Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 17
Maternally Inherited Essential Hypertension May Be Associated with the Mutations in Mitochondrial tRNAGlu Gene
Authors Wang C ![]() , Deng X, Li L, Li M
, Deng X, Li L, Li M
Received 20 August 2023
Accepted for publication 21 December 2023
Published 9 January 2024 Volume 2024:17 Pages 13—26
DOI https://doi.org/10.2147/PGPM.S436235
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Chun Wang,1 Xin Deng,1 Lei Li,2 Mei Li3
1Department of Integrated TCM & Western Medicine, Mengcheng County Second People’s Hospital, Anhui, 233500, People’s Republic of China; 2Department of Cardiology, Mengcheng County Second People’s Hospital, Anhui, 233500, People’s Republic of China; 3Department of Pharmacy, Mengcheng County Second People’s Hospital, Anhui, 233500, People’s Republic of China
Correspondence: Chun Wang, Department of Integrated TCM & Western Medicine, Mengcheng County Second People’s Hospital, Anhui, 233500, People’s Republic of China, Tel +86-0558-7623307, Email [email protected]
Background: Mitochondrial DNA (mtDNA) mutations are associated with essential hypertension (EH), but the molecular mechanism remains largely unknown.
Objective: The aim of this study is to explore the association between mtDNA mutations and EH.
Methods: Two maternally inherited families with EH are underwent clinical, genetic and biochemical assessments. mtDNA mutations are screened by PCR-Sanger sequencing and phylogenetic, and bioinformatics analyses are performed to evaluate the pathogenicity of mtDNA mutations. We also generate cytoplasmic hybrid (cybrid) cell lines to analysis mitochondrial functions.
Results: Matrilineal relatives exhibit variable degree of clinical phenotypes. Molecular analysis reveals the presence of m.A14693G and m.A14696G mutations in two pedigrees. Notably, the m.A14693G mutation occurs at position 54 in the TψC loop of tRNAGlu, a position which is critical for post-transcriptionally modification of tRNAGlu. While the m.A14696G mutation creates a novel base-pairing (51C-64G). Bioinformatic analysis shows that these mutations alter tRNAGlu secondary structure. Additionally, patients with tRNAGlu mutations exhibit markedly decreased in mtDNA copy number, mitochondrial membrane potential (MMP) and ATP, whereas the levels of reactive oxygen species (ROS) increase significantly.
Conclusion: The m.A14696G and m.A14693G mutations lead to failure in tRNAGlu metabolism and cause mitochondrial dysfunction that is responsible for EH.
Keywords: EH, mt-tRNAGlu m.A14693G m.A14696G, mitochondrial dysfunction, Chinese families
Introduction
EH was a very common chronic disease which was becoming an urgent public health problem worldwide, it had been estimated that ~9.4 million hypertensive patients died each year.1 EH was regarded as a risk factor for coronary heart disease (CHD), stroke and renal failure,2 thus, understanding its pathophysiology had become a major research focus. Since the landmark discovery of the draft sequences of human genome, experts announced that within 10 years they expected to determine the important of the genome as related to EH.3 Some great advance had been made toward the molecular basis of EH, for example, nuclear genes such as Nr2f2,4 CUL3,5 and EIF2AK46 had been identified to be associated with EH. However, the detailed molecular mechanism of EH was still unknown.
