Back to Archived Journals » Metalloproteinases In Medicine » Volume 3
Matrix metalloproteinase protein inhibitors: highlighting a new beginning for metalloproteinases in medicine
Authors Mohan V, Talmi-Frank D, Arkadash V, Papo N, Sagi I
Received 12 January 2016
Accepted for publication 17 March 2016
Published 12 July 2016 Volume 2016:3 Pages 31—47
DOI https://doi.org/10.2147/MNM.S65143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yoshifumi Itoh
Vishnu Mohan,1 Dalit Talmi-Frank,1 Valeria Arkadash,2 Niv Papo,2 Irit Sagi1
1Department of Biological Regulation, Weizmann Institute of Science, Rehovot, 2Department of Biotechnology Engineering, Faculty of Engineering Sciences, Ben-Gurion University of the Negev, Beer-Sheva, Israel
Abstract: The development of therapeutic matrix metalloproteinase (MMP) inhibitors has evolved from broad-spectrum peptidomimetic inhibitors with deleterious side effects, to highly selective agents. These range from small molecules to antibodies, antisense inhibitors, and engineered N-terminal tissue inhibitors of metalloproteinase domain. The advances in inhibitor design along with promising new global molecular insights into MMP structures, the protease web, and the role of extracellular matrix in diseases have contributed toward a renewed interest in using MMPs as valid drug targets. This review aims to address the advances and challenges concerning the design, development, and current status of anti-MMP agents in this new era of post-broad-spectrum MMP inhibitors. Highly selective inhibitors of MMPs promise to usher in an era of specific targeting of diseased tissue proteolysis networks, with markedly reduced negative repercussions, and to uncover the molecular and mechanistic roles of MMP isoforms in cancer, inflammation, and infection.
Keywords: matrix metalloproteinase inhibitors, MMPI, small molecules, MMP-inhibiting monoclonal antibodies, engineered N-TIMP, miRNA
A Letter to the Editor has been received and published for this article.
Introduction
Matrix metalloproteinases (MMPs)1 play a pivotal role in extracellular matrix (ECM) remodeling during homeostasis as well as in dysregulated diseased tissues. These pleiotropic zinc endopeptidases cleave the core matrisome proteins2 and non-matrix substrates including cytokines, chemokines, adhesion molecules, growth factors, and their receptors. MMPs thus affect cell–cell signaling milieu3 and cellular contacts4 along with their key roles in proteolytic alteration of matrix architecture and its biophysical properties. Upregulation in cancer microenvironment brought MMPs to the limelight as candidate targets, but owing to their evolutionarily conserved central role in tissue proteolysis, they have also been implicated as candidate drug and theranostic (therapeutic as well as diagnostic) targets in diabetes, sepsis, neurodegenerative diseases, dentine erosion, ocular maladies, cardiovascular diseases, skin disorders, arthritis, respiratory tract disorders, glomerulonephritis, and inflammatory bowel disease.5–13
MMPs form a small subgroup of ECM-remodeling proteases within the metzincin family (containing an HEXXHXXGXXH consensus motif in their catalytic site). MMPs can be classified into six groups on the basis of substrate specificity: collagenases (MMP-1, MMP-8, and MMP-13), gelatinases (MMP-2 and MMP-9), stromelysins (MMP-3, MMP-10, and MMP-11), matrilysins (MMP-7 and MMP-26), membrane-type (MT) MMPs (MMP-14, MMP-15, MMP-16, MMP-17, MMP-24, and MMP-25), and others (MMP-12, MMP-19, MMP-20, MMP-21, MMP-23, MMP-27, and MMP-28), and into four groups on the basis of their domain organization (archetypal MMPs, matrilysins, gelatinases, or furin-activatable MMPs).1,14
The extensive research invested in developing their roles as drug targets is reflected by the fact that almost all of MMPs have been knocked down in murine models (17 out of 23). Surprisingly, only a small fraction of these mice present substantive spontaneous phenotypes;15 a probable reason behind it is suggested to be functional redundancy.16 Interestingly, human MMP genes occur in clusters (the most striking of which is at chromosome 11q22, containing nine out of 24 MMPs) pointing toward coevolution. This coevolution of enzymes could further explain the observed structural homology.
The MMPs are tightly regulated at transcriptional, translational, and protein levels (produced as inactive zymogens whose pro-peptides are shed to release active enzymes) and have a delicate balance with levels of their endogenous inhibitors: tissue inhibitors of metalloproteinases (TIMPs).17 Other endogenous inhibitors of MMPs include α2-macroglobulin, thrombospondin 1/2, and RECK (reversion-inducing cysteine-rich protein with Kazal motifs).18 Cell–ECM and cell–cell interactions, as well as ECM bioactive products, all contribute to an additional layer of MMP regulation. Hence, MMP expression and activity in vivo are dynamically coordinated with tissue-specific processes.19 This vast complexity often designated as the protease web, and the lack of knowledge pertaining to implications on targeting one or few members of it, has been blamed as the major reason for negative outcomes in clinical trials of early anti-MMP inhibitors (MMPIs).
MMPs as therapeutic targets
Cancer
The overwhelming volume of tumor stroma is composed of ECM and ECM-sequestered molecules. Protease degradomes comprising 1.7% of the human genome modulate this environment20 and hence are considered prime targets for therapy in diseased tissue, and are almost always accompanied by perturbations in this complex enzymatic web. Proteases help cancer cells to achieve crucial landmarks of tumor progression such as proliferative signaling, evasion of growth suppressors and host defense, resistance of cell death, angiogenesis, and initiation and sustenance of invasion and metastasis.17
Upregulated MMP and tissue inhibitor of metalloproteinases (TIMP) expression has been related to increased aggressiveness and is a predictor of poor prognosis in cancer21,22 MMPs were first noticed as druggable targets after realizing their central role in the degradation of basement membrane by invading cells23 in metastasis. It is clear that irrespective of the tissue type, from which the cancer originates, unregulated MMP expression points toward a tumor in its advanced and aggressive stages. Table 1 summarizes a few of the molecular mechanisms through which major MMPs are known to impact tumor progression.
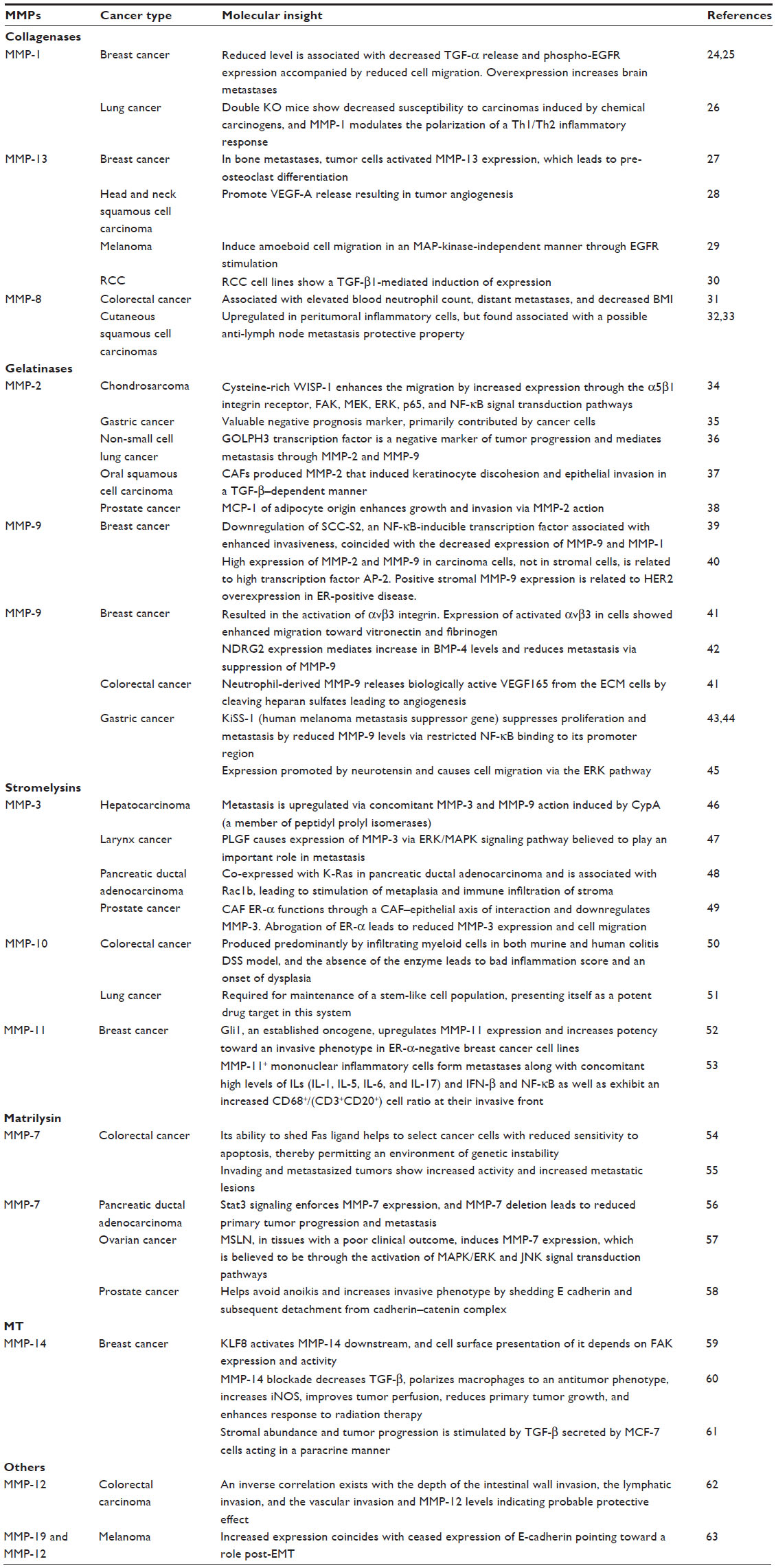 | Table 1 MMPs aid cancer progression and are a quintessential partner for malignancy |
Because of these important roles that MMPs play in cancer, numerous MMPIs, in the form of small molecules and peptidomimetics of natural MMP ligands and inhibitors, have been designed in the past 30 years. The first generation of MMPIs, mainly small molecules and peptides with a hydroxamate zinc-binding group, showed promising broad-spectrum MMP-inhibiting activity in preclinical studies. For example, prinomastat, a small-molecule inhibitor with a zinc-chelating hydroxamic acid group, showed high affinity toward MMP-2, MMP-9, MMP-13, and MMP-14 and exhibited promising tumor-inhibiting activity in several in vivo cancer models, such as malignant glioma,64 colon, lung, breast cancers, and melanoma.65 Despite its preclinical success, prinomastat failed in Phase III clinical trials of non-small cell lung cancer and resulted in musculoskeletal pain66 as a debilitating side effect. The major difference was the targeted tumor stage, ie, the progressive and highly metastatic stage of the patients with cancer vs the early stage tumors in preclinical studies.67,68
Other examples of peptidomimetic broad-spectrum MMPIs include batimastat and marimastat, which showed nanomolar affinity for several MMPs.69 Batimastat showed tumor-reducing activity in preclinical xenograft models, such as breast cancer, pancreas cancer, and melanoma.70 However, due to its poor oral bioavailability, batimastat was superseded by its more bioavailable oral analog marimastat in breast cancer clinical trials. Yet, marimastat-treated patients also did not show clinical improvement and suffered from severe side effects of musculoskeletal pain.71
The overarching reason for the failure of these MMPIs, in addition to inadequate study designs, was that they were designed to chelate Zn2+ within the extended structurally conserved protease cleft that is not exclusive to the MMP family and is abundant in related proteins, such as the ADAM and ADAMTs families.72 It is thus likely that the hydroxamate small-molecule and peptidomimetic inhibitors were not selective enough in vivo. MMPIs that bind non-selectively to many MMP family members are also not desirable, since some MMPs such as MMP-8 and MMP-12 exhibit beneficial effects and even antitumorigenic functions (Table 1). In addition, MMPs have been implicated in both positive and negative regulation of angiogenesis. A prime example is that of the dual role of MMP-9 in angiogenesis. MMP-9 promotes angiogenesis (Table 1), but it is also involved in the generation of angiogenesis inhibitors, such as angiostatin, tumstatin, and endostatin.73,74 The last few years of MMPI development, since the failure of early MMPI clinical trials, hence, have focused on improving selectivity profile, reducing drug toxicity by increasing availability as well as binding efficacy and inventing a new generation of MMPIs that target only active upregulated enzymes in diseased tissues. A number of MMPIs have been showing promising results toward these ends and are discussed in detail in this review.
