Back to Archived Journals » Metalloproteinases In Medicine » Volume 1
Matrix metalloproteinases and their inhibitors in cardiovascular pathologies: current knowledge and clinical potential
Authors Johnson J
Received 2 October 2014
Accepted for publication 23 October 2014
Published 9 December 2014 Volume 2014:1 Pages 21—36
DOI https://doi.org/10.2147/MNM.S50999
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor William Parks
Jason Lee Johnson
Laboratory of Cardiovascular Pathology, School of Clinical Sciences, University of Bristol, Bristol, UK
Abstract: Matrix metalloproteinases (MMPs) constitute a family of endopeptidases that harbor matrix-degrading potential, but also modulate the proliferation, migration, and apoptosis of resident blood-vessel cells and recruited inflammatory cells. Accordingly, they are proposed to play a major regulatory role in numerous cardiovascular pathologies, including restenosis, atherosclerosis, aneurysms, and post-myocardial infarction remodeling. Collectively, clinical and animal studies have begun to unravel the complex and often diverse effects MMPs and their endogenous tissue inhibitors of metalloproteinases (TIMPs) impart during cardiovascular pathologies. It is apparent that some MMPs are beneficial, while others impose detrimental influences on disease progression. This premise is underscored by evidence that broad-spectrum MMP inhibition fails to provide protection from most cardiovascular diseases. However, recent studies in mice using more selective inhibitors have proved promising. Consequently, there is an immediate need to elucidate the precise roles of individual MMPs and TIMPs in the distinct cardiovascular pathologies in order to facilitate the development and translation of new therapeutic approaches to combat cardiovascular disease, the major cause of death worldwide.
Keywords: MMPs, TIMPs, atherosclerosis, aneurysm, myocardial infarction, restenosis
Introduction
Matrix metalloproteinases (MMPs), also known as matrixins, can collectively degrade all of the extracellular matrix (ECM) components that are commonly found within the blood-vessel wall.1 Accordingly, they have been implicated in the pathophysiology of numerous cardiovascular pathologies, including restenosis, atherosclerosis, aneurysms, and post-myocardial infarction (MI) remodeling (Table 1), although MMP-17, -20, -21, -23, -25, -26, and -27 remain unstudied. However, the paradigm of MMP function has recently changed. In addition to ECM digestion, they have also been demonstrated to process a number of cell-surface and soluble mediators that can potently affect cell behavior.2 Consequently, MMPs harbor the potential to modulate the proliferation, migration/invasion, and apoptosis of resident blood-vessel cells, such as vascular smooth-muscle cells (VSMCs),3 endothelial cells,4 and several inflammatory cell types, including monocytes.5
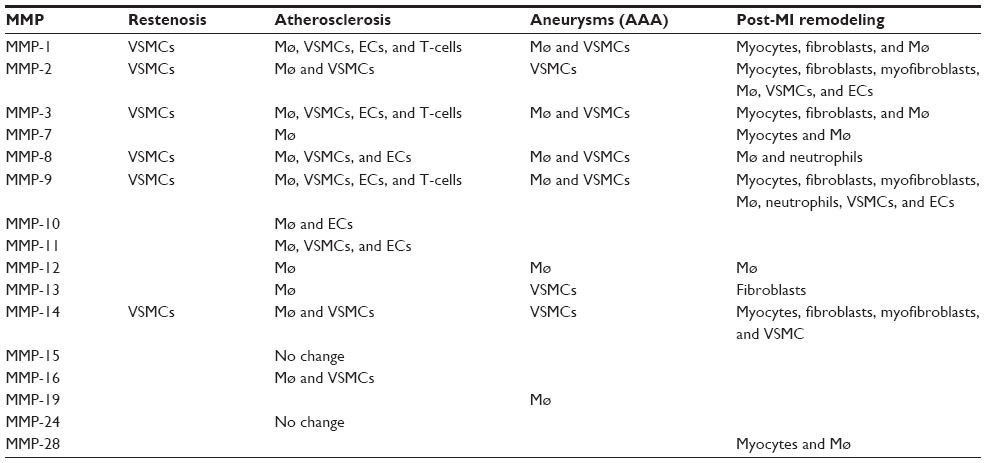 | Table 1 MMPs upregulated in cardiovascular pathologies compared to relevant normal tissues, and cellular sources |
At present, the MMP family consists of 23 members that share a high degree of homology and can be regulated by inflammatory cytokines, growth factors, hormones, and physical cell–cell and cell–matrix interactions.1 They are included in the metzincin family together with ADAMs (a disintegrin and metalloproteinase family) and ADAMTSs (ADAM with thrombospondin motifs), as they all contain a zinc atom and a conserved methionine in the catalytic domain.6 Within the MMP family, subtle differences in their domain structure afford them overlapping substrate specificities (Figure 1). Accordingly, MMPs can be subdivided into interstitial collagenases (MMP-1, -8, -13, and -14) that cleave fibrillar collagens, gelatinases (MMP-2 and -9) that efficiently cleave denatured collagen (ie, gelatin), and stromelysins (MMP-3, -7, -10, and -11) that have broad specificity but do not effectively cleave intact fibrillar collagens. Macrophage metalloelastase (MMP-12), as it name suggests, primarily cleaves elastin.1,7 Moreover, while most MMPs are secreted in a latent form and require activation, numerous MMPs are membrane-bound in an active form (including MMP-14 to -17, -25, and -26) and termed membrane-type MMPs (MT-MMPs).1,7
 | Figure 1 Domain structure for the major classes of matrix metalloproteinases (MMPs). Major domains include the signal peptide (SP), prodomain (Pro), catalytic domain with the active site zinc bound to cysteine residues within this domain and “cysteine-switch” residue in the Pro, the hinge domain (HG), the hemopexin domain, and in some cases either a transmembrane domain (TM) or glycosylphosphatidylinositol (GPI)-anchor domain. A furin-cleavage site (F) between the Pro and the catalytic domain is found in some MMPs. In the gelatinases, fibronectin (FN)-like type II repeats are also present. The schematic within the dotted lines depicts the inhibitory action of tissue inhibitors of metalloproteinases (TIMPs) on MMPs, demonstrating how the N-terminus of the TIMP chelates the active site zinc and blocks MMP activity. |
As such, MT-MMPs are considered potent sheddases due to their pericellular location, and can accordingly dynamically regulate cell activities through the modification of membrane proteins, such as growth-factor and cytokine receptors, integrins, and cell–cell contacts.6 In addition it has also been demonstrated that secreted MMPs can localize to the cell membrane, where their proteolytic actions alongside MT-MMPs can play diverse roles in cell behavior, such as proliferation, phenotypic switching, migration, and apoptosis.8 Conversely, it has been postulated that cells can fine-tune their MMP expression and activity through sensing the mechanical properties imparted by their surrounding ECM components. For instance, integrin9 and syndecan10 family members can relay outside-in signals to modulate MMP expression, localization, and activity. Moreover, growth-factor signaling, such as platelet-derived growth factor, basic fibroblast growth factor, and epidermal growth factor (EGF) can independently or through synergism with integrin signaling mediate vascular cell MMP expression and localization.11 Finally, complex cross talk between cell–cell/cell–matrix interactions and focal adhesions can modulate the activity and localization of MMPs to the migratory edge of vascular cells.12 Taken together, these findings demonstrate the complex regulatory processes involved in MMP regulation, which are discussed in more detail in the associated citations.