While the nuclear genome had been studied extensively with respect to EH, we noticed that mtDNA mutations also played active roles in EH. Mitochondrion was a small symbiotic organelle combined with aerobic bacteria and primordial eukaryotic cells.7 It had its own DNA, called mtDNA, encoding 37 genes spanning tRNAs, rRNAs and the subunits of the respiratory chain.8 As the adaptor that decoded the mRNA sequence into protein, the basic aspects of mt-tRNA structure and function were central to all studies of mitochondrial biomedicine. Unlike canonical tRNAs such as human cytosolic tRNAs, human mt-tRNAs had specific features such as non-classical G-C pairs and mismatches.9 In fact, most mt-tRNAs from all domains of life had a highly conserved cloverleaf structures, consisting of acceptor arm, D-arm, anticodon stem, variable region and TψC loop, with an average length of 73 nucleotides. Mutations in mt-tRNAs had been reported in some cases of maternally inherited EH, such as tRNAIle A4263G,10 tRNASer(UCN) 7471delC,11 and tRNAGln/tRNAMet A4401G mutations.12 These mutations may reduce the steady-state level or aminoacylation ability of corresponding mt-tRNAs, affect the 5’ or 3’ end processing, CCA addition, cause the defects in chemical modification and lead to failure in mt-tRNA metabolism, and subsequently impair mitochondrial translation and function, which was involved in the progression and pathogenesis of EH.13
Most recently, with the aim of exploring the molecular basis of maternally transmitted EH, we carried out a genetic screening program for EH-associated mtDNA mutations in Anhui Province, P.R. China. Herein, we reported clinical, genetic and molecular characterizations of two Chinese pedigrees with EH, sequence analysis of the complete mitochondrial genomes led us to identify two potential pathogenic mutations: m.A14693G and m.A14696G in tRNAGlu gene. To further explore the contributions of mt-tRNAGlu mutations to EH, we analyzed mitochondrial functions in cybrids cell lines derived from six patients with tRNAGlu mutations and four controls without these mutations. Subsequently, we noticed that m.A14693G and m.A14696G mutations affected mitochondrial functions, decreased mtDNA copy number, ATP and MMP, and enhanced ROS production. Therefore, mutations in tRNAGlu caused mitochondrial dysfunction that was responsible for hypertension.
Materials and Methods
Subjects
We enrolled two genetically unrelated Chinese families with EH (Figure 1). These pedigrees were ascertained in the Department of Integrated TCM & Western Medicine, Mengcheng County Second People’s Hospital in Anhui Province of China. This study was complied with the Declaration of Helsinki, and the methodologies for obtaining the blood samples, as well as the clinical examination of all participants from two pedigrees were approved by the Ethic Committees of Mengcheng County Second People’s Hospital. Besides, 268 unrelated healthy controls were obtained from the volunteers in the same area, these subjects were consisted with 160 male and 108 females, aged from 25 to 48 years, with the average of 36 years. The inclusion criteria for these control subjects were as follows: healthy individuals without any diseases or had any family history of cardiovascular and neurological diseases. Exclusion criteria were ongoing maintenance dialysis, a grave acute infectious disease, neoplastic disease, severe liver dysfunction, major surgery, a chronic inflammatory disease and autoimmune disease. Patients who suffered severe life-threatening injury to other organs were also excluded. Both families provided written informed consent, and furthermore, the informed consent to have their case details published were obtained from all subjects enrolled in this study.
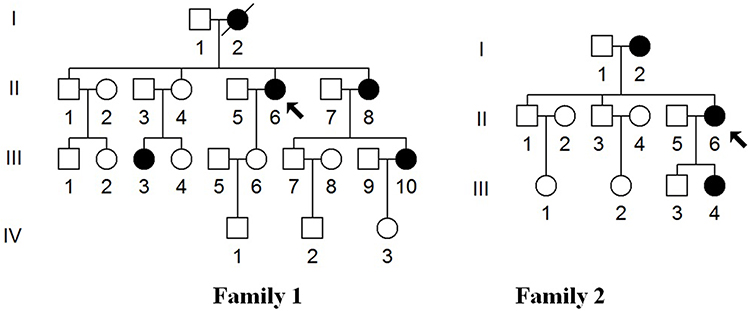 |
Figure 1 Two Han Chinese families with EH, arrows indicate the probands. |
Assessment of BP
The BP of each patient from two families was measured by using a mercury column sphygmomanometer (CARENT Devices, China). The first and the fifth Korotkoff sounds were indicative of systolic and diastolic BP, respectively. The average of three measured BP readings was taken as the examination BP.14 Notably, EH was defined as systolic BP ≥140 mm Hg or diastolic BP ≥90 mm Hg on three consecutive days according to the Seventh Report of the Joint National Committee.15
mtDNA Analysis
