Back to Archived Journals » Metalloproteinases In Medicine » Volume 3
Matrix metalloproteinases in cystic fibrosis: pathophysiologic and therapeutic perspectives
Authors Rojas-Quintero J, Owen C
Received 19 April 2016
Accepted for publication 21 June 2016
Published 22 August 2016 Volume 2016:3 Pages 49—62
DOI https://doi.org/10.2147/MNM.S96916
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yoshifumi Itoh
Joselyn Rojas-Quintero,1,2 Caroline A Owen1-3
1Division of Pulmonary and Critical Care Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, 2Harvard Medical School, Boston, MA, 3The Lovelace Respiratory Research Institute, Albuquerque, NM, USA
Abstract: Cystic fibrosis (CF) is the most common inherited disease in the Caucasian population. CF is caused by loss-of-function mutations in the CFTR gene leading to an altered electrolyte and water movement into the airway surface liquid, dehydration of the mucus layer, defective mucociliary clearance, and increased susceptibility to airway bacterial infections. CF is characterized by airway neutrophilic inflammation and activation of airway epithelial cells leading to increased airway levels of various matrix metalloproteinases (MMPs). Herein, we review MMP structure, regulation, and activity, along with the literature on MMPs expression in CF and the contributions of MMPs to CF pathogenesis. Numerous studies report elevated levels of MMPs (especially MMP-8, MMP-7, and MMP-9) in blood and/or lung samples from CF patients and have correlated MMP levels with measures of lung inflammation and injury and/or clinical parameters. No study has yet investigated the contributions of MMPs to the pathogenesis of CF using MMP gene-targeted murine of CF. Based upon the known activities of MMPs in animal models of other lung diseases, we hypothesize that some MMPs (such as MMP-8 and MMP-9) may amplify airway inflammation, and promote airway injury and airway-remodeling processes, or inhibit chloride epithelial transport by inhibiting activation of the CFTR (MMP-2). Other MMPs (such as MMP-7 and MMP-12) may limit the severity of CF by activating host defense proteins to enhance pathogen clearance from the airways, directly killing pathogens that colonize the airways of CF patients and/or promoting epithelial repair. We discuss potential approaches to selectively target MMPs to develop novel therapies that limit the high morbidity and mortality associated with CF.
Keywords: tissue inhibitor of metalloproteinases, airway inflammation, airway injury, airway infection, airway repair, proteolysis
Introduction
Cystic fibrosis (CF) is one of the most important genetic diseases in the Caucasian population worldwide due to its high prevalence, and high morbidity and mortality rates.1 The current management of CF patients is challenging and includes nutritional recommendations, physical therapy, and pharmacotherapy, including antibiotics, anti-mucolytics, and corticosteroids.2 New classes of therapies that either increase the delivery of the CFTR to the cell surface (CFTR correctors) or increase the activity of CFTR molecules at the cell surface (CFTR potentiators) have recently been shown to slow the rate of decline in lung function in CF patients and been approved for use in CF patients having certain mutations.3 However, the average life expectancy of CF patients is still only ~40 years of age.1 New therapeutics are needed to limit the mortality and morbidity associated with CF.4 It is noteworthy in this respect that recent studies have linked matrix metalloproteinases (MMPs) to the pathogenesis of CF. Herein, the genetics leading to alterations in airway physiology and the clinical features of CF are described. MMPs that have been reported to be upregulated in CF airways are discussed along with the potential mechanisms by which MMPs could promote or limit the progression of CF based upon the known activities of MMPs in other lung diseases. The potential ways by which MMPs could be selectively targeted to develop new therapeutics to limit disease progression in patients with CF are also discussed.
Pathogenesis of CF
Genetics
CF is a monogenic genetic disorder that is inherited in an autosomal recessive manner.5 It is caused by loss-of-function mutations in both copies of the CFTR gene, which is located on human chromosome 7 (in 7q31.2). The CFTR protein belongs to the ATP-Binding Cassette subfamily B (member 7).6 The CFTR functions as an ion channel that controls the flux of halogens (such as chloride [Cl–] ions) and other ions from the cytoplasm to the extracellular space in the apical membrane of epithelia of organs associated with exocrine functions (including salivary and sweat glands, airway and pancreatic ducts).6 Numerous (~2,000) CFTR mutations have been described, and they are categorized into five classes based upon their effects on inducing abnormalities in synthesis of the CFTR protein, mRNA splicing, trafficking of the protein to the plasma membrane, or allosteric control of the ion channel.7 The ∆F508-CFTR is the most common mutation causing a deletion of phenylalanine at position 508 (rs113993960) and is responsible for ~70% of CF cases worldwide8 and 86.4% of cases in the US (46.5% homozygotes and 39.9% heterozygotes).6 The diversity of CFTR polymorphisms leads to variability in phenotype severity, including variability in the age of onset of symptoms, the number of organs affected, and the severity with which organs are affected.9 Most symptomatic patients inherit two defective copies of the CFTR gene, but some individuals with two copies of abnormal genes are asymptomatic.10 While most heterozygous carriers are asymptomatic, some have a mild form of the disease.11
Functional consequences of CFTR mutations
Normal airway function
The most important process altered by the loss of CFTR ion conductance is the biomechanics of the airways.5 Human airways are lined with a highly differentiated and ciliated pseudostratified columnar epithelium, which not only acts as a physical barrier to respiratory pathogens, allergens, and toxins but also actively participates in local immune responses and mucociliary clearance.12 Nonciliated cells in the airway epithelium (including goblet cells and club cells) produce mucus, CC16, and other molecules that have anti-inflammatory and protective activities.13 The airway barrier function is mediated by tight junctions that separate the basolateral and apical compartments. The apical compartment contains the airway surface liquid (ASL), which is a thin film of mucus (~30 mm in depth) in which the cilia associated with the apical epithelial cell surface beat and thereby move the mucus gel toward the mouth to clear secretions from the airways (mucociliary clearance). Mucociliary clearance is a fundamental component of the innate airway defense system.14
Abnormal ion exchange in CF
