Back to Journals » OncoTargets and Therapy » Volume 13
Matrix Stiffness and Colorectal Cancer
Received 14 September 2019
Accepted for publication 4 January 2020
Published 1 April 2020 Volume 2020:13 Pages 2747—2755
DOI https://doi.org/10.2147/OTT.S231010
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Arseniy Yuzhalin
Chongshun Liu, Haiping Pei, Fengbo Tan
Department of Gastrointestinal Surgery, Xiangya Hospital, Central South University, Changsha, People’s Republic of China
Correspondence: Haiping Pei; Fengbo Tan
Department of Gastrointestinal Surgery, Xiangya Hospital, Central South University, No. 87 Xiangya Road, Changsha 410008, People’s Republic of China
Email [email protected]; [email protected]
Abstract: In recent years, a growing consensus is emerging that the mechanical microenvironment of tumors is far more critical in the onset of tumor, tumor progression, invasion, and metastasis. Matrix stiffness, one of the sources of mechanical stimulation, affects tumor cells as well as non-tumor cells in multiple different molecular signaling pathways in solid tumors such as colorectal tumors, which lead to tumor invasion and metastasis, immune evasion and drug resistance. This review will illustrate the relationship between matrix stiffness and colorectal cancer from the following aspects. First, briefly introduce the mechanical microenvironment and colorectal cancer, then explain the origin of colorectal cancer extracellular matrix stiffness, and then synthesize the study of matrix stiffness of colorectal cancer in recent years to elaborate the effects of extracellular matrix stiffness in colorectal cancer’s biological behavior and signaling pathways, and finally we will discuss the transformation treatment for the matrix stiffness of colorectal cancer. An in-depth understanding of matrix stiffness and colorectal cancer can help researchers conduct further experiments to find new targets for the treatment of colorectal cancer.
Keywords: colorectal cancer, mechanical environment, matrix stiffness, signaling pathway, colon fibroblasts, drug resistance
Colorectal Cancer and Mechanical Environment
One of the most striking features of solid tumors is that their internal tumor cells are constantly affected by mechanical stimuli, which are more pronounced in colorectal cancer because such mechanical stimuli come from a variety of different aspects.1,2 A recent review wrote by Ciasca et al,3 attributes mechanical stimuli to three aspects: compression, matrix stiffness, and fluid mechanics.4 The sources of mechanical stimulation of colorectal cells are broadly classified into four categories: First, the gastrointestinal tract itself has undergone a lot of endogenous mechanical stress. These physiological stresses are derived from villus motility, peristalsis, interaction with the contents of the intestinal cavity, and mucosal remodeling and healing.5 Second, in the progression of colorectal cancer, the pathological proliferation of tumor cells combined with the inhibition of apoptosis makes the pressure inside the tumor increase sharply.6 Third, the increased deposition of ECM, as well as collagen cross-linking together with the decrease of matrix degradation mediated by MMRs promotes the ECM stiffness. Fourth, the activation and increased number of fibroblasts produce more endogenous stress. All of these factors ultimately led to the construction of an extremely complex mechanical microenvironment in colorectal cancer.
Mutations in genes initiate the process of colon cancer, and previous studies have focused on the biochemical aspects of colon cancer cells.7,8 The role of biomechanics in tumorigenesis has been underestimated. With the development of methodology in colon cancer research, people gradually realized the indispensable role of mechanical factors in the progression of colorectal cancer. Indeed, the mutation of the gene triggers the onset of tumors, but further progression also depends on the effects of biomechanics. Edwards et al,9 found that the growth of epithelial cells under the activating mutation of the Wnt cascade could produce sufficient compressive stress to cause colonic crypt flexion, which in turn led to subsequent deformation, budding, and crypt fission. Nelson et al,10 built a bilayer model to attach the growing cell layer to a thin compressible elastic beam, further confirmed the hypothesis that the mechanical effects of epithelial growth lead to the flexion of the colonic crypt.
The mechanical microenvironment of colorectal cancer not only participates in tumorigenesis but also plays a vital role in the process of tumor metastasis. As early as 2002, McCarty et al found that collisions in the hydrodynamic environment can increase the platelet-colon cancer cell adhesion, thereby increasing the blood-borne metastasis of colon cancer.11 A recent review3 details the relationship between colon cancer metastasis and mechanical microenvironment. Metastasis of colon cancer requires overcoming two mechanical stresses: the first is migration through the degraded ECM, the second is the shear stress after intravasation. The strategy for colon cancer cells to overcome these two mechanical stresses is to overexpress integrin and integrin E-cadherin, thereby increasing the adhesion of tumor cells and microenvironment.