To screen the EH-associated mtDNA mutations, we first isolated the genomic DNA from matrilineal relatives from two families, as well as 268 control subjects using the Puregene DNA Isolation Kits (Gentra Systems, Minneapolis, MN), according to the manufacturer’s instructions. Briefly, 24 overlapping PCR fragments were generated and amplified the complete mitochondrial genome, according to a previous study.16 The PCR products were then purified and sequenced by the ABI 3700 automated DNA sequencer. The data were analyzed with SeqWeb program GAP (GCG) according to the revised Cambridge reference sequences (GenBank accession number: NC_012920.1).17
Analysis of the Conservation Index (CI)
To assess the potential pathogenic roles of mtDNA mutations, the CI of each variant was calculated by using the phylogenetic conservation analysis.18 The CI ≥75% was considered to be functional potential.19
Bioinformatics Analysis
To see whether m.A14693G and m.A14696G mutations altered the secondary structure of tRNAGlu, the RNA Fold Webserver (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) was used to predict the minimum-free energy (MFE) of tRNAGlu with and without these mutations.20
Analysis of mtDNA Content
The mtDNA content was measured by using real-time PCR as suggested previously.21 Briefly, mtDNA content was normalized to a single copy nuclear β-globin gene. The following primers were used for real-time PCR analysis: for β-globin gene: forward: 5’-CTATgggACgCTTgATgT-3’; reverse: 5’- gCAATCATTCgTCTgTTT-3’. For mtDNA: forward: 5’-CACCAgCCTAACCAg ATTTC-3’; reverse: 5’-gggTTgTATTgATgAgATTAgT-3’. We first generated standard curves for both fragments and calculated their respective amplification efficiencies to test if using the 2−ΔΔCT method was appropriate. The real-time PCR was then conducted for the calibrator mtDNA content. All experiments were duplicated in three times.
Generation of Cybrid Cell Lines
Trans-mitochondrial cybrids were obtained by fusion of mtDNA-less ρ0 human osteosarcoma 143B cells with platelets, which were isolated from the blood of six affected individuals (Family 1: II-6; II-8 and III-10; Family 2: II-1, II-6 and III-4), together with four controls (C1, C2, C3 and C4), as described previously.22 The transformant clones were cultured in DMEM (Sigma-Aldrich) containing 10% FBS (Sigma-Aldrich) at 37°C in a humidified CO2 incubator.
ATP Analysis
The Cell Titer-Glo® Luminescent Cell Viability Assay kit (Promega, Madison, WI, USA) was used for the measurement of ATP levels with some modifications.23 Luminescence intensity was analyzed by using a fluorescence microplate reader (Molecular Devices, CA, USA), and the amount of ATP was calculated from an ATP standard curve. Each experiment was repeated in three times.
Qualification of ROS Levels
The fluorogenic marker 2’, 7’-Dichlorodihydrofluorescein diacetate (H2DCFDA) was live-cell-permeable acetate ester, and upon entry, it was then cleaved by cellular esterases, reacted with cellular ROS and emitted green fluorescence. To analyze the ROS level, a total of 2×106 cells were first incubated with H2DCFDA for 30 min, after which the cells were analyzed using a fluorescence plate reader, as described previously.24 Each experiment was repeated in three times.
MMP Analysis
To determine whether mt-tRNAGlu mutations affected mitochondrial function, the MMP of mutant and control cell lines was performed using JC-1 Assay Kit-Microplate (Abcam). JC-1 was a cationic carbocyanine dye; when it accumulated in the mitochondria of low MMP in monomer form, it produced green fluorescence. At high concentrations in high MMP, it produced red fluorescence.25 Each experiment was repeated in three times.
Evaluation of the Pathogenicity
We further utilized the pathogenicity scoring system to evaluate the potential pathogenic roles of m.A14693G and m.A14696G mutations.26 This pathogenicity scoring system employed a number of weighted criteria covering a range of molecular and genetic data, from which an overall pathogenicity score can be obtained. In particular, a variant was classified as “definitely pathogenic” with a score >11 points, whereas variant was defined as “possible pathogenic” with a core of 7–10 points and a “neutral polymorphism” with a score of <6 points.
Statistical Analyses
Statistical analyses were performed using SPSS 22.0 (SPSS Inc., Chicago, IL, USA). Student’s t-test was used to assess the statistical significance between unpaired samples. P<0.05 was considered to indicate a statistically significant difference.