The abnormal ion exchange occurring in the ASL in CF impairs mucociliary clearance by increasing the sodium content and reducing the volume of the ASL.15 There are at least five functional ion-exchanging transport systems in the healthy airway (Figure 1) including the CFTR, the sodium/potassium ATPase pump, the sodium–potassium–chloride cotransporter, the epithelial sodium channel (ENaC), and other potassium channels.15 The chloride channel regulator, the calcium-activated chloride channel, and two chloride exchangers contribute to sodium homeostasis in the ASL.16 The accumulation of Cl– within epithelial cells allows for the electrochemical passive secretion of this ion when the CFTR is activated, followed by the movement of sodium (and associated water molecules) from the basolateral compartment into the ASL. These events provide sufficient liquid for appropriate cilia beating and mucociliary clearance. In the “high salt” hypothesis, defective Cl– movement across the CF apical membrane impairs movement of sodium from the ASL. Elevated NaCl concentrations in the ASL blunt the bactericidal capacity of airway leukocytes and epithelial cells and inhibit the activity of an endogenous, antimicrobial protein (beta defensin-1), thereby promoting bacterial colonization and infection and associated airway inflammation and injury.17 In the “low volume” hypothesis, loss of CFTR function prevents the normal inhibitory effect of the CFTR on ENaCs, allowing hyperabsorption of Na+ from the ASL, dehydration of the ASL, progressive thickening of the mucus layer, and impaired mucociliary clearance.18 There is also loss of bicarbonate secretion in CF airways resulting in the acidification of mucus19 which, in turn, induces viscosity changes, and increased propensity to bacterial colonization and infection.20
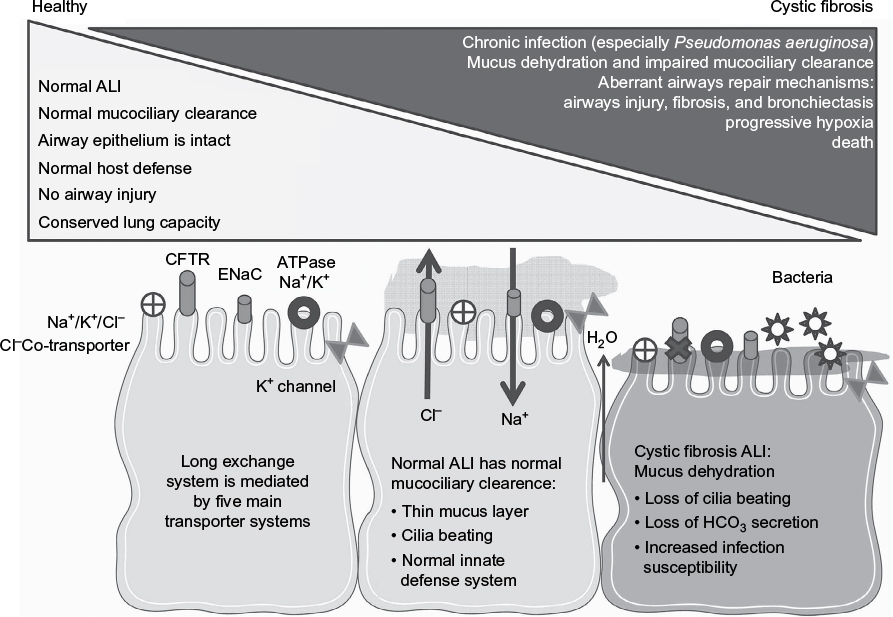 | Figure 1 The pathogenesis of cystic fibrosis. Notes: This figure illustrates the mechanisms by which defective ion exchange in the airway epithelium leads to dehydration of the mucus layer, impaired mucociliary clearance, bacterial colonization and infection, progressive airway injury, lung destruction, and respiratory failure. Abbreviations: ALI, air–liquid interface; ENaC, epithelial sodium channel; HCO3, bicarbonate. |
The two main processes that promote inflammation and injury to CF airways are airway infection (especially with Pseudomonas aeruginosa, Streptococci, Staphylococcus aureus, and anaerobes) and progressively severe airway inflammation.21 Persistent airway inflammation in CF (Figure 1) leads to airway destruction, aberrant remodeling (fibrosis), the development of bronchiectasis, loss of gas exchange units, progressive respiratory failure, and ultimately death (Figure 1).21,22 Airway inflammation in CF is predominantly neutrophilic and associated with increased airway fluid levels of polymorphonuclear neutrophil (PMN)-derived proteinases, oxidants, and proinflammatory mediators.22
Matrix metalloproteinases
The earliest proteinases linked to CF were the neutrophil-derived serine proteinases, especially neutrophil elastase (NE). More recently, MMPs have been shown to be linked to CF.23 MMPs are zinc-dependent proteinases.24,25 There are 24 human MMPs (including duplication of MMP-23) and 23 murine MMPs (Table 1).26 Collectively, MMPs can degrade all components of the extracellular matrix (ECM), and some MMPs proteolytically regulate the biologic activity of cytokines, chemokines, and growth factors, host defense molecules, survival factors, and also cell surface receptors to thereby regulate inflammation, and cell adhesion, migration, and survival.25
 | Table 1 Classification of MMPs, chromosomal localization, cellular sources, and activities Notes: MMPs are classified according to their substrate specificities. The chromosomal location of each human MMP and the murine orthologs and the key substrates known for each MMP are listed in the table. Abbreviations: Chr, chromosome; EMT, epithelial-to-mesenchymal transition; MMPs, matrix metalloproteinases; PMNs, polymorphonuclear neutrophils. |
Structure
Most MMPs have four domains including 1) an N-terminal pro-domain, which maintains pro-matrix metalloproteinases (pro-MMPs) in inactive forms, 2) a catalytic domain containing the active-site zinc atom, 3) a hinge region, and 4) a carboxy-terminal hemopexin-like domain (Figure 2).27 MMPs have a conserved sequence in the catalytic domain (HEXXHXXGXXH) in which the three histidine residues bind the active-site zinc atom, and this sequence controls the zinc-dependent proteolysis of the scissile amide carbonyl in the protein substrate.28 MMPs are classified into six main groups depending on their substrate specificity (Table 1).25
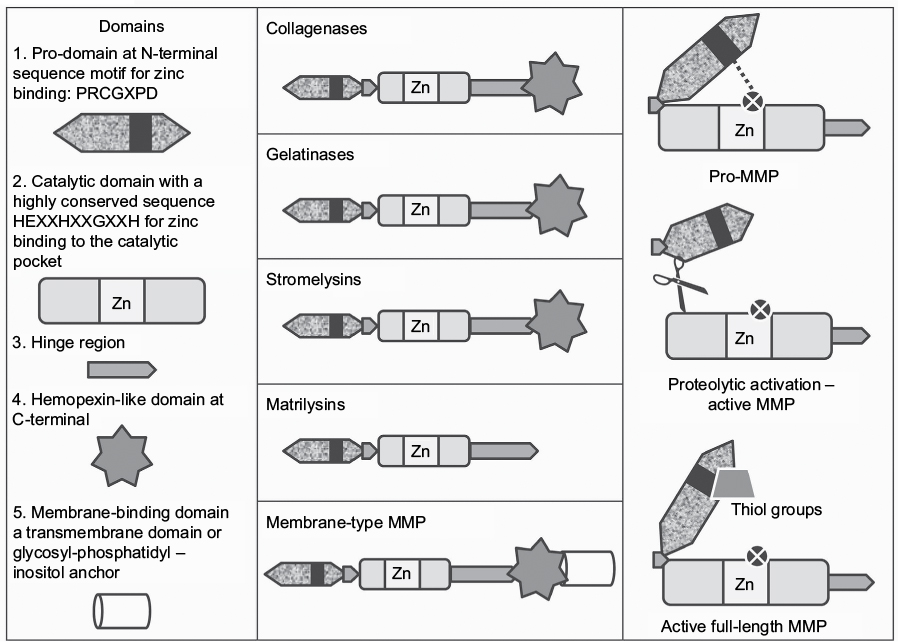 | Figure 2 Structure and activation of pro-MMPs. Notes: The left and middle panels show the multiple domains present in subgroups of pro-MMPs. The “cysteine switch” mechanism by which latency is achieved in pro-MMPs is mediated through an interaction between a cysteine residue in the pro-domain and the active-site zinc atom (right panel). Pro-MMPs are activated by proteolytic cleavage within the pro-domain region or disruption of the cysteine–zinc interaction by thiol groups from donors such as glutathione or reactive oxygen and nitrogen species. Abbreviation: pro-MMPs, pro-matrix metalloproteinases. |
MMP expression
With the exception of MMPs expressed by PMNs, MMPs are generally not constitutively expressed by other cells such as macrophages, epithelial cells, endothelial cells, and fibroblasts (Table 1).29 Rather, MMP expression is induced or repressed by various transcription factors, and MMPs are classified into three groups based on the mechanism by which they are regulated (Figure 3).30 The expression of many MMPs is increased when cells are activated with cytokines or growth factors, which increase the binding of transcription factors to the promoter regions of MMP genes.30
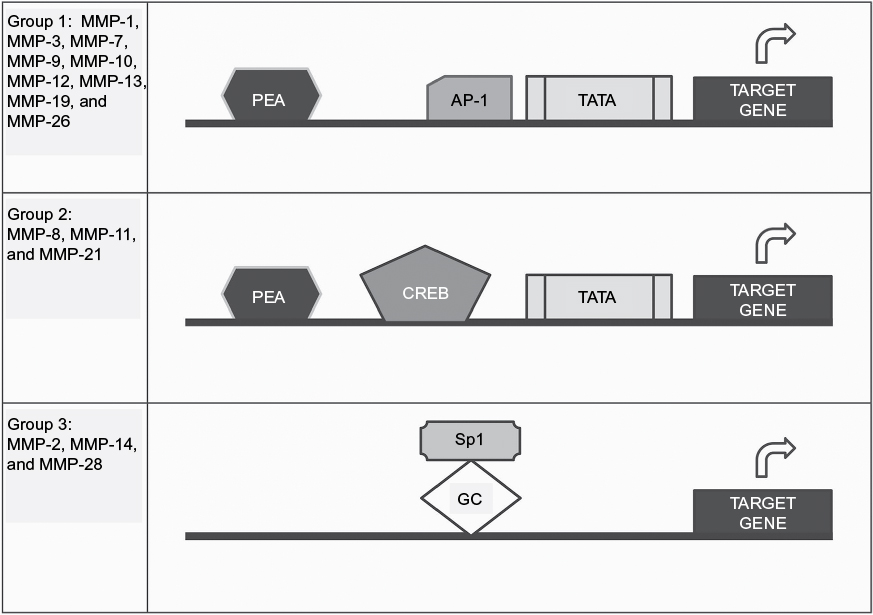 | Figure 3 Regulation of MMP gene expression. Notes: MMPs are classified into three groups by the mechanisms by which gene expression is regulated by transcription factors. Group 1 requires the presence of a TATA box adjacent to AP-1 and PEA-binding sites. Group 2 requires the binding of transcription factors other than AP-1 including the CREB. Group 3 does not require the presence of a TATA box and is mainly constitutively expressed. However, the expression of these group 3 genes is regulated by the members of the Sp1 family of transcription factors binding to GC-rich motifs. Abbreviation: MMPs, metalloproteinases. |
Pro-MMP activation
All MMPs are initially synthesized as inactive proenzymes that need to be activated before they can cleave substrates. Latency of pro-MMPs is achieved by an interaction between a conserved cysteine residue in the pro-domain and the active-site zinc atom, which prevents the active site from accessing the substrate to be cleaved.29 Pro-MMPs are activated when this cysteine–zinc interaction is disrupted, which can be achieved by 1) proteolytic cleavage of the pro-domain or furin-like motif by MMPs and other proteinases and 2) reactive oxygen or nitrogen species.31
Endogenous MMP inhibitors
There are two groups of naturally occurring MMP inhibitors: α2-macroglobulin and the four members of the family of tissue inhibitors of metalloproteinases (TIMPs).29 α2-Macroglobulin is a bait inhibitor that inhibits all four classes of proteinases by trapping them in its homo-tetramer composed of four identical subunits bound by disulfide bonds.32 TIMPs are expressed by both inflammatory cells and lung structural cells such as epithelial cells.33 The expression of TIMPs by most cells generally increases during inflammatory responses.34 TIMPs inhibit MMPs by forming 1:1 complexes with them.34,35
MMP and TIMP expression in CF
MMPs were first linked to CF in 1995 by a seminal publication on sputum MMP-9 levels in CF by Delacourt et al.23 Since then, other MMPs present in blood and lung samples from CF patients have been linked to this disease.