Inflammation and infectious diseases
MMPs known mainly for their role in promoting cancer progression play a central role as regulators of extracellular homeostasis and innate immunity through their role in multiple inflammatory and infectious conditions.75 Inflammatory processes that include the recruitment of multiple cell populations to the infection site and activation of inflammatory cascades result in the eradication of the pathogen, but cause additional tissue damage to the host. The damage is partly a result of multiple MMP cascades involving ECM degradation as well as shedding of cell surface receptors, cytokines, chemokines, cell adhesion molecules, and clotting factors.76
There are major differences between cancer and infection treatment in using MMPs as targets, including: 1) infectious conditions can be discerned as acute or chronic and thus different treatments can be designed for each. 2) Many infections have early emerging symptoms and thus can be treated at the early onset. 3) Short-term treatment of infections can be an advantage vs prolonged treatment and associated side effects. 4) The outcome of an infection depends on the virulence of the pathogen and on the susceptibility or resistance of the host. Considering that bacterial and viral pathogens undergo constant mutations and develop resistance to drugs directly targeting them, it is reasonable to choose MMPs as alternative highly conserved and stable drug targets for viral and bacterial infections.
Infections produce imbalanced MMP expression and activation accompanied by inadequate regulation of TIMPs.77 The imbalanced proteolytic activity is the hallmark of multiple bacterial infections,78–84 viral infections,82,85 spirochete infection,86 protozoan infections,87,88 and combined systemic infections such as sepsis.76,81,89
Treatment of sepsis
Sepsis can be the end result of an ongoing infection or complicated inflammation that leads to barrier disruption and leakage of bacteria or viruses to the systemic circulation. The complexity relies on the immune system functions: 1) eradication of pathogen and 2) maintaining localized and proportional response and maintaining barrier integrity. For this reason, dysregulated proteases, active within the tissue, are critical in the pathogenesis and often define the rates of survival. Levels of active MMP-1 directly correlate with low survival rates in patients. In a colon ligation and puncture-induced sepsis model, MMP-1 was secreted by endothelial cells and high levels of it was detected in circulation shortly after the onset of inflammation. MMP-8 was further shown to be involved in several sepsis-inducing models, and its absence in MMP-8−/− mice significantly improved survival, through NF-κB inhibition.90,91 Another major contributor to inflammation and sepsis in the gut is tumor necrosis factor (TNF)-α, which is cleaved into its active form by another collagenase, MMP-13.92
Lung infectious and inflammatory diseases
In the case of lung diseases, TLR3 activation was found as a driver for MMP production through reactive nitrogen species in human lung fibroblasts.93 Paradoxically, influenza has been found to induce oxidative stress and increased MMP-9 activity associated with severe lung pathology,94 but at the same time, MMP-9 has been found crucial for neutrophil recruitment, viral clearance, and recovery.95 Using the same model in an as-yet unpublished work (Talmi-Frank et al, unpublished data, 2016), it was shown that MMP-14 is a key player in air–blood barrier disruption during influenza infection.
Fibrosis
Fibrotic reactions serve as a natural process that stabilizes the tissue and promotes wound healing. However, persistent insults such as chronic infections, drugs, toxins, and trauma can evoke chronic autoimmune reactions and inflammation leading to the activation of myofibroblast cells and accumulation of mainly collagen types I and III, altering tissue architecture, and ultimately disrupting normal cellular function.96,97 For example, liver fibrosis has been considered an irreversible process. However, recent advances in the molecular understanding of hepatic cellular processes resulted in the accumulation of clinical and experimental evidence of liver recovery with possible remodeling of scar tissue due to liver’s natural regenerative ability.98 Exposure of epithelial cells to MMPs has been shown to lead toward increased levels of cellular reactive oxygen species, which results in trans-differentiation to myofibroblast-like cells in cancer as well.99
As mentioned earlier in the context of infectious diseases, timing and specificity of treatment are crucial and make a substantive difference between beneficial and harmful outcome. MMP attenuation should be specific and localized, since broad-spectrum MMP modulation in the liver during the healing process (from CCl4-induced hepatic injury) was shown to reduce the inflammatory process, but did not reverse, and even exacerbated the scar formation.100
Chronic obstructive pulmonary disease (COPD) is considered a leading cause of death worldwide and is expected to be even deadlier by 2020.101 Numerous cell types in the lung are capable of expressing MMPs, including epithelium, fibroblasts, myofibroblasts, and macrophages. Specifically macrophages secrete various enzymes including MMP-12102 which correlate with the early stages of COPD, which are indeed characterized by elastic fiber degradation and mechanical recoiling of airways.103,104 In line with these observations, some selective inhibitors of MMP-12, such as AS111793, and inhibitors of MMP-9 or MMP-12 reduced the inflammatory process associated with the exposure of mice to cigarette smoke.105,106 Thus, these mild changes that are evident in the early stages of COPD, prior to the disseminated changes in the alveolar compartment, call for an integrated and time-scaled study that will improve our understanding regarding beneficial MMP targets and the disease outcome and prevent further tissue structure and function deterioration.107
Targeting of MMPs in fibrosis may not be straightforward as the MMPs may play a dual role (both beneficial108 and detrimental). MMPs are likely to be necessary to remove the excess collagen deposits present in fibrosis. Naturally, it may be intuitive to target MMPs and TIMPs simultaneously to restore homeostatic balance, eg, TIMP-1 (presumably by attenuating its expressed levels or by sequestering it) and MMP-2 in the case of liver fibrosis.109 Accordingly as in cancer, identifying MMP targets and anti-targets in a particular stage of fibrotic disease is of paramount importance.
Novel MMPIs and inhibition strategies
Small molecules
Recently, attempts have been made to develop more selective small-molecule MMPIs.110 For instance, cis-2-aminocyclohexylcarbamoylphosphonic acid, a zinc-binding inhibitor, was developed by Hoffman et al to selectively target MMP-2 (tested in vitro on recombinant MMPs, with an half maximal inhibitory concentration [IC50] of 4 μM). The compound reduced metastasis in melanoma and prostate in vivo cancer models.111 Another option that has been explored for more selective MMP inhibition is targeting the hemopexin domain of MMPs, which is less conserved than the catalytic site. Examples of inhibitors targeting the hemopexin domain include the compound N-[4-(difluoromethoxy)phenyl]-2-[(4-oxo-6-propyl-1H-pyrimidin-2-yl)sulfanyl]-acetamide, which was identified by Dufour et al112 by using in-silico docking methods. The compound selectively targeted the hemopexin domain of MMP-9, abolished MMP-9 homodimerization (which was shown to promote cell migration), and inhibited lung metastasis in an in vivo MDA-MB-435 xenograft model.113 Another hemopexin-targeting small-molecule, NSC405020 [3,4-dichloro-N-(1-methylbutyl)] was identified by Remacle et al114 by virtual screening of a Developmental Therapeutics Program (National Cancer Institute) compound library with the X-ray crystal structure of the MMP-14 hemopexin domain as a target. The compound inhibited in vivo the tumor growth of MCF7-β3/MT cells (MCF7 cells with overexpressed full-length MMP-14) and did not affect tumors with MMP-14, lacking the hemopexin domain.
The quest to improve these small-molecule inhibitors continues owing to their pharmacological stability and ease of administration. An interesting example of this quest has been the combined topical application of ND-336 (a small molecule that inhibits gelatinases [MMP-2 and MMP-9] and MMP-14 with high selectivity) and the active recombinant MMP-8 enzyme to enhance diabetic wound healing by lowering inflammation and by enhancing angiogenesis as well as reepithelialization of the wound.115
Peptide-based MMPIs
A different approach to developing selective MMPIs is designing selective MMP-inhibiting peptides. Examples include peptide G, a cyclic peptide with the sequence of GACFSIAHECGA, which was developed by Suojanen et al in a phage display system.116 This short peptide was shown to inhibit invasion and migration of fibrosarcoma and melanoma cells by specifically blocking the catalytic activity of MMP-14, both in vitro and in vivo in murine models, albeit at very high concentrations of 150–500 μM. The same research group also developed two peptides against MMP-2 and MMP-9, which were designated CTT1 (CTTHWGFTLC) and CTT2 (GRENYHGCTTHWGFTLC), respectively. The peptides were isolated from a phage display library117 with micromolar affinity and showed inhibitory activity toward both MMP-2 and MMP-9 in vitro in murine mouth squamous carcinoma cells.118
Another MMP-14-targeting peptide is Val-Phe-Asp-Glu-Ala-Ser-Leu-Glu-Pro-NH2, which was derived from blade II, strands 3–4 of the MMP-14 hemopexin domain by Fields. The peptide had an IC50 value of 238 μM toward MMP-14 but did not inhibit MMP-1.119
A number of studies have sought to develop inhibitors for MMP-2. A peptide derived from the MMP-2 collagen-binding domain, which was designated P713 (Cys-Gly-Ala-Hyp-Gly-Ala-Hyp-Gly-Ser-Gln-Gly-Ala, where Hyp stands for 4-hydroxyproline), was identified by Xu et al by screening of a one-bead one-peptide library.120 P713 specifically inhibited the collagenolytic activity of MMP-2, albeit with low affinity (IC50 of ~30 μM), but did not inhibit cleavage of a non-collagen substrate and did not inhibit MMP-8 (also a collagen-cleaving MMP).