Due to the destructive nature of MMPs and their ability to modulate multiple substrates, they are tightly regulated by various nonspecific inhibitors; these include the membrane-anchored reversion-inducing cysteine-rich protein with kazal motifs (RECK), tissue-factor pathway inhibitor-2 (TFPI-2), the pro-collagen C-terminal proteinase enhancer (PCPE), and the plasma protein α2-macroglobulin.13 MMPs are, however, most potently inhibited by endogenous tissue inhibitors of metalloproteinases (TIMPs), which are therefore believed to play the major role in regulating their activity physiologically. In vertebrates, four TIMPs have been identified (TIMP-1, -2, -3, and -4), that can form tight complexes with MMP catalytic domains through inhibitory residues present within their N-terminal domain (Figure 1).13 TIMPs are secreted proteins, but they can also be found on the cell membrane associated with such membrane proteins as MT-MMPs. All the TIMPs can inhibit the activity of all MMPs; however, TIMP-1 has a poor inhibitory effect on MMP-9, -14, -15, -16, and -24. TIMPs also harbor the ability to inhibit members of both the ADAM and ADAMTS families of proteinases, especially TIMP-3.1 Moreover, the C-terminal domain of TIMP-3 displays high affinity for ECM proteins, enabling it to accumulate within the pericellular space and prolong its half-life.13 The balance between MMPs and TIMPs is therefore crucial for homeostasis, and alterations in this balance have been implicated in numerous cardiovascular diseases.11
Intimal formation/restenosis
The development of a vascular smooth muscle cell (VSMC)-rich intimal thickening underlies several occlusive cardiovascular pathologies that form as a result of interventions for preexisting atherosclerosis; these include angioplasty, stent deployment, and saphenous vein bypass grafting. Within the healthy blood-vessel wall, VSMCs are restricted to the media in a quiescent state and regulate vessel contractility. Upon injurious insults, such as the aforementioned interventions, medial VSMCs proliferate and migrate into the intima, where their irregular growth can impede lumen patency or serve as a soil bed for superimposed atherosclerosis.3 It has been proposed that MMPs may directly liberate VSMCs from their normally retentive ECM alongside disruption of cell–cell contacts within the media and therefore facilitate their migration and growth within the developing neointima.3 Numerous MMPs have been shown to be expressed by various cell types within intimal thickenings (Table 1), and accordingly a plethora of in vivo studies have been undertaken to elucidate which MMPs direct intimal formation (Table 2).14–36
 | Table 2 Results of in vivo animal studies evaluating the effects of modulating matrix metalloproteinases (MMPs) or tissue inhibitors of metalloproteinases (TIMPs) on neointimal and vascular smooth-muscle cell behavior, using recombinant adenoviral overexpression, gene knockout, or pharmacological inhibitors of MMPs (MMPis) |
Due to their ability to degrade cellular basement membranes and in vitro evidence implicating them in migration and proliferation of VSMCs,3 initial work focused on gelatinolytic MMPs, including MMP-2, -9, and -14, in neointima formation. Studies utilizing mouse carotid artery-injury models revealed that gene deletion of MMP-2,15,16 MMP-9,17,19,20 and MMP-1421 retarded intimal formation, primarily through effects on VSMC migration, whereas proliferation was less affected. Recently, MMP-3 has been indirectly implicated, due to its ability to activate MMP-9.17 Intimal formation was also abrogated in mice receiving an MMP-1 inhibitor14 or Mmp8-knockout (KO) mice18 after wire-induced injury of the carotid artery. The mechanisms underlying the detrimental effects of MMPs on neointima formation have been discussed elsewhere,3 and chiefly affect VSMC function through the cleavage of cell–cell and cell–matrix contacts.3 In particular, N-cadherin represents a major cell–cell adhesion molecule in VSMCs that has been shown to be cleaved by several MMPs (MMP-7, -9, and -12), altering VSMC behavior, including migration, proliferation, and apoptosis.37,38 Accordingly, controlling N-cadherin function may represent a therapeutic avenue for retarding intimal thickening, as recently intimated in an ex vivo model where inhibition of N-cadherin and its downstream effects retarded intimal thickening via induction of VSMC apoptosis.39
In line with MMPs playing an important role, the overexpression of TIMPs has been demonstrated to negate intima formation in various species. TIMP-1 gene transfer was shown to reduce balloon injury-induced intimal formation in the rat,23–25 while expectedly intimal size was increased in a mouse wire injury model.22 Similar protective effects have been reported for TIMP-2,26,27 TIMP-3,28,29 and TIMP-4,30 primarily through retarded VSMC migration or apoptosis.
Similarly but less specifically, broad-spectrum MMP inhibitors have been utilized, and demonstrated that both hydroxamate-based31–33 and tetracycline-derived compounds34–36 retarded intimal thickening in rodent models. However, the use of broad-spectrum MMP inhibitors in nonhuman primates40 and man (reviewed by Peterson41) has yielded less promising results. Therefore, while other antiproliferative agents have become widely adopted as therapeutic tools to prevent in-stent restenosis, interest in targeting MMPs has waned. Nonetheless, preclinical studies in pigs28,29 have proven that TIMP-3 gene therapy is effective at reducing vein-graft intimal thickening, and is consequently moving toward a Phase II clinical trial. The use of TIMPs affords a level of specificity above and beyond that of broad-spectrum MMP inhibitors, targeting select MMPs. Accordingly, the development and employment of specific-MMP inhibitors may prove more fruitful as a therapeutic approach for restenosis. Such a tactic may also circumvent the possibility that MMPs may play beneficial and detrimental roles during restenosis and a more selective inhibitory course is required, as recently shown for atherosclerosis.42,43
Atherosclerosis
The rupture of an atherosclerotic plaque with associated luminal thrombosis is the major underlying cause of acute coronary syndromes (such as coronary artery disease and ensuing MI) and stroke. Clinically relevant atherosclerotic lesions develop primarily within the coronary and carotid arteries of humans over many decades, and are widely accepted as an inflammatory disease,44 although VSMCs and endothelial cells also play prominent roles.3,45 In particular, VSMCs appear to play a divergent role in atherosclerosis; while their phenotypic modulation and growth can precipitate early lesion formation, in advanced plaques they take on a beneficial role through the generation and maintenance of a protective fibrous cap that shields them from plaque rupture.3,12 Nonetheless inflammatory cells, especially monocytes/macrophages, are considered pivotal to atherosclerotic plaque progression and destabilization (rupture) through their roles in fibrous cap thinning, VSMC loss, lipid/necrotic core expansion and microcalcification, all characteristics associated with rupture-prone plaques.44,46 A wealth of biochemical and histological studies have highlighted a prominent role for MMPs in atherosclerotic plaque progression and vulnerability (Table 1), particularly those derived from macrophages,11 except for MMP-15 and -24, whose expression has been shown to be unaltered in human atherosclerotic coronary artery plaques.47 Accordingly, researchers have turned to animal models to elucidate the contribution of MMPs in atherogenesis and the formation of advanced plaques.
Both transgenic and KO animal models have been utilized to investigate the role of MMPs and TIMPs in the pathogenic steps of atherosclerotic plaque formation, progression, and rupture, notably rabbits and mice. When these animals are rendered hypercholesterolemic, they develop atherosclerotic lesions at multiples sites that resemble the human analogs and progress similarly from fatty streaks to complex advanced lesions.48 Accordingly, there are numerous empirical factors that can be assessed in addition to plaque size; these include characteristics associated with plaque vulnerability in humans, such as VSMC and macrophage number, collagen content, lipid-core size, neovascularization and intraplaque hemorrhage, and elastin fragmentation.49 Recent studies have highlighted the potential detrimental role of neovascularization (a form of angiogenesis) in human atherosclerosis,50 which is mediated in part by MMPs.4 However, plaque neovascularization is uncommon in plaques from animal models, and their incidence is rarely reported.50 Utilizing mainly apolipoprotein E (Apoe)-KO mice, and to a lesser degree low-density lipoprotein receptor (Ldlr)-KO animals, a large number of studies have been conducted where these mice are either bred with transgenic or KO mice, or treated with potential therapeutic agents. These studies have illuminated the potential roles of MMPs and TIMPs in atherosclerotic plaque progression and stability (summarized in Table 3).33,42,43,51–67
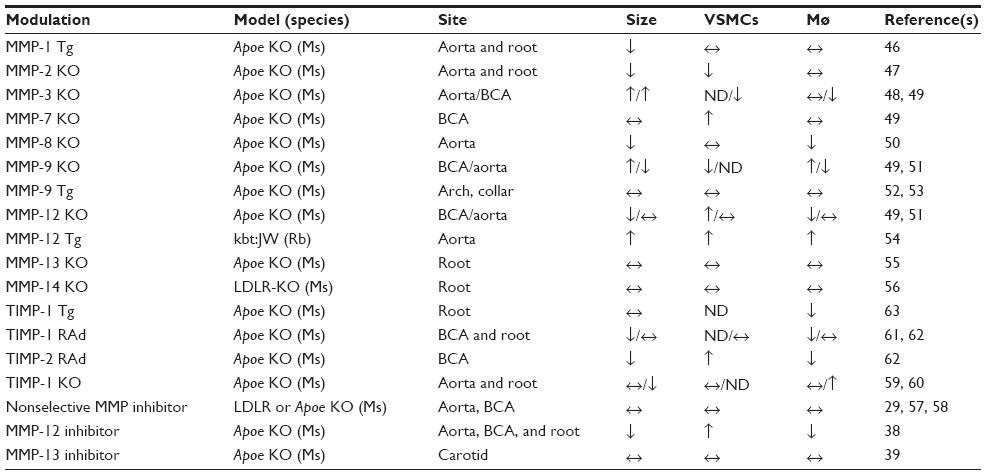 | Table 3 Results of in vivo animal studies evaluating the effects of modulating matrix metalloproteinases (MMP) or tissue inhibitors of metalloproteinases (TIMPs) on atherosclerotic plaque size and cellular composition, using transgenic (Tg) or recombinant adenoviral (RAd) overexpression, gene knockout (KO), or pharmacological inhibitors of MMPs |
Unlike humans, mice do not actively express MMP-1. Nonetheless, a study where human MMP-1 was overexpressed selectively in macrophages of Apoe-KO mice unexpectedly showed a reduction in plaque size and collagen content.51 Mmp-KO studies in Apoe-KO mice have indicated both protective and detrimental effects. MMP-2-KO mice presented a reduction in plaque size, potentially through a decrease in VSMC accumulation and suggesting plaque stability may be compromised.52 MMP-3 deletion resulted in larger aortic and brachiocephalic plaques53,54 associated with fewer medial elastin breaks,53 but lowered VSMC content and concomitant increase in buried fibrous layers (a surrogate marker of mouse brachiocephalic plaque instability),54 implying MMP-3 affords greater stability. In agreement, reduced VSMC migration and associated neointimal formation was observed in Mmp3-KO mice.17 In contrast, an increase in VSMC content was observed within brachiocephalic plaques of Mmp7-KO mice,54 consistent with a proapoptotic role of MMP-7 on VSMCs.37 Lesion size and macrophage numbers were reduced in Mmp8-KO mice, suggesting improved stability.55 Conflicting observations have been reported in Mmp9-KO mice; whereas aortic plaque burden and macrophage number were reduced in one study examining the aortic root,56 increased plaque size, macrophage content, and buried fibrous layers were detected in another focusing on the brachiocephalic artery.54 In agreement with promoting plaque stability, reduced VSMC migration and associated neointimal formation was observed in Mmp9-KO mice.17 Moreover, macrophage-specific overexpression of pro-MMP-9 did not influence lesion instability in arterial plaques.57 Conversely, intraluminal overexpression of pro-MMP-9 triggered intraplaque hemorrhage in advanced lesions (but not intermediate plaques) induced in carotid arteries by collar implantation in Apoe-KO mice.58 Additionally, nonphysiological overexpression of a fully autoactivated form of MMP-9 augmented plaque progression.57
Mmp12 KO reduced brachiocephalic artery lesion area and number of buried fibrous layers,54 while also decreasing aortic elastin fragmentation.56 Additionally, the intraplaque ratio between VSMCs and macrophages was favorably increased toward VSMC content, implying heightened stability, in part through modulation of monocyte/macrophage invasion and apoptosis.42 In support, macrophage overexpression of an active form of MMP-12 in rabbits increased plaque size and increased inflammation,59 suggesting that MMP-12 promotes plaque progression and instability. Deletion of MMP-13 or MMP-14 had negligible effects on plaque size and composition, but increased fibrillar collagen content, indicating a role in plaque instability.60,61 Taken together, these findings indicate that some MMPs (such as MMP-2, -3, and -9) may exert a protective role in atherosclerosis by promoting VSMC growth and associated fibrous cap formation, and therefore enhance plaque stability. Conversely, other MMPs (including MMP-7, -8, -12, -13, and -14) may promote plaque progression through increased matrix degradation, inflammation, and susceptibility to apoptosis, and thus participate in triggering of plaque rupture.