Matrix stiffness is one of the most important mechanical microenvironments faced by colorectal cancer cells. It regulates the progression, proliferation, invasion, and metastasis of colorectal cancer cells from a variety of cellular pathways. Recent article wrote by Despotovic et al,12 has found remodeling of the matrix in the uninvolved rectal mucosa 10 cm, 20 cm away from the neoplasm, which is accompanied by an increase in matrix stiffness, indicated that the increase in matrix stiffness in colorectal cancer is not limited to the primary lesion but also to the uninvolved site, suggests an extremely complex effect of matrix stiffness in colorectal cancer.
The Origin of Matrix Stiffness of Colorectal Cancer
During the formation of colorectal tumors, changes in extracellular matrix (ECM) are mainly concentrated in two aspects: increased stiffness and degradation of the matrix. Overexpression of collagen, pathological collagen cross-linking, and fiber arrangement are three major causes of the increased stiffness of the colorectal cancer ECM. Recent experiments conducted by Brauchle et al,13 have shown that the collagen-rich area in CRC is much stiffer than normal colon tissue and an increased alignment of collagen fibers by comparing colon tissue from the same patient, which suggested that the changes in amount and structure of collagen likely play an important role in the increase of ECM stiffness. Further, the experiment also found an elevated level of glycosaminoglycans (GAGs) activity within the collagen network of colon carcinoma tissues, which imply a potential role of GAG, as a major non-fibrillar ECM component, in ECM remodeling and stiffness.
Lysyl oxidase (LOX) is an enzyme that catalyzes the cross-linking of collagen in extracellular matrices and plays a key role in tissue stabilization and remodeling wound healing and other processes.14 As early as 2009, Levental et al found the role of LOX in breast cancer. It was found that LOX can increase the matrix stiffness of breast tumors by increasing the cross-linking of collagen. The increased matrix stiffness induces focal adhesions assembly and up-regulates GFR-dependent PI3K signaling, which ultimately led to tumor progression.15 However, in colorectal cancer, the role of LOX is controversial.16,17 In 2013, Baker et al explored the role of LOX in colorectal cancer using the cell line expressing a catalytically inactive mutation form of LOX. SW480 tumors overexpressing active LOX were much stiffer than those expressing catalytically inactive mutant LOX, indicating that LOX activity increased matrix stiffness in colorectal cancer.18
Cancer-associated fibroblasts interact with almost all cells in tumor microenvironments and play a key role in the formation of matrix stiffness.19 Cancer-associated fibroblasts promote m2 cell polarization by releasing fibroblast growth factor, and polarized m2 cells will increase matrix stiffness by expressing large amounts of TGFβ, promoting fibroblasts to become cancer-associated fibroblasts and forming a positive feedback loop.20 Cancer-associated fibroblasts can also increase contractility by expressing α-SMA, causing further shrink and stiffen in the tumor microenvironment.21
Effect of Extracellular Matrix Stiffness on Colorectal Cancer Biological Behavior and Signaling Pathway
Effect of Matrix Stiffness on the Signaling Pathway of Colorectal Cancer Cells
Matrix stiffness affects tumor cells as well as non-tumor cells in multiple different molecular signaling pathways in colorectal tumors, which lead to tumor invasion and metastasis, immune evasion and drug resistance (Table 1). Whitehead et al, demonstrated that mechanical strains produced by external forces in a mechanical deformation device can initiate the expression of tumorlike genes in preneoplastic tissues in CRC. In APC knockout healthy mice, mechanical stimulation induces b-catenin phosphorylation, and b-catenin is relocated to the nucleus. Reduction of the linkage between E-cadherin weakened the adhesion within tumor cells as well as cellular adhesion to specific components of ECM, the expression of myc and twist1, two oncogenes regulated through beta-catenin, are triggered.22 This suggests that the mechanical stimulation of matrix stiffness on colorectal cancer cells can regulate tumor progression through the Wnt/b-catenin pathway. It also implied that matrix stiffness can accelerate the carcinogenesis of precancerous tissues.
 |
Table 1 List of Matrix Stiffness Affecting Colorectal Cancer |
Similarly, Fernandez-Sanchez et al,23 inserted a magnet near the colon in the model mouse, the implanted magnet generated a magnetic force on ultra-magnetic liposomes which quantitatively mimicked ECM stiffness, it was found a rapid Ret activation and downstream phosphorylation of b-catenin on Tyr654, decreasing its interaction with the E-cadherin in adherens junctions. And one month later, an up-regulation of the b-catenin target gene expression was discovered.
We reviewed the relevant literature and found that matrix stiffness affects the Wnt pathway in many types of cells. For example, an increase in matrix stiffness promotes the differentiation of mesenchymal stem cells through activation of the Wnt pathway and maintains the phenotype of chondrocytes24. It has also been pointed out that the extracellular matrix can increase the concentration of β-catenin in uterine fibroids through activation of Wnt pathway.25 These studies suggest that the effect of matrix stiffness on the Wnt pathway of cells is a ubiquitous phenomenon and therefore needs to be taken seriously.