Higashi et al, showed that fusion of TIMP-2 with ten amino acid residues derived from β-amyloid precursor protein, namely APP inhibitory peptide (APP-IP), increases the affinity and selectivity of the chimeric protein toward MMP-2 (with Ki value of 0.68 pM).121 The ISYGNDALMP sequence of the APP-IP-derived peptide is a selective MMP-2-catalytic-site-binding peptide.122 By fusing the peptide to the N-terminus of TIMP-2, the binding of TIMP-2 to the MMP-2 catalytic site is prevented. Simultaneously, the C-terminal domain of TIMP-2 specifically binds to the hemopexin domain of MMP-2 to form a complex, and the APP-IP peptide then inhibits the catalytic site.121 The fusion protein inhibited HT1080 fibrosarcoma cell line invasion in vitro.
Burg-Roderfeld et al developed MMP-9 hemopexin-like domain as an MMP-9 inhibitor. The recombinant MMP-9 hemopexin domain had relatively high affinities toward gelatin (Kd=295 nM), collagen type I (Kd=444 nM), and collagen type IV (Kd=655 nM).123 The recombinant MMP-9 hemopexin domain inhibited melanoma124 and colon cancer cell123 invasion in a Boyden chamber migration assay and also inhibited angiogenesis and tumor growth in a glioblastoma xenograft in vivo model.125
The phosphinic peptide RXP470.1, a selective murine MMP-12 inhibitor, was found to significantly reduce the atherosclerotic plaque cross-sectional area at vascular sites in apolipoprotein E knockout mice, and MMP-12 inhibition was found to result in a more fibrous and less complex plaque with characteristically reduced macrophage/monocyte invasion.126
Targeting MMPs with antibodies
In light of the aforementioned information, it has become clear that inhibitors with narrow or single MMP specificity hold much greater therapeutic promise; nevertheless, such specific inhibitors are difficult to obtain because of the high homology of the catalytic sites of the MMP family members. The size of small molecules and peptides limits them to interact with the catalytic site alone and does not allow them to target more distant sites as well. It is, however, likely that larger protein-based agents would be able to interact with residues that are distant from the catalytic site and would thus exhibit improved specificity.
In this context, monoclonal antibodies (mAbs) directed against MMPs to detect and treat primary and metastatic cancers have been developed. As of now, none of them have been approved by the US Food and Drug Administration. These antibodies can be designed to be highly specific due to their large surface that enables interaction not only with the highly conserved catalytic site of MMPs but also with a variety of surrounding residues. Antibodies, such as the full-size mAb REGA-3G12, which have been raised against MMP-9, target and inhibit the catalytic domain of MMP-9 but not MMP-2.127 Another MMP-9-targeting antibody, GS-5745, is a highly selective allosteric full-length mAb that decreased tumor growth and metastasis in a colorectal carcinoma model. Importantly, GS-5745 did not trigger the side effect of musculoskeletal pain in a rat model.128 Therapeutic promise has also been shown by DX-2400, an antibody fragment (Fab) isolated from a phage display library to selectively target MMP-14. The selectivity was confirmed by measuring the Ki of DX-2400 toward various MMPs. DX-2400 displayed anti-invasive, antitumor, and anti-angiogenic properties and blocked proMMP-2 processing in several preclinical models; for example, in an MDA-MB-231 xenograft model, treatment with the selective DX-2400 antibody significantly reduced tumor growth and metastasis.129
A selective MMP-12 inhibitor was recently patented for the treatment of respiratory syncytial virus and Coxacivirus.82 Specifically, this inhibitor acts on extracellular MMP-12 rather than intracellular MMP-12. It is based on the dual role of MMP-12 in interferon (IFN)-α regulation. Intracellular MMP-12 is necessary for IFN-α secretion, while extracellular MMP-12 is involved in the clearance of circulating IFN-α. Therefore, by inhibiting the activity of the extracellular MMP-12, it is possible to decrease the clearance of circulating IFN-α, and thereby to increase the amount of IFN-α, while not impacting on its intracellular effect.
Sela-Passwell et al have been successful in utilizing the natural TIMP–MMP-binding protein–protein interaction (PPI) to develop specific function-blocking inhibitory anti-MMP mAbs and demonstrated that these mAbs called SDS3 and SDS4 inhibited MMP-9 and its close analog MMP-2 (Ki values of 1±0.1 μM and 0.054±0.006 μM for SDS3 and SDS4, respectively). They were also found effective in reducing inflammatory phenotype and tissue damage in rodent models of inflammatory bowel disease.130
To further improve the selectivity, inhibitors possessing molecular interactions with unique surface epitopes with improved drug selectivity have also been developed by several groups. The antibody LEM-2/15131 showed CAT-MT1-MMP (MMP-14) inhibition with an impressive IC50 value of 45±2.4 nM. Conformational rearrangements of the enzyme scaffold were found to enable binding to the Fab fragment and helped control the enzymatic activity. LEM-2/15 binding to MMP-14 was demonstrated to belong to an allosteric mechanism of inhibition.132
Using a Fab fragment of anti-MMP-14 inhibitor Ab, it was shown in our laboratory that blocking of MMP-14 catalytic activity with aforementioned LEM-2/15 (Figure 1) in macrophages and neutrophils caused rescue of tissue morphology and composition during single influenza infection as well as coinfection with Streptococcus pneumoniae. In this case, using a Fab fragment ligated to nanoparticles targeted directly at the lung could benefit in reducing side effects (if any) and dosage. In line with these findings, a recent study has shown the extreme expression levels of MMP-14 in human primary monocytes as well as tuberculosis-infected human lung samples.83 Moreover, using an MMP-14 inhibitor further showed the potential of such inhibition on maintaining lung structure and architecture.
 | Figure 1 MMPs contributing to disease progression in inflammation as well as cancer of lungs (A) and the gut (B) can be targeted with specific selective inhibitors of high affinity. |
Another interesting application of anti-MMP-14 antibody, DX-2400, in collagen-induced arthritis DBA/1 mice leads to reduced cartilage degradation. In addition, a synergistic effect of anti-MMP-14 inhibition along with TNF blockade contributing toward beneficial effects such as synovial inflammation and joint damage was also observed. MMP-14 blockade also inhibited the increase in interleukins caused due to polarization toward an M phenotype in bone-marrow-derived macrophages.133 A recent report by Murphy et al demonstrated the construction of minimized antibodies in the form of Fc-scFv format, which was screened utilizing phage display library. This inhibitory anti-MMP-14 antibody fragment was shown to be effective in attenuating tumor growth and progression by binding the catalytic domain of MMP-14 and abrogating specifically its cell surface functions.134
Despite their advantages, antibodies have several well-known limitations, including high production costs,135 long serum half-lives (causing background noise for imaging purposes and toxicity in therapy136), and difficulties in renal clearance. In addition, antibodies are often associated with a side effect of an immune system response137 and with potentially undesirable effector functions.138 Using Fab fragments alone or encapsulated in nanobodies with improved delivery and site specificity is a solution being explored to overcome these difficulties.
Natural inhibitors of MMPs and engineering N-terminal TIMPS as specific high affinity MMPIs
From the abovementioned examples, we now know that it is possible to design protein-based MMPIs with a single specificity and high affinity. A potent candidate’s starting point for engineering affinity and specificity is the natural broad-spectrum MMPI, TIMP-2. This inhibitor possesses a high affinity for various MMPs (10−10–10−9 M) and is nontoxic and nonimmunogenic to humans.139 It has been shown that TIMP-2 has therapeutic potential for the treatment of several cancers. For example, in in vivo murine models of ovarian and hepatocellular cancers, gene therapy with a conditionally replicating TIMP-2-expressing adenovirus (Ad5/3-CXCR4-TIMP-2) delayed tumor growth and significantly increased survival.140,141
There are four homologous mammalian TIMPs (TIMP-1–4) that show 40%–50% sequence identity to one another and exhibit slightly different preferences for various MMPs.142 For example, TIMP-1 inhibits most of the MMPs, with the exception of MMP-14, MMP-16, MMP-19, and MMP-24.143 TIMPs (184–194 amino acids in size) consist of two domains folding as separate units, each having three conserved disulfide bonds.2 Three-dimensional structures of TIMP–MMP complexes have revealed that binding of a TIMP to an MMP active site takes place largely through the wedge-shaped ridge of the N-terminal TIMP (N-TIMP) domain, consisting of ~125 amino acids. The interaction occurs through the α-amino and carbonyl groups of the cysteine at position 1 of the TIMP, which serves to chelate the Zn2+ of the MMP’s catalytic site. In addition, the serine/threonine group at position 2 of the N-TIMPs interacts with the nucleophilic glutamine of the MMP catalytic site, thus displacing a water molecule that is crucial for substrate hydrolysis.142
The full-length TIMP-2, despite the inhibitory function of its N-domain, plays a role in MMP-2 activation by bringing MMP-14 and MMP-2 in close proximity, forming a MMP-14/TIMP-2/MMP-2 complex (Figure 2A). The N-terminus of TIMP-2 binds to and inhibits the catalytic domain of MMP-14 on the cell surface, while the C-terminus of the same TIMP-2 binds to the hemopexin domain of the secreted pro-MMP-2 zymogen. Binding of TIMP-2 triggers dimerization of the inhibited MMP-14 with a neighboring active MMP-14 (Figure 2A). The MMP-14 that is not inhibited can cleave and activate a bound pro-MMP-2. Increased expression of MMP-2 has been implicated in several types of cancer and has been shown to be associated with poor prognosis.144,145 TIMP-2 secreted from cancer cells activates MMP-2 from endothelial cells.146 The activation of MMP-2 induced the transmigration of the endothelial cells, whereas TIMP-2 knockdown cells did not migrate. Thus, the activation of MMP-2 by the TIMP-2/MMP-14 complex is crucial for cancer cell migration.