In advocacy, transgenic or gene therapy-directed systemic overexpression of TIMP-1 failed to retard plaque progression,66,68 although atherogenesis was retarded in another study.67 Similarly TIMP-1 deficiency had no effect on plaque burden in one study,64 while increasing lesion size in another.65 Conversely, both short-term and long-term overexpression of TIMP-2 but not TIMP-1 retarded progression of advanced plaques, in part through inhibiting monocyte/macrophage invasion and their susceptibility to apoptosis.66 These studies lend further support for pro- and antiatherosclerotic effects of MMPs, as TIMP-1 and -2 display salient differences in their inhibitory efficacies toward specific MMPs, such as MT-MMPs.13 These findings may explain why the results of in vivo studies utilizing synthetic compounds containing zinc-chelating groups (such as thiol or hydroxamate groups or tetracycline derivates), which serve as broad-spectrum MMP inhibitors, have been so disappointing. Indeed, a hydroxamic acid-based, nonselective MMP inhibitor exhibited no beneficial effects on plaque development or progression in LDLR-KO mice33 or Apoe-KO animals.63 Similarly, the widely used antibiotic doxycycline, which also displays broad-spectrum MMP-inhibitory properties, failed to prevent plaque development in Apoe-KO mice.62 Moreover, treatment with doxycycline in two independent, randomized, double-blind, and placebo-controlled clinical trials in patients with symptomatic coronary and carotid artery disease failed to have any positive effects on plaque phenotype or clinical outcome.69,70
Accordingly, selectively targeting MMPs with a clear detrimental role represents a more effective approach for retarding atherosclerotic plaque progression. In point of fact, two recent studies employing selective inhibitors to MMP-1242 or MMP-1343 have supported this tactic. Selective MMP-12 inhibition blocked plaque progression and improved stability through reduction of lipid-core expansion and macrophage apoptosis, increased VSMC-to-macrophage ratio, decreased plaque calcification, and attenuated elastinolysis.42 All these effects, together with a reduction of buried fibrous layers in brachiocephalic plaques, mirrored those observed in Mmp12/Apoe double-KO mice.54 Similarly a highly specific inhibitor toward MMP-1343 retarded intraplaque collagenolytic activity, preserving fibrillar collagen levels in plaques, which would confer increased plaque stability, and resembled the effects previously observed in Mmp13-KO mice.60 Therefore, these proof-of-principle studies in mice provide motivation to translate selective MMP-inhibitor treatment to human patients harboring atherosclerosis. Two more recent studies have highlighted the potential of targeting micro-ribonucleic acids that modulate MMP/TIMP expression as novel therapeutic avenues for preventing plaque progression, miR-24 regulation of macrophage MMP-14 expression,71 and miR-712 modulation of endothelial cell TIMP-3 levels.72
An array of studies in cells, animal models, and humans has decisively established that MMPs play a central role in the development, progression, and rupture of atherosclerotic plaques. Additionally, the functions of MMPs in all vascular cell types and the subsequent consequences for atherosclerosis have been further elucidated. Together, this panoply of work has highlighted that modulation of MMP activity can reverse atherosclerosis, but that broad-spectrum MMP inhibition cannot replicate these properties, possibly owing to effects on both beneficial and detrimental MMPs. Consequently, it is now accepted that inhibitors with restricted specificity toward individual MMPs, such as MMP-12 and -13, are desirable for translation to man, particularly in the context of atherosclerotic plaque stabilization.
Abdominal aortic aneurysms
Although found at various locations throughout the vascular tree, abdominal aortic aneurysms (AAAs) demonstrate predominance, and it is estimated that up to 8% of men aged over 65 years and up to 2.2% of women of the same age harbor aneurysms.73 Patients with AAAs frequently have atherosclerosis, and associations have been shown in meta-analysis studies.73 Moreover, intimal atherosclerosis is commonly present in AAA lesions,74 although the composition is different compared to coronary and carotid plaques, and medial elastin fragmentation is more prevalent.73 Consequently, AAAs are considered a form of atherosclerosis with subtle differences in etiology to those observed in nascent atherosclerosis, and are regularly referred to as “atherosclerotic aneurysms”.73,75,76 Pathological observations suggest that ECM remodeling in unison with inflammatory cell infiltration, including macrophages at both the adventitial and medial aspects is a striking feature of human atherosclerotic AAAs,74,77 particularly the transition of small “silent” aortic dilatations to large clinically relevant AAAs.76 Similarly, macrophage accumulation is a common feature in animal models of AAAs, irrespective of species or stimuli, and throughout all stages of development.78 Moreover, imaging of the abdominal aorta of angiotensin (Ang)-II-infused Apoe-KO mice demonstrated an association between persistent macrophage accumulation and AAA pathogenesis.79
It is considered that the principal detrimental role of macrophages in AAAs is their release of proteolytic enzymes, which are responsible for the excessive loss of ECM, especially elastin and fibrillar collagens, which characterize AAA progression and rupture.77 MMPs have been demonstrated to be abundantly expressed by macrophage infiltrates within AAAs.80 There is already substantial evidence that increased expression and activity of various MMPs play an important role in the pathogenesis of AAAs.78,81 For instance, expression of MMP-1, -2, -3, -8, -9, -12, -13, and -14 is increased in aneurysmal tissues compared to control nondiseased arteries (Table 1).82 Furthermore, plasma levels of MMP-9 are elevated in patients with AAAs compared with controls.83
Accordingly, a plethora of in vivo studies have been undertaken to elucidate which MMPs modulate AAA formation (Table 4).33,53,84–104 It was shown that MMP-2,84 MMP-3,53 MMP-9,84–86 MMP-12,85,87,88 MMP-13,89 and MMP-14 KO mice90 exhibit attenuated experimental aneurysm formation (assessed principally as aortic dilatation) compared to wild-type controls. This evidence suggests that MMPs are vitally important in the formation of aneurysms, and that MMP inhibition may be a potential therapeutic strategy for aneurysms. In fact, a multitude of studies have evaluated the effects of MMP inhibitors or pleiotropic compounds with MMP-inhibitory properties in human aneurysmal samples, organ culture, or animal models of AAAs.81 For instance, animal studies with doxycycline, a tetracycline analog that reduces both MMP-9 expression and activity attenuated aneurysm formation in elastase-induced,85,95–97 calcium chloride application,98,99 and AngII-induced100–102 rodent models. Results from three preliminary human studies suggest that these experimental findings may also apply to human disease.105–107 CGS 27023A, batimastat, and RS132908, all hydroxamate-based broad-spectrum MMP inhibitors, also suppressed the expansion of experimental AAAs in murine models.33,103,104 However, other studies have reported more modest effects or no reduction in AAAs after MMP inhibition.78,81