In the previous experiments by Baker et al,18 it was not only proved that LOX played a key role in the increase of matrix stiffness in colorectal cancer, the experiment also found that LOX-mediated matrix stiffness promoted the invasion and metastasis through b1-integrin activation and FAK/SRC signaling.
Krndija et al,26 found that receptor-type tyrosine-protein phosphatase alpha (RPTPα) capable of sensing mechanical stimulation was expressed in colon cancer cell SW480. It was found that RPTPα regulates cell contraction by SFK and myosin light chain kinase (MLCK) and leads to a more metastatic property of colon cancer cells. This indicates that matrix stiffness can also regulate the metastasis of colorectal cancer cells by SFK and MLCK.
However, in a 2013 study, Luca et al27 cultured different colon cancer cell lines in a 3D laminin-rich-extracellular matrix and found low expression of EGFR at the gene level as well as at the protein level, the research also detected an impair of the regulatory genes involved in proliferation. This indicates that the regulation of matrix stiffness for colorectal cancer cells is multi-faceted, it also shows that the effect of ECM stiffness on the molecular pathway of colorectal cancer is extremely complicated.
Effect of Matrix Stiffness on Biological Behavior of Colorectal Cancer Cells
The effect of increased matrix stiffness on colorectal cancer is reflected in different aspects, one of the most important aspects is distant metastasis. In a 2010 study, Tang et al2 found that when cultured HCT-8 cells on a medium stiffness matrix (Polyacrylamide gels with stiffness 21–47 kPa), the epithelial-like phenotype cells (E-cell) gradually changed into a rounded, separated, morphologically-like phenotype (R-cell), and the E-Cadherin between cells showed a significant decrease, indicating that the cells were more likely metastasis. When these isolated R-cells were cultured on medium of different stiffness, they still retained metastasis-like properties. This suggests that matrix stiffness is likely to have an irreversible effect on colon cancer cells, and this effect is ultimately reflected in colon cancer cells that are more likely to metastasize.
Tang et al,28 further conducted experiments related to E-R conversion. It was found that not only the E-R transformation of HCT-8 cells was present, but also the SW480 and HCT116 cell lines undergo E-R transformation in 1.0 kPa, 10 kPa gels, respectively. Further RNA sequence analysis revealed that the metastasis-related genes in R cells were overexpressed. The experiment found that 11 genes were significantly up-regulated, and most of them were related to tumor proliferation, motility, metabolism. Finally, the experiment tested the deformability of R cells by contact mode of atomic force microscopy and found that R cells have higher deformability, which means that R cells are more likely to penetrate the epithelium of blood vessels. This further demonstrates the extremely complex effects of matrix stiffness on the biological behavior of colorectal cancer cells.
Effect of Matrix Stiffness on the Characteristics of Cancer Stem Cells in Colorectal Cancer Cells
Matrix stiffness can not only increase the possibility of distant metastasis of colorectal cancer but also regulate the characteristics of cancer stem cells (CSC) in colorectal cancer. In a previously listed literature,28 Tang et al, found that colorectal cancer cells cultured under appropriate matrix stiffness can produce E-R conversion, and R cells are more aggressive. Experiments have also found that more than 90% of R form cells express an identified enzyme –ALDH3A1, which is a marker of various types of cancer stem cells, suggesting that proper matrix stiffness can promote colon cancer tumors to produce cancer stem cells.
Jabbari et al, found that the matrix stiffness of 25kpa was optimum matrix stiffness for colon cancer tumor stem cell growth and molecular marker expression.29 At this optimum stiffness, the expression level of the tumor Yes-associated protein (YAP/TAZ) transcription factor is also the highest. This experiment only found this interesting phenomenon, it did not further explore the relationship between matrix stiffness and CSC/YAP. In fact, as early as 2011, Dupont et al30 explored the relationship between YAP/TAZ transcription factors and matrix stiffness and found that YAP/TAZ is regulated by matrix stiffness and cell shape, Rho GTPase and the tension of the actomyosin skeleton were involved in the regulatory process. Subsequent experiments confirmed that matrix stiffness can maintain the tension of actomyosin in tumor cells through the mechanical perception of integrin, and this high tension can be applied to F-actin, which causes F-actin to be stretched, thereby enhancing YAP/TAZ transcription factor activity.31
In order to explore the effects of matrix stiffness on cancer stem cells and whether YAP activity was involved, we have recently conducted a series of experiments.32 In our experiment, we found that ECM stiffness remarkably upregulates the expression of CSC markers. Further experiment showed that matrix stiffness can increase the amount of dephosphorylated YAP and its entry into the nucleus. Through the knockout of YAP, we found that there was a significant decrease in the number of clone formation on a matrix with a higher stiffness, but on a substrate with a small stiffness, the number of clone formation did not change much. Thus, we first established a link between YAP and tumor stem cell characteristics in colorectal cancer. After the usage of anti-integrin antibodies and a FAK inhibitor, we finally found a significant decrease of CSC markers on stiff ECM. Although we are the first to propose that matrix stiffness can regulate the stemness characteristics through integrin b1/FAK/YAP pathway, we are not yet able to conclude that the b1/FAK/YAP pathway is a colorectal-specific pathway, we think more experiments are needed to validate it.