Results
Clinical and Genetic Characterizations of Two Chinese Families with EH
In Family 1, the proband (II-6) was a 75-year-old woman who came from Mengcheng City of Anhui Province. She developed EH at the age of 70, her average BP was 150/100 mmHg. She went to our hospital for regular treatment of hypertension. After comprehensive history examination, we noticed that several members (II-8, III-3 and III-10) were also hypertensive persons. Therefore, the transmission of EH was a typical maternally inheritance. In addition, all members in Family 1 did not suffer from vision or hearing loss, cancer, diabetes, neurological disorders, suggested that they manifested hypertension as a sole clinical phenotype.
In Family 2, the proband (II-6) was a 66-year-old woman who lived in Mengcheng City of Anhui Province. After the genetic counseling, we found that she suffered from EH when she was 60, and her BP was 180/100 mmHg. In addition, her mother (I-2) and daughter (III-4) were hypertensive individuals. Moreover, all members in Family 2 did not exhibit other mitochondrial disorders such as hearing impairment, vision loss, neurological diseases, cancer and other endocrine diseases, indicating that they expressed the EH as sole clinical phenotypes. The clinical characterizations of these members from two families were listed in Table 1.
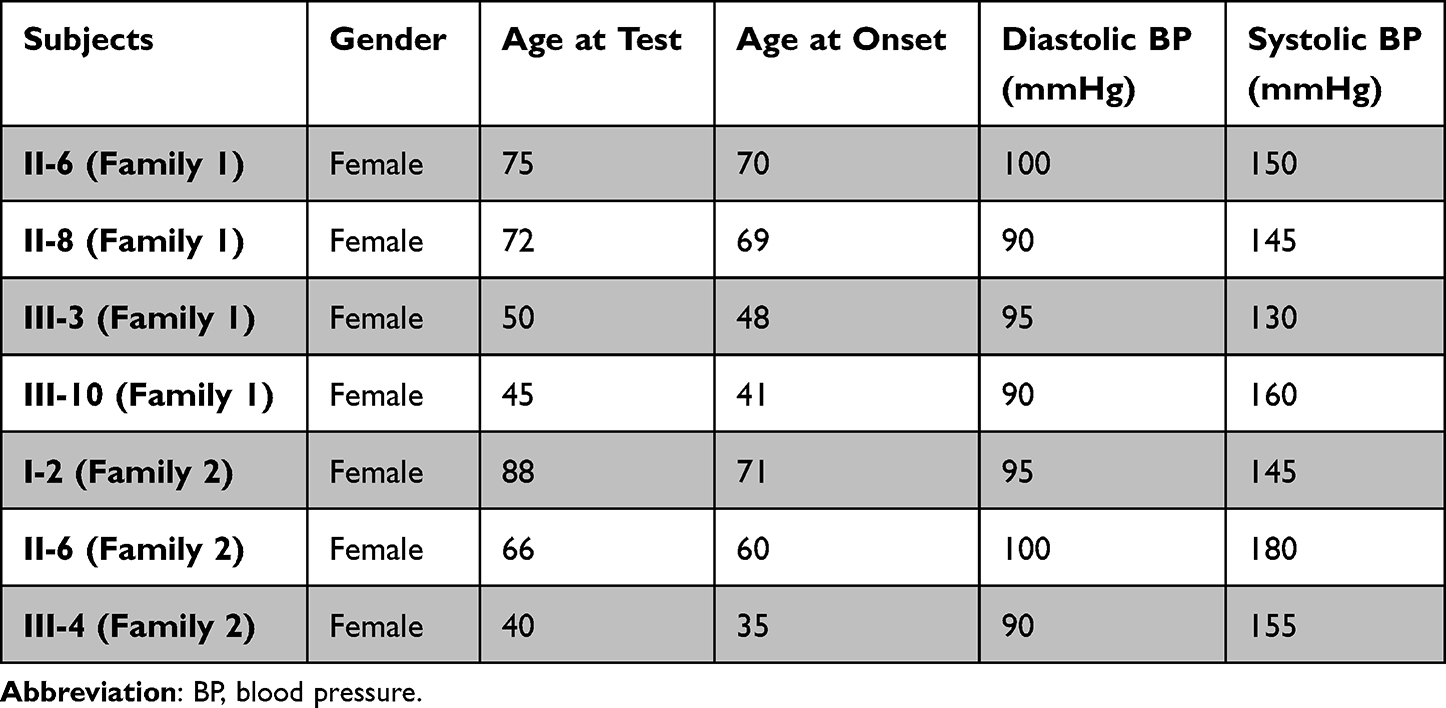 |
Table 1 Summary of Clinical Data for Some Members in Two Families with EH |
Screening for mtDNA Mutations
As these pedigrees were maternally transmitted, which suggested the involvement of mtDNA mutations in the phenotypic manifestation of EH. For this purpose, we amplified the whole mitochondrial genomes from the matrilineal relatives of Family 1 (II-6, II-8, III-3 and III-10) and Family 2 (I-2, II-6 and III-4). After PCR amplification, the products were sequenced by automatic DNA sequencer and subsequently analyzed by using DNA Star software to detect mtDNA variants. The data were listed in Table 2, sequence analysis revealed a set of genetic polymorphisms, in addition to the tRNAGlu A14693G and A14696G mutations (Figures 2 and 3). Note that, there were 14 variants in D-loop region, four known mutations in 12S rRNA and three mutations in 16S rRNA, whereas other genetic polymorphisms were mainly localized at oxidative phosphorylation (OXPHOS)-related genes. Moreover, 12 missense mutations were identified, including the ND2 G4924A (Ser to Asn) and C5178A (Leu to Met), A8 C8414T (Leu to Phe), A6 A8701G (Thr to Ala) and A8860G (Thr to Ala), CO3 A9327G (Thr to Ala), ND3 A10398G (Thr to Ala), ND5 G13708A (Ala to Thr) and G13928C (Ser to Thr), CytB A15326G (Thr to Ala) and A15851G (Ile to Val).