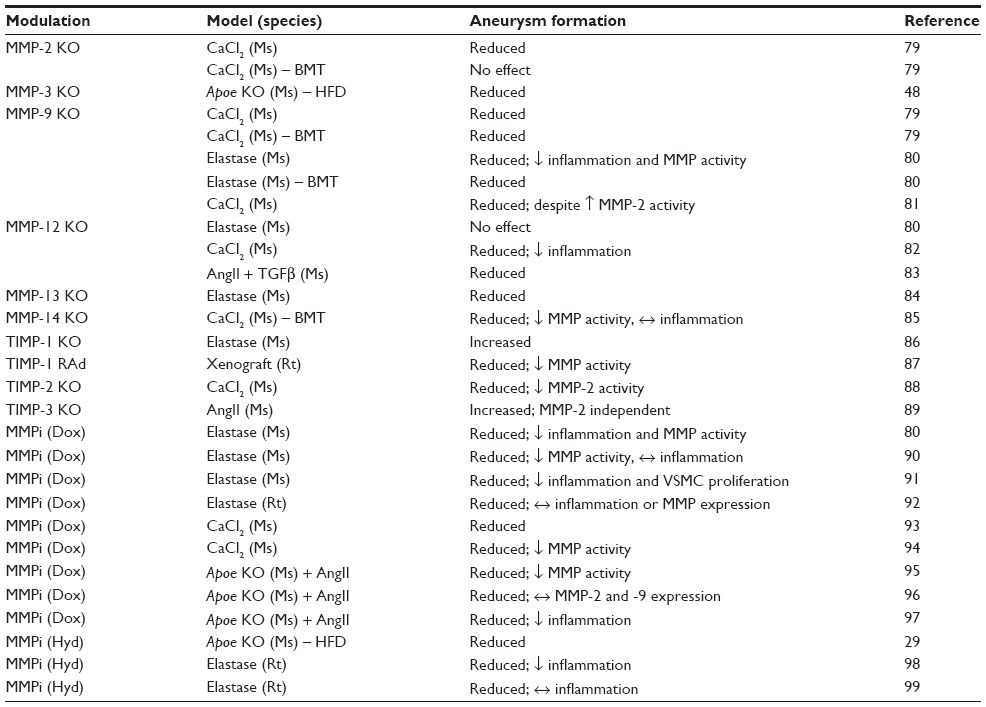 | Table 4 Results of in vivo animal studies evaluating the effects of modulating matrix metalloproteinases (MMPs) or tissue inhibitors of metalloproteinases (TIMPs) on abdominal aortic aneurysm formation and cellular composition, using recombinant adenoviral (RAd) overexpression, gene knockout (KO), or pharmacological inhibitors of MMPs (MMPis) |
Nonetheless, an endogenous tissue inhibitor of metalloproteinases, TIMP-1, appears to have a protective effect on AAA formation, since Timp1-KO mice have larger aneurysms than wild-type mice,91 and overexpression of TIMP-1 prevents elastin degradation, aneurysm formation, and rupture in a rat model of AAAs.92 Conversely, KO of TIMP-2 reduced AAA formation, although this may have been through reduced MMP-2 activation.93 Interestingly, TIMP-3 expression has been shown to be elevated in plasma from AAA patients108 and aneurysmal tissues, primarily in macrophages,109 and is considered a positive-feedback mechanism that reflects an effort to counteract MMP activity.110 In support, a recent study in nonatherosclerotic mice demonstrated that Timp3 gene deletion triggered adverse remodeling of the abdominal aorta in response to AngII, in part through increased inflammation and associated proteolytic activity,94 suggesting that restoring TIMP-3 levels may be protective against AAA progression and rupture.
The promising results observed in single-MMP or TIMP-modified animal models, alongside the more ambivalent findings seen with broad-spectrum MMP inhibitors in both mice and humans, again suggest that the roles of individual MMPs are of more relevance, and thus selective MMP inhibition is required for therapeutic modulation of AAA progression and rupture. It must also be noted that the majority of the clinical trials reported were underpowered and exhibited other ambiguities in experimental design.111 Nonetheless, two large, pivotal trials are in progress, and their outcomes are eagerly anticipated.112
Post-myocardial infarction remodeling
Patients who have had a previous MI have an increased susceptibility to heart failure, due primarily to aberrant structural and functional changes to the left ventricle, termed post-MI remodeling. The myocardial damage rendered subsequently to an MI and ensuing ischemic injury triggers a number of cellular and ECM events. These include the expression of a multitude of inflammatory mediators and chemokines, recruitment and infiltration of inflammatory cells (such as neutrophils and monocytes), proliferation and transdifferentiation of cardiac fibroblasts, and the initiation of ECM synthesis and proteolysis pathways.113 Moreover, at the injury site, macrophages themselves orchestrate a myriad of responses, including removal of necrotic cardiac myocytes and apoptotic neutrophils; secretion of cytokines, chemokines, and growth factors; and modulation of phases of the angiogenic response.114 Indeed, neovascularization is an important aspect of effective post-MI remodeling, and consequently promoting angiogenesis is considered a therapeutic strategy to prevent heart failure. Intriguingly, MMP activity is considered a vital determinant during the initiation and growth of angiogenic vessels.4 Together, these processes are considered a homeostatic wound-healing response that becomes maladaptive due to dysregulated ECM turnover and defective inflammation resolution, resulting in adverse post-MI remodeling and progression to heart failure.115 In particular, recruited inflammatory cells secrete numerous proteases, including MMPs, which facilitates their margination while also degrading the recently formed ECM at the periphery of the infarct, and it is this overlapping balance between inflammation-mediated proteolysis and fibrosis-driven remodeling that is fundamental for efficient healing and scar formation.114 Accordingly, there has been considerable interest in the spatial and temporal expression patterns of MMPs during post-MI remodeling. Indeed, a myriad of MMPs are upregulated post-MI by both resident cardiac cells and newly recruited inflammatory cells (Table 1), and are also detecTable in the circulation.116 More recently, researchers have utilized salient animal models to elucidate the contribution of MMPs during post-MI remodeling (Table 5).117–137
 | Table 5 Results of in vivo animal studies evaluating the effects of modulating matrix metalloproteinases (MMPs) or tissue inhibitors of metalloproteinases (TIMPs) on post-MI remodeling, using recombinant adenoviral (RAd) overexpression (Ovex), gene knockout (KO [KO* < heterozygote]), gene knockdown (KD), transgenic (Tg), or pharmacological inhibitors of MMPs (Inhib) |
Inhibition of MMP-2 activity through gene deletion was shown to improve survival rates after left anterior descending coronary artery ligation-induced MI, compared to wild-type control animals.117 This benefit was afforded primarily due to inhibition of cardiac rupture, despite negligible effects on infarct size and heart function. Similar findings were reported by Matsumura et al118 employing either Mmp2-KO mice or an MMP-2-selective inhibitor. However, it was noted that MMP-2 loss/inhibition reduced the inflammatory infiltrate at early time points, potentially resulting in failed clearance of necrotic cardiomyocytes. It should also be highlighted that the MMP-2 inhibitor used was not selective and also targeted other MMPs.118 Deletion of Mmp3 had insignificant effects on post-MI remodeling and survival.119 Cardiac rupture and mortality were attenuated in Mmp7-KO mice alongside an improvement in cardiac function, potentially due to a novel role for MMP-7 in modulating connexin-43 levels.120
While modulation of MMP-9 failed to impact on infarct size, benefits on heart function and reduced inflammation have been reported,119,121–123 although improved survival rates were observed in only one study.119 The beneficial effects of reduced MMP-9 levels are considered to be through modulating fibrosis121 and improving post-MI angiogenesis,122 although MMP-9 has also been shown to promote angiogenesis after ischemic injury.138 Intriguingly, macrophage-specific overexpression did not influence macrophage accumulation at the infarct site, but did favorably alter the inflammatory milieu and limit ECM synthesis to improve left ventricular (LV) function post-MI.123 Mmp12 deletion did not influence infarct size, inflammation, or survival subsequent to left anterior descending coronary artery ligation.119
Several studies utilizing either Mmp14 heterozygote gene-knockdown mice124,125 or cardiac-restricted overexpression of MMP-14124,126 have collectively demonstrated that perturbed MMP-14 expression and activity affords a distinct survival advantage associated with improved cardiac function through preservation of the fibrillar collagen network in a TGFβ-dependent manner, although reduced inflammation and scar size was also noted.125 Mmp28 deficiency aggravated post-MI remodeling and resulted in decreased survival, due to increased cardiac rupture rates and worse function compared with wild-type controls. Mmp28 deletion also resulted in a reduced inflammatory response, as well as decreases in cardiac fibroblast numbers, ECM deposition, and collagen cross-linking, attribuTable to impaired M2 macrophage activation in the absence of MMP-28.127 Intriguingly, this study suggests that similar to other cardiovascular pathologies, MMPs play divergent roles in the MI setting, and MMP-28 may represent a beneficial protease.