In addition, our experiments have some limitations: we only chose one cell line, HCT-116, and we only did cell experiments, lacking validation in vivo experiments, we only did 2D experiments, which made our experiment less general. Regarding the effect of matrix stiffness on colorectal cancer, we believe that more genetic research should be done in the future, and genetic research is also lacking in this field. In the future, we should also focus on the clinical impact of matrix stiffness on the prognosis of patients with colorectal cancer.
Effect of Matrix Stiffness on Colorectal Cancer Fibroblasts
Colorectal tumors are solid masses, not only the extracellular matrix and tumor cells, but also fibroblasts, inflammatory cells, and neovascularization are within the tumor. Therefore, matrix stiffness does not only affect tumor cell but also it affects various non-tumor cells such as fibroblasts. In a 2013 study,33 the researchers found that the morphology of fibroblasts changed significantly as the matrix stiffness increased, from a circular shape to an activated satellite morphology. Increased matrix stiffness can significantly increase the expression of αSMA protein, while αSMA protein expression up-regulates the contractile activity of fibroblasts. Moreover, matrix stiffness also increases the amount of mature FAK in fibroblasts, indicating that matrix stiffness will also change the biological activity of fibroblasts via the FAK signaling pathway.
Johnson et al,34 also found increased expression of fibrotic genes, reduction of expression of MMPs when increase ECM stiffness. Fiber contraction is mediated by myosin light chain kinase (MLCK), MYLK expression was up-regulated by increased ECM stiffness, thereby promoting fiber contraction. IL-1b is an inflammatory cytokine that induces catabolism by activating MMPs (matrix metalloproteinases) when wounds heal.35 MMPs mediate the degradation of extracellular matrices and so maintain the balance of extracellular matrix under physiological conditions. When the matrix stiffness increased, the IL-1b and IL-1b reactive inflammatory genes in fibroblasts were inhibited, resulted in a significant decrease in the expression of MMPs, further increased the matrix stiffness. Moreover, the increase in matrix stiffness can significantly repress the expression of the PTGS2 gene, resulted in a decrease in the content of the COX-2 enzyme, which further activated the function of fibroblasts. The increased nuclear localization of the myocardin-related transcription factor (MRTF-A), which activates the αSMA promoter,36 was also found during the study.
Although the authors have discovered the increased nuclear localization of MRTF-A, there is no specific pathway to explain this phenomena, so in 2014, Johnson et al37 conducted a series of experiments and found a novel pathway which can eventually explain the preceding phenomena in colon fibroblasts in the presence of increased ECM stiffness. Matrix stiffness, as well as TGFβstimulation, lead to Rho GTPases and ROCK activation, which allows free globular actin (G-actin) to aggregate into filamentous actin (F-actin) in the cytoplasm, and the globular actin bound to MRTF-A also detached. In the meantime, MRTF-A enters the nucleus and interacts with the transcription factor SRF to induce expression of αSMA and MYLK, thereby aggravating fibrosis. These studies have jointly demonstrated that matrix stiffness affects fibroblasts through multiple signal pathways, such as FAK, Rho/ROCK (Rho-associated protein kinase), and MYLK. It also reveals that the effect of matrix stiffness on fibroblasts is extremely complex.
Transformation Therapy for Colorectal Cancer Matrix Stiffness
There are three main approaches to transformation therapy for matrix stiffness: first, targeting the sources of matrix stiffness, preventing matrix stiffness from the outset. Second, the reversible treatment of matrix stiffness. Third, inhibition of downstream pathways caused by matrix stiffness. All three approaches can effectively reduce the effect of matrix stiffness on the tumor.38 For the sources of matrix stiffness, LOX39 and cancer-associated fibroblasts40 can be used as targets to prevent increased matrix stiffness. Growth factors can also be targeted, such as TGFβ41 PDGF, VEGF.42,43 For the downstream pathway of matrix stiffness, integrin, FAK,44 Rho-GTPase,45 NF-κB46 can be used as targets. Although many therapeutic targets for matrix stiffness have emerged, most of these studies have focused on tumors such as breast cancer, and few studies have been aimed at colorectal cancer.
Judah Folkman47,48 proposed that angiogenesis plays a crucial role in tumor growth and proliferation, then anti-angiogenic therapy was widely accepted in the following decades. As an anti-angiogenic drug, the anti-VEGF antibody is widely used in the treatment of metastatic colorectal cancer in combination with chemotherapy,49 but the survival benefit is modest. The mechanism of resistance is also unclear.
