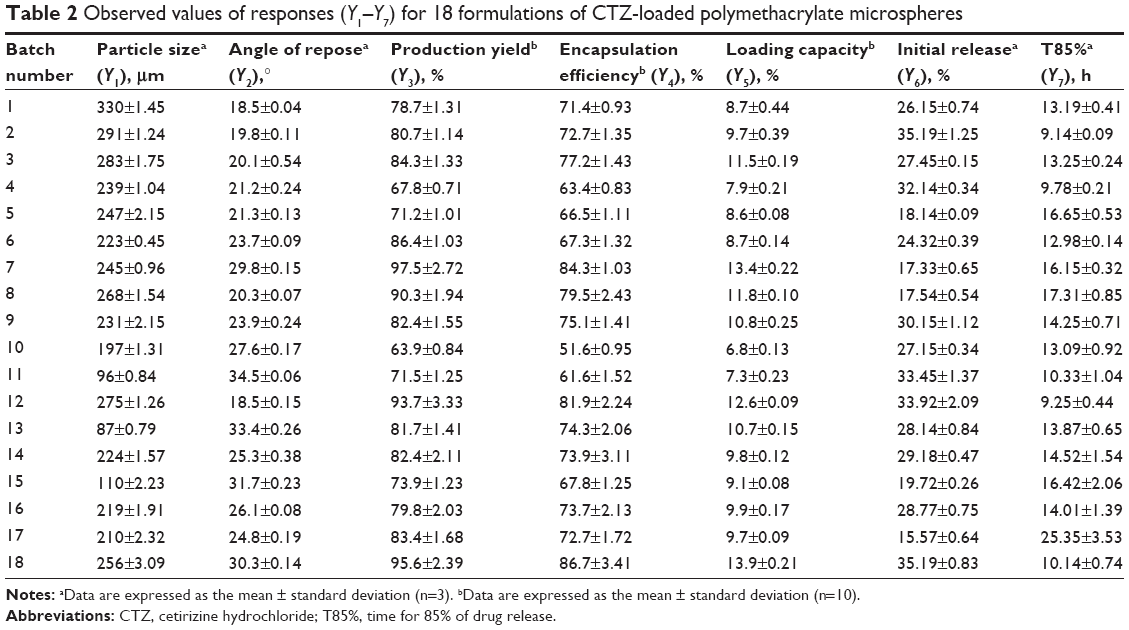 | Table 2 Observed values of responses (Y1–Y7) for 18 formulations of CTZ-loaded polymethacrylate microspheres |
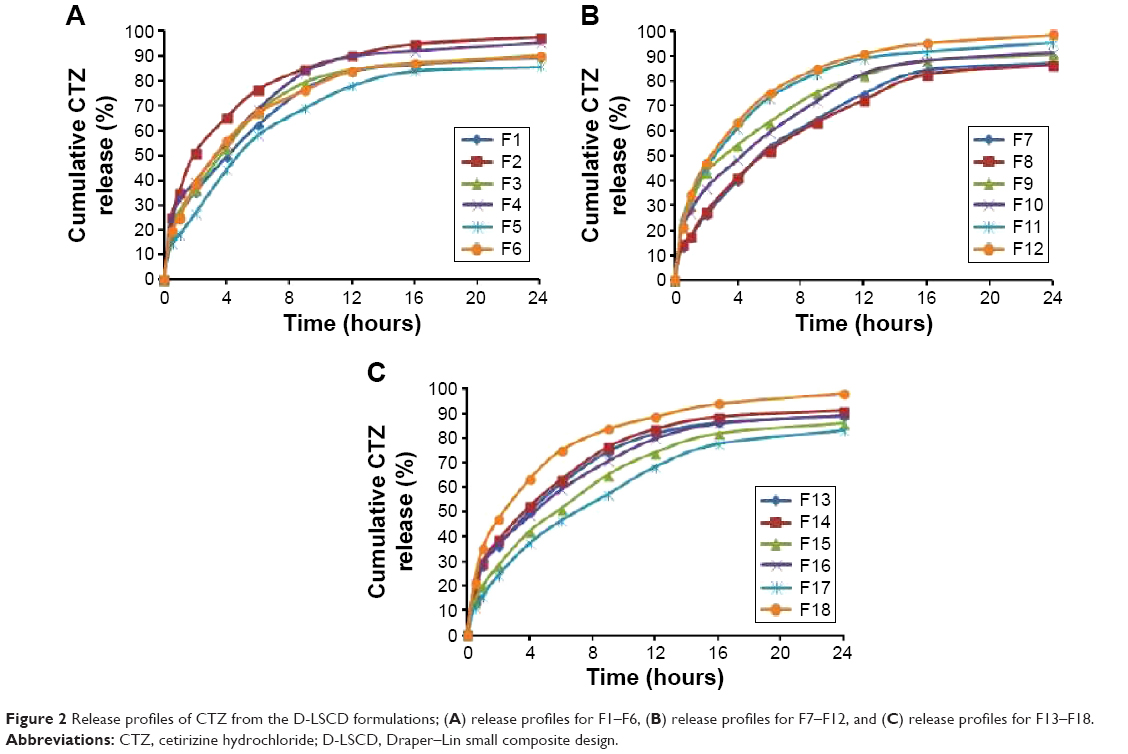 | Figure 2 Release profiles of CTZ from the D-LSCD formulations; (A) release profiles for F1–F6, (B) release profiles for F7–F12, and (C) release profiles for F13–F18. |
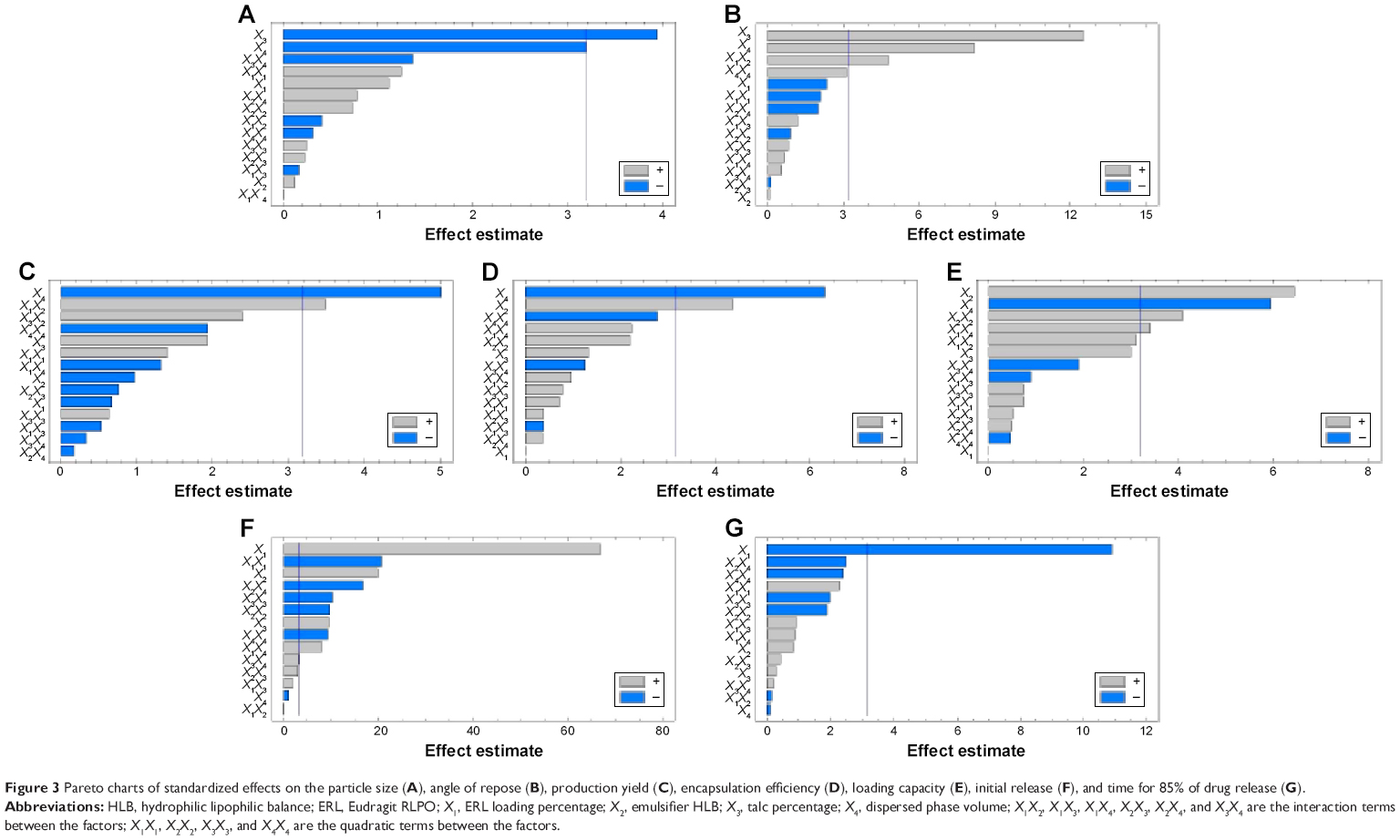 | Figure 3 Pareto charts of standardized effects on the particle size (A), angle of repose (B), production yield (C), encapsulation efficiency (D), loading capacity (E), initial release (F), and time for 85% of drug release (G). |
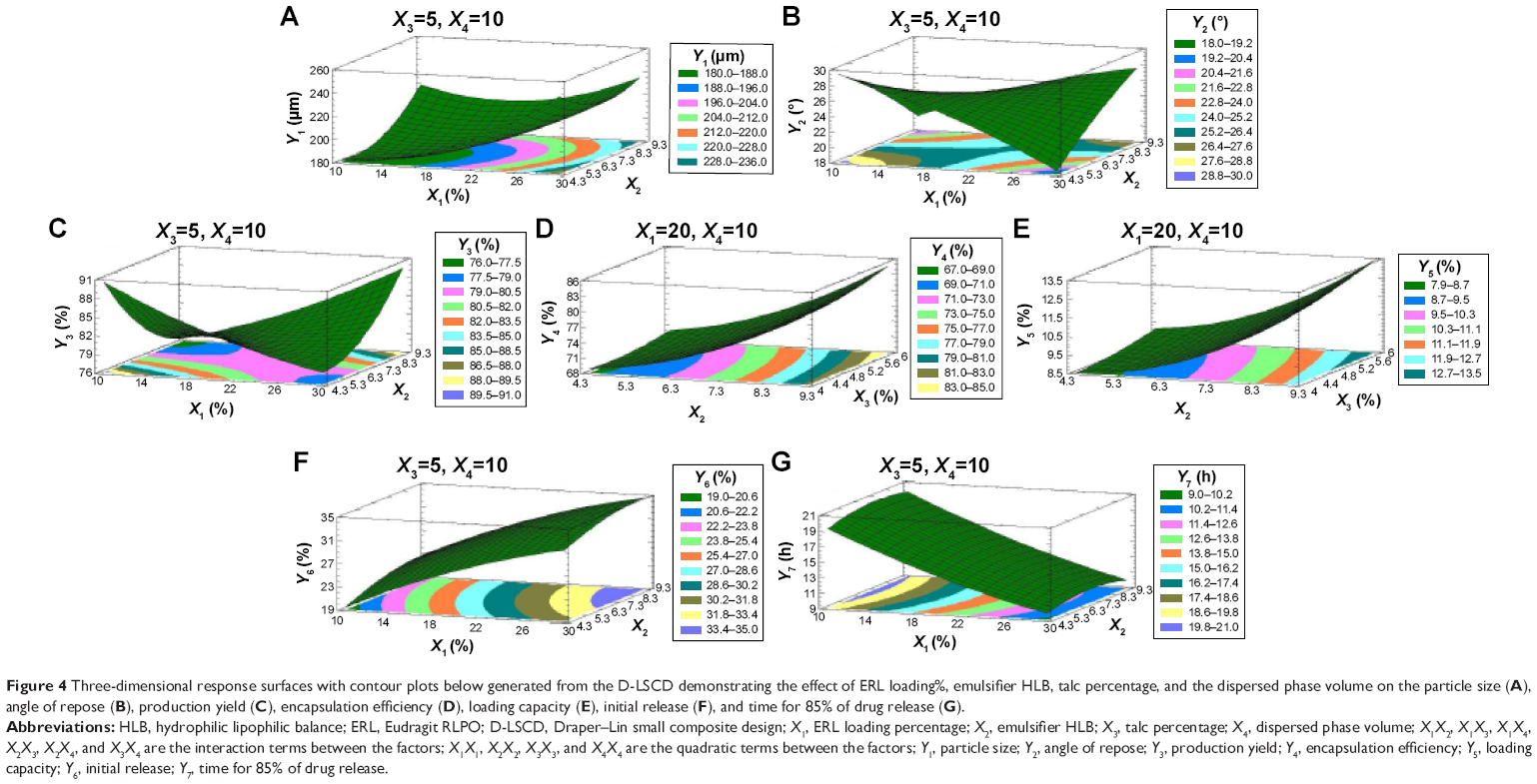 | Figure 4 Three-dimensional response surfaces with contour plots below generated from the D-LSCD demonstrating the effect of ERL loading%, emulsifier HLB, talc percentage, and the dispersed phase volume on the particle size (A), angle of repose (B), production yield (C), encapsulation efficiency (D), loading capacity (E), initial release (F), and time for 85% of drug release (G). |
 | Figure 5 Correlations of the factors and responses. |
Drug release kinetics
The release data of CTZ were fitted to various equations of release kinetics. Most formulations were fit better with Higuchi model (r=0.9563–0.9908) and Korsmeyer–Peppas (r=0.9756–0.9964) equations than other equations (zero order, first order). Moreover, the values of r for the rest of the formulations that fit the first order were very close to the Higuchi model that explains the diffusion control kinetic. Based on the Korsmeyer–Peppas equation, formulations (2–4, 9–12, 16, and 18) with exponent (n) values <0.43 indicated that the release mechanism followed the pure diffusion, Fickian or case I release. On the other hand, formulations (1, 5–8, 13–15, and 17) that have exponent value of 0.43<n<0.85 indicated pure case II, non-Fickian or anomalous release. In general, case II refers to erosion of the polymeric chain, while anomalous transport refers to a combination of both diffusion and erosion controlled-drug release.38 Finally, the release behavior of CTZ from the optimized microspheres was best explained by the first-order equation (r=0.9909) and Korsmeyer–Peppas model (r=0.9874), which supports erosion mechanism. This erosion mechanism for the release is preferable as it sustains the release and may be helpful in avoiding release failure.
Statistical data analysis and model validation
Fitting of data to the model
Four variables with three levels are listed in Table 1. All the response variables were observed experimentally for 18 batches as proposed by the D-LSCD and are expressed as the mean ± standard deviation in Table 2. Here, the models for different responses were attained using Statgraphics Centurion XV software. Meanwhile, the statistical ANOVA for the measured responses (Y1–Y7) is listed in Table 3. The values of probability (P-value), F-ratio, R2, adjusted R2, standard error of estimate, and the mean absolute error values for each response are listed in Table 3 along with their ANOVA results. After analysis of data, the following correlations for response variables were obtained in terms of coded factors. To quantify response values, model equations (Equations 5–11) that carry the variables along with coefficients were obtained. Positive sign of the coefficient indicates synergistic effects, while negative sign represents an antagonistic effect.39
|
|
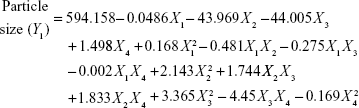
|
|
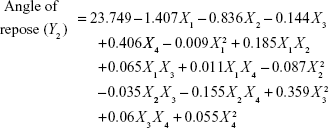
|
|
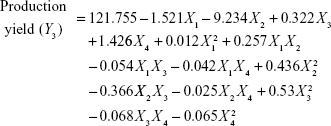
|
|

|
|
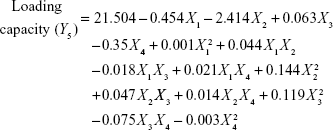
|
|
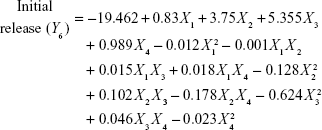
|
|
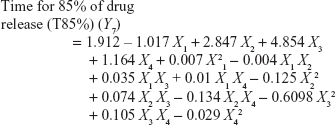
 | Table 3 Statistical ANOVA of the responses (Y1–Y7) |
All the regression equations were found to be statistically significant (P<0.05), as determined using ANOVA (Table 3).
Three-dimensional response surface with two-dimensional contour plot analysis