Studies employing animal models of post-MI remodeling where TIMPs were modulated have further supported a deleterious role for MMPs, although differences in changes in myocardial structure were observed. Experimental MI induction in Timp1-deficient mice accelerated adverse LV remodeling, resulting in abrogated heart function,128,129 due in part to decreased ECM structural integrity linked to increased MMP activity. Indeed, broad-spectrum MMP inhibition could partially rescue the Timp1-KO phenotype,129 while adenoviral-directed increased plasma levels of TIMP-1 improved post-MI survival alongside retardation of the infarct inflammatory response.139 Accelerated post-MI remodeling was observed in Timp2-KO mice compared to wild-type controls, which was associated with increased inflammation and infarct size, although no effects on survival were observed.130 Moreover, Timp2 deficiency resulted in decreased MMP-2 activity after MI induction, while overall myocardial collagenolysis was increased due to heightened MMP-14 activity, underscoring the selective regulation of adverse MMP-14 remodeling by TIMP-2.130 In support, adenoviral-mediated myocardial delivery of the TIMP-2 gene reduced MMP activity and improved post-MI survival, while limiting inflammatory cell recruitment and adverse LV remodeling.131
Three independent studies have demonstrated that MI induction in Timp3-KO mice was associated with increased cardiac rupture and reduced survival.132–134 The mechanisms underlying these profound effects were postulated as accelerated ECM degradation and inflammatory cytokine activation,132 heightened and persistent myocardial inflammation,133 and via EGF/EGF receptor (EGFR) signaling and downregulation of TGFβ1 expression and collagen synthesis.134 Indeed, it was demonstrated that inhibition of EGFR by cetuximab, an EGFR-neutralizing antibody, protects against cardiac rupture and improves survival in Timp3-KO mice.134 Eckhouse et al exploited the high affinity TIMP-3 has to the ECM by delivering to the myocardium recombinant TIMP-3 that was encapsulated in a hyaluronic acid-based gel.135 This approach retarded infarct expansion and LV remodeling, providing proof of principle that targeted delivery of TIMPs may have therapeutic potential for adverse post-MI remodeling. Finally, Timp4-KO mice were more susceptible to MI-induced dysfunctional LV remodeling and increased mortality, in part through elevated inflammation and MMP activity, as coadministration of a broad-spectrum MMP inhibitor rescued the observed phenotype.136 Concomitantly, myocardial targeted adenoviral or transgenic TIMP-4 overexpression perturbed adverse LV remodeling post-MI in mice, which was associated with reduced MMP activity, while fibrillar collagen synthesis and content was increased.137
Due to the overwhelming evidence outlined implicating MMPs as orchestrators of adverse post-MI remodeling, the hypothesized beneficial effects of MMP inhibition have been explored. A multitude of animal studies have been conducted where global MMP inhibition has been assessed pre-, during, and post-MI, demonstrating in most studies that broad-spectrum MMP inhibition afforded protection from adverse post-MI LV remodeling, while infarct size and mortality were rarely affected.140 Despite the plethora of in vivo findings in relevant animal models, only a limited number of approaches have been advanced to the clinical arena. Firstly, the PREMIER study evaluated the potential beneficial effect of a hydroxamate-based broad-spectrum MMP inhibitor (PG11680) in patients who had suffered an MI.141 Disappointingly, adverse LV remodeling and clinical outcomes were unaffected, despite previously observed beneficial effects in a preclinical animal model with a similar compound.142 The inefficacy of the PREMIER trial may be attribuTable to a myriad of factors, including an inadequate dosing regime and a poorly selected end point. Indeed, the plasma profiles achieved in patients receiving PG11680 were not reported, casting further doubt on the validity of the study. Further potentially confounding factors include inhibition of possible beneficial MMPs, spatial and/or temporal effects on MMP activity, and indiscriminate effects on different resident and infiltrating cell types. Encouragingly, a very recent trial (TIPTOP) has yielded more promising results.143 Administration of submicrobial doses of doxycycline given to patients postreperfusion subsequent to an MI improved heart function and reduced infarct size compared to placebo controls.143 Although encouraging, larger clinical trials are warranted to further validate these preliminary findings and to test chemically modified tetracycline derivatives, which express a safer clinical profile.
Taken together, the numerous findings discussed highlight that potential therapeutic strategies to modulate adverse post-MI remodeling may be more effective when inhibiting select subsets or individual MMPs rather than global inhibition, with MMP-28 a noTable exception.127 Moreover, stratagems to direct inhibition toward specific sites within the myocardium at precise time points and within select cell types may prove most efficacious.
Conclusion
MMPs and their endogenous inhibitors play fundamental roles in the progression of pertinent cardiovascular pathologies, including intimal formation, atherosclerosis, AAAs, and adverse post-MI remodeling (Figure 2). Genetic manipulation studies primarily performed in mice, alongside associative studies in human pathological tissues, have established the roles and functions of individual MMPs. Accordingly, varying methodologies to restore or overexpress TIMPs have proven effective in reducing MMP activity in vivo and rescuing pathological phenotypes. Lamentably, the utilization of broad-spectrum MMP inhibitors has not consistently replicated the effects observed with TIMPs in pertinent preclinical models or in clinical trials in man. Such poor translation may be due in part to the beneficial as well as detrimental effects MMPs exert in numerous cardiovascular pathologies, alongside divergent effects on differing cell types. Accordingly, prominence has moved toward the development of MMP inhibitors that display restricted specificity, as recently demonstrated in the atherosclerosis-research field with inhibitors directed toward MMP-1242 and MMP-13.43 Additionally, consideration of the temporal and spatial expression of MMPs and their activity in the differing cardiovascular pathologies is necessary to ensure maximal efficacy of any potential inhibitor-based therapeutic approaches. Furthermore, it may be advantageous to target specific cell types, such as macrophages, which may spare the positive actions of certain MMPs within beneficial cell types, although attention to the stage of disease progression is paramount, as some cell-specific MMPs will have divergent effects dependent on the disease stage. Consequently, in spite of caveats in the translation of findings in animal models to human pathologies, the reviewed studies instill a renewed impetus for future clinical trials of selective MMP inhibitors in prominent cardiovascular pathologies. Indeed, there are several clinical trials under way assessing the potential of elective MMP inhibitors in several cardiovascular diseases, including atherosclerosis, AAAs, and post-MI remodeling.
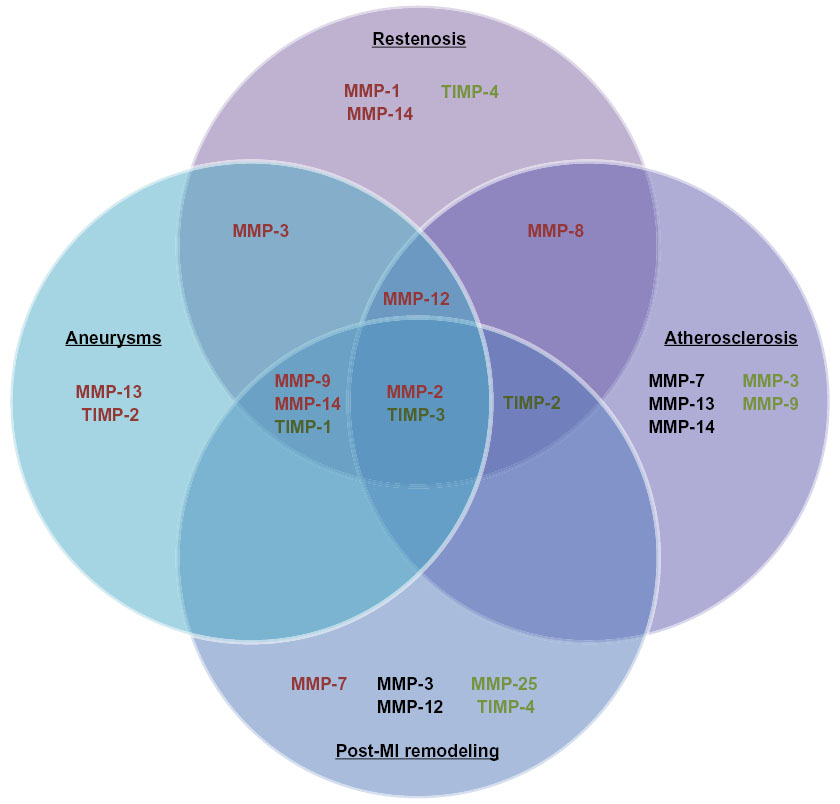 | Figure 2 This diagram illustrates the matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) determined detrimental (red), neutral (black), or beneficial (green) in animal models of intimal formation, abdominal aortic aneurysms, atherosclerosis, and post-myocardial infarction (MI) remodeling. |
Disclosure
The author reports no conflicts of interest in this work.
