Back to Archived Journals » Metalloproteinases In Medicine » Volume 4
Matrix metalloproteinases in pancreatitis: current insights
Authors Sri Manjari K, Nallari P, Venkateshwari A
Received 21 September 2016
Accepted for publication 22 November 2016
Published 31 January 2017 Volume 2017:4 Pages 15—23
DOI https://doi.org/10.2147/MNM.S105774
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yoshifumi Itoh
Kavutharapu Sri Manjari, Pratibha Nallari, Ananthapur Venkateshwari
Department of Cell Biology, Institute of Genetics and Hospital for Genetic Diseases, Osmania University, Begumpet, Hyderabad, India
Abstract: Matrix metalloproteinases (MMPs) belong to a family of zinc-dependent endopeptidases belonging to the metzincin superfamily of metalloproteinases. MMPs can degrade most of the constituents of the extracellular matrix, basement membrane, and inflammatory mediators. Most of the MMPs are not expressed in normal tissue, but their expression and activity increase dramatically during wound healing as a result of a change in matrix composition, inflammation, and repair. Pancreatitis refers to inflammation of the pancreas resulting in an imbalance in its normal function. MMPs are involved in events like chronic inflammation, fibrosis, and resolution of the disease. There have been substantial advances in understanding the pathogenesis of acute and chronic pancreatitis, because of the recent scientific progress in the underlying genetic, cellular, and molecular pathophysiology. This study will provide an evidence-based review of the importance of MMPs in pancreatitis.
Keywords: pancreatic stellate cells, acinar cells, extracellular matrix, inflammation, matrix remodeling
Introduction
The extracellular matrix (ECM), a noncellular component, is present in all tissues and organs providing vital somatic scaffolding for the cellular components. In addition, ECM is crucial to initiate biochemical and biomechanical signals that are essential for tissue morphogenesis, differentiation, and homeostasis.1,2 The biochemical and mechanical properties of an organ are characterized by the physical and biochemical characteristics of the ECM. The ECM in the normal pancreas is distributed throughout the gland but is modest in volume. The ECM is a complex mixture of diverse macromolecules whose structural integrity and functional composition are important to maintain normal tissue architecture and development.3 The ECM consists of two main classes of macromolecules: fibrous proteins and proteoglycans.4,5 The main fibrous proteins are collagens, elastins, fibronectins, and laminins.6 Collagen, the most abundant fibrous protein within the ECM of the pancreas, constitutes up to 30% of the total protein mass. The main and interlobular ducts of the pancreas have thick dense collagenous walls. The pancreatic stroma that surrounds acini and ducts has both a noncellular component composed of ECM proteins and a cellular component containing different cells, such as neurons, endothelial cells, resident macrophages, adipocytes, fibroblasts, and pancreatic stellate cells (PSCs). PSCs have cytoplasmic lipid droplets rich in vitamin A and are a rare cell population discovered in close proximity to pancreatic acini.7,8 Interstitial collagen and other structural components of the ECM are secreted by pancreatic fibroblasts. New evidence suggests that PSCs are present sparsely in a healthy adult pancreas where they are considered “quiescent”, and their normal function in the healthy organ is still a matter of examination. The basement membrane of the pancreas, a noncellular component which surrounds acini and ducts, as well as local blood vessels, plays an important role to support the architecture of these epithelial cells by separating them from the interstitial stroma.9 Thus the pancreatic ECM is composed mainly of collagen fibers and to a lesser extent elastic fibers.
The tissue microenvironment can be reshaped by degrading the ECM by extracellular proteases which can also cleave and activate signaling molecules such as growth factors and their receptors. ECM remodeling is a crucial process for any normal physiological tissue development and repair. On the flip side, many pathological conditions are characterized by irregular extracellular proteolysis resulting in the tissue malfunction and destruction. In particular, most of the chronic inflammatory disorders are a result of extensive extracellular proteolytic processes. Matrix metalloproteinases (MMPs) are an important family of metal ion-dependent enzymes that represent a major class of endopeptidases responsible for degradation and remodeling of ECM components. MMPs are structurally related and they are divided according to their primary substrate specificities: collagenases (MMP1, -8, -13, -18), gelatinases (MMP2, -9), stromelysins (MMP3, -7, -10, -11), elastase (MMP12), membrane types (MMP14, -15, -16, -17, -24, -25), and others (MMP19, -20, -23, -26, -27, -28) based on their putative substrate specificity and internal homologies.10 Various epithelial and stromal cells, such as T cells, neutrophils, mesenchymal cells, macrophages, monocytes, neutrophils, myofibroblasts, keratinocytes, etc., secrete MMPs as latent, inactive zymogens. Activation of the MMPs usually occurs in the extracellular space. The ECM proteolysis by MMPs results in pericellular space into which cells can migrate, produce specific fragments of ECM proteins with independent biological activity, regulate tissue architecture, activate, deactivate, and modify the activity of signaling molecules.11 The destruction of the ECM not only plays a crucial role in pancreatic diseases but also has been seen in disease progression and severity in other complex disorders, such as atherosclerosis, cardiomyopathies, rheumatoid arthritis, kidney disorders, brain stroke, skin disorders, pulmonary disorders, different types of cancers, etc.12
Pancreatitis is inflammation of the pancreas which is a serious condition that establishes in either acute or chronic form. Acute pancreatitis (AP) is a short-term inflammation of the pancreas where the normal function is usually resolved when the primary causes or factors are eliminated, whereas chronic pancreatitis (CP) gradually develops over time, resulting in permanent pancreatic damage.13 In modern paradigms of pathogenesis, AP is a necessary first step to developing into CP. It is most accurate to think that AP which is a distinguishable event from that of CP are two distinct entities of the same pole.14 Fibroinflammatory changes to the pancreatic tissue are a characteristic feature of pancreatitis, which may develop in association with alcohol abuse, smoking, gene mutations, autoimmune syndromes, metabolic disturbances, environmental conditions, and anatomical abnormalities. The regenerative response of the damaged pancreas is assumed to be determined by a balance between newly synthesized and deposited ECM and degradation of ECM. The major contributor to the pathogenesis of pancreatitis, especially CP is pancreatic fibrosis which is the deposition of excess connective tissue. The role of MMPs in fibrosis is both inhibitory and stimulating, as some MMPs act to degrade ECM and others aid in the accumulation of ECM in fibrosis. The end result would be the accumulation of ECM in fibrosis, which would be, in part, a condition of deficient proteolysis which would differ from organ to organ because of different composition of ECM.15 The present review aims at highlighting the importance of matrix metalloproteinases in acute and chronic pancreatitis.