MMP levels in lung samples
Sputum samples
A number of studies have measured MMP and TIMP levels in sputum samples from CF patients versus controls and related levels of these molecules to acute pulmonary exacerbations, lung function, bacterial colonization, and NE levels. Most of these studies focused on the sputum soluble (sol) phase and have reported increased sputum levels of several MMPs in CF patients versus controls, and greater increases in sputum MMP levels during pulmonary exacerbations. The studies generally report that sputum MMP levels are indirectly related to lung function but not related to airway pathogen burdens.
Delacourt et al23 measured sputum MMP-9 and TIMP-1 levels in 27 children with CF who were hospitalized for systemic antibiotic treatment of an acute pulmonary exacerbation versus nine children hospitalized with acute asthma as controls. Sputum levels of MMP-9 and TIMP-1 were measured immediately before antibiotic therapy was initiated and 2 weeks later. CF severity was assessed by measuring pulmonary function 2 months later in 20 of these children who were over 6 years of age and in a clinically stable state. Sputum pro- and active MMP-9 levels were 3.7-fold and 23-fold higher in the CF patients with exacerbations, respectively. Sputum total MMP-9 levels were tenfold higher than sputum TIMP-1 levels. Sputum total and active MMP-9 levels decreased following antibiotic therapy. Direct correlations between sputum active MMP-9 levels and measurements of type IV collagen degradation, and total MMP-9 levels and PMN degranulation were reported, but MMP-9 levels were not related to airway bacterial burdens. In CF children in a clinically stable state, sputum active MMP-9 levels correlated indirectly with disease severity as assessed by forced expiratory volume in 1 second (FEV1) and forced vital capacity. Interestingly, urinary desmosine levels (a readout of elastin degradation) increased during an acute pulmonary exacerbation,36 suggesting that increased release of elastin-degrading proteinases (including MMP-9) from inflammatory cells during acute exacerbations contributes to airway injury and loss of lung function in CF patients.
Another study measured sputum levels of PMN-derived proteinases (NE, MMP-8, and MMP-9) in 33 adult CF patients hospitalized for an acute pulmonary exacerbation, eleven clinically stable adult CF outpatients, 26 children intubated for acute respiratory failure (ARF), and 14 outpatient controls without lung disease undergoing elective surgery.37 Elevated sputum MMP-9 levels were detected in CF inpatients and ARF patients (but not stable CF outpatients) versus healthy controls, lower sputum MMP-8 levels in CF inpatients and stable outpatients versus controls, and lower sputum levels of TIMP-1 in the CF inpatients and stable outpatients versus controls. Sputum NE levels were increased in CF outpatients versus controls and even higher in CF inpatients. In contrast to CF outpatients and ARF patients, in the CF inpatients, sputum MMP-9 was fully active and correlated with sputum NE levels. However, sputum MMP-9 levels did not correlate with FEV1 or forced vital capacity in CF inpatients or outpatients. As NE cleaves and inactivates TIMP-1, the increased sputum NE levels may have contributed to the increased sputum MMP-9 activity levels by NE degrading TIMP-1 present in sputum samples.37 Another study confirmed elevated sputum MMP-9 levels in 17 CF patients with an acute pulmonary exacerbation and reported that MMP-9 levels were reduced following intravenous antibiotic therapy.38
A subsequent study of nine nonexacerbating CF patients versus five subjects without disease reported elevated gelatinase levels, lower TIMP-1 levels, and higher MMP-9:TIMP-1 ratios in sputum samples from the CF patients versus healthy controls.39 As NE cleaves and activates pro-MMP-9 and NE cleaves and inactivates TIMP-1, the increases in sputum NE levels may have contributed to the increased MMP-9/TIMP-1 sputum ratios in stable CF patients and CF patients with acute pulmonary exacerbations.39 Another study confirmed elevated MMP-9 levels in sputum samples from stable CF patients but found no relationship between sputum MMP-9 and TIMP-1 levels and lower airway colonization with P. aeruginosa, S. aureus, or both.40
Not all studies of MMPs in CF samples have yielded positive results. One prospective, 3-year longitudinal study of sputum biomarkers (including MMP-2, MMP-9, and TIMP-1) was conducted on 35 children with CF (mean age ~11 years) having normal or only mildly impaired lung function.41 While sputum TIMP-1 levels increased over time, MMP-9 levels did not increase over time, and MMP-2 was not detectable in sputum samples. Sputum MMP-9 levels did not correlate with reductions of FEV1 over time. The reasons for the lack of a relationship between sputum MMP-9 levels and lung function over time in the latter study versus the other studies described earlier are not clear but may be related to the selection of CF patients at an earlier stage in the disease process (having normal or only mildly impaired lung function) in the latter longitudinal study.
Overall, these studies on sputum levels of MMPs suggest that CF patients have increased production of MMP-8 and MMP-9 by airway PMNs and possibly other cells such as airway epithelial cells, macrophages, and monocytes recruited into inflammed airways (MMP-9).42 Moreover, sputum MMP levels increase further during acute pulmonary exacerbations due to increases in the numbers and activation states of inflammatory cells, and activation of epithelial cells. Increased sputum levels of MMPs (especially when they exceed sputum TIMP levels) lead to airway injury and loss of lung function, at least in part, by MMPs degrading structural ECM components of the airway walls such as elastin and collagens. However, these studies of sputum samples from CF patients were only correlative. Additional mechanistic studies (eg, in animal models of CF) are needed to dissect the contributions of individual MMP to the disease processes occurring in CF airways.
Airway cell MMP expression
MMP and TIMP expression by airway macrophages and epithelial cells has been evaluated and related to CF therapies, clinical outcomes, and repair processes occurring in CF airways.