References
Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–573. | |
Somerville RP, Oblander SA, Apte SS. Matrix metalloproteinases: old dogs with new tricks. Genome Biol. 2003;4:1–11. | |
Johnson JL. Matrix metalloproteinases: influence on smooth muscle cells and atherosclerotic plaque stability. Expert Rev Cardiovasc Ther. 2007;5:265–282. | |
van Hinsbergh VW, Koolwijk P. Endothelial sprouting and angiogenesis: matrix metalloproteinases in the lead. Cardiovasc Res. 2008;78:203–212. | |
Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat Rev Immunol. 2013;13:649–665. | |
Murphy G. Fell-Muir Lecture: Metalloproteinases: from demolition squad to master regulators. Int J Exp Pathol. 2010;91:303–313. | |
Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases. Circ Res. 2003;93:827–839. | |
Murphy G, Nagase H. Localizing matrix metalloproteinase activities in the pericellular environment. FEBS J. 2011;278:2–15. | |
Stefanidakis M, Koivunen E. Cell-surface association between matrix metalloproteinases and integrins: role of the complexes in leukocyte migration and cancer progression. Blood. 2006;108:1441–1450. | |
Kwon MJ, Jang B, Yi JY, Han IO, Oh ES. Syndecans play dual roles as cell adhesion receptors and docking receptors. FEBS Lett. 2012;586: 2207–2211. | |
Newby AC. Dual role of matrix metalloproteinases (matrixins) in neointima formation and atherosclerotic plaque rupture. Physiol Rev. 2005;85:1–31. | |
Johnson JL. Emerging regulators of vascular smooth muscle cell function in the development and progression of atherosclerosis. Cardiovasc Res. 2014;103:452–460. | |
Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci. 2002;115: 3719–3727. | |
Austin KM, Nguyen N, Javid G, Covic L, Kuliopulos A. Noncanonical matrix metalloprotease-1-protease-activated receptor-1 signaling triggers vascular smooth muscle cell dedifferentiation and arterial stenosis. J Biol Chem. 2013;288:23105–23115. | |
Kuzuya M, Kanda S, Sasaki T, et al. Deficiency of gelatinase a suppresses smooth muscle cell invasion and development of experimental intimal hyperplasia. Circulation. 2003;108:1375–1381. | |
Johnson C, Galis ZS. Matrix metalloproteinase-2 and -9 differentially regulate smooth muscle cell migration and cell-mediated collagen organization. Arterioscler Thromb Vasc Biol. 2004;24:54–60. | |
Johnson JL, Dwivedi A, Somerville M, George SJ, Newby AC. Matrix metalloproteinase (MMP)-3 activates MMP-9 mediated vascular smooth muscle cell migration and neointima formation in mice. Arterioscler Thromb Vasc Biol. 2011;31:e35–e44. | |
Xiao Q, Zhang F, Grassia G, et al. Matrix metalloproteinase-8 promotes vascular smooth muscle cell proliferation and neointima formation. Arterioscler Thromb Vasc Biol. 2014;34:90–98. | |
Cho A, Reidy MA. Matrix metalloproteinase-9 is necessary for the regulation of smooth muscle cell replication and migration after arterial injury. Circ Res. 2002;91:845–851. | |
Galis ZS, Johnson C, Godin D, et al. Targeted disruption of the matrix metalloproteinase-9 gene impairs smooth muscle cell migration and geometrical arterial remodeling. Circ Res. 2002;91:852–859. | |
Filippov S, Koenig GC, Chun TH, et al. MT1-matrix metalloproteinase directs arterial wall invasion and neointima formation by vascular smooth muscle cells. J Exp Med. 2005;5:663–671. | |
Lijnen HR, Soloway P, Collen D. Tissue inhibitor of matrix metalloproteinases-1 impairs arterial neointima formation after vascular injury in mice. Circ Res. 1999;85:1186–1191. | |
Forough R, Koyama N, Hasenstab D, et al. Overexpression of tissue inhibitor of matrix metalloprotienase-1 inhibits vascular smooth muscle cell functions in vitro and in vivo. Circ Res. 1996;79:812–820. | |
Dollery CM, Humphries SE, McClelland A, Latchman DS, McEwan JR. Expression of tissue inhibitor of matrix metalloproteinases 1 by use of an adenoviral vector inhibits smooth muscle cell migration and reduces neointimal hyperplasia in the rat model of vascular balloon injury. Circulation. 1999;99:3199–3205. | |
Furman C, Luo Z, Walsh K, et al. Systemic tissue inhibitor of metalloproteinase-1 gene delivery reduces neointimal hyperplasia in balloon-injured rat carotid artery. FEBS Lett. 2002;531:122–126. | |
Cheng L, Mantile G, Pauly R, et al. Adenovirus-mediated gene transfer of the human tissue inhibitor of metalloproteinase-2 blocks vascular smooth muscle cell invasiveness in vitro and modulates neointimal development in vivo. Circulation. 1998;98:2195–2201. | |
Hu YH, Baker AH, Zou YP, Newby AC, Xu QB. Local gene transfer of tissue inhibitor of metalloproteinase-2 influences vein graft remodeling in a mouse model. Arterioscler Thromb Vasc Biol. 2001;21: 1275–1280. | |
George SJ, Lloyd CT, Angelini GD, Newby AC, Baker AH. Inhibition of late vein graft neointima formation in human and porcine models by adenovirus-mediated overexpression of tissue inhibitor of metalloproteinase-3. Circulation. 2000;101:296–304. | |
George SJ, Wan S, Hu J, MacDonald R, Johnson JL, Baker AH. Sustained reduction of vein graft neointima formation by ex vivo TIMP-3 gene therapy. Circulation. 2011;124:S135–S142. | |
Guo YH, Gao W, Li Q, Li PF, Yao PY, Chen KH. Tissue inhibitor of metalloproteinases-4 suppresses vascular smooth muscle cell migration and induces cell apoptosis. Life Sci. 2004;75:2483–2493. | |
Zempo N, Koyama N, Kenagy RD, Lea HJ, Clowes AW. Regulation of vascular smooth muscle cell migration and proliferation in vitro and in injured rat arteries by a synthetic matrix metalloproteinase inhibitor. Arterioscler Thromb Vasc Biol. 1996;16:28–33. | |
Bendeck MP, Irvin C, Reidy MA. Inhibition of matrix metalloproteinase activity inhibits smooth muscle cell migration but not neointimal thickening after arterial injury. Circ Res. 1996;78:38–43. | |
Prescott MF, Sawyer WK, Von Linden-Reed J, et al. Effect of matrix metalloproteinase inhibition on progression of atherosclerosis and aneurysm in LDL receptor-deficient mice overexpressing MMP3, MMP-12, and MMP-13 and on restenosis in rats after balloon injury. Ann N Y Acad Sci. 1999;878:179–190. | |
Bendeck MP, Conte M, Zhang M, Nili N, Strauss BH, Farwell SM. Doxycyline modulates smooth muscle cell growth, migration, and matrix remodeling after arterial injury. Am J Pathol. 2002;160:1089–1095. | |
Islam MM, Franco CD, Courtman DW, Bendeck MP. A nonantibiotic chemically modified tetracycline (CMT-3) inhibits intimal thickening. Am J Pathol. 2003;163:1557–1566. | |
Pinney SP, Chen HJ, Liang DX, Wang XY, Schwartz A, Rabbani LE. Minocycline inhibits smooth muscle cell proliferation, migration and neointima formation after arterial injury. J Cardiovasc Pharmacol. 2003;42:469–476. | |
Williams H, Johnson JL, Jackson CL, White SJ, George SJ. MMP-7 mediates cleavage of N-cadherin and promotes smooth muscle cell apoptosis. Cardiovasc Res. 2010;87:137–146. | |
Dwivedi A, Slater SC, George SJ. MMP-9 and -12 cause N-cadherin shedding and thereby β-catenin signalling and vascular smooth muscle cell proliferation. Cardiovasc Res. 2009;81:178–186. | |
Lyon CA, Koutsouki E, Aguilera CM, Blaschuk OW, George SJ. Inhibition of N-cadherin retards smooth muscle cell migration and intimal thickening via induction of apoptosis. J Vasc Surg. 2010;52: 1301–1309. | |
Cherr GS, Motew SJ, Travis JA, et al. Metalloproteinase inhibition and the response to angioplasty and stenting in atherosclerotic primates. Arterioscler Thromb Vasc Biol. 2002;22:161–166. | |
Peterson JT. The importance of estimating the therapeutic index in the development of matrix metalloproteinase inhibitors. Cardiovasc Res. 2006;69:677–687. | |
Johnson JL, Devel L, Czarny B, et al. A selective matrix metalloproteinase-12 inhibitor retards atherosclerotic plaque development in apolipoprotein E-knockout mice. Arterioscler Thromb Vasc Biol. 2011;31:528–535. | |
Quillard T, Tesmenitsky Y, Croce K, et al. Selective inhibition of matrix metalloproteinase-13 increases collagen content of established mouse atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:2464–2472. | |
Libby P, Tabas I, Fredman G, Fisher EA. Inflammation and its resolution as determinants of acute coronary syndromes. Circ Res. 2014;114: 1867–1879. | |
Hergenreider E, Heydt S, Tréguer K, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. 2012;14:249–256. | |
Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. 2014;114:1852–1866. | |
Horozoglu C, Özdeş T, Turgay E, Ünaltuna NE. Expression of MMP-15 and MMP-24 in atherosclerotic and nonatherosclerotic coronary arteries. Metalloproteinases Med. 2014;1:15–20. | |
Van der Laan PA, Reardon CA, Getz GS. Site specificity of atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:12–22. | |
Getz GS, Reardon CA. Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:1104–1115. | |