Origin of pancreatitis
What do we really know about the cause of pancreatitis? First, an injury to the pancreatic acinar, ductal, or mesenchymal cells in a person causes a normal pancreas to progressively deteriorate into an end-stage, sclerotic pancreatic remnant over some period of time. Which of the pancreatic cells are affected depends on the type of etiological factor. The change of resident fibroblasts/PSCs into myofibroblasts is caused by the damage to any one of these tissue compartments of the pancreas, which is associated with subsequent production and deposition of ECM, mainly consisting of collagen types I and III and fibronectin.16
Alcoholism tops the list of different etiological factors causing pancreatitis (both AP and CP) in developed countries17–21 but several studies suggest that alcohol per se may not cause pancreatitis. There is a marked heterogeneity in susceptibility to alcoholic CP (ACP) and clinically significant pancreatic disease occurs only in 10% of heavy alcohol users.22,23 Ethanol feeding to most animal species (in particular, rats and mice) even for a long time, with either liquid diet or continuous intragastric infusion24,25 does not result in a noticeable injury to the pancreas.25–27 Although alcohol consumption and tobacco smoking are clearly associated with CP, the likelihood of an average person developing pancreatitis after exposure to either factor is very low. Therefore, it can be conclusive to say that alcohol is a cofactor in the development of ACP in susceptible humans.19,21–23 Therefore other genetic or environmental factors may contribute to the development of alcoholic pancreatitis.
Hereditary pancreatitis is a result of germ-line mutations in the cationic trypsinogen gene, also known as protease, serine1 (PRSS1) which is one of the most abundant molecules produced by pancreatic acinar cells. The most common mutation in PRSS1 results due to an arginine-to-histidine substitution, which removes the key site crucial for the rapid self-destruction of trypsin in solutions. Therefore, trypsin becomes resistant to inactivation, and the abnormally active trypsin in the acinar cells results in the progression of AP and its relapse can lead to CP.28 The serine protease inhibitor Kazal type 1 (SPINK1; also known as pancreatic secretory trypsin inhibitor, PSTI), as the name suggests, inhibits any abnormal activation of trypsin. The mutation of the SPINK1 gene results in loss of this inhibitory function which in turn escalates the risk of recurrent AP and CP.29 Therefore, the abnormal early activation of trypsinogen within the acinar cell initiates the zymogen activation cascade resulting in pancreatic autodigestion and pancreatitis. Another mechanism by which bicarbonate secretion in the pancreas may be impaired is a defect in the cystic fibrosis gene product. Cystic fibrosis is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. CFTR is a key molecule expressed in the pancreatic ducts in the pathway of bicarbonate secretion wherein mutations in CFTR gene may impair this secretion in the ducts which may lead to obstruction resulting in recurrent AP and CP.30 Ductal obstruction is the primary event and another etiological factor causing idiopathic CP and cystic fibrosis. Protein precipitation due to abnormal bicarbonate secretion leads to plugs within the interlobular and intralobular ducts and are one of the earliest findings in pancreatitis. If not the initiating event, protein plugs at least perpetuate inflammation of the gland through repeated obstruction of the pancreatic duct system. Plugs are also the nidus for calcification.31 Their clinical importance is highlighted by the fact that endoscopic removal of plugs transiently improves the pain in patients with CP.32
A variety of causes can be related to pancreatic inflammation in AP and CP, such as chronic ethanol consumption, tobacco smoking, duct obstruction, mutations in genes namely trypsinogen, SPINK, CFTR, chymotrypsin C, and other genes. If any of the various etiological factors stimuli are sufficiently strong, an inflammatory response results in loss of acinar cells and activation and recruitment of PSCs resulting in the formation of histological CP. Several reiterations of this cycle lead to full-blown clinical CP.27
Depending upon the morphological characteristics, like the pattern of fibrosis and the composition of the inflammatory infiltrate, we may try to identify or correlate the etiology. For example, the development of interlobular fibrosis or intralobular fibrosis is determined by the initial site of injury in the pancreas which in turn is strongly correlated to the etiological factor.28 Taken together, a number of mechanisms that are not mutually exclusive may lead to perturbations in acinar cell function which predispose the gland to autodigestive injury and acute necroinflammation. These changes sensitize the cell such that in the presence of an appropriate trigger/cofactor, the overt injury is initiated which in turn activates PSCs. Pancreatitis whether acute or chronic is an individualized disease that although driven by the same etiology, may progress rapidly in one patient while developing slowly and being clinically insignificant in another patient, presenting the complexities involved in the disease.