 |
Table 2 mtDNA Sequence Variations in Two Families with EH |
 |
Figure 2 Identification of tRNAGlu A14693G and A14696G mutations by direct sequencing. Abbreviations: MT, mutant; WT, wild type. |
 |
Figure 3 Secondary structure of tRNAGlu gene with and without the m.A14693G and m.A14696G mutations. |
We used the following criteria to classify mtDNA pathogenic mutations: 1) occurred in <1% of the controls; 2) evolutionary conservation (CI ≥ 75%); 3) potential structural and functional alterations; 4) pathogenicity scoring system. Phylogenetic conservation analysis including the mtDNA sequences from mouse,27 bovine28 and Xenopus laevis29 was also performed. We found that besides the m.A14693G and m.A14696G mutations (Figure 4), others were not well conserved from different species. Moreover, further genetic analysis revealed that the m.A14693G and m.A14696G mutations were not detected in 268 control subjects (P<0.05 for all), suggesting that the m.A14693G and m.A14696G mutations may be involved in the pathogenesis of EH in these families.
 |
Figure 4 Alignment of tRNAGlu gene from various species, arrows indicated the positions of 51 and 54, corresponding to the m.A14696G and m.A14693G mutations. |
In fact, the m.A14693G mutation occurred at position 54 in TψC loop of tRNAGlu, which was extremely conserved from bacteria to human mitochondria.30 In addition, the m.A14696G mutation was localized at position 51 in TψC loop of tRNAGlu, creating a novel base pairing (51C-64G).31
The m.A14693G and m.A14696G Mutations Altered tRNAGlu Structure
The secondary structures of tRNAGlu with and without A14696G and A14693G mutations were predicted by RNA Fold Webserver.20 As shown in Figure 5, we noticed that m.A14696G and m.A14693G mutations significantly altered tRNAGlu structure, highlighting the impact of these mutations on tRNA functions.32
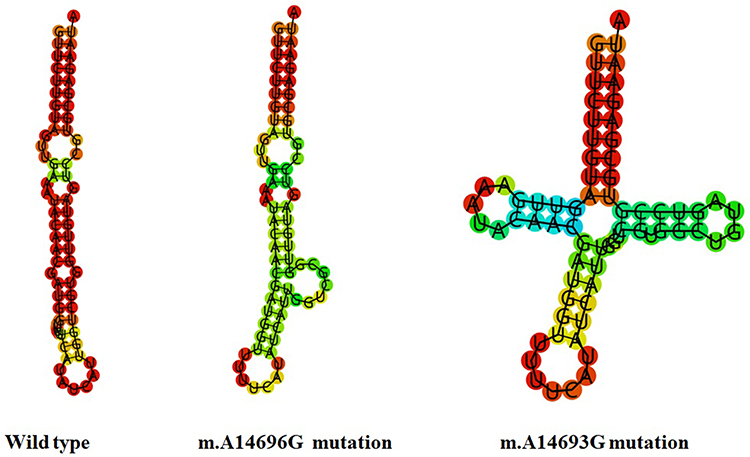 |
Figure 5 Prediction of tRNAGlu secondary structure with and without the m.A14696G and m.A14693G mutations. |
The m.A14693G and m.A14696G Mutations Decreased mtDNA Copy Number
As shown in Figure 6A, patients with m.A14696G and m.A14693G mutations exhibited much lower levels of mtDNA copy number when comparing with the controls (P=0.0030).