Sluimer JC, Daemen MJ. Novel concepts in atherogenesis: angiogenesis and hypoxia in atherosclerosis. J Pathol. 2009;218:7–29. | |
Lemaître V, O’Byrne TK, Borczuk AC, Okada Y, Tall AR, D’Armiento J. ApoE knockout mice expressing human matrix metalloproteinase-1 in macrophages have less advanced atherosclerosis. J Clin Invest. 2001;107:1227–1234. | |
Kuzuya M, Nakamura K, Sasaki T, Cheng XW, Itohara S, Iguchi A. Effect of MMP-2 deficiency on atherosclerotic lesion formation in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:1120–1125. | |
Silence J, Lupu F, Collen D, Lijnen HR. Persistence of atherosclerotic plaque but reduced aneurysm formation in mice with stromelysin-1 (MMP-3) gene inactivation. Arterioscler Thromb Vasc Biol. 2001;21: 1440–1445. | |
Johnson JL, George SJ, Newby AC, Jackson CL. Divergent effects of matrix metalloproteinases -3, -7, -9 and -12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc Natl Acad Sci U S A. 2005;102:15575–15580. | |
Laxton RC, Hu Y, Duchene J, et al. A role of matrix metalloproteinase-8 in atherosclerosis. Circ Res. 2009;105:921–929. | |
Luttun A, Lutgens E, Manderveld A, et al. Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E-deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation. 2004;109: 1408–1414. | |
Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in ApoE-deficient mice. J Clin Invest. 2006;116:59–69. | |
de Nooijer R, Verkleij CJ, von der Thuesen JH, et al. Lesional overexpression of matrix metalloproteinase-9 promotes intraplaque hemorrhage in advanced lesions, but not at earlier stages of atherogenesis. Arterioscler Thromb Vasc Biol. 2006;26:340–346. | |
Liang J, Liu E, Yu Y, et al. Macrophage metalloelastase accelerates the progression of atherosclerosis in transgenic rabbits. Circulation. 2006;113:1993–2001. | |
Deguchi JO, Aikawa E, Libby P, et al. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation. 2005;112:2708–2715. | |
Schneider F, Sukhova GK, Aikawa M, et al. Matrix metalloproteinase-14 deficiency in bone marrow-derived cells promotes collagen accumulation in mouse atherosclerotic plaques. Circulation. 2008;117:931–939. | |
Manning MW, Cassis LA, Daugherty A. Differential effects of doxycycline, a broad-spectrum matrix metalloproteinase inhibitor, on angiotensin II-induced atherosclerosis and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2003;23:483–488. | |
Johnson JL, Fritsche-Danielson R, Behrendt M, et al. Effect of broad-spectrum matrix metalloproteinase inhibition on atherosclerotic plaque stability. Cardiovasc Res. 2006;71:586–595. | |
Lemaître V, Soloway PD, D’Armiento J. Increased medial degradation with pseudo-aneurysm formation in apolipoprotein E-knockout mice deficient in tissue inhibitor of metalloproteinases-1. Circulation. 2003;107:333–338. | |
Silence J, Collen D, Lijnen HR. Reduced atherosclerotic plaque but enhanced aneurysm formation in mice with inactivation of the tissue inhibitor of metalloproteinase-1 (TIMP-1) gene. Circ Res. 2002;90: 897–903. | |
Johnson JL, Baker AH, Oka K, et al. Suppression of atherosclerotic plaque progression and instability by tissue inhibitor of metalloproteinase-2: involvement of macrophage migration and apoptosis. Circulation. 2006;113:2435–2444. | |
Rouis M, Adamy C, Duverger N, et al. Adenovirus-mediated overexpression of tissue inhibitor of metalloproteinase-1 reduces atherosclerotic lesions in apolipoprotein E-deficient mice. Circulation. 1999;100:533–540. | |
Cuaz-Pérolin C, Jguirim I, Larigauderie G, et al. Apolipoprotein E knockout mice over-expressing human tissue inhibitor of metalloproteinase 1 are protected against aneurysm formation but not against atherosclerotic plaque development. J Vasc Res. 2006;43:493–501. | |
Brown DL, Desai KK, Vakili BA, Nouneh C, Lee HM, Golub LM. Clinical and biochemical results of the metalloproteinase inhibition with subantimicrobial doses of doxycycline to prevent acute coronary syndromes (MIDAS) pilot trial. Arterioscler Thromb Vasc Biol. 2004; 24:733–738. | |
Axisa B, Loftus IM, Naylor AR, et al. Prospective, randomized, double-blind trial investigating the effect of doxycycline on matrix metalloproteinase expression within atherosclerotic carotid plaques. Stroke. 2002;33:2858–2864. | |
Di Gregoli K, Jenkins N, Salter R, White S, Newby AC, Johnson JL. MicroRNA-24 regulates macrophage behavior and retards atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34:1990–2000. | |
Son DJ, Kumar S, Takabe W, et al. The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nat Commun. 2013;4:3000. | |
Golledge J, Norman PE. Atherosclerosis and Abdominal aortic aneurysm: cause, response, or common risk factors? Arterioscler Thromb Vasc Biol. 2010;30:1075–1077. | |
Davies MJ. Aortic aneurysm formation. Lessons from human studies and experimental models. Circulation. 1998;98:193–195. | |
Michel JB, Martin-Ventura JL, Egido J, et al. Novel aspects of the pathogenesis of aneurysms of the abdominal aorta in humans. Cardiovasc Res. 2011;90:18–27. | |
Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2011;8:92–102. | |
Shimizu K, Mitchell RN, Libby P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2006;26:987–994. | |
Lu H, Rateri D, Bruemmer D, Cassis L, Daugherty A. Novel mechanisms of abdominal aortic aneurysms. Curr Atheroscler Rep. 2012;14:402–412. | |
Nahrendorf M, Keliher E, Marinelli B, et al. Detection of macrophages in aortic aneurysms by nanoparticle positron emission tomography-computed tomography. Arterioscler Thromb Vasc Biol. 2011;31:750–757. | |
Reeps C, Pelisek J, Seidl S, et al. Inflammatory infiltrates and neovessels are relevant sources of MMPs in abdominal aortic aneurysm wall. Pathobiology. 2009;76:243–252. | |
Newby AC. Matrix metalloproteinase inhibition therapy for vascular diseases. Vasc Pharmacol. 2012;56:232–244. | |
Aziz F, Kuivaniemi H. Role of matrix metalloproteinase inhibitors in preventing abdominal aortic aneurysm. Ann Vasc Surg. 2007;21: 392–401. | |
Hovsepian DM, Ziporin SJ, Sakurai MK, Lee JK, Curci JA, Thompson RW. Elevated plasma levels of matrix metalloproteinase-9 in patients with abdominal aortic aneurysms: a circulating marker of degenerative aneurysm disease. J Vasc Interv Radiol. 2000;11:1345–1352. | |
Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinase 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–632. | |
Pyo R, Lee JK, Shipley JM, et al. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–1649. | |
Ikonomidis JS, Barbour JR, Amani Z, et al. Effects of deletion of the matrix metalloproteinase 9 gene on development of murine thoracic aortic aneurysms. Circulation. 2005;112:I242–I248. | |
Longo GM, Buda SJ, Fiotta N, et al. MMP-12 has a role in abdominal aortic aneurysms in mice. Surgery. 2005;137:457–462. | |
Wang Y, Ait-Oufella H, Herbin O, et al. TGF-β activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J Clin Invest. 2010;120:422–432. | |
Lizarbe TR, TarÍn C, GÓmez M, et al. Nitric oxide induces the progression of abdominal aortic aneurysms through the matrix metalloproteinase inducer EMMPRIN. Am J Pathol. 2009;175:1421–1430. | |
Xiong W, Knispel R, MacTaggart J, Greiner TC, Weiss SJ, Baxter BT. Membrane-type 1 matrix metalloproteinase regulates macrophage-dependent elastolytic activity and aneurysm formation in vivo. J Biol Chem. 2009;284:1765–1771. | |
Eskandari MK, Vijungco JD, Flores A, Borensztajn J, Shively V, Pearce WH. Enhanced abdominal aortic aneurysm in TIMP-1-deficient mice. J Surg Res. 2005;123:289–293. | |
Allaire E, Forough R, Clowes M, Starcher B, Clowes AW. Local overexpression of TIMP-1 prevents aortic aneurysm degeneration and rupture in a rat model. J Clin Invest. 1998;102:1413–1420. | |
Xiong WF, Knispel R, Mactaggart J, Baxter BT. Effects of tissue inhibitor of metalloproteinase 2 deficiency on aneurysm formation. J Vasc Surg. 2006;44:1061–1066. | |
Basu R, Fan D, Kandalam V, et al. Loss of Timp3 gene leads to abdominal aortic aneurysm formation in response to angiotensin II. J Biol Chem. 2012;287:44083–44096. | |
Petrinec D, Liao S, Holmes DR, Reilly JM, Parks WC, Thompson RW. Doxycycline inhibition of aneurysmal degeneration in an elastase-induced rat model of abdominal aortic aneurysm: preservation of aortic elastin associated with suppressed production of 92 kD gelatinase. J Vasc Surg. 1996;23:336–346. | |
Sho E, Chu J, Sho M, et al. Continuous periaortic infusion improves doxycycline efficacy in experimental aortic aneurysms. J Vasc Surg. 2004;39:1312–1321. | |
Curci JA, Petrinec D, Liao S, Golub LM, Thompson RW. Pharmacologic suppression of experimental abdominal aortic aneurysms: a comparison of doxycycline and four chemically modified tetracyclines. J Vasc Surg. 1998;28:1082–1093. | |