PSCs, MMPs, and pancreatitis
PSCs are resident cells of the pancreas, located at the basolateral part of acinar cells. The steps involved in pancreatic damage, activation of PSCs, and progression to pancreatic fibrosis have been termed as a necroinflammatory response. There are three basic mechanisms by which acinar injury might activate PSCs: mechanical stress, cytokine production, and matrix changes. The position of PSCs suggests that they will be predisposed to mechanical stress if there is any injury to acinar cells like swelling or deformity of the acinus. Biochemical response to stretch (mechanotransduction) is a widely distributed cellular phenomenon33 and has been described in skeletal, vascular, hepatic, and pulmonary tissue. Stretch modifies transforming growth factor beta (TGF-β) production in hepatic stellate cells34 and PSCs have been shown to trigger and proliferate in response to a change in pressure in vitro.35 PSCs in vitro are activated by a varied range of proinflammatory cytokines including TGF-β, platelet-derived growth factor (PDGF), tumor necrosis factor alpha (TNF-α), interleukin 1, and interleukin 6. Injured acinar cells, therefore, have the capacity to activate PSCs both directly and indirectly, via inflammatory cell recruitment. It is interesting to find that acinar cells in CP (but not in the normal pancreas) express death domain receptors for TNF-related apoptosis-inducing ligand (TRAIL) and activated PSCs express TRAIL. So it can be hypothesized that TRAIL binds to the receptors on acinar cells in the process of inflammation and thus induces them to apoptosis, thereby reducing their number in areas of pancreatic fibrosis. Conversely, PSCs also express the death domain receptor P75, the ligand for which, nerve growth factor, is expressed in acinar cells in CP. There is, therefore, evidence of extensive crosstalk between the injured acinar cells and activated PSCs.36
PSCs are sensitive to subtle matrix changes. The release of digestive enzymes due to the disruption of acinar cells disturbs the normal matrix as a result of exposure of collagen fibers, which effectively triggers the activation of PSCs. They assume a myofibroblast-like appearance in response to stimulation by a plethora of inflammatory infiltrates, such as pro- and anti-inflammatory cytokines, growth factors (TGF-β and PDGF), angiotensin II, and reactive oxygen species, which are released by damaged acinar and ductal cells and recruited leukocytes.37 Further, activated PSCs in turn produce autocrine factors, such as TGF-β, PDGF, TRAIL, and proinflammatory molecules (e.g., cyclooxygenase2).38 Thus PSCs are adept at performing several biological functions, such as proliferation, synthesis, migration, and secretion of ECM, as well as synthesis and secretion of matrix-degrading enzymes and their inhibitors.
PSCs are particularly important in the remodeling of the ECM as they are not only involved in the synthesis of ECM but also in the degradation of its components as well, thus, implying their key role in the sustenance of the normal architecture of the healthy pancreas.39 PSCs secrete different types of MMPs, which aid in ECM degradation.40 Especially, MMP1, MMP2, MMP3, MMP9, and MMP13 are shown to be secreted by PSCs.41–43 MMPs have different substrate specificities; MMP2 and MMP9 primarily degrade gelatin (type IV collagen), the main collagen present in the basement membrane, whereas MMP1 and MMP13 degrade type-1 collagen. The secretion of MMP2 is far greater than MMP13 when induced by ethanol and acetaldehyde. PSCs have also been shown to secrete tissue inhibitors of metalloproteinases (TIMPs) that inhibit the activity of MMPs. MMP2 in the pancreas is primarily inhibited by TIMP2. However, during inflammation the increase in TIMP2 was considerably less than the increase in MMP2, resulting in the net increase in MMP2 activity.43 Thus, the amassing fibrotic type 1 collagen in pancreatitis could be due to these changes in MMPs secretion and inhibition by PSCs.
Types of MMPs
All MMPs have related modular architecture having a prodomain which is involved in latency, a catalytic domain important for proteolytic activity, a linker or hinge region which helps in better flexibility of catalytic region, and a C-terminal hemopexin-like domain which helps in further interactions of MMPs with molecules like fibronectin.44 Different MMPs vary structurally in small variations in these regions resulting in distinct individual functions, according to which they are categorized into collagenases (MMP1, MMP8, MMP13, MMP18), gelatinases (MMP2, MMP9), stromelysins (MMP3, MMP10, MMP11), matrilysins (MMP7, MMP26), membrane type MMPs (MMP14, MMP15, MMP16, MMP17, MMP24, MMP25), and others (MMP12, MMP19, MMP20, MMP21, MMP23, MMP27, MMP28). These MMPs are naturally inhibited by the TIMPs.45 Although collagenases and stromelysins have similar domain arrangements, stromelysins do not cleave interstitial collagens. Matrilysin lacks the linker and hemopexin-like domain. Furthermore, gelatinases possess an additional domain with homology similar to that of the gelatin-binding domain of fibronectin and membrane type-matrix metalloproteinases (MT-MMPs) have a furin recognition sequence at the C-terminus of the pro-peptide.46 Although the primary structures of MMPs regions have little homology among the families, the general protein folds are analogous.