 |
Figure 6 Analysis of mitochondrial functions in cybrids. (A) mtDNA copy number; (B) ATP analysis; (C) ROS analysis; (D) MMP analysis. |
ATP Decreased in Cells Carrying tRNAGlu Mutations
Since mitochondria generated ATP through OXPHOS, impairment in ATP synthesis was thought to be involved in mitochondrial function.33 As shown in Figure 6B, approximately 33% reduction of ATP was observed in patients carrying the mt-tRNAGlu mutations when compared with the healthy subjects (P<0.05).
Enhancement of ROS Production
As shown in Figure 6C, patients with m.A14693G or m.A14696G mutations showed an approximately 37% increased in ROS production as compared with controls without these mutations (P<0.05).
Decreased in MMP
Decreased in MMP was an early event for apoptosis and critical for mitochondrial function.34 As shown in Figure 6D, subjects with mt-tRNAGlu mutation showed an approximately 31% reduction in MMP as compared with controls without these mutations (P=0.0001).
The m.A14693G and m.A14696G Mutations May Be Risk Factors for EH
According to the revised pathogenicity scoring system by Yarham et al,26 the total scores of m.A14693G and m.A14696G mutations were 11 and 9 points, respectively (Table 3), suggesting that they belonged to “definitely pathogenic” and “possibly pathogenic” at this stage.
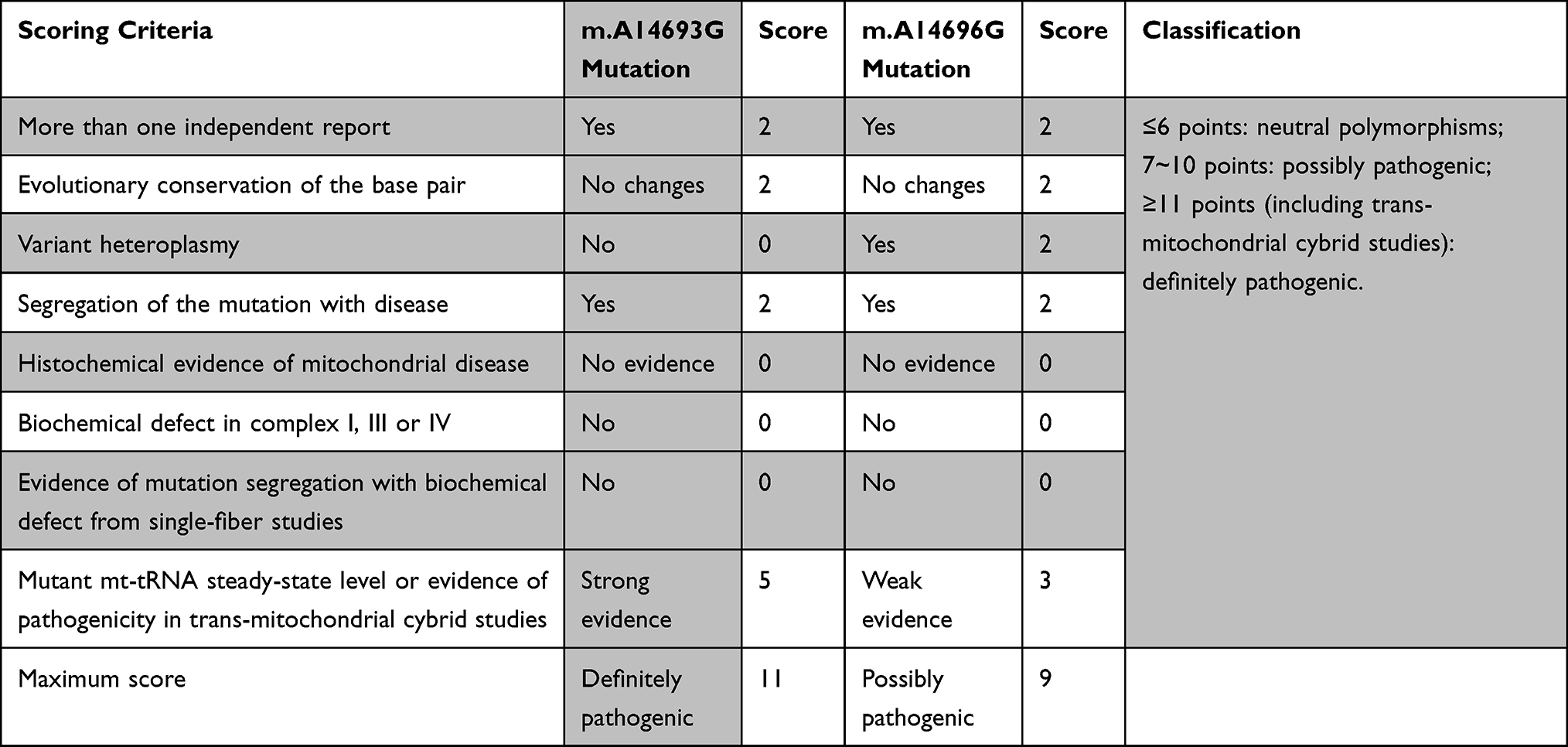 |
Table 3 The Pathogenicity Scoring System for m.A14693G and m.A14696G Mutations |
Discussion
In the current study, we carried out clinical and genetic assessments of two Chinese families with EH, and further investigated the contributions of mtDNA mutations to EH. Notably, members of these pedigrees expressed variable degrees of BP and different age at onset of EH. Interestingly, the age of onset of EH in Family 1 ranged from 41 to 70 years, with the average of 57 years. Meanwhile, the age of onset of EH in Family 2 ranged from 35 to 71 years, with the average of 55 years. In addition, compared with the first and second generations of this pedigree, members in the third generation had an earlier age of onset of EH, indicating that screening for the mtDNA pathogenic mutations was necessary for early diagnosis and prevention of EH.
Analysis of the entire mitochondrial genomes from the matrilineal relatives revealed the presence of homoplasmic mt-tRNAGlu A14693G and A14696G mutations, as well as sets of polymorphisms belonging to human mitochondrial haplogroup Y2 and D4a,35 respectively. Notably, nucleotide at position 54 was more prone to be modified than other positions of tRNA.36 The nucleotide modification at this position had been shown to have a pivotal role in the stabilization of tertiary structure and the biochemical function of tRNAGlu.37 Therefore, the m.A14693G mutation caused an impairment of tRNA modification and mitochondrial translation.38 Moreover, the m.A14693G mutation had been reported to be related to Leber’s hereditary optic neuropathy (LHON) and acted as a modifier for increasing the penetrance and expressivity of m.A1555G-induced deafness.39,40