Prall AK, Longo GM, Mayhan WG, et al. Doxycycline in patients with abdominal aortic aneurysms and in mice: comparison of serum levels and effect on aneurysm growth in mice. J Vasc Surg. 2002;35:923–929. | |
Sheth RA, Maricevich M, Mahmood U. In vivo optical molecular imaging of matrix metalloproteinase activity in abdominal aortic aneurysms correlates with treatment effects on growth rate. Atherosclerosis. 2010;212:181–187. | |
Turner GH, Olzinski AR, Bernard RE, et al. In vivo serial assessment of aortic aneurysm formation in apolipoprotein E-deficient mice via MRI. Circ Cardiovasc Imaging. 2008;1:220–226. | |
Vinh A, Gaspari TA, Liu HB, Dousha LF, Widdop RE, Dear AE. A novel histone deacetylase inhibitor reduces abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. J Vasc Res. 2008;45:143–152. | |
Tedesco MM, Terashima M, Blankenberg FG, et al. Analysis of in situ and ex vivo vascular endothelial growth factor receptor expression during experimental aortic aneurysm progression. Arterioscler Thromb Vasc Biol. 2009;29:1452–1457. | |
Bigatel DA, Elmore JR, Carey DJ, Cizmeci-Smith G, Franklin DP, Youkey JR. The matrix metalloproteinase inhibitor BB-94 limits expansion of experimental abdominal aortic aneurysms. J Vasc Surg. 1999;29:130–139. | |
Moore G, Liao S, Curci JA, et al. Suppression of experimental abdominal aortic aneurysms by systemic treatment with a hydroxamate-based matrix metalloproteinase inhibitor (RS 132908). J Vasc Surg. 1999;29:522–532. | |
Mosorin M, Juvonen J, Biancari F, et al. Use of doxycycline to decrease the growth rate of abdominal aortic aneurysms: a randomized, double-blind, placebo-controlled pilot study. J Vasc Surg. 2001;34:606–610. | |
Baxter BT, Pearce WH, Waltke EA, et al. Prolonged administration of doxycycline in patients with small asymptomatic abdominal aortic aneurysms: report of a prospective (phase II) multicenter study. J Vasc Surg. 2002;36:1–12. | |
Lindeman JH, Abdul-Hussien H, van Bockel JH, Wolterbeek R, Kleemann R. Clinical trial of doxycycline for matrix metalloproteinase-9 inhibition in patients with an abdominal aneurysm: doxycycline selectively depletes aortic wall neutrophils and cytotoxic T cells. Circulation. 2009;119:2209–2216. | |
Lamblin N, Ratajczak P, Hot D, et al. Profile of macrophages in human abdominal aortic aneurysms: a transcriptomic, proteomic, and antibody protein array study. J Proteome Res. 2010;9:3720–3729. | |
Lipp C, Lohoefer F, Reeps C, et al. Expression of a disintegrin and metalloprotease in human abdominal aortic aneurysms. J Vasc Res. 2012;49:198–206. | |
Tsarouhas K, Soufla G, Apostolakis S, et al. Transcriptional regulation of TIMPs in ascending aorta aneurysms. Thromb Res. 2010;126: 399–405. | |
Dodd BR, Spence RA. Doxycycline inhibition of abdominal aortic aneurysm growth: a systematic review of the literature. Curr Vasc Pharmacol. 2011;9:471–478. | |
Golledge J, Norman PE. Current status of medical management for abdominal aortic aneurysm. Atherosclerosis. 2011;217:57–63. | |
Spinale FG, Villarreal F. Targeting matrix metalloproteinases in heart disease: lessons from endogenous inhibitors. Biochem Pharmacol. 2014;90:7–15. | |
Lambert JM, Lopez EF, Lindsey ML. Macrophage roles following myocardial infarction. Int J Cardiol. 2008;130:147–158. | |
Thorp E. Contrasting inflammation resolution during atherosclerosis and post myocardial infarction at the level of monocyte/macrophage phagocytic clearance. Front Immunol. 2012;3:39. | |
Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007;87:1285–1342. | |
Hayashidani S, Tsutsui H, Ikeuchi M, et al. Targeted deletion of MMP-2 attenuates early LV rupture and late remodeling after experimental myocardial infarction. Am J Physiol Heart Circ Physiol. 2003; 285:H1229–H1235. | |
Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest. 2005;115:599–609. | |
Heymans S, Luttun A, Nuyens D, et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5:1135–1142. | |
Lindsey ML, Escobar GP, Mukherjee R, et al. Matrix metalloproteinase-7 affects connexin-43 levels, electrical conduction, and survival after myocardial infarction. Circulation. 2006;113:2919–2928. | |
Ducharme A, Frantz S, Aikawa M, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. | |
Lindsey ML, Escobar GP, Dobrucki LW, et al. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;290:H232–H239. | |
Zamilpa R, Ibarra J, de Castro Brás LE, et al. Transgenic overexpression of matrix metalloproteinase-9 in macrophages attenuates the inflammatory response and improves left ventricular function post-myocardial infarction. J Mol Cell Cardiol. 2012;53:599–608. | |
Zavadzkas JA, Mukherjee R, Rivers WT, et al. Direct regulation of membrane type 1 matrix metalloproteinase following myocardial infarction causes changes in survival, cardiac function, and remodeling. Am J Physiol Heart Circ Physiol. 2011;301:H1656–H1666. | |
Koenig GC, Rowe RG, Day SM, et al. MT1-MMP-dependent remodeling of cardiac extracellular matrix structure and function following myocardial infarction. Am J Pathol. 2012;180:1863–1878. | |
Spinale FG, Mukherjee R, Zavadzkas JA, et al. Cardiac restricted overexpression of membrane type-1 matrix metalloproteinase causes adverse myocardial remodeling following myocardial infarction. J Biol Chem. 2010;285:30316–30327. | |
Ma Y, Halade GV, Zhang J, et al. Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circ Res. 2013;112:675–688. | |
Creemers EE, Davis JN, Parkhurst AM, et al. Deficiency of TIMP-1 exacerbates LV remodeling after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2003;284:H364–H371. | |
Ikonomidis JS, Hendrick JW, Parkhurst AM, et al. Accelerated LV remodeling after myocardial infarction in TIMP-1-deficient mice: effects of exogenous MMP inhibition. Am J Physiol Heart Circ Physiol. 2005;288:H149–H158. | |
Kandalam V, Basu R, Abraham T, et al. TIMP2 deficiency accelerates adverse post-myocardial infarction remodeling because of enhanced MT1-MMP activity despite lack of MMP2 activation. Circ Res. 2010;106:796–808. | |
Ramani R, Nilles K, Gibson G, et al. Tissue inhibitor of metalloproteinase-2 gene delivery ameliorates postinfarction cardiac remodeling. Clin Transl Sci. 2011;4:24–31. | |
Tian H, Cimini M, Fedak PW, et al. TIMP-3 deficiency accelerates cardiac remodeling after myocardial infarction. J Mol Cell Cardiol. 2007;43:733–743. | |
Kandalam V, Basu R, Abraham T, et al. Early activation of matrix metalloproteinases underlies the exacerbated systolic and diastolic dysfunction in mice lacking TIMP3 following myocardial infarction. Am J Physiol Heart Circ Physiol. 2010;299:H1012–H1023. | |
Hammoud L, Lu X, Lei M, Feng Q. Deficiency in TIMP-3 increases cardiac rupture and mortality post-myocardial infarction via EGFR signaling: beneficial effects of cetuximab. Basic Res Cardiol. 2011;106:459–471. | |
Eckhouse SR, Purcell BP, McGarvey JR, et al. Local hydrogel release of recombinant TIMP-3 attenuates adverse left ventricular remodeling after experimental myocardial infarction. Sci Transl Med. 2014;6:223ra21. | |
Koskivirta I, Kassiri Z, Rahkonen O, et al. Mice with tissue inhibitor of metalloproteinases 4 (Timp4) deletion succumb to induced myocardial infarction but not to cardiac pressure overload. J Biol Chem. 2010;285:24487–24493. | |
Zavadzkas JA, Stroud RE, Bouges S, et al. Targeted overexpression of tissue inhibitor of matrix metalloproteinase-4 modifies post-myocardial infarction remodeling in mice. Circ Res. 2014;114:1435–1445. | |
Johnson C, Sung HJ, Lessner SM, Fini ME, Galis ZS. Matrix metalloproteinase-9 is required for adequate angiogenic revascularization of ischemic tissues: potential role in capillary branching. Circ Res. 2004;94:262–268. | |
Heymans S, Lupu F, Terclavers S, et al. Loss or inhibition of uPA or MMP-9 attenuates LV remodeling and dysfunction after acute pressure overload in mice. Am J Pathol. 2005;166:15–25. | |
Hughes BG, Schulz R. Targeting MMP-2 to treat ischemic heart injury. Basic Res Cardiol. 2014;109:1–19. | |
Hudson MP, Armstrong PW, Ruzyllo W, et al. Effects of selective matrix metalloproteinase inhibitor (PG-116800) to prevent ventricular remodeling after myocardial infarction: results of the PREMIER (Prevention of Myocardial Infarction Early Remodeling) Trial. J Am Coll Cardiol. 2006;48:15–20. | |
Yarbrough WM, Mukherjee R, Escobar GP, et al. Selective targeting and timing of matrix metalloproteinase inhibition in post-myocardial infarction remodeling. Circulation. 2003;108:1753–1759. | |
Cerisano G, Buonamici P, Valenti R, et al. Early short-term doxycycline therapy in patients with acute myocardial infarction and left ventricular dysfunction to prevent the ominous progression to adverse remodelling: the TIPTOP trial. Eur Heart J. 2014;35:184–191. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.