Collagenases (MMP1, MMP8, MMP13, MMP18) in pancreatitis
The major component of ECM in interstitial tissue is fibrillar type I collagen. The major content of connective tissue is type I collagen, the archetypal fibrillar collagen which constitutes ~90% of the protein. Collagenases are characterized for their abilities to cleave triple-helical regions of interstitial collagens (types I, II, and III) at a site approximately three-fourths away from the N-terminus. Four MMPs belong to this subgroup: MMP1 (interstitial collagenase/collagenase I), MMP8 (neutrophil collagenase/collagenase II), MMP13 (collagenase III), and MMP18 (collagenase IV).44
MMP1, degrades types I and III collagens, and is one of the vital enzymes in fibrolysis, a process closely linked to tissue remodeling. In a study by Tang et al in rat pancreas, MMP1 positive fibroblasts were observed in the connective tissues surrounding interlobular ducts, but few of the positive cells could be observed elsewhere in connective tissues.43 The pancreatic acini and islets are believed to originate from common ductal precursors. The cells around ducts with MMP1 positive staining indicated that they could release MMP and dissolve ECM. Therefore, they may play a central role in the development and differentiation of pancreatic acini. Additionally, the expression of MMP1 was found in some acinar cells of the exocrine gland, especially in those of the peripheral areas of pancreatic lobules. In view of the MMP1 expressing cells localizing in the peripheral area pancreatic lobules, it could be inferred that the MMP1 expression in these cells is related to proliferation and remodeling of the pancreas. During these processes, the acinar cells may dissolve the fibers of ECM through synthesis and release of MMP1, thereby promoting acinar proliferation and remodeling of pancreatic lobules (Figure 1).47
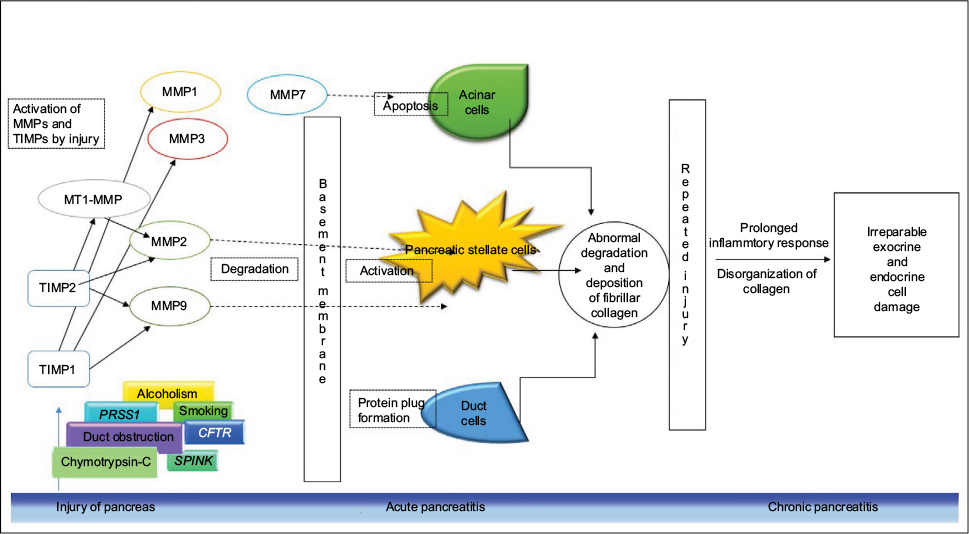 | Figure 1 Matrix metalloproteinases in pancreatitis. Abbreviations: CFTR, cystic fibrosis transmembrane conductance regulator; MMP, matrix metalloproteinase; MT1-MMP, membrane type-1 matrix metalloproteinase; PRSS1, protease, serine1; TIMPs, tissue inhibitors of metalloproteinases; SPINK, serine protease inhibitor Kazal. |
Tasaki et al investigated MMP1 secretion from human pancreatic periacinar myofibroblasts in response to proinflammatory cytokines IL-1β and TNF-α and found to be regulated by the proinflammatory cytokines via the mitogen-activated protein/extracellular signal-regulated kinase cascade.48 A study by Li et al on the effect of pancreatic-associated protein on human PSCs showed an inhibitory effect of MMP1.49 In an experimental rat model of AP study by Müller-Pillasch et al, the level of MMP1 remained constant before and after induction of AP.50 A study by Nakae et al found a significant association of MMP1 levels in AP. Higher levels of MMP1 were found in patients with more severity.51 However, our studies in CP patients revealed not only significantly increased levels of MMP1, but also a higher expressive 2G genotype at position −1607 of the MMP1 gene promoter.52
A study by Giachino et al revealed high circulating levels of MMP8 in very few patients with pancreatitis.53 Another study by Phillips et al showed MMP13 expression in cultured PSCs in the medium containing TNF-α and ethanol. MMP13 is known to degrade fibrillar collagen.54
The available information concerning the role of these collagenases MMP1, MMP8, and MMP13 in pancreatitis is, however, limited and would require further studies to understand their functions in the disease pathogenesis.
Stromelysins (MMP3, MMP10, MMP11) in pancreatitis
MMP3 (stromelysin 1), MMP10 (stromelysin 2), and MMP11 (stromelysin 3) are assigned to this subgroup which degrades proteoglycans and glycoproteins. MMP3 and MMP10 have a domain organization analogous to that of collagenases, but they do not cleave fibrillar collagens. Both enzymes are secreted from the cell as inactive zymogens and participate in proMMP activation and digestion of a number of ECM molecules. MMP11 has a very weak activity towards ECM molecules.55
An increased MMP3 level was found in the earlier stages of the disease in a rat model of cerulein-induced pancreatitis compared to later stage onset in a study by Gress et al, in the later stages, it was found that MMP3 levels were decreased.56 Treiber et al showed MMP10 as one potential protective factor attenuating fibrosis on the transcriptional and posttranscriptional level in CP by using microarray gene expression analysis.57 Yet further analyses are needed to identify the exact role of MMP3 and MMP10 in pancreatic fibrogenesis.