While the homoplasmic m.A14696G mutation occurred at position 51 in the TψC loop of tRNAGlu, which was also very conserved from various species.41 Interestingly, the m.A14696G mutation created a novel base pairing (51C-64G) and may result a failure in tRNA metabolism. Importantly, the stem of TψC loop harbored a wobble composed of nucleotides 50 to 51 and 63 to 64 in human mt-tRNAGlu,42 therefore, the mutant 14,696 reduced the wobble and may lead to a failure in tRNA metabolism. Furthermore, bioinformatic analysis revealed that the m.A14693G and m.A14696G mutations caused obviously change of tRNAGlu secondary structure, indicating that the alternation of tRNA structure may affect its steady-state level, as well as its aminoacylation ability, as in the case of tRNALeu(UUR) A3243G and ND6 T14502C mutations.43
To see the contributions of m.A14693G and m.A14696G mutations to EH, we analyzed mitochondrial functions including mtDNA content, ATP, MMP and ROS in hypertension patients and healthy controls. As a result, markedly decreased in mtDNA copy number, ATP and MMP were observed in patients carrying tRNAGlu mutations, whereas ROS increased significantly. In fact, mtDNA copy number was a mitochondrial function marker that reflected its depletion, energy reserves and oxidative stress.44 Recent experimental studies indicated that decreased peripheral mtDNA copy number was associated with the risk of heart failure and long-term outcomes.45 MMP reflected the pumping of hydrogen ions across the inner membrane during the process of electron transport and OXPHOS.46 The defects in MMP may be due to strongly decreased efficiency of respiratory chain-mediated proto extrusion for the matrix, as in the case of tRNAHis T12201C mutation.47
Additionally, a decreased mtDNA copy number had been demonstrated to lead to increased ROS levels; ROS induced by mitochondrial dysfunction can increase mitochondrial Ca2+ accumulation and may act as potential pathophysiological mechanism in hypertension.48 Furthermore, the ATP dropped significantly indicated that the OXPHOS complexes were impaired in subjects with m.A14693G and m.A14696G mutations. Through the application of the pathogenicity scoring system,26 the total scores of m.A14693G and m.A14696G mutations were 11 and 9 points, respectively, belonged to “definitely” and “possibly” pathogenic at this stage (Table 3).