Statgraphics software generated the three-dimensional response surface with two-dimensional contour plots, which visualized the effects of the process variables on the response variables: particle size (Y1), angle of repose (Y2), production yield (Y3), encapsulation efficiency (Y4), loading capacity (Y5), initial CTZ release (Y6), and the time for 85% of drug release (Y7). Mean particle size (Y1) was decreased with increasing levels of X3 and X4. The crystalline flat-shaped talc molecules might render the microsphere particle with high percentage that is a more compact matrix than with low percentage, leading to decrease in particle size of the microspheres, which is in good concordance with the previously published finding.40 In contrast, the angle of repose (Y2) was decreased at higher levels of X3 and X4 as evidenced in the Pareto chart (Figure 3A and B) and response surface plots (Figure 4A and B). Meanwhile, production yield (Y3), encapsulation efficiency (Y4), and loading capacity (Y5) were increased with the decrease of levels X4 from 15 mL to 5 mL and were decreased with decreasing HLB value of the emulsifier as observed in Figures 3C–E and 4C–E. On the other hand, both initial drug release (Y6) and the time for 85% of drug release (Y7) were significantly affected by X1 as depicted in Figures 3F and G and 4F and G. With the increase in levels of X1, the initial drug release and the time for 85% of drug release increased and decreased, respectively.