Matrilysins (MMP7, MMP26) in pancreatitis
The name matrilysin denotes lysis of (ECM) matrix. Matrilysin-1 enzyme is the smallest member of the MMP family that is encoded by MMP7 gene (matrilysin). Previous studies in CP highlighted the role of MMP7 based on expression data and immunohistochemical analyses.11 MMP7 is exclusively expressed in the metaplastic ductal epithelium of CP patients and has been shown to control acinar cell apoptosis (Figure 1) through proteolytic release of its pro-apoptotic molecule Fas ligand.58 Sires et al have reported that MMP7 efficiently cleaves the basement membrane protein entactin, which bridges laminin and collagen type IV, thereby proposing the disruption of basement membranes by inflammatory cells by the potentially imperative MMP7 during inflammation.59 Crawford et al tested the effects of MMP7 in a mouse model of human CP and suggested that CP phenotype requires MMP7 for the transition to pancreatic ductal adenocarcinoma.58 In a preliminary study, we did not find any significant difference in the serum levels of MMP7 in CP patients from that of controls, though we found an association with the higher expressive GG genotype of the −181 (A/G) promoter polymorphism of MMP7.60 No studies were found which reported empirical data regarding MMP26/Matrilysin-2 to be associated with AP or CP.
Gelatinases (MMP2, MMP9) in pancreatitis
MMP2 (gelatinase A) and MMP9 (gelatinase B) belong to this subgroup. MMP2 (72 kDa type IV collagenase) is known to cleave type IV collagen and fibronectin as well as types V, VII, and XI collagen, and it also degrades gelatin and laminin. MMP2 is well-documented in most of the studies till now in pancreatitis. The prime MMP secreted by PSCs is latent MMP2 which may be profibrogenic in two ways. First, increased MMP2 would result in abnormal degradation of normal basement membrane collagen (type IV) which in turn would enable the deposition of pathological fibrillar collagen in the acinar gland (Figure 1). Second, at the site of injury MMP2 may activate quiescent PSCs leading to abnormal collagen synthesis.41,54 Trypsin which is secreted by acinar cells has been shown to be an efficient activator of most proMMPs in vitro.61,62
In an experimental model of acute necrotizing pancreatitis using arginine in rats, Kihara et al observed that MMP2 mRNA was deposited around disrupted acinar cells and interstitial spaces during regeneration of acinar cells and not observed in the regenerated acinar cells after healing, which was followed by the disappearance of fibronectin. MMP1 mRNA expression did not change throughout the regeneration period.63 An immunolocalization study by Ishihara et al in surgical specimens of the fibrotic pancreas from patients with CP showed that MMP2 decomposed type IV collagen of the basement membrane of the pancreatic ducts in CP. Release and activation of MMP2 may result in degradation of the normal basement membrane. However, this may simply reflect a response to injury as a phase of regeneration and repair process, as cells responding to injury may need to degrade the ECM to migrate. In CP, the expression of MMP2 in ductal epithelia may also reflect the response of injury. However, repeated injury and prolonged inflammatory response may lead to aggravation of the injuries of pancreatic ducts in CP. Histologically, the basement membrane of the pancreas exists between the interstitium and the duct, is mainly composed of type IV collagen and laminin. Any abnormal disturbances in the basement membrane result in increased permeability, which stimulates diffusion of active enzymes into the interstitial spaces and diffusion of calcium into the ducts. As a result of which protein plugs are formed within pancreatic ducts which are eventually calcified, thereby obstructing the pancreatic ducts leading to periductal inflammation and fibrosis.64
In another study by Choi et al, the intensity of MMP2 staining tended to be lower in the pancreatic ductal epithelial cells in autoimmune pancreatitis (AIP) than in ACP, which may lead to the relatively preserved collagen type IV of the basement membrane in AIP, which may lead to regression of pancreatic fibrosis after steroid therapy in an earlier stage of the disease.65 The results of a study by Yamaguchi et al suggest that long-term increase of gelatinolytic activity of active MMP2 cause continuous disorganization of type IV collagen in basement membrane during progressive atrophy of the pancreatic gland in oleic acid-induced pancreatitis, and that membrane type 1 (MT1)-MMP and TIMP2 act as activators during proMMP2 activation.66
MMP9 (92 kDa type IV collagenase) is also known to cleave Col-IV. The study by Chen et al found that serum MMP9 levels were significantly increased in patients with severe AP when compared to those with mild AP and control subjects, immediately after admission to the hospital. The MMP9 levels decreased after 2 days after admission.67 In the study by Ishihara et al in CP model, MMP9 seemed to be less involved in the degradation of Col-IV and laminin than MMP2.64
Serine proteases such as chymase and trypsin stimulate both MMP2 and MMP9 in vivo thereby revealing a biological significance. In mice with AP, trypsin induced the activation of proMMP2 and proMMP9. ProMMP9, when activated by endogenous trypsin, was reported to be a permissive factor for insulin degradation and diabetes.68 Keck et al have revealed that MMP9 levels correlated with the severity of AP.69 The elevated MMP9 may reflect a neutrophilic inflammatory response in the pancreas, as the major source of MMP9 are neutrophils.70
MT-MMPs (MMP14, MMP15, MMP16, and MMP24) in pancreatitis
MT-MMPs are membrane associated proteases that majorly result in recruitment of receptors to the site of injury. There are six members that belong to this subgroup. Four of them (MMP14, MMP15, MMP16, and MMP24) are type I transmembrane proteins whereas the other two are glycosylphosphatidylinositol-anchored proteins (MMP17 and MMP25). Among them, MT1-MMP (MMP14) is the most extensively studied and plays a key role in cell migration and matrix invasion and angiogenesis. MT1 and TIMP2 work as activating factors during proMMP2 activation (Figure 1), which was shown in a study by Yamaguchi et al in oleic acid-induced pancreatitis in rats. The formation of trimolecular complex, MT1-MMP/TIMP2/proMMP2, was important for cell-mediated activation of proMMP2,66 thereby suggesting a unique role of regulation of MMP2 in cell–matrix interactions.