During the chemotherapy of solid tumors, the drug is absorbed into the bloodstream and then enters the interior of the tumor. Therefore, blood perfusion is one of the key factors determining the drug transport efficiency. When the drug reaches the tumor tissue from the blood vessel, free diffusion acts as the main mechanism of drug transport. In a review by Jain et al,50 summarized the barriers to delivery of cancer therapeutics and concluded that the dense and heterogeneous structure of ECM in solid tumor are critical determinants of interstitial transport.
Recently, an article wrote by Rahbari et al,51 explored the role of antiangiogenic therapy in the deposition of the extracellular matrix of mCRC and conducted research in which regarded tumor vascular perfusion as an acquired resistance mechanism in liver metastasis of colorectal cancer. It was found that the expression of hyaluronic acid (HA) and sulfated glycosaminoglycans (sGAG) was significantly elevated in liver mCRC tissues from patients treated with preoperative bevacizumab and chemotherapy. Anti-VEGF treatment was also found to increase tumor stiffness. Due to Immune cells such as myeloid-derived suppressor cell (MDSC) play a key role in liver fibrosis, tumor progression, and resistance to anti-angiogenic therapy,52 researchers investigate the potential function of MDSC in ECM deposition after anti-VEGF therapy by selectively depleted those immune cell populations and found that accumulation of HA and sGAG is independent of the tumor infiltration by MDSC. Since hepatic satellite cells (HSC) are the main source of liver matrix synthesis, experiments explored the relationship between HSC and HA deposition. The experiment found that the deposition of HA is probably due to the activation of HSC caused by hypoxia. The researcher then deleted HA by treated the model mice with intravenous administration of polyethylene glycol conjugated (PEGylated) hyaluronidase (PEG-HAse), found that targeting HA increases perfusion and improves the efficacy of chemotherapy in liver metastases CRC after anti-VEGF therapy. This experiment found a mechanism that can partially explain acquired resistance, provides a better perspective for the treatment of advanced colorectal cancer and provides ECM-related therapeutic targets for the treatment of colorectal cancer.
In a further study, HSCs can be used as a direct target to explore new ways to improve drug delivery efficiency in colorectal cancer. In previous experiments,53 Ding et al, found that vitamin D receptor ligands inhibit the activation of HSC cells through the TGFβ pathway, so vitamin receptor ligands may be an additional strategy. In another study conducted by Dou et al,54 ECM stiffness can increase of nuclear targeting of p300, a histone acetyltransferase that regulates transcription, thereby increasing p300-dependent transcription of αSMA, ultimately leading to activation of HSC. Therefore, p300 can be used as a new therapeutic target to reduce the activation of HSC, thereby reducing the acquired resistance of liver metastasis colorectal cancer.
Future Expectations
Although there are many studies that concentrate on matrix stiffness, and the researchers also found the effect of matrix stiffness on many different pathways in CRC (Figure 1), the perception of this effect is still at a relatively shallow level. At present, it is only roughly known matrix stiffness affects which pathway, and which molecules participate in that process, the more complete and concrete analysis is very rare. For example, Johnson et al proposed a complete pathway in their experiments, and this pathway is not an existing classical pathway, which provides a new perspective for this field, rather than just verified the classical pathway. Moreover, most of the current researches are limited to the influence of the mechanical microenvironment on colorectal cancer, few experiments can indicate how mechanical stimulation works together with biochemical factors to promote the progression of colorectal cancer. Therefore, the future direction should be 1) to explore the more concrete signal pathways about the effect of matrix stiffness on CRC. 2) to explore the combined effects of matrix stiffness as well as other mechanical stimuli and biochemical factors on the progression of colorectal cancer. Only when a very clear mechanism of these pathways is discovered can new therapeutic targets for the mechanical microenvironment be found from these concrete mechanisms. In addition, the study of matrix stiffness in colorectal cancer is relatively less compared to solid tumors such as breast cancer. Since colorectal cancer cells face an extremely complex mechanical environment, future research directions should include searching for a new approach that can mimic the real mechanical microenvironment of colorectal cancer cells.