For instance, response surface optimization was performed to get the optimum levels of the process variables: Eudragit RLPO loading percentage in total polymer (X1), the emulsifier HLB (X2), the antitacking percentage (X3), and the dispersed phase volume (X4) and develop Cetirizine hydrochloride controlled release (CTZ-CR) microspheres with minimum particle size, highest possible encapsulation efficiency, and extended-release pattern. Upon “trading-off” various response variables, the following criteria were accepted: mean particle size <300 μm, entrapment efficiency >60%, and T85% >540 minutes.41 Accordingly, formulation 13 was ranked as the best batch that achieved the maximum desirability.
Validation of optimized CTZ-CR formulation
To validate the reliability of the D-LSCD results, a new formulation was prepared according to the predicted model and evaluated for the responses as listed in Table 4. The observed values of the responses were compared with the predicted values that demonstrated no considerable residuals, and the predicted error percentage of the responses was <6%, indicating that the employed design was quite valuable for optimizing CTZ-CR microspheres.42
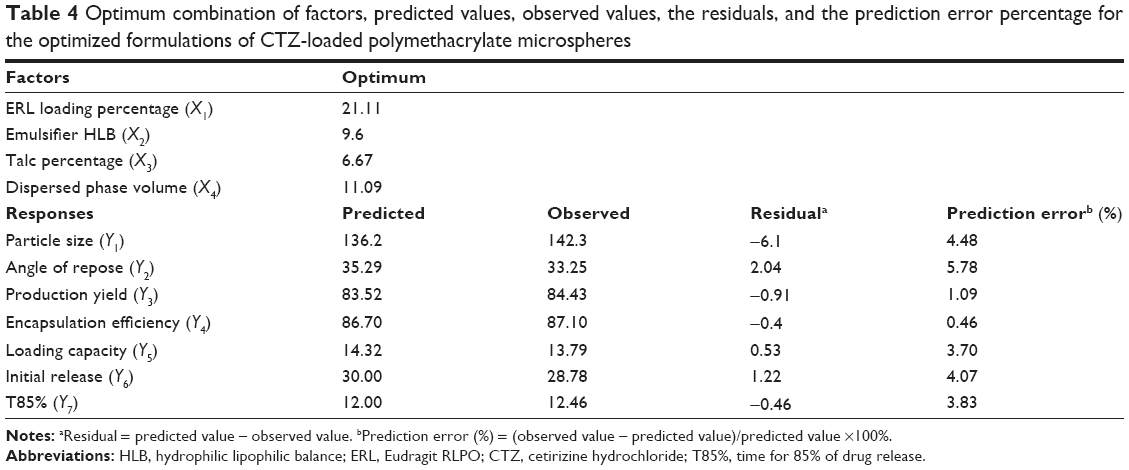 | Table 4 Optimum combination of factors, predicted values, observed values, the residuals, and the prediction error percentage for the optimized formulations of CTZ-loaded polymethacrylate microspheres |
Surface morphology analysis
Scanning electron photomicrographs (SEM) of the optimized CTZ-loaded polymethacrylate microspheres were adapted at various magnification powers to display the surface morphology of microspheres (Figure 1). SEM photographs exhibited spherical particles with smooth even surface, which indicated the absence of any drug crystal on the surface and confirmed the even distribution of drug in the polymeric matrix.