TIMPs in pancreatitis
Although TIMPs are mainly characterized as inhibitors of MMPs, they are also known to have a wide range of activities. TIMP1 is a 28.5 kDa glycoprotein that binds with all activated MMPs and with the proenzyme form of MMP9.71,72 TIMP2 is a 21 kDa protein consisting of 194 amino acids and it binds to proMMP2 and proMMP9, to active MMP2. TIMP2 is considered to play a key role in controlling the activity of MMP2 in pancreatitis than does TIMP1. Both TIMPI and TIMP2 bind stoichiometrically with the activated form of the MMPs in a 1:1 ratio and any small changes in their level may result in major biological change in proteolytic activity. TIMPI binds to the activated forms of MMP1, MMP3, and MMP9 whereas TIMP2 binds to activated proMMP2 (Figure 1).73,74 Bramhall et al tested the immune reactivity of acinar and duct epithelial cells for MMP2, MMP3, and TIMP1 in CP sections and showed MMP2 and MMP3 to be moderately strong in three out of five sections, whereas for TIMP1 the epithelial staining was significant in only one out of five.75 TIMP2 was evident in only PSC secretions at the protein level. Ethanol and acetaldehyde also significantly increased the production of TIMP2 by PSCs as seen by Phillips et al.54
Conclusion
Many studies have demonstrated that MMP-dependent functions are not only limited to ECM turnover during pancreatic fibrosis but can also stimulate the activation of PSCs, which in turn results in increased inflammation by activating multiple cascades of other ECM molecules. Thus understanding these complex pathways of MMPs in pancreatitis paves the way to comprehend and analyze the quantitative variation of MMPs with risk genotype identification in the predisposition of the disease. This also helps in the correlation of genotypes to the signals transduced in the regulation of apoptotic and immune mechanisms. MMPs may thus help in the resolution of pancreatic fibrosis similar to that found in liver, thus there is a potential importance in understanding the role of MMPs in AP and CP.
Acknowledgments
The authors acknowledge financial support from University Grants Commission (UGC), New Delhi. Authors are also thankful to the Department of Biotechnology, New Delhi for providing the laboratory facilities. Funding from Council of Scientific and Industrial Research (CSIR), India, is highly acknowledged (grant number: 09/132 (0828)/2014-EMR-I).
Disclosure
The authors report no conflicts of interest in this work.
References
Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123(24):4195–4200. | ||
Venkatasubramanian PN. Imaging the pancreatic ECM. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment. Trivandrum, India: Transworld Research Network; 2012. Chapter 2. | ||
Sengupta N, MacDonald TT. The role of matrix metalloproteinases in stromal/epithelial interactions in the gut. Physiology (Bethesda). 2007;22:401–409. | ||
Jarvelainen H, Sainio A, Koulu M, Wight TN, Penttinen R. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacol Rev. 2009;61(2):198–223. | ||
Schaefer L, Schaefer RM. Proteoglycans: from structural compounds to signaling molecules. Cell Tissue Res. 2010;339(1):237–246. | ||
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. 4th ed. London, England: Garland; 2002. | ||
Apte MV, Haber PS, Applegate TL, et al. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43(1):128–133. | ||
Bachem MG, Schneider E, Gross H, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115(2):421–432. | ||
Yuen A, Díaz B. The impact of hypoxia in pancreatic cancer invasion and metastasis. Hypoxia. 2014;2:91–106. | ||
Zhang J, Jin X, Fang S, et al. The functional polymorphism in the matrix metalloproteinase-7 promoter increases susceptibility to eosophageal squamous cell carcinoma, gastric cardiac adenocarcinoma and non-small cell lung carcinoma. Carcinogenesis. 2005;26(10):1748–1753. | ||
Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. | ||
Nagase H, Woessner JF Jr. Matrix metalloproteinases. J Biol Chem. 1999;274:21491–21494. | ||
Banks PA, Conwell DL, Toskes PP. The management of acute and chronic pancreatitis. Gastroenterol Hepatol. 2010;6(2 Suppl 5):1–16. | ||
Fosmark CE. Chronic pancreatitis. In: Feldman M, Friedman LS, Brandt LJ, editors. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. 10th ed. Philadelphia, PA: Elsevier Saunders; 2016. Chapter 59. | ||
Giannandrea M, Parks WC. Diverse functions of matrix metalloproteinases during fibrosis. Dis Model Mech. 2014;7(2):193–203. | ||
Glasbrenner B, Dominguez-Muñoz JE, Nelson DK, Riepl RL, Büchler M, Malfertheiner P. Relationship between postprandial release of CCK and PP in health and in CP. Regul Pept. 1994;50(1):45–52. | ||
Etemad B, Whitcomb DC. Chronic pancreatitis: diagnosis, classification, and new genetic developments. Gastroenterology. 2001;120(3):682–707. | ||
Apte MV, Wilson JS. Alcohol-induced pancreatic injury. Best Pract Res Clin Gastroenterol. 2003;17(4):593–612. | ||
Oruc N, Whitcomb DC. Theories, mechanisms, and models of alcoholic chronic pancreatitis. Gastroenterol Clin North Am. 2004;33(4):733–750. | ||