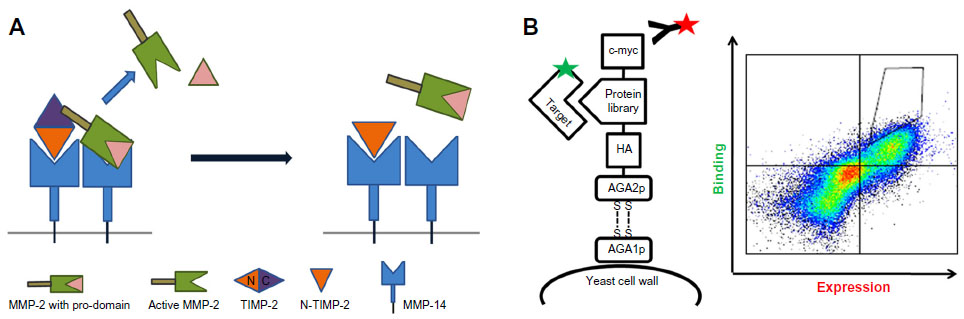 | Figure 2 Engineering approaches for MMPs inhibition with TIMPs. |
The isolated N-TIMP (~126 amino acids) is stable, retains its structure, and is a potent inhibitor of various MMPs.147 It has thus been repeatedly used as a substitute for the full-length protein in various experimental in vitro and in vivo studies. For example, the angiogenesis-inhibiting activity of N-TIMP-2 is the same as that of the intact full-length TIMP-2.148 One of the advantages of N-TIMP-2 is its relatively small size, which facilitates its production by microbial cultures, allowing for the site-specific conjugation of imaging probes, radioisotopes, or small-molecule inhibitors for direct delivery to the surface of cancer cells. Yet, another advantage is that the in vivo pharmacokinetic properties of N-TIMP-2 can be chemically tailored to diverse clinical applications.149 N-TIMP-2 can thus be used as a versatile platform for the development of targeted cancer therapies.
Other scaffolds (15–60 residues) with a motif corresponding to four N-terminal TIMP-2 residues were identified by a protein data bank screened using STAMPS software (search for three-dimensional atom motifs in protein structures), which identifies motifs in protein structures.150 Testing for MMP binding of the ten scaffolds that were produced in the study revealed affinities ranging from 450 nM to 450 μM.85
Various rational design studies exploiting knowledge of the structures of the proteins and of TIMP/MMP PPIs have been performed over the years. The shared feature of the results of all these studies is that single point mutations at the binding interface of TIMPs have a marked influence on their binding affinity and selectivity.151,152 For example, introducing a single point mutation (Thr98Leu) to N-TIMP-1, which did not bind to MMP-14, increased its affinity for MMP-14 by two orders of magnitude.152 In a different study, it was found that single point mutations introduced to the binding interface altered the specificity of N-TIMP-2 toward several MMPs: N-TIMP-2 variants containing the mutations Tyr36Gly or Tyr36Thr decreased the affinity of N-TIMP-2 toward MMP-14 while retaining its affinity to other MMPs.153 Although these protein-engineering studies highlighted the potential of designing high-affinity and selective TIMP variants, they were all based on the mutation of just one amino acid. Addressing the issue of simultaneously introducing more than one single-point mutation, a study on N-TIMP-1 showed that a combination of mutations has greater effect on affinity and selectivity than just one mutation. A triple N-TIMP-1 mutant, namely Thr2Ser/Val4Ala/Ser68Tyr, combining three single point mutations of the N-TIMP-1-binding interface showed picomolar affinity toward MMP-3 compared to much lower affinities obtained for the single mutation variants.154 Therefore, the way forward appears to lie in combining a few mutations at the binding interface of TIMPs to develop potent high affinity and selectivity among the MMPIs.
Two different strategies are commonly used to engineer protein-based inhibitors. The first is combinatorial protein engineering, also known as directed evolution, which relies on the development of mutant libraries. Mutations are introduced into the libraries either randomly or by a semi-rational method, only to the regions that are known as important for the binding. The libraries are then expressed in a display system (phage, bacterial, yeast, etc) and screened with high-throughput technologies.155 There are two advantages to this approach: previous knowledge of the protein structure is not required and a phenotype–genotype link facilitates recovery of the DNA with the desired feature for the purposes of identifying the mutation and further use of the material.156
This combinatorial protein engineering approach has been used for the generation of a number of TIMP-based MMPIs. One such example is the generation of TIMP-2 (full-length) mutants with a phage display system.157 In the study, TIMP-2 mutant libraries were created with randomization of positions 2, 4, 5, and 67–70 of the N-TIMP-2-binding interface. TIMP-2 mutants were screened for specific binding to MMP-1 over binding to MMP-3 and MMP-14 in a phage enzyme-linked immunosorbent assay. One of the mutants identified (namely TM8) had high binding affinity for MMP-1 (10 nM) and undetectable affinity for MMP-3 and MMP-14.
The second approach to PPI engineering is computational protein design, which facilitates the exploration of an astronomically large number of protein sequences in silico to find the best protein binders, which are then verified experimentally by site-directed mutagenesis. This approach requires knowledge of the structures and the PPIs. An excellent example of the use of computational methods for the prediction of interactions of MMP-14/N-TIMP-2-binding interface is the recent work of the Shifman laboratory.158,159 The available high-resolution structure of the MMP-14/TIMP-2 complex was applied to define the binding interface and the residues directly involved in binding. Then, each position in the binding interface was exhaustively mutated computationally, the surrounding residues were repacked, and the energy of the complex was calculated. The change in the free energy of binding (ΔΔGbind) was determined by subtracting the intermolecular energy of the wild-type complex from that of the mutant complex. By this computational method, seven positions at the N-TIMP-2 interface that are tolerant to mutations that increase the affinity for MMP-14 were identified.
Both combinatorial and computational methods have been used in recent years to enhance binding affinity and specificity of various PPIs and to obtain novel binders and inhibitors from various scaffold proteins. While both methods have their advantages and limitations, several recent works have shown that a combination of computational and combinatorial techniques produces superior results.160 In these studies, computational design was used to narrow down the choices (for the construction of smaller and focused combinatorial libraries), and then in vitro evolution methods were used to quickly assay the binding of millions of protein variants.
In a recent study by Arkadash et al ( Arkadash et al, unpublished data, 2016), N-terminal domain of TIMP-2 was designed for high affinity and specificity for MMP-14. In this work, we combine the computational and combinatorial techniques by creating a focused library with seven randomized positions at the N-TIMP-2/MMP-14-binding interface previously identified using computational methods. The focused library was then expressed in a yeast surface display (YSD) screening system, which is a eukaryotic display system that allows some eukaryotic posttranslational modifications. YSD, which is used for engineering proteins with improved ligand binding affinity, stability, expression, and enzymatic activity,161 exploits glycosylphosphatidylinositol-anchored proteins on the yeast cell surface and protein secretion pathways.162 In this system, which was developed by Boder and Wittrup,163 AGA1 is an a-agglutinin subunit anchored to the cell surface and bound by a disulfide bond to an AGA2 subunit. To enable the expression of the protein of interest on the yeast surface, the protein is fused to the AGA2 protein (Figure 2B), which is flexible in a way that allows fusion to its either C terminus or N terminus; this flexibility may be important for expression and is critical for binding of the displayed protein of interest to its soluble target.164,165 The YSD system allows the characterization of isolated high-affinity mutants directly in the display form, prior to soluble expression and purification.161 High binding affinity mutants are then identified by a flow cytometry technique, in which the ligand is fluorescently labeled, and high binding variants are identified and selected (Figure 2B).
In the aforementioned study, high-throughput flow cytometry screening of the N-TIMP-2 focused library against the catalytic domain of MMP-14 revealed four high-affinity clones. These clones possessed combinations of seven mutations, where most of these mutations predicted as affinity enhancing. The combinatorial effect of the library and the selective pressure produced variants with picomolar values of Ki for MMP-14, compared to the nanomolar values of Ki published for the wild-type N-TIMP.151,159 Importantly, the four clones did not exhibit significantly increased affinity toward any of the other MMPs tested so far in kinetic inhibition experiments.
Targeting MMP gene expression using MicroRNAs
MicroRNAs (miRNAs) are small noncoding RNAs (17–25 nucleotides) involved in posttranscriptional regulation of gene expression. miRNAs control mRNA stability and translation through complementary base pairing at specific untranslated 3′ region. MMPs are evidently directly regulated by these miRNAs, and they are considered theranostic targets in various disease types such as glioblastoma multiforme (GBM),166 osteoarthritis,167 and aortic aneurysms.168 A complicated interactive network of the MMPs, TIMPs, RECK, and their respective miRNAs exists, adding an additional layer of MMP regulation. MMP-16 was found to be a downstream target of miR-146b in U373 glioma cells, an miRNA implicated in GBM cell migration and invasion.169 miR-21 is significantly elevated also in GBM, and the inhibition of it leads to elevated levels of RECK and TIMP-3. Antisense inhibition of miR-21 has been considered a potential and novel anticancer therapy.170 Intravenously administered chlorotoxin-coupled (targeted) stable-nucleic-acid-lipid-particle-formulated anti-miR-21 oligonucleotides have been shown to promote efficient miR-21 silencing, preferentially in GBM.171
Effects of miRNA on activity can be multiplied by indirect effects produced by upstream signaling protein regulation. For example, Hwang et al isolated brain-trophic metastatic MDA-MB-435-LvBr2 (LvBr2) breast cancer cells via left ventricle injection of MDA-MB-435 cells into immunodeficiency (NOD/SCID) mice, which were shown to express negligent levels of miR-146a. The overexpression of miR-146a leads to the firing up of β-catenin expression and heterogeneous nuclear ribonucleoprotein C1/C2 levels with suppressed invasive potential with concomitant decrease in MMP-1 levels.172
Targeting MMP activity using miRNAs is plagued by possible lack of specificity, due to sequence homology in closely related MMPs. miR-143 expression decreased protein levels of MMP-2 and MMP-9 in pancreatic cancer,173 directly targeted MMP-13, and indirectly MMP-9 in osteosarcoma, reflecting the complexity of using the overexpression of miR-143 as a therapeutic strategy.174,175
miRNAs have also been implicated as important modulators of fibrosis. Aforementioned miRNA 146a was found to be involved in transforming growth factor (TGF) -β1- induced hepatic stellate cell activation by potentially targeting SMAD4.176 The miR-29 family has also been reported as crucial regulators of liver fibrosis. miR-29a/b is negatively regulated by TGF-β, and increased levels of miR-29 family lead to reduced collagen deposition.177 miR-29 was also shown positively correlated with TIMP-1 levels and negatively correlated with MMP-9 levels in diabetic nephropathy.178
Targeting MMP activity through miRNAs in infectious diseases has also been proposed, keeping in mind that MMPs and their endogenous regulators play a crucial role in infectious disease progression. miR-17p was found significantly upregulated post-infection with human cytomegalovirus in C373/MICA*019 cells. The infection leads to TIMP-3 downregulation, and miR-17p is believed to contribute toward the same.179
A number of patents have been filed for the use of miRNAs as theranostic tools. A complete understanding of the complexity of these posttranscriptional regulators of MMP activity is yet to be elucidated. The quest to tap into miRNAs as effective tools continues to generate future interests and meanwhile the focus is on deciphering the possible mechanism(s) by which the effects of putative miRNA targets affect cellular and ECM phenotype.