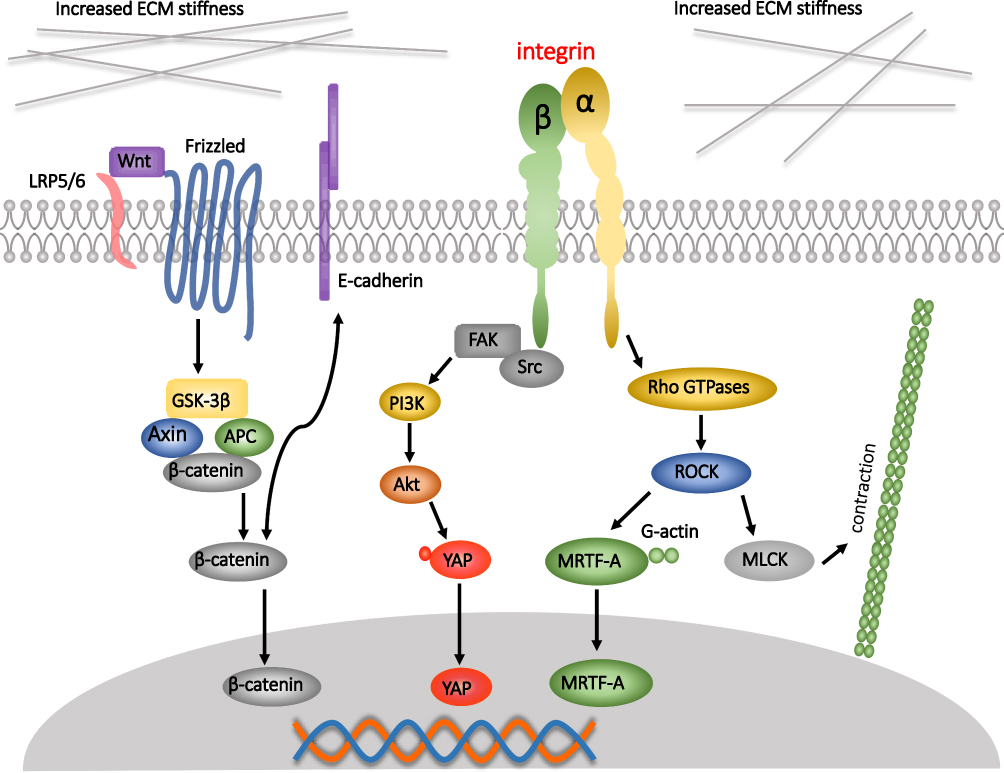 |
Figure 1 Increased matrix stiffness can affect cells in different signaling pathways in colorectal cancer (CRC). Increased matrix stiffness can induce the activation of Wnt/β-catenin pathway in CRC cells, then 1) promote the translocation of β-catenin into the nucleus, and initiate a series of changes in gene level, leading to the proliferation and metastasis of CRC cells, 2) Promote the detachment of β-catenin from E-cadherin, resulting in instability of cell-to-cell connections and promoting metastasis. An increase in matrix stiffness can also increase the translocation of YAP to the nucleus via the FAK/Src pathway, thereby altering the characteristics of the tumor stem cells. The increase in matrix stiffness also promotes the conversion of Rho GDP to Rho GTPases. The activation of Rho 1) induces myocardin-related transcription factor A (MRTF-A) to remove G-actin, enter the nucleus to participate in a series of regulation, and 2) increase the myosin light chain kinase (MLCK) and promote cell contraction. Black arrows indicate events in which a series of effects of matrix stiffness on the cellular pathway occur. |
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
The study was sponsored by the Nature Scientific Foundation of China (grant number: 81702956); The Strategy-Oriented Special Project of Central South University in China; (grant number: ZLXD2017003); The XiangYa-Peking University Wei Ming Clinical and Rehabilitation Research Fund (xywm2015I21).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Palmieri V, Lucchetti D, Maiorana A, et al. Mechanical and structural comparison between primary tumor and lymph node metastasis cells in colorectal cancer. Soft Matter. 2015;11(28):5719–5726. doi:10.1039/C5SM01089F
2. Tang X, Kuhlenschmidt TB, Zhou J, et al. Mechanical force affects expression of an in vitro metastasis-like phenotype in HCT-8 cells. Biophys J. 2010;99(8):2460–2469. doi:10.1016/j.bpj.2010.08.034
3. Ciasca G, Papi M, Minelli E, Palmieri V, De Spirito M. Changes in cellular mechanical properties during onset or progression of colorectal cancer. World J Gastroenterol. 2016;22(32):7203–7214. doi:10.3748/wjg.v22.i32.7203
4. Avvisato CL, Yang X, Shah S, et al. Mechanical force modulates global gene expression and beta-catenin signaling in colon cancer cells. J Cell Sci. 2007;120(Pt 15):2672–2682. doi:10.1242/jcs.03476
5. Basson MD. Paradigms for mechanical signal transduction in the intestinal epithelium. Category: molecular, cell, and developmental biology. Digestion. 2003;68(4):217–225. doi:10.1159/000076385
6. Li XL, Zhou J, Chen ZR, Chng WJ. P53 mutations in colorectal cancer - molecular pathogenesis and pharmacological reactivation. World j Gastroenterol. 2015;21(1):84–93. doi:10.3748/wjg.v21.i1.84
7. Valle L, Vilar E, Tavtigian SV, Stoffel EM. Genetic predisposition to colorectal cancer: syndromes, genes, classification of genetic variants and implications for precision medicine. J Pathol. 2018;247:574–588.