In vivo study and pharmacokinetics evaluation
In this study, the mean plasma concentration–time profiles after the oral administration of 10 mg single dose of the optimized CTZ-loaded polymethacrylate microspheres formulation and the marketed CTZ tablets are displayed in Figure 6. The relevant pharmacokinetic parameters were calculated using Kinetica version 5.0.11 software (Thermo Fisher Scientific) and listed in Table 5. The absorption from the marketed tablets was faster, reaching Cmax in 1.5 hours, whereas, the mean tmax was 6 hours following administration of the optimized formulation. The Cmax was lower following the administration of the optimized formulation of CTZ-loaded polymethacrylate microspheres (257.288 ng/mL) as compared with the marketed tablets (285.413 ng/mL). Besides that, statistical difference (P<0.05) was found between the two formulations at all time intervals of the study, which is depicted with respect to the AUC curve of the two treatments, indicating a comparable extent of absorption. Also, the absorption rate is considered as a good parameter for the assessment of extended-release formulations, which can be calculated by the division of Cmax by AUC.43 Moreover, the absorption rate of CTZ from the optimized microspheres (0.0641 h−1) was slower than that obtained from the marketed tablet (0.1177 h−1), which was in good agreement with the study by El-Kamel et al.44 The extended-release behavior of the microspheres was also reflected by the mean residence time, which had a higher value (11.098±0.93 hours) as compared with the marketed tablet (8.877±1.91 hours). Also, the relative bioavailability of the optimized formulation of CTZ-loaded polymethacrylate microspheres was 165.5% with respect to the marketed CTZ tablet indicating a significant enhancement of CTZ bioavailability. Thus, there is a 1.65-fold increase in the bioavailability of CTZ, with an expectation to decrease the administered dose and the frequency of administration, and subsequently minimize the adverse effects present with the administration of a large dose that maximizes patient compliance.
 | Figure 6 Mean plasma concentration–time profiles for CTZ after the oral administration of a single dose (10 mg) of the marketed tablet and the optimized CTZ-loaded polymethacrylate microspheres. |
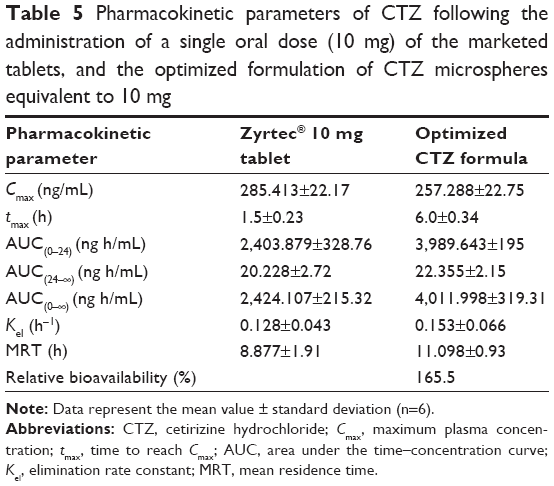 | Table 5 Pharmacokinetic parameters of CTZ following the administration of a single oral dose (10 mg) of the marketed tablets, and the optimized formulation of CTZ microspheres equivalent to 10 mg |
Conclusion
The results of this study revealed that CTZ extended-release microspheres were prepared successfully using ESE method. Furthermore, the D-LSCD was a highly efficient tool in the optimization of the four process variables on the quality attributes of the prepared microspheres. The optimized formulation showed a mean particle size of 142.3 μm, production yield of 84.43%, encapsulation efficiency of 87.1%, sufficient initial release of 28.78%, and release of 85% of its drug content in >12 hours. Besides that, the surface morphology of microspheres was spherical with smooth surface that improves the flowability and ensures the uniformity of dosage amounts during capsule filling. In addition, the pharmacokinetic parameters of the developed formulation provide a longer period of residence time required for achieving once daily dosing and enhancing drug bioavailability relative to the marketed product that could improve patient compliance.
Acknowledgments
This work was supported by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under Grant No 166-670-D1435. Therefore, the authors gratefully acknowledge the DSR technical and financial support.
Disclosure
The authors report no conflicts of interest in this work.