Schenker S, Montalvo R. Alcohol and the pancreas. Recent Dev Alcohol. 1998;14:41–65. | ||
Whitcomb DC. Hereditary pancreatitis: new insights into acute and chronic pancreatitis. Gut. 1999;45(3):317–322. | ||
Lieber CS, DeCarli LM. The feeding of ethanol in liquid diets. Alcohol Clin Exp Res. 1986;10(5):550–553. | ||
Tsukamoto H, Towner SJ, Yu GS, French SW. Potentiation of ethanol induced pancreatic injury by dietary fat. Induction of chronic pancreatitis by alcohol in rats. Am J Pathol. 1988;131(2):246–257. | ||
Kono H, Nakagami M, Rusyn I, et al. Development of an animal model of chronic alcohol-induced pancreatitis in the rat. Am J Physiol Gastrointest Liver Physiol. 2001;280(6):G1178–G1186. | ||
Schneider A, Whitcomb DC, Singer MV. Animal models in alcoholic pancreatitis—what can we learn? Pancreatology. 2002;2(3):189–203. | ||
Witt H, Apte MV, Keim V, Wilson JS. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology. 2007;132(4):1557–1573. | ||
Klöppel G, Detlefsen S, Feyerabend B. Fibrosis of the pancreas: the initial tissue damage and the resulting pattern. Virchows Arch. 2004;445(1):1–8. | ||
Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14(2):141–145. | ||
Witt H, Luck W, Hennies HC, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25(2):213–216. | ||
Sharer N, Schwarz M, Malone G, Howarth A, Painter J, Super M, Braganza J. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med. 1998;339(10):645–652. | ||
Tsurumi T, Fujii Y, Takeda M, Tanaka J, Harada H, Oka H. A case of chronic pancreatitis successfully treated by endoscopic removal of protein plugs. Acta Med Okayama. 1984;38(2):169–174. | ||
Harada H, Miyake H, Miki H, Kobayashi T, Sasaki T, Kimura I. Role of endoscopic elimination of protein plugs in the treatment of chronic pancreatitis. Gastroenterol Jpn. 1982;17(5):463–468. | ||
Han B, Bai XH, Lodyga M, et al. Conversion of mechanical force into biochemical signaling. J Biol Chem. 2004;279(52):54793–54801. | ||
Sakata R, Ueno T, Nakamura T, Ueno H, Sata M. Mechanical stretch induces TGF-beta synthesis in hepatic stellate cells. Eur J Clin Invest. 2004;34(2):129–136. | ||
Watanabe S, Nagashio Y, Asaumi H, et al. Pressure activates rat pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2004;287(6):G1175–G1181. | ||
Hasel C, Dürr S, Rau B, et al. In chronic pancreatitis, widespread emergence of TRAIL receptors in epithelia coincides with neoexpression of TRAIL by pancreatic stellate cells of early fibrotic areas. Lab Invest. 2003;83(6):825–836. | ||
Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest. 2007;117(1):50–59. | ||
Stevens T, Conwell DL, Zuccaro G. Pathogenesis of chronic pancreatitis: an evidence-based review of past theories and recent developments. Am J Gastroenterol. 2004;99(11):2256–2270. | ||
Bhanot UK, Möller P. Mechanisms of parenchymal injury and signaling pathways in ectatic ducts of chronic pancreatitis: implications for pancreatic carcinogenesis. Lab Invest. 2009;89(5):489–497. | ||
Phillips PA, Wu MJ, Kumar RK, et al. Cell migration: a novel aspect of pancreatic stellate cell biology. Gut. 2003;52(5):677–682. | ||
Shek FW, Benyon RC, Walker FM, et al. Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am J Pathol. 2002;160(5):1787–1798. | ||
Yokota T, Denham W, Murayama K, Pelham C, Joehl R, Bell RH Jr. Pancreatic stellate cell activation and MMP production in experimental pancreatic fibrosis. J Surg Res. 2002:104(2):106–111. | ||
Tang L, Liu S, Wang F, Liu Z, Xu Y, Wang X, Li Z. Observation on in situ hybridization and immunocytochemistry of matrix metalloproteinases in rat pancreas. J Huazhong Univ Sci Technolog Med Sci. 2003;23(4):332–334. | ||
Bode W. A helping hand for collagenases: the haemopexin-like domain. Structure. 1995;3(6):527–530. | ||
Nagase, Hideaki. Substrate specificity of MMPs. In: Neil JC, Krzysztof A, editors. Matrix Metalloproteinase Inhibitors in Cancer Therapy. New Jersey: Humana Press; 2001:39–66. | ||
Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69(3):562–573. | ||
Murphy GJ, Murphy G, Reynolds JJ. The origin of matrix metalloproteinases and their familial relationships. FEBS Lett. 1991;289(1):4–7. | ||
Tasaki K, Shintani Y, Saotome T, Andoh A, Fujiyama Y, Hozawa S, Bamba T. Pro-inflammatory cytokine-induced matrix metalloproteinase-1 (MMP-1) secretion in human pancreatic periacinar myofibroblasts. Pancreatology. 2003;3(5):414–421. | ||
Li L, Bachem MG, Zhou S, et al. Pancreatitis-associated protein inhibits human pancreatic stellate cell MMP-1 and -2, TIMP-1 and -2 secretion and RECK expression. Pancreatology. 2009;9(1–2):99–110. | ||
Müller-Pillasch F, Gress TM, Yamaguchi H, Geng M, Adler G, Menke A. The influence of transforming growth factor beta 1 on the expression of genes coding for matrix metalloproteinases and tissue inhibitors of metalloproteinases during regeneration from cerulein-induced pancreatitis. Pancreas. 1997;15(2):168–175. | ||