Conclusion: are we facing new horizons in the era of post-broad range inhibitors for metalloproteinases in medicine?
MMPs are unquestionably potent drug targets in cancers, infections, and inflammations. Their role in advancing these disease conditions has been well established thus far. In cancer, lessons have been learnt from the past regarding specificity of MMPIs and stages of the disease amenable to meaningful treatment with MMPIs. Several new protein-based novel inhibitors, which result in increased overall survival and reduced primary and metastatic tumor burden, are being tested in animal models. Going forward, the exciting molecular insights would come from characterizations of MMP activity profile in different stages of disease, eg, cancer, and in adapting this knowledge to use selective inhibitors against those specific MMPs in relevant temporo-spatial windows, for maximum therapeutic results.
Novel MMPIs inspired by high-resolution protein structures of MMPs and their natural inhibitors such as TIMPs including engineered N-TIMPs and anti-MMP antibodies are proving to be of great advantage. Other innovative approaches, such as reconstructing the zymogen form with stabilized form of enzyme akin to the work performed in the Sagi laboratory by Wong et al (unpublished data, 2015) with ADAM17 pro-domain specifically attenuating ADAM17 enzymatic activity in vitro, in situ, and in vivo, could also be used for the development of MMPIs.
It is also clear from activity profiles of MMPs and molecular studies about their roles that they have contradictory disease supporting and attenuating effects in tissues. In addition, the new frontier of MMP inhibition might not lie within the paradigm of direct MMP inhibition. Owing to their contradictory protective and disease-promoting roles in tissues, it might be prudent also to target specific pathways, which get disrupted by increased MMP activity. Pioneering attempts are being made using protein engineering and directed evolution, to produce inactive mutants with improved binding specificity (decoys) that will target key MMP substrates. These are designed to compete on molecular pathways governed by selective enzyme–substrate interactions and restore homeostasis.
Concerning inflammation and infectious diseases, in addition to structural ECM substrates, MMP-mediated cleavage and release of signaling molecules produce a positive feedback loop, which exacerbates the inflammatory reaction, making increased proteolysis a prime target for drug design. Moreover, the outcome of an infection depends on the pathogenicity or virulence of the pathogen and on the susceptibility or resistance of the host to that pathogen. Neither of them are consistent druggable factors in contrast to the MMPs. In addition, however, it is crucial to remember that MMPs that serve as therapeutic targets in some conditions may be anti-targets in other conditions.
The treatment of infectious diseases is dominated by the tussle between the need to mount normal immunologic response, resulting in induced MMP activity aimed at pathogen clearance, and the need to reduce immune-related collateral tissue damage. Moreover, it is important to specifically target key MMPs that are responsible for the exacerbated damage without interfering with their roles in normal tissue homeostasis and repair. One way to target MMP activity focuses on targeting the pathogen invasion such as antiviral drugs used in clinical trials to treat hepatitis C180 or HIV181, which proved partial efficacy. The other is targeting the host-derived proteases that are up regulated in multiple infection models. These protease subject the tissue to overwhelming ECM remodeling resulting in detrimental outcome.83,182
Moreover, patients are often fighting off high titers of pathogens, which requires an acute and a drastic inflammatory reaction; thus it is not always clear to what extent the inflammation should be blocked. Therefore, partial or local inhibition of inflammatory mediators such as elements of the MMPs proteolytic network may decrease inflammation while maintaining an appropriate level of immune activation.
After many years of trials and tribulation with hydroxamate-, thiol-, pyrimidine-, hydroxypyrone-, phosphorus-, and tetracycline-based inhibitors, there seems to be an impetus toward MMPIs inspired by the three-dimensional structure of MMPs. This new class of MMPIs promises to negate several of the negative drug interactions responsible for poor outcome of earlier broad-spectrum inhibitors that underwent clinical trial. Considering the long drawn-out and resource-intensive clinical trial process, it would be quite a while before these novel strategies are successfully adopted and tested for clinical applications and a new sense of confidence in using MMPs as druggable targets arises in the pharma fraternity. Nevertheless, it would be safe to say that exciting new generations of MMPIs have arrived, and once again MMPs are being considered as potent drug targets thanks to their central role in dysregulated tissue mechanisms.
Acknowledgments
IS is supported by the Israel Science Foundation, Rising Tides Foundation, Memorial Sloan Kettering Cancer Center-Weizmann Fund, the Thompson Foundation, and the Kimmelman Center at the Weizmann Institute of Science. NP is supported by the Prostate Cancer Foundation, Friends for an Earlier Breast Cancer Test Fund, Israel Cancer Research Fund, European Research Council (ERC), Israel Science Foundation, and the DKFZ-MOST cooperation in Cancer Research Fund.
Disclosure
The authors report no conflicts of interest in this work.
References
Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov. 2014;13(12):904–927. | |
Hynes RO, Naba A. Overview of the matrisome – an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol. 2012;4(1):a004903. | |
Rodriguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803(1):39–54. | |
Harisi R, Jeney A. Extracellular matrix as target for antitumor therapy. Onco Targets Ther. 2015;8:1387–1398. | |
Galliera E, Tacchini L, Corsi Romanelli MM. Matrix metalloproteinases as biomarkers of disease: updates and new insights. Clin Chem Lab Med. 2015;53(3):349–355. | |
Zarella BL, Cardoso CA, Pelá VT, Kato MT, Tjäderhane L, Buzalaf MA. The role of matrix metalloproteinases and cysteine-cathepsins on the progression of dentine erosion. Arch Oral Biol. 2015;60(9):1340–1345. | |
Sivak JM, Fini ME. MMPs in the eye: emerging roles for matrix metalloproteinases in ocular physiology. Prog Retin Eye Res. 2002;21(1):1–14. | |
Lenglet S, Mach F, Montecucco F. Role of matrix metalloproteinase-8 in atherosclerosis. Mediators Inflamm. 2013;2013:659282. | |
Kahari VM, Saarialho-Kere U. Matrix metalloproteinases in skin. Exp Dermatol. 1997;6(5):199–213. | |
Itoh T, Matsuda H, Tanioka M, Kuwabara K, Itohara S, Suzuki R. The role of matrix metalloproteinase-2 and matrix metalloproteinase-9 in antibody-induced arthritis. J Immunol. 2002;169(5):2643–2647. | |
Ohnishi K, Takagi M, Kurokawa Y, Satomi S, Konttinen YT. Matrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysema. Lab Invest. 1998;78(9):1077–1087. | |
Hayashi K, Horikoshi S, Osada S, Shofuda K, Shirato I, Tomino Y. Macrophage-derived MT1-MMP and increased MMP-2 activity are associated with glomerular damage in crescentic glomerulonephritis. J Pathol. 2000;191(3):299–305. | |
Shimshoni E, Yablecovitch D, Baram L, Dotan I, Sagi I. ECM remodelling in IBD: innocent bystander or partner in crime? The emerging role of extracellular molecular events in sustaining intestinal inflammation. Gut. 2015;64(3):367–372. | |
Duarte S, Baber J, Fujii T, Coito AJ. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015;44-46:147–156. | |
Wilson D. Support for writers. Aust Vet J. 1998;76(5):331. | |
Gjorevski N, Nelson CM. Bidirectional extracellular matrix signaling during tissue morphogenesis. Cytokine Growth Factor Rev. 2009;20(5–6):459–465. | |
Cathcart J, Pulkoski-Gross A, Cao J. Targeting matrix metalloproteinases in cancer: bringing new life to old ideas. Genes Dis. 2015;2(1):26–34. | |
Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–174. | |
Gaffney J, Solomonov I, Zehorai E, Sagi I. Multilevel regulation of matrix metalloproteinases in tissue homeostasis indicates their molecular specificity in vivo. Matrix Biol. 2015;44-46:191–199. | |
Overall CM, Kleifeld O. Tumour microenvironment – opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6(3):227–239. | |
Hadler-Olsen E, Winberg JO, Uhlin-Hansen L. Matrix metalloproteinases in cancer: their value as diagnostic and prognostic markers and therapeutic targets. Tumour Biol. 2013;34(4):2041–2051. | |
Kuvaja P, Wurtz SØ, Talvensaari-Mattila A, Brunner N, Pääkkö P, Turpeenniemi-Hujanen T. High serum TIMP-1 correlates with poor prognosis in breast carcinoma – a validation study. Cancer Biomark. 2007;3(6):293–300. | |
Liotta LA, Tryggvason K, Garbisa S, Hart I, Foltz CM, Shafie S. Metastatic potential correlates with enzymatic degradation of basement membrane collagen. Nature. 1980;284(5751):67–68. | |
Liu H, Kato Y, Erzinger SA, et al. The role of MMP-1 in breast cancer growth and metastasis to the brain in a xenograft model. BMC Cancer. 2012;12:583. | |
Okuyama N, Matsumine A, Kosugi R, Wakabayashi H, Uchida A. Matrix metalloproteinase-1 is a crucial bone metastasis factor in a human breast cancer-derived highly invasive cell line. Oncol Rep. 2008;20(6):1497–1504. | |