Airway macrophages
The phenotype of sputum macrophages (a key source of MMPs) from CF patients being treated chronically with azithromycin has been evaluated. These cells have reduced expression of proinflammatory M1 macrophage markers suggesting that these cells had a alternatively activated macrophage (M2-like anti-inflammatory and pro-repair) phenotype.43 Treating alveolar macrophages ex vivo with azithromycin induced an M2 associated with increased secretion of MMP-9 by the cells,44 and azithromycin therapy in CF patients increased macrophage MMP-9 expression.43 Clinical trials of long-term azithromycin treatment of CF patients have confirmed that this therapy induces M2 polarization of macrophages and shown that this is associated with reduced clinical severity and ECM turnover, lung function improvement, and lower infection risk with P. aeruginosa.45
Airway epithelial cells
A humanized airway xenograft model of regenerating human CF epithelium has been developed in which human CF epithelial cells are seeded onto denuded rat tracheas which were then transplanted into the subcutaneous space of nude mice. This model has been used to assess the expression of MMPs during various stages of epithelial regeneration (including epithelial cell adhesion, migration, proliferation, pseudostratification, and mucociliary differentiation). Airway epithelial MMP-7 and MMP-9 expression and secretion increased especially in the early stages of airway epithelial regeneration in CF grafts.46 Moreover, chemical inhibition of MMP-9 led to an immature monostratified epithelium composed mainly of goblet cells, and inhibition of MMP-7 led to a hyperplastic epithelium. Thus, epithelial cell-derived MMP-7 and MMP-9 may participate in epithelial repair in CF airways. If these findings are confirmed by other studies, novel therapeutic approaches could be developed to increase epithelial production of these MMPs to enhance epithelial repair in CF to limit loss of lung function. Thus, these studies indicate that, unlike MMPs expressed by PMNs, some MMPs (MMP-7 and MMP-9) expressed by other airway cells (eg, macrophages and epithelial cells) may promote resolution of inflammation and epithelial repair.
MMP expression in bronchoalveolar lavage fluid samples from CF patients
Some studies have measured MMP and TIMP levels in bronchoalveolar lavage fluid (BALF) samples from CF patients, linked BALF MMP and TIMP levels to lung injury, and shown that BALF MMP levels are modulated by medical therapies for CF. BALF MMP-8 and MMP-9 levels and MMP-9/TIMP-1 ratios were elevated in untreated children with mild CF and correlated positively with BALF lactoferin levels (a readout of PMN degranulation) and BALF α2-macroglobulin (a measure of alveolar–capillary barrier injury).47 These results suggested that PMNs were a source of MMP-8 and MMP-9, which contribute to airway injury in CF patients. Also, BALF MMP-8 and MMP-9 levels were reduced by Dornase Alpha therapy indicating that this therapy also induces anti-inflammatory activities in CF airways, which may limit further loss of lung function.47 Hilliard et al48 reported that BALF MMP-9 levels directly correlated with NE, elastin, glycosaminoglycans, and collagen BALF levels in children with recently diagnosed CF. Bergin et al49 reported that BALF MMP-2 MMP-9, and IL-8 levels were also higher in adult CF patients versus healthy controls and patients with non-CF-related bronchiectasis. Moreover, sputum MMP-9 (and NE) levels correlated indirectly with CF severity as assessed by the percent predicted FEV1. A recent study reported that elevated MMP-9/TIMP-1 ratios in BALF were associated with elevated BALF NE activity levels and progression of structural airway injury (bronchiectasis) in children with CF.50
MMP expression in human lungs and related models
Surprisingly, only one study has immunostained lung sections from CF patients to localize the cellular sources of MMPs detected in lung samples. This study reported that in CF lung sections, MMP-7 expression was increased in airway epithelial cells and induced in type II alveolar epithelial cells, but expression of MMP-1, MMP-3, or MMP-9 was not detected.51
MMP levels in CF blood samples
Serum and plasma MMP-8, MMP-9, TIMP-1, and TIMP-2 levels have been measured in CF patients versus controls and related to acute pulmonary exacerbations, lung function, and extrapulmonary disease. Plasma MMP levels were measured in a cohort of 54 adult CF patients in a clinically stable state and also in seven of these patients before and after acute pulmonary exacerbations were treated with systemic antibiotics.52 Plasma MMP-1, MMP-8, and MMP-9 levels were higher in CF patients than healthy controls; levels of these MMPs increased further during exacerbations, and decreased following antibiotic therapy. In contrast, MMP-2 levels were lower in CF plasma samples versus controls, and levels declined further during acute exacerbations.52 Only plasma MMP-8 levels correlated (indirectly) with FEV1. Another study of adults and children with moderate-to-severe CF lung disease reported elevated serum MMP-8, MMP-9, and TIMP-1 levels in clinically stable CF patients, and higher serum MMP-8 and MMP-9 levels in adult CF patients having acute pulmonary exacerbations.53 However, serum MMP-8, MMP-9, and TIMP-1 levels were not correlated with CF liver disease or pancreatic insufficiency in this cohort.
A cross-sectional survey of 73 CF patients and 40 control subjects measured plasma MMP-9 and TIMP-1 levels and related them to lung function decline and mortality over a median 49-month follow-up period.54 Plasma MMP-9 levels were elevated in CF patients and indirectly associated with percent predicted FEV1 and rate of FEV1 decline. Plasma TIMP-1 levels were inversely associated with mortality. CF-associated liver disease develops in ~30% of CF patients and correlates with poor outcomes. Two studies linked elevated plasma TIMP-1 and TIMP-2 levels to the presence of liver disease in CF patients.55,56 Thus, plasma levels of MMP-8, MMP-9, and TIMPs may have utility as prognostic biomarkers for CF.
CFTR and MMPs
MMPs have potential to contribute to the pathogenesis of CF by proteolytically modifying the CFTR or other epithelial ion channels to promote dehydration of the ASL. However, to date, only one MMP (MMP-2) has been reported to modulate chloride flux across airway epithelial cells by regulating the CFTR. Active MMP-2 endogenously expressed by airway epithelial cell lines (or exogenous enzyme added to cell cultures) inhibits activation of the CFTR in vitro.57 If MMP-2 is confirmed to inhibit the CFTR in vivo, it may further reduce chloride and bicarbonate secretion in CF airways and promote colonization of the airways with bacteria. It is not known whether MMP-2 inhibits activation of CFTR directly by proteolytically cleaving this receptor or indirectly by cleaving another proteinase that regulates the CFTR.
Serine proteinases expressed by epithelial cells (prostasin58) or PMNs (NE59), and acidic proteinases expressed by macrophages and epithelial cells (cathepsins B and S60,61) proteolytically activate airway ENaC to increase sodium hyperabsorption and ASL dehydration. However, to date, there are no reports in the literature that MMPs proteolytically regulate ENaC or other sodium channels in the airway epithelium. If MMPs linked to CF are confirmed to regulate chloride and sodium channels in vivo, therapeutic approaches inhibiting MMP expression or activity could be tested for their efficacy in CF patients.
Functions of MMPS in CF
The contributions of individual MMPs to pathologic processes occurring in CF airways have not been evaluated in animal models of CFs.62 However, studies of MMP gene-targeted mice or murine models of pulmonary infections show that MMPs regulate host responses to pathogens implicated in CF pathogenesis. The latter studies provide indirect evidence that some MMPs may regulate disease progression in CF patients. The following section describes the potential activities of MMPs that are upregulated in CF airways in regulating airway infections relevant to CF pathogenesis. In addition, hypotheses are generated on the potential contributions of these MMPs to regulating airway inflammation, injury, and remodeling in CF airways based upon the known functions of MMPs in other disease models. MMPs have been arbitrarily divided into two categories: those with potentially deleterious versus beneficial activities in CF airways based upon the reported positive versus negative correlations between their levels and clinical measures of disease severity, activity, or adverse outcomes, respectively. However, it is important to note that individual MMPs are subject to temporal and spatial regulation and are likely to be expressed by different cell types at different times in the course of the disease. Thus, individual MMPs could have both beneficial activities (such as promoting epithelial repair) and deleterious activities in the airways depending on the cell type that is producing them, the timing of their expression during the disease process, and the availability of susceptible substrates for their active sites at a given point in the disease process.
MMPs with potentially deleterious functions
Three MMPs (MMP-2, MMP-8, and MMP-9) are increased in blood and/or lung samples in CF patients, and MMP-8 and MMP-9 have been linked to readouts of lung injury (increased ECM degradation and vascular permeability) and adverse clinical outcomes (acute pulmonary exacerbations, bronchiectasis, and reductions in pulmonary function).