Nakae H, Endo S, Inoue Y, Fujino Y, Wakabayashi G, Inada K, Sato S. Matrix metalloproteinase-1 and cytokines in patients with acute pancreatitis. Pancreas. 2003;26(2):134–138. | ||
Sri Manjari K, Nallari P, Balakrishna N, Vidyasagar A, Prabhakar B, Jyothy A, Venkateshwari A. Influence of matrix metalloproteinase-1 gene -1607 (1G/2G) (rs1799750) promoter polymorphism on circulating levels of MMP-1 in chronic pancreatitis. Biochem Genet. 2013;51(7–8):644–654. | ||
Giachino F, Loiacono M, Lucchiari M, et al. Rule out of acute aortic dissection with plasma matrix metalloproteinase 8 in the emergency department. Critical Care. 2013;17(1):R33. | ||
Phillips PA, McCarroll JA, Park S, et al. Rat pancreatic stellate cells secrete matrix metalloproteinases: implications for extracellular matrix turnover. Gut. 2003;52(2):275–282. | ||
Nagase H, Visse R. Matrix metalloproteinases: an overview. In: Supuran T, editor. Drug Design of Zinc-Enzyme Inhibitors. Hoboken, NJ: John Wiley & Sons, Inc.; 2009:487–519. | ||
Gress TM, Menke A, Bachem M, et al. Extracellular matrix and pancreatic diseases. Digestion. 1998;59(6):625–637. | ||
Treiber M, Neuhöfer P, Anetsberger E, et al. Myeloid, but not pancreatic, RelA/p65 is required for fibrosis in a mouse model of chronic pancreatitis. Gastroenterology. 2011;141(4):1473–1485. | ||
Crawford HC, Scoggins CR, Washington MK, Matrisian LM, Leach SD. Matrix metalloproteinase 7 is expressed by pancreatic cancer precursors and regulates acinar-to-ductal metaplasia in exocrine pancreas. J Clin Invest. 2002;109(11):1437–1444. | ||
Sires UI, Griffin GL, Broekelmann TJ, et al. Degradation of entactin by matrix metalloproteinases. Susceptibility to matrilysin and identification of cleavage sites. J Biol Chem. 1993;268(3):2069–2074. | ||
Manjari KS, Jyothy A, Kumar PS, Prabhakar B, Nallari P, Venkateshwari A. Association of matrix metalloproteinase-7 (−181A/G) promoter polymorphism in chronic pancreatitis. Indian J Med Res. 2014;140(5):609–615. | ||
Woessner JF Jr, Nagase H. Matrix Metlloprotienases and TIMPS. New York, NY: Oxford University Press; 2000. | ||
Okada Y, Morodomi T, Enghild JJ, et al. Matrix metalloproteinase 2 from human rheumatoid synovial fibroblasts. Purification and activation of the precursor and enzymic properties. Eur J Biochem. 1990;194(3):721–730. | ||
Kihara Y, Tashiro M, Nakamura H, Yamaguchi T, Yoshikawa H, Otsuki M. Role of TGF-β1, extracellular matrix, and matrix metalloproteinase in the healing process of the pancreas after induction of acute necrotizing pancreatitis using arginine in rats. Pancreas. 2001;23(3):288–295. | ||
Ishihara T, Hayasaka A, Yamaguchi T, Kondo F, Saisho H. Immunohistochemical study of transforming growth factor-beta 1, matrix metalloproteinase-2,9, tissue inhibitors of metalloproteinase-1,2, and basement membrane components at pancreatic ducts in chronic pancreatitis. Pancreas. 1998;17(4):412–418. | ||
Choi EK, Kim MH, Jang SE et al. Differences in pancreatic immunohistochemical staining profiles of TGF-b1, MMP2, and TIMP-2 between autoimmune and alcoholic chronic pancreatitis. Pancreas. 2009;38(7):739–745. | ||
Yamaguchi T, Nakamura H, Kihara Y, Taguchi M, Yoshikawa H, Otsuki M. Long-term overexpression of membrane type-1 matrix metalloproteinase and matrix metalloproteinase-2 in oleic acid-induced pancreatitis in rats. Pancreas. 2002;24(4):348–356. | ||
Chen P, Yuan Y, Wang S, Zhan L, Xu J. Serum matrix metalloproteinase 9 as a marker for the assessment of severe acute pancreatitis. Tohoku J Exp Med. 2006;208(3):261–266. | ||
Hadler-Olsen E, Fadnes B, Sylte I, Uhlin-Hansen L, Winberg JO. Regulation of matrix metalloproteinase activity in health and disease. FEBS J. 2011;278(1):28–45. | ||
Keck T, Jargon D, Klunsch A, et al. MMP-9 in serum correlates with the development of pulmonary complications in experimental acute pancreatitis. Pancreatology. 2006;6(4):316–322. | ||
Shiomi T, Lemaître V, D’Armiento J, Okada Y. Matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs in non-neoplastic diseases. Pathol Int. 2010;60(7):477–496. | ||
Welgus HG, Stricklin GP. Human skin fibroblast collagenase inhibitor. J Biol Chem. 1983;258(20):12259–12264. | ||
Welgus HG, Jeffrey JJ, Eisen AZ, Roswit WT, Stricklin GP. Human skin fibroblast collagenase: interaction with substrate and inhibitor. Coll Relat Res. 1985;5(2):167–179. | ||
Wilhelm SM, Collier IE, Marmer BL, Eisen AZ, Grant GA, Goldberg GI. SV40 –transformed human lung fibroblasts secrete a 92-kDa type IV collagenase which is identical to that secreted by normal human macrophages. J Biol Chem. 1989;264(29):17213–17221. | ||
Stetler-Stevenson WG, Brown PD, Onisto M, Levy AT, Liotta LA. Tissue inhibitor of metalloproteinases-2 (TIMP-2) mRNA expression in tumor cell lines and human tumor tissues. J Biol Chem. 1990;265(23):13933–13938. | ||
Bramhall SR, Stamp GW, Dunn J, Lemoine NR, Neoptolemos JP. Expression of collagenase (MMP2), stromelysin (MMP3) and tissue inhibitor of the metalloproteinases (TIMP1) in pancreatic and ampullary disease. Br J Cancer. 1996;73(8):972–978. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.