Fanjul-Fernandez M, Folgueras AR, Fueyo A, et al. Matrix metalloproteinase Mmp-1a is dispensable for normal growth and fertility in mice and promotes lung cancer progression by modulating inflammatory responses. J Biol Chem. 2013;288(20):14647–14656. | |
Pivetta E, Scapolan M, Pecolo M, et al. MMP-13 stimulates osteoclast differentiation and activation in tumour breast bone metastases. Breast Cancer Res. 2011;13(5):R105. | |
Kudo Y, Iizuka S, Yoshida M, et al. Matrix metalloproteinase-13 (MMP-13) directly and indirectly promotes tumor angiogenesis. J Biol Chem. 2012;287(46):38716–38728. | |
Meierjohann S, Hufnagel A, Wende E, et al. MMP13 mediates cell cycle progression in melanocytes and melanoma cells: in vitro studies of migration and proliferation. Mol Cancer. 2010;9:201. | |
Kominsky SL, Doucet M, Thorpe M, Weber KL. MMP-13 is over-expressed in renal cell carcinoma bone metastasis and is induced by TGF-beta1. Clin Exp Metastasis. 2008;25(8):865–870. | |
Vayrynen JP, Vornanen J, Tervahartiala T, et al. Serum MMP-8 levels increase in colorectal cancer and correlate with disease course and inflammatory properties of primary tumors. Int J Cancer. 2012;131(4):E463–E474. | |
Lempinen M, Lyytinen I, Nordin A, et al. Prognostic value of serum MMP-8, -9 and TIMP-1 in patients with hepatocellular carcinoma. Ann Med. 2013;45(7):482–487. | |
Decock J, Hendrickx W, Vanleeuw U, et al. Plasma MMP1 and MMP8 expression in breast cancer: protective role of MMP8 against lymph node metastasis. BMC Cancer. 2008;8:77. | |
Hou CH, Chiang YC, Fong YC, Tang CH. WISP-1 increases MMP-2 expression and cell motility in human chondrosarcoma cells. Biochem Pharmacol. 2011;81(11):1286–1295. | |
Łukaszewicz-Zajac M, Mroczko B, Guzińska-Ustymowicz K, et al. Matrix metalloproteinase 2 (MMP-2) and their tissue inhibitor 2 (TIMP-2) in gastric cancer patients. Adv Med Sci. 2013;58(2):235–243. | |
Wang R, Ke ZF, Wang F, et al. GOLPH3 overexpression is closely correlated with poor prognosis in human non-small cell lung cancer and mediates its metastasis through upregulating MMP-2 and MMP-9. Cell Physiol Biochem. 2015;35(3):969–982. | |
Hassona Y, Cirillo N, Heesom K, Parkinson EK, Prime SS. Senescent cancer-associated fibroblasts secrete active MMP-2 that promotes keratinocyte dis-cohesion and invasion. Br J Cancer. 2014;111(6):1230–1237. | |
Ito Y, Ishiguro H, Kobayashi N, et al. Adipocyte-derived monocyte chemotactic protein-1 (MCP-1) promotes prostate cancer progression through the induction of MMP-2 activity. Prostate. 2015;75(10):1009–1019. | |
Wang L, Zhang ZG, Zhang RL, et al. Matrix metalloproteinase 2 (MMP2) and MMP9 secreted by erythropoietin-activated endothelial cells promote neural progenitor cell migration. J Neurosci. 2006;26(22):5996–6003. | |
Pellikainen JM, Ropponen KM, Kataja VV, Kellokoski JK, Eskelinen MJ, Kosma VM. Expression of matrix metalloproteinase (MMP)-2 and MMP-9 in breast cancer with a special reference to activator protein-2, HER2, and prognosis. Clin Cancer Res. 2004;10(22):7621–7628. | |
Rolli M, Fransvea E, Pilch J, Saven A, Felding-Habermann B. Activated integrin alphavbeta3 cooperates with metalloproteinase MMP-9 in regulating migration of metastatic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(16):9482–9487. | |
Shon SK, Kim A, Kim JY, Kim KI, Yang Y, Lim JS. Bone morphogenetic protein-4 induced by NDRG2 expression inhibits MMP-9 activity in breast cancer cells. Biochem Biophys Res Commun. 2009;385(2):198–203. | |
Li N, Wang HX, Zhang J, Ye YP, He GY. KISS-1 inhibits the proliferation and invasion of gastric carcinoma cells. World J Gastroenterol. 2012;18(15):1827–1833. | |
Yan C, Wang H, Boyd DD. KiSS-1 represses 92-kDa type IV collagenase expression by down-regulating NF-kappa B binding to the promoter as a consequence of Ikappa Balpha -induced block of p65/p50 nuclear translocation. J Biol Chem. 2001;276(2):1164–1172. | |
Akter H, Park M, Kwon OS, Song EJ, Park WS, Kang MJ. Activation of matrix metalloproteinase-9 (MMP-9) by neurotensin promotes cell invasion and migration through ERK pathway in gastric cancer. Tumour Biol. 2015;36(8):6053–6062. | |
Zhang M, Dai C, Zhu H, et al. Cyclophilin A promotes human hepatocellular carcinoma cell metastasis via regulation of MMP3 and MMP9. Mol Cell Biochem. 2011;357(1–2):387–395. | |
Zhou X, Qi Y. PLGF inhibition impairs metastasis of larynx carcinoma through MMP3 downregulation. Tumour Biol. 2014;35(9):9381–9386. | |
Mehner C, Miller E, Khauv D, et al. Tumor cell-derived MMP3 orchestrates Rac1b and tissue alterations that promote pancreatic adenocarcinoma. Mol Cancer Res. 2014;12(10):1430–1439. | |
Slavin S, Yeh CR, Da J, et al. Estrogen receptor alpha in cancer- associated fibroblasts suppresses prostate cancer invasion via modulation of thrombospondin 2 and matrix metalloproteinase 3. Carcinogenesis. 2014;35(6):1301–1309. | |
Koller FL, Dozier EA, Nam KT, et al. Lack of MMP10 exacerbates experimental colitis and promotes development of inflammation-associated colonic dysplasia. Lab Invest. 2012;92(12):1749–1759. | |
Justilien V, Regala RP, Tseng IC, et al. Matrix metalloproteinase-10 is required for lung cancer stem cell maintenance, tumor initiation and metastatic potential. PLoS One. 2012;7(4):e35040. | |
Kwon YJ, Hurst DR, Steg AD, et al. Gli1 enhances migration and invasion via up-regulation of MMP-11 and promotes metastasis in ERalpha negative breast cancer cell lines. Clin Exp Metastasis. 2011;28(5):437–449. | |
Eiró N, Fernandez-Garcia B, González LO, Vizoso FJ. Cytokines related to MMP-11 expression by inflammatory cells and breast cancer metastasis. Oncoimmunology. 2013;2(5):e24010. | |
Strand S, Vollmer P, van den Abeelen L, et al. Cleavage of CD95 by matrix metalloproteinase-7 induces apoptosis resistance in tumour cells. Oncogene. 2004;23(20):3732–3736. | |
Adachi Y, Yamamoto H, Itoh F, Hinoda Y, Okada Y, Imai K. Contribution of matrilysin (MMP-7) to the metastatic pathway of human colorectal cancers. Gut. 1999;45(2):252–258. | |
Fukuda A, Wang SC, Morris JP 4th, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 2011;19(4):441–455. | |
Chang MC, Chen CA, Chen PJ, et al. Mesothelin enhances invasion of ovarian cancer by inducing MMP-7 through MAPK/ERK and JNK pathways. Biochem J. 2012;442(2):293–302. | |
Davies G, Jiang WG, Mason MD. Matrilysin mediates extracellular cleavage of E-cadherin from prostate cancer cells: a key mechanism in hepatocyte growth factor/scatter factor-induced cell-cell dissociation and in vitro invasion. Clin Cancer Res. 2001;7(10):3289–3297. | |
Lu H, Hu L, Yu L, et al. KLF8 and FAK cooperatively enrich the active MMP14 on the cell surface required for the metastatic progression of breast cancer. Oncogene. 2014;33(22):2909–2917. | |
Ager EI, Kozin SV, Kirkpatrick ND, et al. Blockade of MMP14 activity in murine breast carcinomas: implications for macrophages, vessels, and radiotherapy. J Natl Cancer Inst. 2015;107(4):djv017. | |
Tobar N, Avalos MC, Mendez N, et al. Soluble MMP-14 produced by bone marrow-derived stromal cells sheds epithelial endoglin modulating the migratory properties of human breast cancer cells. Carcinogenesis. 2014;35(8):1770–1779. | |
Yang W, Arii S, Gorrin-Rivas MJ, Mori A, Onodera H, Imamura M. Human macrophage metalloelastase gene expression in colorectal carcinoma and its clinicopathologic significance. Cancer. 2001;91(7):1277–1283. | |
Muller M, Beck IM, Gadesmann J, et al. MMP19 is upregulated during melanoma progression and increases invasion of melanoma cells. Mod Pathol. 2010;23(4):511–521. | |
Price A, Shi Q, Morris D, et al. Marked inhibition of tumor growth in a malignant glioma tumor model by a novel synthetic matrix metalloproteinase inhibitor AG3340. Clin Cancer Res. 1999;5(4):845–854. | |
Shalinsky DR, Brekken J, Zou H, et al. Broad antitumor and antiangiogenic activities of AG3340, a potent and selective MMP inhibitor undergoing advanced oncology clinical trials. Ann N Y Acad Sci. 1999;878:236–270. | |
Bissett D, O’Byrne KJ, von Pawel J, et al. Phase III study of matrix metalloproteinase inhibitor prinomastat in non-small-cell lung cancer. J Clin Oncol. 2005;23(4):842–849. | |
Zucker S, Cao J, Chen WT. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene. 2000;19(56):6642–6650. | |
Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295(5564):2387–2392. | |
Millar AW, Brown PD, Moore J, et al. Results of single and repeat dose studies of the oral matrix metalloproteinase inhibitor marimastat in healthy male volunteers. Br J Clin Pharmacol. 1998;45(1):21–26. | |
Rasmussen HS, McCann PP. Matrix metalloproteinase inhibition as a novel anticancer strategy: a review with special focus on batimastat and marimastat. Pharmacol Ther. 1997;75(1):69–75. | |
Sparano JA, Bernardo P, Stephenson P, et al. Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: Eastern Cooperative Oncology Group trial E2196. J Clin Oncol. 2004;22(23):4683–4690. | |
Stone AL, Kroeger M, Sang QX. Structure-function analysis of the ADAM family of disintegrin-like and metalloproteinase-containing proteins (review). J Protein Chem. 1999;18(4):447–465. | |
Hamano Y, Zeisberg M, Sugimoto H, et al. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 2003;3(6):589–601. | |
Heljasvaara R, Nyberg P, Luostarinen J, et al. Generation of biologically active endostatin fragments from human collagen XVIII by distinct matrix metalloproteases. Exp Cell Res. 2005;307(2):292–304. | |
McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289(5482):1202–1206. | |
Vanlaere I, Libert C. Matrix metalloproteinases as drug targets in infections caused by Gram-negative bacteria and in septic shock. Clin Microbiol Rev. 2009;22(2):224–239. Table of Contents. | |