References
Tayade PT, Kale RD. Encapsulation of water-insoluble drug by a cross-linking technique: effect of process and formulation variables on encapsulation efficiency, particle size, and in vitro dissolution rate. AAPS PharmSci. 2004;6(1):112–119. | ||
Dey NS, Majumdar S, Rao MEB. Multiparticulate drug delivery systems for controlled release. Trop J Pharm Res. 2008;7(September):1067–1075. | ||
Kim BK, Hwang SJ, Park JB, Park HJ. Preparation and characterization of drug-loaded polymethacrylate microspheres by an emulsion solvent evaporation method. J Microencapsul. 2002;19(6):811–822. | ||
Behera B, Sahoo S, Dhal S, Barik BB, Gupta BK. Characterization of glipizide-loaded polymethacrylate microspheres prepared by an emulsion solvent evaporation method. Trop J Pharm Res. 2008;7(March):879–885. | ||
Obeidat W, Price J. Viscosity of polymer solution phase and other factors controlling the dissolution of theophylline microspheres prepared by the emulsion solvent evaporation method. J Microencapsul. 2003;20(1):57–65. | ||
Soppimath K, Kulkarni A, Aminabhavi T, Bhaskar C. Cellulose acetate microspheres prepared by o/w emulsification and solvent evaporation method. J Microencapsul. 2001;18(6):811–817. | ||
Palmieri G, Grifantini R, Di Martino P, Martelli S. Emulsion/solvent evaporation as an alternative technique in pellet preparation. Drug Dev Ind Pharm. 2000;26(11):1151–1158. | ||
Abd El-Bary A, Aboelwafa AA, Al Sharabi IM. Influence of some formulation variables on the optimization of pH-dependent, colon-targeted, sustained-release mesalamine microspheres. AAPS PharmSciTech. 2012;13(1):75–84. | ||
Song M, Li N, Sun S, Tiedt LR, Liebenberg W, de Villiers MM. Effect of viscosity and concentration of wall former, emulsifier and pore-inducer on the properties of amoxicillin microcapsules prepared by emulsion solvent evaporation. Farmaco. 2005;60(3):261–267. | ||
Yang C, Tsay S, Tsiang R. Encapsulating aspirin into a surfactant-free ethyl cellulose microsphere using non-toxic solvents by emulsion solvent-evaporation technique. J Microencapsul. 2001;18(2): 223–236. | ||
Hariharan M, Price J. Solvent, emulsifier and drug concentration factors in poly(D,L-lactic acid) microspheres containing hexamethylmelamine. J Microencapsul. 2002;19(1):95–109. | ||
Nilkumhang S, Basit AW. The robustness and flexibility of an emulsion solvent evaporation method to prepare pH-responsive microparticles. Int J Pharm. 2009;377(1–2):135–141. | ||
Miyazaki Y, Onuki Y, Yakou S, Takayama K. Effect of temperature-increase rate on drug release characteristics of dextran microspheres prepared by emulsion solvent evaporation process. Int J Pharm. 2006;324(2):144–151. | ||
Douroumis DD, Gryczke A, Schminke S. Development and evaluation of cetirizine HCl taste-masked oral disintegrating tablets. AAPS PharmSciTech. 2011;12(1):141–151. | ||
Montgomery DC. Design and Analysis of Experiments. Hoboken, NJ: John Wiley & Sons; 2008. | ||
Martin A, Bustamante P, Chun AHC. Micromeritics. In: Mundorff GH, editor. Physical Pharmacy; Physical Chemical Principles in the Pharmaceutical Sciences. Philadelphia, PA: Lippincott Williams & Wilkins; 1993:423–452. | ||
Maiti S, Dey P, Kaity S, Ray S, Maji S, Sa B. Investigation on processing variables for the preparation of fluconazole-loaded ethyl cellulose microspheres by modified multiple emulsion technique. AAPS PharmSciTech. 2009;10(3):703–715. | ||
Palanisamy M, Khanam J. Cellulose-based matrix microspheres of prednisolone inclusion complex: preparation and characterization. AAPS PharmSciTech. 2011;12(1):388–400. | ||
El-Say KM, El-Helw AR, Ahmed OA, et al. Statistical optimization of controlled release microspheres containing cetirizine hydrochloride as a model for water soluble drugs. Pharm Dev Technol. 2015;20(6):738–746. | ||
Wagner JG. Interpretation of percent dissolved-time plots derived from in vitro testing of conventional tablets and capsules. J Pharm Sci. 1969;58(10):1253–1257. | ||
Desai S, Singh P, Simonelli A, Higuchi WI. Investigation of factors influencing release of solid drug dispersed in inert matrices III. Quantitative studies involving the polyethylene plastic matrix. J Pharm Sci. 1966;55(11):1230–1234. | ||