8. Vacante M, Borzi AM, Basile F, Biondi A. Biomarkers in colorectal cancer: current clinical utility and future perspectives. World j Clin Cases. 2018;6(15):869–881. doi:10.12998/wjcc.v6.i15.869
9. Edwards CM, Chapman SJ. Biomechanical modelling of colorectal crypt budding and fission. Bull Math Biol. 2007;69(6):1927–1942. doi:10.1007/s11538-007-9199-8
10. Nelson MRHD, Jensen OE, King JR, Rose FR, Waters SL. Growth-induced buckling of an epithelial layer. Biomech Model Mechanobiol. 2011;10:883–900. doi:10.1007/s10237-010-0280-0
11. McCarty OJ, Jadhav S, Burdick MM, Bell WR, Konstantopoulos K. Fluid shear regulates the kinetics and molecular mechanisms of activation-dependent platelet binding to colon carcinoma cells. Biophys J. 2002;83(2):836–848. doi:10.1016/S0006-3495(02)75212-0
12. Despotovic SZ, Milicevic NM, Milosevic DP, et al. Remodeling of extracellular matrix of the lamina propria in the uninvolved human rectal mucosa 10 and 20 cm away from the malignant tumor. Tumour Biol. 2017;39(7):1010428317711654. doi:10.1177/1010428317711654
13. Brauchle E, Kasper J, Daum R, et al. Biomechanical and biomolecular characterization of extracellular matrix structures in human colon carcinomas. Matrix Biol. 2018;68–69:180–193. doi:10.1016/j.matbio.2018.03.016
14. Wang TH, Hsia SM, Shieh TM. Lysyl oxidase and the tumor microenvironment. Int J Mol Sci. 2016;18:1. doi:10.3390/ijms18010001
15. Levental KR, Yu H, Kass L, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906. doi:10.1016/j.cell.2009.10.027
16. Csiszar K, Fong SF, Ujfalusi A, et al. Somatic mutations of the lysyl oxidase gene on chromosome 5q23.1 in colorectal tumors. Int j Cancer. 2002;97(5):636–642. doi:10.1002/ijc.10035
17. Kim Y, Roh S, Park JY, Kim Y, Cho DH, Kim JC. Differential expression of the LOX family genes in human colorectal adenocarcinomas. Oncol Rep. 2009;22(4):799–804. doi:10.3892/or_00000502
18. Baker AM, Bird D, Lang G, Cox TR, Erler JT. Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene. 2013;32(14):1863–1868. doi:10.1038/onc.2012.202
19. Najafi M, Farhood B, Mortezaee K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J Cell Biochem. 2019;120(3):2782–2790. doi:10.1002/jcb.v120.3
20. Zhao P, Wang Y, Kang X, et al. Dual-targeting biomimetic delivery for anti-glioma activity via remodeling the tumor microenvironment and directing macrophage-mediated immunotherapy. Chem Sci. 2018;9(10):2674–2689. doi:10.1039/C7SC04853J
21. Follonier Castella L, Gabbiani G, McCulloch CA, Hinz B. Regulation of myofibroblast activities: calcium pulls some strings behind the scene. Exp Cell Res. 2010;316(15):2390–2401. doi:10.1016/j.yexcr.2010.04.033
22. Whitehead J, Vignjevic D, Futterer C, Beaurepaire E, Robine S, Farge E. Mechanical factors activate beta-catenin-dependent oncogene expression in APC mouse colon. HFSP J. 2008;2(5):286–294. doi:10.2976/1.2955566
23. Fernandez-Sanchez ME, Barbier S, Whitehead J, et al. Mechanical induction of the tumorigenic beta-catenin pathway by tumour growth pressure. Nature. 2015;523(7558):92–95. doi:10.1038/nature14329
24. Du J, Zu Y, Li J, et al. Extracellular matrix stiffness dictates Wnt expression through integrin pathway. Sci Rep. 2016;6:20395. doi:10.1038/srep20395
25. Ko YA, Jamaluddin MFB, Adebayo M, et al. Extracellular matrix (ECM) activates beta-catenin signaling in uterine fibroids. Reproduction. 2018;155(1):61–71. doi:10.1530/REP-17-0339
26. Krndija D, Schmid H, Eismann JL, et al. Substrate stiffness and the receptor-type tyrosine-protein phosphatase alpha regulate spreading of colon cancer cells through cytoskeletal contractility. Oncogene. 2010;29(18):2724–2738. doi:10.1038/onc.2010.25
27. Luca AC, Mersch S, Deenen R, et al. Impact of the 3D microenvironment on phenotype, gene expression, and EGFR inhibition of colorectal cancer cell lines. PLoS One. 2013;8(3):e59689. doi:10.1371/journal.pone.0059689
28. Tang X, Kuhlenschmidt TB, Li Q, et al. A mechanically-induced colon cancer cell population shows increased metastatic potential. Mol Cancer. 2014;13:131. doi:10.1186/1476-4598-13-131
29. Jabbari E, Sarvestani SK, Daneshian L, Moeinzadeh S. Optimum 3D matrix stiffness for maintenance of cancer stem cells is dependent on tissue origin of cancer cells. PLoS One. 2015;10(7):e0132377. doi:10.1371/journal.pone.0132377
30. Dupont SML, Aragona M, Enzo E, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179–183. doi:10.1038/nature10137
31. Dupont S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp Cell Res. 2016;343(1):42–53. doi:10.1016/j.yexcr.2015.10.034
32. Tan F, Huang Y, Pei Q, Liu H, Pei H, Zhu H. Matrix stiffness mediates stemness characteristics via activating the yes-associated protein in colorectal cancer cells. J Cell Biochem. 2018;120:2213–2225.
33. Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12(9):2730–2741. doi:10.1091/mbc.12.9.2730
34. Johnson LA, Rodansky ES, Sauder KL, et al. Matrix stiffness corresponding to strictured bowel induces a fibrogenic response in human colonic fibroblasts. Inflamm Bowel Dis. 2013;19(5):891–903. doi:10.1097/MIB.0b013e3182813297
35. Hiramitsu T, Yasuda T, Ito H, et al. Intercellular adhesion molecule-1 mediates the inhibitory effects of hyaluronan on interleukin-1beta-induced matrix metalloproteinase production in rheumatoid synovial fibroblasts via down-regulation of NF-kappaB and p38. Rheumatology (Oxford). 2006;45(7):824–832. doi:10.1093/rheumatology/kel026
36. Zhao XH, Laschinger C, Arora P, Szaszi K, Kapus A, McCulloch CA. Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J Cell Sci. 2007;120(Pt 10):1801–1809. doi:10.1242/jcs.001586
37. Johnson LA, Rodansky ES, Haak AJ, Larsen SD, Neubig RR, Higgins PD. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-beta-induced fibrogenesis in human colonic myofibroblasts. Inflamm Bowel Dis. 2014;20(1):154–165. doi:10.1097/01.MIB.0000437615.98881.31
38. Lampi MC, Reinhart-King CA. Targeting extracellular matrix stiffness to attenuate disease: from molecular mechanisms to clinical trials. Sci Transl Med. 2018;10:422. doi:10.1126/scitranslmed.aao0475
39. Chan N, Willis A, Kornhauser N, et al. Influencing the tumor microenvironment: a Phase II study of copper depletion using tetrathiomolybdate in patients with breast cancer at high risk for recurrence and in preclinical models of lung metastases. Clin Cancer Res. 2017;23(3):666–676. doi:10.1158/1078-0432.CCR-16-1326
40. O’Reilly S. MicroRNAs in fibrosis: opportunities and challenges. Arthritis Res Ther. 2016;18:11. doi:10.1186/s13075-016-0929-x
41. Morris JC, Tan AR, Olencki TE, et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One. 2014;9(3):e90353. doi:10.1371/journal.pone.0090353
42. Heldin CH. Targeting the PDGF signaling pathway in the treatment of non-malignant diseases. J Neuroimmune Pharmacol. 2014;9(2):69–79. doi:10.1007/s11481-013-9484-2
43. Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123(5):1887–1901. doi:10.1172/JCI66028
44. Golubovskaya VM. Targeting FAK in human cancer: from finding to first clinical trials. Front Biosci. 2014;19:687–706. doi:10.2741/4236
45. Wang CY, Liu PY, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med. 2008;14(1):37–44. doi:10.1016/j.molmed.2007.11.004
46. Muz B, Ghazarian RN, Ou M, Luderer MJ, Kusdono HD, Azab AK. Spotlight on ixazomib: potential in the treatment of multiple myeloma. Drug Des Devel Ther. 2016;10:217–226. doi:10.2147/DDDT.S93602
47. Folkman J, Parris EE, Folkman J. Tumor angiogenesis: therapeutic implications. N Engl j Med. 1971;285(21):1182–1186. doi:10.1056/NEJM197111182852108
48. Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29(6 Suppl 16):15–18. doi:10.1053/sonc.2002.37263
49. Fakih MG. Metastatic colorectal cancer: current state and future directions. J clin oncol. 2015;33(16):1809–1824. doi:10.1200/JCO.2014.59.7633
50. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. doi:10.1038/nrclinonc.2010.139
51. Rahbari NN, Kedrin D, Incio J, et al. Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases. Sci Transl Med. 2016;8(360):360ra135. doi:10.1126/scitranslmed.aaf5219
52. Y HY C, Reiberger T, Duyverman AM, et al. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology. 2014;59:1435–1447. doi:10.1002/hep.26790
53. Ding N, Yu RT, Subramaniam N, et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell. 2013;153(3):601–613. doi:10.1016/j.cell.2013.03.028
54. Dou C, Liu Z, Tu K, et al. P300 acetyltransferase mediates stiffness-induced activation of hepatic stellate cells into tumor-promoting myofibroblasts. Gastroenterology. 2018;154(8):2209–2221 e2214. doi:10.1053/j.gastro.2018.02.015
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.