Matrix metalloproteinase-2
BALF MMP-2 levels are increased in CF patients and likely macrophages, epithelial cells, and fibroblasts produce MMP-2 in CF airways. MMP-2 may promote airway injury and aberrant remodeling processes and dampen effective airway inflammatory responses to bacterial colonization and infection in CF airways. First, MMP-2 degrades several basement membrane proteins,29 which promotes angiogenesis63 and fibrotic responses.64 MMP-2 has also been implicated in epithelial-to-mesenchymal transition in which airway epithelial cells express markers of myofibroblasts and contribute to collagen deposition.65 Interestingly, epithelial-to-mesenchymal transition has been linked to CF by gene expression studies66 and could contribute to the remodeling in CF airways. MMP-2 could also promote CF progression by dampening the airway inflammatory response to pathogens as it cleaves CCL7, a chemokine for monocytes, to generate a form that still binds to its receptors on monocytes without activating the cells.67 If MMP-2 is confirmed to inhibit activation of the CFTR in vivo (described in the “CFTR and MMPs” section), this proteinase may promote disease progression by increasing dehydration of the ALS and increasing bacterial colonization and infection of the airways.
Matrix metalloproteinase-8
MMP-8 levels are elevated in blood and lung samples and correlate with readouts of lung injury suggesting that MMP-8 promotes CF pathogenesis. Likely PMNs, activated macrophages, and bronchial epithelial cells produce MMP-8 in CF airways.68 MMP-8 is a potent interstitial collagenase in vitro69 and could promote degradation of interstitial collagens and thus structural changes in CF airways including the development of bronchiectasis. However, in murine models of pulmonary fibrosis, MMP-8 promotes fibroproliferative responses, in part, by cleaving and inactivating the chemokine, CCL3, and reducing lung levels of anti-fibrotic CXCL10.70 Thus, MMP-8 may promote aberrant remodeling processes in CF airways. MMP-8 may promote neutrophilic lung inflammation and injury by participating (along with MMP-9) in a proteolytic cascade that degrades interstitial collagen in the airways generating proline–leucine–proline (PGP) fragments of collagen that have been detected in CF lung samples and stimulate airway PMN recruitment.71,72 MMP-8 also inactivates CCL3, a key chemokine for monocytes/macrophages and could limit the recruitment of monocytes into CF airways.70 MMP-8 also inactivates α1-antitrypsin (the main inhibitor of NE69), and could increase NE-mediated injury to CF airways.
Matrix metalloproteinase-9
Leukocytes, epithelial cells, endothelial cells, and fibroblasts are likely key sources of MMP-9 in CF lung and blood samples.29 MMP-9 is likely to promote CF pathogenesis and progression as blood and lung MMP-9 levels increase further during acute pulmonary exacerbations, and correlate positively with rates of degradation of basement membrane (type IV) collagen, reductions in lung function, and progression of structural injury (bronchiectasis) in CF patients. As MMP-9 is an elastase,29 it could contribute to the degradation of lung elastin (as well as basement membrane collagen) reported in CF patients.36 MMP-9 also inactivates α1-antitrypsin,73 which could increase NE-mediated airway injury. MMP-9 cleaves and activates IL-8.74 As IL-8 is a potent PMN chemokine, MMP-9 may amplify neutrophilic airway inflammation and thus injury in CF airways by activating IL-8.
MMP-9 also activates latent TGF-β in vitro and may thereby contribute to remodeling processes in CF airways.75 MMP-9 may reduce host responses to pathogens as it cleaves SP-D to reduce the capacity of SP-D to opsonize bacteria and thereby reduce phagocytosis of bacteria by airway phagocytes.76 MMP-9-mediated cleavage of SP-D in CF airways may increase the susceptibility of CF patients to recurrent airway infections and disease progression. However, as outlined earlier, MMP-9 expressed by airway epithelial cells during epithelial repair processes is required for normal repair in a humanized animal model system.46
MMPs with potentially protective activities
Two MMPs (MMP-7 and MMP-12) may have protective activities in CF airways by controlling the inflammatory response to airway infection in CF airways, activating host defense proteins, and direct bacterial-killing activities.
Matrix metalloproteinase-7
MMP-7 expression is rapidly upregulated in the early phase after epithelial injury, and MMP-7 has beneficial activities in regulating airway inflammation and repair in models of other lung diseases. Following airway injury, early MMP-7-mediated shedding of syndecan-1 releases keratinocyte-derived chemokine that is bound to syndecan-1 into the alveolar space. This process activates keratinocyte-derived chemokine and promotes transepithelial migration of PMNs into the alveolar space where these cells phagocytose and kill bacteria.77 MMP-7-mediated shedding of syndecan-1 also promotes migration of epithelial cells and epithelial repair by activating α2β1 integrins on epithelial cells.78 MMP-7-mediated shedding of E-cadherin occurs later than syndecan-1 shedding in murine models and contributes to the resolution of the inflammatory response by promoting the influx of immunosuppressive dendritic cells.79 MMP-7 may promote airway repair or remodeling by activating growth factors as it degrades IGFBPs thereby releasing active IGF,80 and directly cleaves and activates pro-HB-EGF.81
MMP-7 may also have protective activities during airway infections with P. aeruginosa. MMP-7 expression is upregulated in the lungs of wild-type (WT) mice soon after infection with P. aeruginosa, and Mmp-7–/– mice have more severe pneumonia and lung injury when infected with P. aeruginosa than wild-type mice.82 Although the mechanisms by which Mmp-7 protects mice from P. aeruginosa pneumonia are not clear, MMP-7 cleaves and thereby activates α-defensins, which are small cationic peptides involved in bacterial killing.83 Mmp7–/– mice are more susceptible to experimental models of gastrointestinal infections.83 Human bronchial epithelial cells secrete human α-defensin-5, and it is possible that MMP-7 activates this defensin in CF airways.
Matrix metalloproteinase-12
MMP-12 has not been well studied in human CF samples, but activated macrophages are likely to be key sources of MMP-12 in CF airways.29 Infection of epithelial cell cultures with P. aeruginosa induces MMP-12 expression by the cells.84 Although MMP-12 degrades ECM proteins including elastin29 and could contribute to airway injury, it is also required for the recruitment of monocytes/macrophages into the lungs by generating fragments of ECM (matrikines) that are chemotactic for these cells85 that play key roles in clearance of airway pathogens. MMP-12 may increase pathogen killing in CF airways as it promotes killing of Gram-positive and Gram-negative bacteria that colonize CF airways in mice with pneumonia induced by these pathogens via its carboxy-terminal domain loop disrupting the outer membrane integrity of these bacteria.86
Targeting MMPS to limit disease progression in CF
The current literature suggests that MMP-8 and MMP-9 promote excessive airway inflammation and structural injury in CF patients, but MMP-7 and MMP-12 may enhance clearance of bacteria from CF airways. If deleterious activities of MMP-8 and MMP-9 are confirmed in CF airways, they could be selectively targeted to limit CF progression while preserving the activities of MMPs having beneficial activities. Selective inhibition of individual MMPs using small-molecule active-site inhibitors is challenging as the active sites of MMPs are structurally very similar, and MMP inhibitors generated to date have been associated with limiting side effects, including musculoskeletal syndrome, when tested in clinical trials for other diseases.87 Thus, other approaches that selectively reduce MMP levels or activity in the lung might be effective therapeutic approaches for CF (Table 2). In particular, monoclonal antibodies and single-domain antibodies (nanobodies) to MMPs (including MMP-8) are being developed and tested to selectively target individual MMPs to limit the progression of other lung diseases.88 However, MMPs that have deleterious activities in some lung disease have protective activities in other diseases.25 Thus, it will be necessary to test these alternatives and more selective approaches to reduce MMP levels or activity in cell culture and animal models of CF to determine whether they have efficacy and lack significant toxicity.