Lorente L, Martin MM, Solé-Violán J, et al. Association of sepsis-related mortality with early increase of TIMP-1/MMP-9 ratio. PLoS One. 2014;9(4):e94318. | |
O’Donoghue AJ, Knudsen GM, Beekman C, et al. Destructin-1 is a collagen-degrading endopeptidase secreted by Pseudogymnoascus destructans, the causative agent of white-nose syndrome (vol 112, pg 7478, 2015). Proc Natl Acad Sci U S A. 2015;112(24):7478–7483. | |
Gao N, Kumar A, Yu FS. Matrix metalloproteinase-13 as a target for suppressing corneal ulceration caused by Pseudomonas aeruginosa infection. J Infect Dis. 2015;212(1):116–127. | |
Ricci S, Grandgirard D, Wenzel M, et al. Inhibition of matrix metalloproteinases attenuates brain damage in experimental meningococcal meningitis. BMC Infect Dis. 2014;14:726. | |
Sivula M, Hastbacka J, Kuitunen A, et al. Systemic matrix metalloproteinase-8 and tissue inhibitor of metalloproteinases-1 levels in severe sepsis-associated coagulopathy. Acta Anaesthesiol Scand. 2015;59(2):176–184. | |
Marchant DJ, Bellac CL, Moraes TJ, et al. A new transcriptional role for matrix metalloproteinase-12 in antiviral immunity. Nat Med. 2014;20(5):493–502. | |
Sathyamoorthy T, Tezera LB, Walker NF, et al. Membrane type 1 matrix metalloproteinase regulates monocyte migration and collagen destruction in tuberculosis. J Immunol. 2015;195(3):882–891. | |
Kim MP, Gaydos CA, Wood BJ, Hardick JP, Zhang YH, Wahl LM. Chlamydia pneumoniae enhances cytokine-stimulated human monocyte matrix metalloproteinases through a prostaglandin E-2-Dependent mechanism. Infect Immun. 2005;73(1):632–634. | |
Zuo X, Pan W, Feng T, Shi X, Dai J. Matrix metalloproteinase 3 promotes cellular anti-dengue virus response via interaction with transcription factor NF kappa B in cell nucleus. PLoS One. 2014;9(1):e84748. | |
Heilpern AJ, Wertheim W, He J, Perides G, Bronson RT, Hu LT. Matrix metalloproteinase 9 plays a key role in lyme arthritis but not in dissemination of Borrelia burgdorferi. Infect Immun. 2009;77(7):2643–2649. | |
Prato M, D’Alessandro S, Van den Steen PE, et al. Natural haemozoin modulates matrix metalloproteinases and induces morphological changes in human microvascular endothelium. Cell Microbiol. 2011;13(8):1275–1285. | |
Harbut MB, Velmourougane G, Dalal S, et al. Bestatin-based chemical biology strategy reveals distinct roles for malaria M1-and M17-family aminopeptidases. Proc Natl Acad Sci U S A. 2011;108(34):E526–E534. | |
Elkington PT, O’Kane CM, Friedland JS. The paradox of matrix metalloproteinases in infectious disease. Clin Exp Immunol. 2005;142(1):12–20. | |
Solan PD, Dunsmore KE, Denenberg AG, Odoms K, Zingarelli B, Wong HR. A novel role for matrix metalloproteinase-8 in sepsis. Crit Care Med. 2012;40(2):379–387. | |
Hoffmann U, Brueckmann M, Borggrefe M. Matrix metalloproteinases and their inhibitors: promising novel biomarkers in severe sepsis? Crit Care. 2009;13(6):1006. | |
Vandenbroucke RE, Dejonckheere E, Van Hauwermeiren F, et al. Matrix metalloproteinase 13 modulates intestinal epithelial barrier integrity in inflammatory diseases by activating TNF. EMBO Mol Med. 2013;5(7):1000–1016. | |
Ichikawa T, Sugiura H, Koarai A, et al. TLR3 activation augments matrix metalloproteinase production through reactive nitrogen species generation in human lung fibroblasts. J Immunol. 2014;192(11):4977–4988. | |
Lee YH, Lai CL, Hsieh SH, Shieh CC, Huang LM, Wu-Hsieh BA. Influenza A virus induction of oxidative stress and MMP-9 is associated with severe lung pathology in a mouse model (vol 178, pg 411, 2013). Virus Res. 2014;183:123. | |
Bradley LM, Douglass MF, Chatterjee D, Akira S, Baaten BJ. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathog. 2012;8(4):e1002641. | |
Zhou WC, Zhang QB, Qiao L. Pathogenesis of liver cirrhosis. World J Gastroenterol. 2014;20(23):7312–7324. | |
Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209–218. | |
Fallowfield JA, Kendall TJ, Iredale JP. Reversal of fibrosis: no longer a pipe dream? Clin Liver Dis. 2006;10(3):481–497, viii. | |
Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem. 2007;101(4):830–839. | |
de Meijer VE, Sverdlov DY, Popov Y, et al. Broad-spectrum matrix metalloproteinase inhibition curbs inflammation and liver injury but aggravates experimental liver fibrosis in mice. PLoS One. 2010;5(6):e11256. | |
Mannino DM, Kiriz VA. Changing the burden of COPD mortality. Int J Chron Obstruct Pulmon Dis. 2006;1(3):219–233. | |
Lagente V, Le Quement C, Boichot E. Macrophage metalloelastase (MMP-12) as a target for inflammatory respiratory diseases. Expert Opin Ther Targets. 2009;13(3):287–295. | |
Merrilees MJ, Ching PST, Beaumont B, Hinek A, Wight TN, Black PN. Changes in elastin, elastin binding protein and versican in alveoli in chronic obstructive pulmonary disease. Respir Res. 2008;9:41. | |
Eurlings IMJ, Dentener MA, Cleutjens JPM, et al. Similar matrix alterations in alveolar and small airway walls of COPD patients. BMC Pulm Med. 2014;14:90. | |
Le Quément C, Guénon I, Gillon JY, et al. The selective MMP-12 inhibitor, AS111793 reduces airway inflammation in mice exposed to cigarette smoke. Br J Pharmacol. 2008;154(6):1206–1215. | |
Churg A, Wang R, Wang X, Onnervik PO, Thim K, Wright JL. Effect of an MMP-9/MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax. 2007;62(8):706–713. | |
Bidan CM, Veldsink AC, Meurs H, Gosens R. Airway and extracellular matrix mechanics in COPD. Front Physiol. 2015;6:346. | |
Han YP. Matrix metalloproteinases, the pros and cons, in liver fibrosis. J Gastroenterol Hepatol. 2006;21(suppl 3):S88–S91. | |
Afdhal NH, Nunes D. Evaluation of liver fibrosis: a concise review. Am J Gastroenterol. 2004;99(6):1160–1174. | |
Jacobsen JA, Major Jourden JL, Miller MT, Cohen SM. To bind zinc or not to bind zinc: an examination of innovative approaches to improved metalloproteinase inhibition. Biochim Biophys Acta. 2010;1803(1):72–94. | |
Hoffman A, Qadri B, Frant J, et al. Carbamoylphosphonate matrix metalloproteinase inhibitors 6: cis-2-aminocyclohexylcarbamoylphosphonic acid, a novel orally active antimetastatic matrix metalloproteinase-2 selective inhibitor – synthesis and pharmacodynamic and pharmacokinetic analysis. J Med Chem. 2008;51(5):1406–1414. | |
Dufour A, Sampson NS, Li J, et al. Small-molecule anticancer compounds selectively target the hemopexin domain of matrix metalloproteinase-9. Cancer Res. 2011;71(14):4977–4988. | |
Dufour A, Sampson NS, Zucker S, Cao J. Role of the hemopexin domain of matrix metalloproteinases in cell migration. J Cell Physiol. 2008;217(3):643–651. | |
Remacle AG, Golubkov VS, Shiryaev SA, et al. Novel MT1-MMP small-molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res. 2012;72(9):2339–2349. | |
Gao M, Nguyen TT, Suckow MA, et al. Acceleration of diabetic wound healing using a novel protease-anti-protease combination therapy. Proc Natl Acad Sci U S A. 2015;112(49):15226–15231. | |
Suojanen J, Salo T, Koivunen E, Sorsa T, Pirilä E. A novel and selective membrane type-1 matrix metalloproteinase (MT1-MMP) inhibitor reduces cancer cell motility and tumor growth. Cancer Biol Ther. 2009;8(24):2362–2370. | |
Koivunen E, Arap W, Valtanen H, et al. Tumor targeting with a selective gelatinase inhibitor. Nat Biotechnol. 1999;17(8):768–774. | |
Heikkilä P, Suojanen J, Pirilä E, et al. Human tongue carcinoma growth is inhibited by selective antigelatinolytic peptides. Int J Cancer. 2006;118(9):2202–2209. | |
Fields GB. New strategies for targeting matrix metalloproteinases. Matrix Biol. 2015;4(4–46):239–246. | |
Xu X, Chen Z, Wang Y, Bonewald L, Steffensen B. Inhibition of MMP-2 gelatinolysis by targeting exodomain-substrate interactions. Biochem J. 2007;406(1):147–155. | |
Higashi S, Hirose T, Takeuchi T, Miyazaki K. Molecular design of a highly selective and strong protein inhibitor against matrix metalloproteinase-2 (MMP-2). J Biol Chem. 2013;288(13):9066–9076. | |
Hashimoto H, Takeuchi T, Komatsu K, Miyazaki K, Sato M, Higashi S. Structural basis for matrix metalloproteinase-2 (MMP-2)-selective inhibitory action of beta-amyloid precursor protein-derived inhibitor. J Biol Chem. 2011;286(38):33236–33243. | |
Burg-Roderfeld M, Roderfeld M, Wagner S, Henkel C, Grotzinger J, Roeb E. MMP-9-hemopexin domain hampers adhesion and migration of colorectal cancer cells. Int J Oncol. 2007;30(4):985–992. | |
Roeb E, Schleinkofer K, Kernebeck T, et al. The matrix metalloproteinase 9 (mmp-9) hemopexin domain is a novel gelatin binding domain and acts as an antagonist. J Biol Chem. 2002;277(52):50326–50332. | |
Ezhilarasan R, Jadhav U, Mohanam I, Rao JS, Gujrati M, Mohanam S. The hemopexin domain of MMP-9 inhibits angiogenesis and retards the growth of intracranial glioblastoma xenograft in nude mice. Int J Cancer. 2009;124(2):306–315. | |
Johnson JL, Devel L, Czarny B, et al. A selective matrix metalloproteinase-12 inhibitor retards atherosclerotic plaque development in apolipoprotein E-knockout mice. Arterioscler Thromb Vasc Biol. 2011;31(3):528–535. | |
Paemen L, Martens E, Masure S, Opdenakker G. Monoclonal antibodies specific for natural human neutrophil gelatinase B used for affinity purification, quantitation by two-site ELISA and inhibition of enzymatic activity. Eur J Biochem. 1995;234(3):759–765. | |
Marshall DC, Lyman SK, McCauley S, et al. Selective allosteric inhibition of MMP9 is efficacious in preclinical models of ulcerative colitis and colorectal cancer. PLoS One. 2015;10(5):e0127063. | |