Higuchi T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52(12):1145–1149. | ||
Korsmeyer R, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of potassium chloride release from compressed, hydrophilic, polymeric matrices: effect of entrapped air. J Pharm Sci. 1983;15(1): 1189–1191. | ||
Ritger PL, Peppas NA. A simple equation for description of solute release. II. Fickian and anomalous release from swellable devices. J Control Release. 1987;5(1):37–42. | ||
Tan Z-R, Ouyang D-S, Zhou G, et al. Sensitive bioassay for the simultaneous determination of pseudoephedrine and cetirizine in human plasma by liquid-chromatography-ion trap spectrometry. J Pharm Biomed Anal. 2006;42(2):207–212. | ||
Viswanathan NB, Thomas P, Pandit J, Kulkarni MG, Mashelkar RA. Preparation of non-porous microspheres with high entrapment efficiency of proteins by a (water-in-oil)-in-oil emulsion technique. J Control Release. 1999;58(1):9–20. | ||
Das SK, Yuvaraja K, Khanam J, Nanda A. Formulation development and statistical optimization of ibuprofen-loaded polymethacrylate microspheres using response surface methodology. Chem Eng Res Des. 2015;96:1–14. | ||
Mandenius C, Brundin A. Review: biocatalysts and bioreactor design optimization, bioprocess methodology, using design-of-experiments. Biotechnol Progr. 2008;24:1191–1203. | ||
Draper NR, Lin DK. Small response- surface designs. Technometrics. 1990;32(2):187–194. | ||
Mastiholimath VS, Dandagi PM, Jain SS, Gadad AP, Kulkarni AR. Time and pH dependent colon specific, pulsatile delivery of theophylline for nocturnal asthma. Int J Pharm. 2007;328(1):49–56. | ||
Dinarvand R, Mirfattahi S, Atyabi F. Preparation, characterization and in vitro drug release of isosorbide dinitrate microspheres. J Microencapsul. 2002;19(1):73–81. | ||
Dandagi PM, Mastiholimath VS, Gadad AP, Kulkarni AR, Konnur K. pH-sensitive mebeverine microspheres for colon delivery. Indian J Pharm Sci. 2009;71(4):464–468. | ||
Haznedar S, Dortunc B. Preparation and in vitro evaluation of Eudragit microspheres containing acetazolamide. Int J Pharm. 2004;269(1):131–140. | ||
Pandit SS, Hase DP, Bankar MM, Patil AT, Gaikwad NJ. Ketoprofen-loaded Eudragit RSPO microspheres: an influence of sodium carbonate on in vitro drug release and surface topology. J Microencapsul. 2009;26(3):195–201. | ||
Das SK, Das NG. Preparation and in vitro dissolution profile of dual polymer (Eudragit RS100 and RL100) microparticles of diltiazem hydrochloride. J Microencapsul. 1998;15(4):445–452. | ||
Perumal D, Dangor CM, Alcock R, Hurbans N, Moopanar KR. Effect of formulation variables on in vitro drug release and micromeritic properties of modified release ibuprofen microspheres. J Microencapsul. 1999;16(4):475–487. | ||
Lewis L, Boni R, Adeyeye CM. Effect of emulsifier blend on the characteristics of sustained release diclofenac microspheres. J Microencapsul. 1998;15(3):283–298. | ||
Shato H, Miyagawa Y, Okabe T, Miyajima M, Sunada H. Dissolution mechanism of diclofenac sodium from wax matrix granules. J Pharm Sci. 1997;86:929–934. | ||
Mahajan HS, Tatiya BV, Nerkar PP. Ondansetron loaded pectin based microspheres for nasal administration: in vitro and in vivo studies. Powder Technol. 2012;221:168–176. | ||
Islam S, Jalil R. Effects of release modifier and annealing on polyvinyl acetate-polyvinyl pyrrolidone microspheres containing diclofenac sodium. Dhaka Univ J Pharm Sci. 2013;12(1):41–50. | ||
Dey S, Pramanik S, Malgope A. Formulation and optimization of sustained release stavudine microspheres using response surface methodology. ISRN Pharm. 2011;2011:1–7. | ||
Mujtaba A, Ali M, Kohli K. Statistical optimization and characterization of pH-independent extended-release drug delivery of cefpodoxime proxetil using Box–Behnken design. Chem Eng Res Des. 2014;92(1): 156–165. | ||
Gibaldi M, editor. Compartmental and noncompartmental pharmacokinetics. Biopharmaceutices and Clinical Pharmacokinetics. 4th ed. Philadelphia, PA: Lea & Febiger; 1991:14–23. | ||
El-Kamel AH, Al-Gohary OMN, Hosny EA. Alginate-diltiazem hydrochloride beads: optimization of formulation factors, in vitro and in vivo availability. J Microencapsul. 2003;20(2):211–225. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.