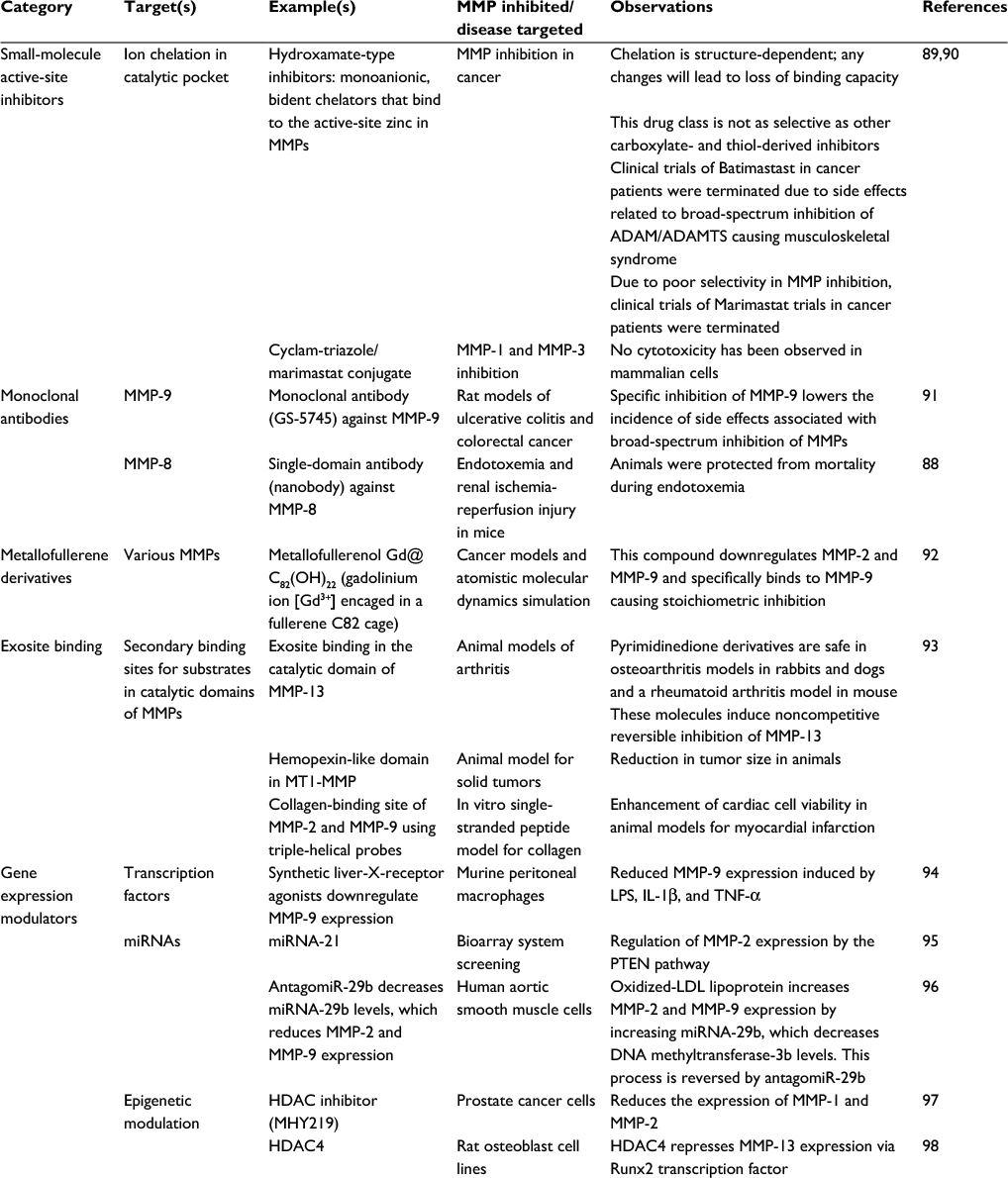 | Table 2 Potential approaches to reduce the expression or activity of deleterious MMPs in CF Notes: Potential therapeutic approaches for targeting MMPs include small-molecule active-site inhibitors, monoclonal antibodies, metallofullerene derivatives, and exosite binding which targets domains of the MMP other than the active-site zinc atom that contributes to binding of the MMP to the substrate, and approaches for regulating MMP gene expression include targeting transcription factors, miRNAs, and epigenetic modifications that regulate MMP expression. Abbreviations: ADAM, proteinase with a disintegrin and a metalloproteinase domain; ADAMTS, proteinase with a disintegrin, a metalloproteinase, and a thrombospondin domain; CF, cystic fibrosis; HDAC, histone deacetylase; LDL, low-density lipoprotein; LPS, lipopolysaccharide; miRNAs, microRNAs; MMPs, matrix metalloproteinases; PTEN, Phosphatase and tensin homolog |
Conclusion
Evidence is mounting that CF airways have increased levels of MMP-2, MMP-8, and MMP-9 (that likely overwhelm lung levels of inhibitory TIMPs, especially TIMP-1) and these changes may promote the progression of CF. However, the literature to date is mainly descriptive, and studies have focused on a small number of MMPs and TIMPs. These studies generally report only elevated levels of a limited number of MMPs (or altered MMP:TIMP ratios) in blood and lung samples from CF patients and correlations between MMP or TIMP levels and a limited number of readouts of CF severity and clinical outcomes. Additional studies are needed to determine whether other MMPs are upregulated in CF airways and to identify the cellular sources of these MMPs and contributions to the disease process. Future studies should validate MMPs and TIMPs as prognostic biomarkers for CF in larger cohorts. Studies of gene-targeted in murine of CF are also urgently needed to dissect the role of MMPs and TIMPs in the disease processes in CF airways. If elevated levels of MMPs in CF airways are confirmed to promote CF progression, future studies could determine whether therapeutic approaches that selectively inhibit the expression or activity of deleterious MMPs are effective at limiting the progression of CF in animal models of CF, and subsequently in randomized clinical trials in CF patients.
Acknowledgments
This work was supported by the Public Health Service, the National Heart, Lung, and Blood Institute Grants HL111835, PO1 HL105339, P01 HL114501, and AI111475-01, the Flight Attendants Medical Research Institute grant CIA123046, and the Brigham and Women’s Hospital–Lovelace Respiratory Research Institute Consortium.
Disclosure
The authors report no conflicts of interest in this work.