Devy L, Huang L, Naa L, et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009;69(4):1517–1526. | |
Sela-Passwell N, Kikkeri R, Dym O, et al. Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat Med. 2012;18(1):143–147. | |
Gálvez BG, Matias-Román S, Albar JP, Sánchez-Madrid F, Arroyo AG. Membrane type 1-matrix metalloproteinase is activated during migration of human endothelial cells and modulates endothelial motility and matrix remodeling. J Biol Chem. 2001;276(40):37491–37500. | |
Udi Y, Grossman M, Solomonov I, et al. Inhibition mechanism of membrane metalloprotease by an exosite-swiveling conformational antibody. Structure. 2015;23(1):104–115. | |
Kaneko K, Williams RO, Dransfield DT, Nixon AE, Sandison A, Itoh Y. Selective inhibition of membrane type 1 matrix metalloproteinase abrogates progression of experimental inflammatory arthritis: synergy With tumor necrosis factor blockade. Arthritis Rheumatol. 2016;68(2):521–531. | |
Botkjaer KA, Kwok HF, Terp MG, et al. Development of a specific affinity-matured exosite inhibitor to MT1-MMP that efficiently inhibits tumor cell invasion in vitro and metastasis in vivo. Oncotarget. Epub 2016 Feb 27. | |
Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157(2):220–233. | |
Chung CH. Managing premedications and the risk for reactions to infusional monoclonal antibody therapy. Oncologist. 2008;13(6):725–732. | |
Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–558. | |
Romer T, Leonhardt H, Rothbauer U. Engineering antibodies and proteins for molecular in vivo imaging. Curr Opin Biotechnol. 2011;22(6):882–887. | |
Kulasegaram R, Giersing B, Page CJ, et al. In vivo evaluation of 111In-DTPA-N-TIMP-2 in Kaposi sarcoma associated with HIV infection. Eur J Nucl Med. 2001;28(6):756–761. | |
Tran PL, Vigneron JP, Pericat D, et al. Gene therapy for hepatocellular carcinoma using non-viral vectors composed of bis guanidinium-tren-cholesterol and plasmids encoding the tissue inhibitors of metalloproteinases TIMP-2 and TIMP-3. Cancer Gene Ther. 2003;10(6):435–444. | |
Yang SW, Cody JJ, Rivera AA, et al. Conditionally replicating adenovirus expressing TIMP2 for ovarian cancer therapy. Clin Cancer Res. 2011;17(3):538–549. | |
Murphy G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011;12(11):233. | |
Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 2010;1803(1):55–71. | |
Bauvois B. New facets of matrix metalloproteinases MMP-2 and MMP-9 as cell surface transducers: outside-in signaling and relationship to tumor progression. Biochim Biophys Acta. 2012;1825(1):29–36. | |
Têtu B, Brisson J, Wang CS, et al. The influence of MMP-14, TIMP-2 and MMP-2 expression on breast cancer prognosis. Breast Cancer Res. 2006;8(3):R28. | |
Shen Q, Lee ES, Pitts RL, Wu MH, Yuan SY. Tissue inhibitor of metalloproteinase-2 regulates matrix metalloproteinase-2-mediated endothelial barrier dysfunction and breast cancer cell transmigration through lung microvascular endothelial cells. Mol Cancer Res. 2010;8(7):939–951. | |
Murphy G, Houbrechts A, Cockett MI, Williamson RA, O’Shea M, Docherty AJ. The N-terminal domain of tissue inhibitor of metalloproteinases retains metalloproteinase inhibitory activity. Biochemistry. 1991;30(33):8097–8102. | |
Fernandez CA, Butterfield C, Jackson G, Moses MA. Structural and functional uncoupling of the enzymatic and angiogenic inhibitory activities of tissue inhibitor of metalloproteinase-2 (TIMP-2): loop 6 is a novel angiogenesis inhibitor. J Biol Chem. 2003;278(42):40989–40995. | |
Batra J, Robinson J, Mehner C, et al. PEGylation extends circulation half-life while preserving in vitro and in vivo activity of tissue inhibitor of metalloproteinases-1 (TIMP-1). PLoS One. 2012;7(11):e50028. | |
Tlatli R, Nozach H, Collet G, et al. Grafting of functional motifs onto protein scaffolds identified by PDB screening – an efficient route to design optimizable protein binders. FEBS J. 2013;280(1):139–159. | |
Hamze AB, Wei S, Bahudhanapati H, Kota S, Acharya KR, Brew K. Constraining specificity in the N-domain of tissue inhibitor of metalloproteinases-1; gelatinase-selective inhibitors. Protein Sci. 2007;16(9):1905–1913. | |
Grossman M, Tworowski D, Dym O, et al. The intrinsic protein flexibility of endogenous protease inhibitor TIMP-1 controls its binding interface and affects its function. Biochemistry. 2010;49(29):6184–6192. | |
Butler GS, Hutton M, Wattam BA, et al. The specificity of TIMP-2 for matrix metalloproteinases can be modified by single amino acid mutations. J Biol Chem. 1999;274(29):20391–20396. | |
Wei S, Chen Y, Chung L, Nagase H, Brew K. Protein engineering of the tissue inhibitor of metalloproteinase 1 (TIMP-1) inhibitory domain. In search of selective matrix metalloproteinase inhibitors. J Biol Chem. 2003;278(11):9831–9834. | |
Banta S, Dooley K, Shur O. Replacing antibodies: engineering new binding proteins. Annu Rev Biomed Eng. 2013;15:93–113. | |
Jones DS, Silverman AP, Cochran JR. Developing therapeutic proteins by engineering ligand-receptor interactions. Trends Biotechnol. 2008;26(9):498–505. | |
Bahudhanapati H, Zhang Y, Sidhu SS, Brew K. Phage display of tissue inhibitor of metalloproteinases-2 (TIMP-2): identification of selective inhibitors of collagenase-1 (metalloproteinase 1 (MMP-1)). J Biol Chem. 2011;286(36):31761–31770. | |
Sharabi O, Shirian J, Shifman JM. Predicting affinity- and specificity-enhancing mutations at protein-protein interfaces. Biochem Soc Trans. 2013;41(5):1166–1169. | |
Sharabi O, Shirian J, Grossman M, Lebendiker M, Sagi I, Shifman J. Affinity- and specificity-enhancing mutations are frequent in multispecific interactions between TIMP2 and MMPs. PLoS One. 2014;9(4):e93712. | |
Gebauer M, Skerra A. Engineered protein scaffolds as next-generation antibody therapeutics. Curr Opin Chem Biol. 2009;13(3):245–255. | |
Wittrup KD. Protein engineering by cell-surface display. Curr Opin Biotechnol. 2001;12(4):395–399. | |
Pepper LR, Cho YK, Boder ET, Shusta EV. A decade of yeast surface display technology: where are we now? Comb Chem High Throughput Screen. 2008;11(2):127–134. | |
Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15(6):553–557. | |
Boder ET, Bill JR, Nields AW, Marrack PC, Kappler JW. Yeast surface display of a noncovalent MHC class II heterodimer complexed with antigenic peptide. Biotechnol Bioeng. 2005;92(4):485–491. | |
Wang Z, Mathias A, Stavrou S, Neville DM Jr. A new yeast display vector permitting free scFv amino termini can augment ligand binding affinities. Protein Eng Des Sel. 2005;18(7):337–343. | |
Yan W, Zhang W, Sun L, et al. Identification of MMP-9 specific microRNA expression profile as potential targets of anti-invasion therapy in glioblastoma multiforme. Brain Res. 2011;1411:108–115. | |
Yu C, Chen WP, Wang XH. MicroRNA in osteoarthritis. J Int Med Res. 2011;39(1):1–9. | |
Maegdefessel L, Dalman RL, Tsao PS. Pathogenesis of abdominal aortic aneurysms: microRNAs, proteases, genetic associations. Annu Rev Med. 2014;65:49–62. | |
Xia H, Qi Y, Ng SS, et al. microRNA-146b inhibits glioma cell migration and invasion by targeting MMPs. Brain Res. 2009;1269:158–165. | |
Gabriely G, Wurdinger T, Kesari S, et al. MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol Cell Biol. 2008;28(17):5369–5380. | |
Costa PM, Cardoso AL, Custódia C, Cunha P, Pereira de Almeida L, Pedroso de Lima MC. MiRNA-21 silencing mediated by tumor-targeted nanoparticles combined with sunitinib: a new multimodal gene therapy approach for glioblastoma. J Control Release. 2015;207:31–39. | |
Hwang SJ, Seol HJ, Park YM, et al. MicroRNA-146a suppresses metastatic activity in brain metastasis. Mol Cells. 2012;34(3):329–334. | |
Hu Y, Ou Y, Wu K, Chen Y, Sun W. miR-143 inhibits the metastasis of pancreatic cancer and an associated signaling pathway. Tumour Biol. 2012;33(6):1863–1870. | |
Osaki M, Takeshita F, Sugimoto Y, et al. MicroRNA-143 regulates human osteosarcoma metastasis by regulating matrix metalloprotease-13 expression. Mol Ther. 2011;19(6):1123–1130. | |
Wang Q, Cai J, Wang J, Xiong C, Zhao J. MiR-143 inhibits EGFR-signaling-dependent osteosarcoma invasion. Tumour Biol. 2014;35(12):12743–12748. | |
He Y, Huang C, Sun X, Long XR, Lv XW, Li J. MicroRNA-146a modulates TGF-beta1-induced hepatic stellate cell proliferation by targeting SMAD4. Cell Signal. 2012;24(10):1923–1930. | |
He Y, Huang C, Lin X, Li J. MicroRNA-29 family, a crucial therapeutic target for fibrosis diseases. Biochimie. 2013;95(7):1355–1359. | |
Wang J, Gao Y, Ma M, et al. Effect of miR-21 on renal fibrosis by regulating MMP-9 and TIMP1 in kk-ay diabetic nephropathy mice. Cell Biochem Biophys. 2013;67(2):537–546. | |
Esteso G, Luzón E, Sarmiento E, et al. Altered microRNA expression after infection with human cytomegalovirus leads to TIMP3 downregulation and increased shedding of metalloprotease substrates, including MICA. J Immunol. 2014;193(3):1344–1352. | |
Kwo PY, Lawitz EJ, McCone J, et al; SPRINT-1 investigators. Efficacy of boceprevir, an NS3 protease inhibitor, in combination with peginterferon alfa-2b and ribavirin in treatment-naive patients with genotype 1 hepatitis C infection (SPRINT-1): an open-label, randomised, multicentre phase 2 trial. Lancet. 2010;376(9742):705–716. | |
Eron JJ Jr. HIV-1 protease inhibitors. Clin Infect Dis. 2000;30(suppl 2):S160–S170. | |
Takahashi S, Ishii M, Namkoong H, et al. Pneumococcal infection aggravates elastase-induced emphysema via MMP-12 overexpression. J Infect Dis. 2016;213(6):1018–1030. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.