References
O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373(9678):1891–1904. | ||
Solomon M, Bozic M, Mascarenhas MR. Nutritional issues in cystic fibrosis. Clin Chest Med. 2016;37(1):97–107. | ||
Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373(18):220–231. | ||
Mogayzel PJ Jr, Naureckas ET, Robinson KA, et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2013;187(7):680–689. | ||
Clunes MT, Boucher RC. Cystic fibrosis: the mechanisms of pathogenesis of an inherited lung disorder. Drug Discov Today Dis Mech. 2007;4(2):63–72. | ||
Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. 1999;79(1 Suppl):S23–S45. | ||
Bombieri C, Seia M, Castellani C. Genotypes and phenotypes in cystic fibrosis and cystic fibrosis transmembrane regulator-related disorders. Semin Respir Crit Care Med. 2015;36(2):180–193. | ||
Schroeder SA, Gaughan DM, Swift M. Protection against bronchial asthma by CFTR delta F508 mutation: a heterozygote advantage in cystic fibrosis. Nat Med. 1995;1(7):703–705. | ||
Cutting GR. Modifier genes in Mendelian disorders: the example of cystic fibrosis. Ann N Y Acad Sci. 2010;1214:57–69. | ||
Gan KH, Veeze HJ, van den Ouweland AM, et al. A cystic fibrosis mutation associated with mild lung disease. N Engl J Med. 1995;333(2):95–99. | ||
van de Vosse E, Ali S, de Visser AW, et al. Susceptibility to typhoid fever is associated with a polymorphism in the cystic fibrosis transmembrane conductance regulator (CFTR). Hum Genet. 2005;118(1):138–140. | ||
Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology. 2003;8(4):432–446. | ||
Laucho-Contreras ME, Polverino F, Tesfaigzi Y, Pilon A, Celli BR, Owen CA. Club Cell Protein 16 (CC16) augmentation: a potential disease-modifying approach for chronic obstructive pulmonary disease (COPD). Expert Opin Ther Targets. 2016;20(7):869–883. | ||
Kurbatova P, Bessonov N, Volpert V, et al. Model of mucociliary clearance in cystic fibrosis lungs. J Theor Biol. 2015;372:81–88. | ||
Li H, Cai Z, Chen JH, Ju M, Xu Z, Sheppard DN. The cystic fibrosis transmembrane conductance regulator Cl(-) channel: a versatile engine for transepithelial ion transport. Sheng Li Xue Bao. 2007;59(4):416–430. | ||
Sala-Rabanal M, Yurtsever Z, Berry KN, Brett TJ. Novel roles for chloride channels, exchangers, and regulators in chronic inflammatory airway diseases. Mediators Inflamm. 2015;2015:497387. | ||
Smith JJ, Travis SM, Greenberg EP, Welsh MJ. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell. 1996;85(2):229–236. | ||
Matsui H, Grubb BR, Tarran R, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95(7):1005–1015. | ||
Coakley RD, Grubb BR, Paradiso AM, et al. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc Natl Acad Sci U S A. 2003;100(26):16083–16088. | ||
Tang XX, Ostedgaard LS, Hoegger MJ, et al. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J Clin Invest. 2016;126(3):879–891. | ||
Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. 2015;372(4):1574–1575. | ||
Cantin AM, Hartl D, KonstanMW, Chmiel JF. Inflammation in cystic fibrosis lung disease: pathogenesis and therapy. J Cyst Fibros. 2015;14(4):419–430. | ||
Delacourt C, Le Bourgeois M, D’Ortho MP, et al. Imbalance between 95 kDa type IV collagenase and tissue inhibitor of metalloproteinases in sputum of patients with cystic fibrosis. Am J Respir Crit Care Med. 1995;152(2):765–774. | ||
Greenlee KJ, Werb Z, Kheradmand F. Matrix metalloproteinases in lung: multiple, multifarious, and multifaceted. Physiol Rev. 2007;87(1):69–98. | ||
Craig VJ, Zhang L, Hagood JS, Owen CA. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2015;53(5):585–600. | ||
Jackson BC, Nebert DW, Vasiliou V. Update of human and mouse matrix metalloproteinase families. Hum Genomics. 2010;4(3):194–201. | ||
Gomis-Ruth FX. Structural aspects of the metzincin clan of metalloendopeptidases. Mol Biotechnol. 2003;24(2):157–202. | ||
Whittaker M, Floyd CD, Brown P, Gearing AJ. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem Rev. 1999;99(9):2735–2776. | ||
Owen CA, Campbell EJ. The cell biology of leukocyte-mediated proteolysis. J Leukoc Biol. 1999;65(2):137–150. | ||
Yan C, Boyd DD. Regulation of matrix metalloproteinase gene expression. J Cell Physiol. 2007;211(1):19–26. | ||
Zhang X, Shen YH, LeMaire SA. Thoracic aortic dissection: are matrix metalloproteinases involved? Vascular. 2009;17(3):147–157. | ||
Sottrup-Jensen L, Stepanik TM, Kristensen T, et al. Primary structure of human alpha 2-macroglobulin. V. The complete structure. J Biol Chem. 1984;259(13):8318–8327. | ||
Yamamoto K, Murphy G, Troeberg L. Extracellular regulation of metalloproteinases. Matrix Biol. 2015;44–46:255–263. | ||
Arpino V, Brock M, Gill SE. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015;44–46:247–254. | ||
Schuster A, Haarmann A, Wahn V. Cytokines in neutrophil-dominated airway inflammation in patients with cystic fibrosis. Eur Arch Otorhinolaryngol. 1995;252 Suppl 1:S59–S60. | ||
Laguna TA, Wagner BD, Starcher B, et al. Urinary desmosine: a biomarker of structural lung injury during CF pulmonary exacerbation. Pediatr Pulmonol. 2012;47:856–863. | ||
Gaggar A, Li Y, Weathington N, et al. Matrix metalloprotease-9 dysregulation in lower airway secretions of cystic fibrosis patients. Am J Physiol Lung Cell Mol Physiol. 2007;293:L96–L104. | ||
Muller U, Hentschel J, Janhsen WK, et al. Changes of proteases, antiproteases, and pathogens in cystic fibrosis patients’ upper and lower airways after IV-antibiotic therapy. Mediators Inflamm. 2015;2015:626530. | ||
Jackson PL, Xu X, Wilson L, et al. Human neutrophil elastase-mediated cleavage sites of MMP-9 and TIMP-1: implications to cystic fibrosis proteolytic dysfunction. Mol Med. 2010;16(5–6):159–166. | ||
Fischer N, Hentschel J, Markert UR, Keller PM, Pletz MW, Mainz JG. Non-invasive assessment of upper and lower airway infection and inflammation in CF patients. Pediatr Pulmonol. 2014;49(11):1065–1075. | ||
Sagel SD, Kapsner RK, Osberg I. Induced sputum matrix metalloproteinase-9 correlates with lung function and airway inflammation in children with cystic fibrosis. Pediatr Pulmonol. 2005;39(3):224–232. | ||
Owen CA, Campbell MA, Boukedes SS, Campbell EJ. Monocytes recruited to sites of inflammation express a distinctive proinflammatory (P) phenotype. Am J Physiol. 1994;267(6 Pt 1):L786–L796. | ||
Cory TJ, Birket SE, Murphy BS, et al. Impact of azithromycin treatment on macrophage gene expression in subjects with cystic fibrosis. J Cyst Fibros. 2014;13(12):164–171. | ||
Cory TJ, Birket SE, Murphy BS, Mattingly C, Breslow-Deckman JM, Feola DJ. Azithromycin increases in vitro fibronectin production through interactions between macrophages and fibroblasts stimulated with Pseudomonas aeruginosa. J Antimicrob Chemother. 2013;68(4):840–851. | ||
Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA. 2003;290(13):1749–1756. | ||
Hajj R, Lesimple P, Nawrocki-Raby B, Birembaut P, Puchelle E, Coraux C. Human airway surface epithelial regeneration is delayed and abnormal in cystic fibrosis. J Pathol. 2007;211(3):340–350. | ||
Ratjen F, Hartog CM, Paul K, Wermelt J, Braun J. Matrix metalloproteases in BAL fluid of patients with cystic fibrosis and their modulation by treatment with dornase alpha. Thorax. 2002;57(11):930–934. | ||
Hilliard TN, Regamey N, Shute JK, Nicholson AG, Alton EW, Bush A, Davies JC. Airway remodelling in children with cystic fibrosis. Thorax. 2007;62(12):1074–1080. | ||
Bergin DA, Hurley K, Mehta A, et al. Airway inflammatory markers in individuals with cystic fibrosis and non-cystic fibrosis bronchiectasis. J Inflamm Res. 2013;6:1–11. | ||
Garratt LW, Sutanto EN, Ling KM, et al. Matrix metalloproteinase activation by free neutrophil elastase contributes to bronchiectasis progression in early cystic fibrosis. Eur Respir J. 2015;46(2):384–394. | ||
Dunsmore SE, Saarialho-Kere UK, Roby JD, et al. Matrilysin expression and function in airway epithelium. J Clin Invest. 1998;102(7):1321–1331. | ||
Roderfeld M, Rath T, Schulz R, et al. Serum matrix metalloproteinases in adult CF patients: relation to pulmonary exacerbation. J Cyst Fibros. 2009;8(5):338–347. | ||
Rath T, Zwaschka L, Hage L, et al. Identification of neutrophil activation markers as novel surrogate markers of CF lung disease. PLoS One. 2014;9:e115847. | ||
Devereux G, Steele S, Jagelman T, et al. An observational study of matrix metalloproteinase (MMP)-9 in cystic fibrosis. J Cyst Fibros. 2014;13(5):557–563. | ||
Pereira TN, Lewindon PJ, Smith JL, et al. Serum markers of hepatic fibrogenesis in cystic fibrosis liver disease. J Hepatol. 2004;41(4):576–583. | ||
Rath T, Menendez KM, Kugler M, et al. TIMP-1/-2 and transient elastography allow non invasive diagnosis of cystic fibrosis associated liver disease. Dig Liver Dis. 2012;44(9):780–787. | ||
Duszyk M, Shu Y, Sawicki G, Radomski A, Man SF, Radomski MW. Inhibition of matrix metalloproteinase MMP-2 activates chloride current in human airway epithelial cells. Can J Physiol Pharmacol. 1999;77(7):529–535. | ||
Nimishakavi S, Besprozvannaya M, Raymond WW, Craik CS, Gruenert DC, Caughey GH. Activity and inhibition of prostasin and matriptase on apical and basolateral surfaces of human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2012;303(2):L97–L106. | ||
Caldwell RA, Boucher RC, Stutts MJ. Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am J Physiol Lung Cell Mol Physiol. 2005;288(5):L813–L819. | ||
Haerteis S, Krappitz M, Bertog M, et al. Proteolytic activation of the epithelial sodium channel (ENaC) by the cysteine protease cathepsin-S. Pflugers Arch. 2012;464(4):353–365. | ||
Tan CD, Hobbs C, Sameni M, Sloane BF, Stutts MJ, Tarran R. Cathepsin B contributes to Na+ hyperabsorption in cystic fibrosis airway epithelial cultures. J Physiol. 2014;592(23):5251–5268. | ||
Fisher JT, Zhang Y, Engelhardt JF. Comparative biology of cystic fibrosis animal models. Methods Mol Biol. 2011;742:311–334. | ||
Bergers G, Brekken R, McMahon G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2(10):737–744. | ||
Keane MP, Arenberg DA, Lynch JP III, et al. The CXC chemokines, IL-8 and IP-10, regulate angiogenic activity in idiopathic pulmonary fibrosis. J Immunol. 1997;159(3):1437–1443. | ||
Cheng S, Pollock AS, Mahimkar R, Olson JL, Lovett DH. Matrix metalloproteinase 2 and basement membrane integrity: a unifying mechanism for progressive renal injury. FASEB J. 2006;20(11):1898–1900. | ||
Clarke LA, Botelho HM, Sousa L, Falcao AO, Amaral MD. Transcriptome meta-analysis reveals common differential and global gene expression profiles in cystic fibrosis and other respiratory disorders and identifies CFTR regulators. Genomics. 2015;106(5):268–277. | ||
McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289(5482):1202–1206. | ||
Craig VJ, Polverino F, Laucho-Contreras ME, et al. Mononuclear phagocytes and airway epithelial cells: novel sources of matrix metalloproteinase-8 (MMP-8) in patients with idiopathic pulmonary fibrosis. PLoS One. 2014;9:e97485. | ||
Owen CA, Hu Z, Lopez-Otin C, Shapiro SD. Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J Immunol. 2004;172(12):7791–7803. | ||
Craig VJ, Quintero PA, Fyfe SE, et al. Profibrotic activities for matrix metalloproteinase-8 during bleomycin-mediated lung injury. J Immunol. 2013;190(8):4283–4296. | ||
Gaggar A, Jackson PL, Noerager BD, et al. A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J Immunol. 2008;180(8):5662–5669. | ||
Gaggar A, Rowe SM, Matthew H, Blalock JE. Proline-glycine-proline (PGP) and high mobility group box protein-1 (HMGB1): potential mediators of cystic fibrosis airway inflammation. Open Respir Med J. 2010;4:32–38. | ||
Owen CA, Hu Z, Barrick B, Shapiro SD. Inducible expression of tissue inhibitor of metalloproteinases-resistant matrix metalloproteinase-9 on the cell surface of neutrophils. Am J Respir Cell Mol Biol. 2003;29(3 Pt 1):283–294. | ||
Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. 2000;96(8):2673–2681. | ||
Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14(2):163–176. | ||
Bratcher PE, Weathington NM, Nick HJ, Jackson PL, Snelgrove RJ, Gaggar A. MMP-9 cleaves SP-D and abrogates its innate immune functions in vitro. PLoS One. 2012;7:e41881. | ||
Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111(5):635–646. | ||
Chen P, Abacherli LE, Nadler ST, Wang Y, Li Q, Parks WC. MMP7 shedding of syndecan-1 facilitates re-epithelialization by affecting alpha(2)beta(1) integrin activation. PLoS One. 2009;4:e6565. | ||
Manicone AM, Huizar I, McGuire JK. Matrilysin (Matrix Metalloproteinase-7) regulates anti-inflammatory and antifibrotic pulmonary dendritic cells that express CD103 (alpha(E)beta(7)-integrin). Am J Pathol. 2009;175(6):2319–2331. | ||
Nakamura M, Miyamoto S, Maeda H, et al. Matrix metalloproteinase-7 degrades all insulin-like growth factor binding proteins and facilitates insulin-like growth factor bioavailability. Biochem Biophys Res Commun. 2005;333(3):1011–1016. | ||
Yu WH, Woessner JF Jr, McNeish JD, Stamenkovic I. CD44 anchors the assembly of matrilysin/MMP-7 with heparin-binding epidermal growth factor precursor and ErbB4 and regulates female reproductive organ remodeling. Genes Dev. 2002;16(3):307–323. | ||
Kassim SY, Gharib SA, Mecham BH, Birkland TP, Parks WC, McGuire JK. Individual matrix metalloproteinases control distinct transcriptional responses in airway epithelial cells infected with Pseudomonas aeruginosa. Infect Immun. 2007;75(12):5640–5650. | ||
Wilson CL, Ouellette AJ, Satchell DP, et al. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286(5437):113–117. | ||
Park JW, Shin IS, Ha UH, Oh SR, Kim JH, Ahn KS. Pathophysiological changes induced by Pseudomonas aeruginosa infection are involved in MMP-12 and MMP-13 upregulation in human carcinoma epithelial cells and a pneumonia mouse model. Infect Immun. 2015;83(12):4791–4799. | ||
Houghton AM, Quintero PA, Perkins DL, et al. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116(3):753–759. | ||
Houghton AM, Hartzell WO, Robbins CS, Gomis-Ruth FX, Shapiro SD. Macrophage elastase kills bacteria within murine macrophages. Nature. 2009;460(7255):637–641. | ||
Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295(5564):2387–2392. | ||
Demeestere D, Dejonckheere E, Steeland S, et al. Development and validation of a small single-domain antibody that effectively inhibits matrix metalloproteinase 8. Mol Ther. 2016;24(5):890–902. | ||
Serra P, Bruczko M, Zapico JM, et al. MMP-2 selectivity in hydroxamate-type inhibitors. Curr Med Chem. 2012;19:1036–1064. | ||
Yu M, Lim NH, Ellis S, et al. Incorporation of bulky and cationic cyclam-triazole moieties into marimastat can generate potent MMP inhibitory activity without inducing cytotoxicity. ChemistryOpen. 2013;2:99–105. | ||
Marshall DC, Lyman SK, McCauley S, et al. Selective allosteric inhibition of MMP9 is efficacious in preclinical models of ulcerative colitis and colorectal cancer. PLoS One. 2015;10:e0127063. | ||
Kang SG, Araya-Secchi R, Wang D, Wang B, Huynh T, Zhou R. Dual inhibitory pathways of metallofullerenol Gd@C(8)(2)(OH)(2)(2) on matrix metalloproteinase-2: molecular insight into drug-like nanomedicine. Sci Rep. 2014;4:4775. | ||
Fields GB. New strategies for targeting matrix metalloproteinases. Matrix Biol. 2015;44–46:239–246. | ||
Castrillo A, Joseph SB, Marathe C, Mangelsdorf DJ, Tontonoz P. Liver X receptor dependent repression of matrix metalloproteinase-9 expression in macrophages. J Biol Chem. 2003;278:10443–10449. | ||
Roy S, Khanna S, Hussain SR, et al. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res. 2009;82:21–29. | ||
Chen KC, Wang YS, Hu CY, et al. OxLDL up-regulates microRNA-29b, leading to epigenetic modifications of MMP-2/MMP-9 genes: a novel mechanism for cardiovascular diseases. FASEB J. 2011;25:1718–1728. | ||
De U, Kundu S, Patra N, et al. A new histone deacetylase inhibitor, MHY219, inhibits the migration of human prostate cancer cells via HDAC1. Biomol Ther. (Seoul.) 2015;23:434–441. | ||
Shimizu E, Selvamurugan N, Westendorf JJ, Olson EN, Partridge NC. HDAC4 represses matrix metalloproteinase-13 transcription in osteoblastic cells, and parathyroid hormone controls this repression. J Biol Chem. 2010;285:9616–9626. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.