However, the homoplasmic form, late onset and incomplete penetrance of EH observed in these Chinese families carrying the tRNAGlu mutations suggested that the m.A14693G and m.A14696G mutations were involved in the development of EH but may be insufficient to produce a clinical phenotype; hence, other factors such as nuclear genes (ADD1; ALDH1A3),49,50 environmental components (air pollution), epigenetic modification (histone modification, DNA methylation)51,52 and personal lifestyle (smoking or high salt intake) may contribute to EH expression in these two families.53,54 In particular, mito-nuclear communication played a putative role in the pathogenesis of cardiovascular disease.55 The main limitations of this study were the relatively small sample size, further studies including more EH patients and controls, as well as the examinations of tRNA functions in the cybrids were needed to verify this conclusion.
Conclusion
Our study indicated that mt-tRNAGlu A14693G and A14696G mutations altered the tRNA structure and functions, led to mitochondrial dysfunction that was involved in EH, screening for tRNA mutations was recommended for early diagnosis and detection of EH.
Abbreviations
EH, essential hypertension; mtDNA, mitochondrial DNA; cybrid, cytoplasmic hybrid; MMP, mitochondrial membrane potential; ROS, reactive oxygen species; mt-tRNA, mitochondrial tRNA; CHD, coronary heart disease; BP, blood pressure; CI, conservation index; MFE, minimum-free energy; H2DCFDA, 2’, 7’-Dichlorodihydrofluorescein diacetate; OXPHOS, oxidative phosphorylation; LHON, Leber’s hereditary optic neuropathy.
Data Sharing Statement
The datasets used and analyzed during the current study are available from corresponding author (Chun Wang, E-mail: [email protected]) on reasonable request.
Ethics Approval and Consent to Participate
This study was approved by the Ethics Committee of Mengcheng County Second People’s Hospital and conformed to the ethical principles of the Declaration of Helsinki. All participants from two pedigrees signed informed consent forms, as well as their cases to be published were obtained before participating in this study.
Acknowledgments
We are grateful to the members of two pedigrees for participating in this study.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
No potential conflict of interest was reported by the author(s).
References
1. Marian AJ. Mitochondrial genetics and human systemic hypertension. Circ Res. 2011;108(7):784–786. doi:10.1161/CIRCRESAHA.111.242768
2. Zhang Y, Xu G, Smoking WP. Hypertension, and GG Genotype of the IL-6 rs1800796 Polymorphism are Independent Risk Factors for Abdominal Aortic Aneurysm in Han Population. Pharmgenomics Pers Med. 2021;14:1115–1121. doi:10.2147/PGPM.S328894
3. Oliveira-Paula GH, Pereira SC, Tanus-Santos JE, et al. Pharmacogenomics and Hypertension: current Insights. Pharmgenomics Pers Med. 2019;12:341–359. doi:10.2147/PGPM.S230201
4. Kumarasamy S, Waghulde H, Gopalakrishnan K, et al. Mutation within the hinge region of the transcription factor Nr2f2 attenuates salt-sensitive hypertension. Nat Commun. 2015;6:6252. doi:10.1038/ncomms7252
5. Cornelius RJ, Yang CL, Ellison DH. Hypertension-causing cullin 3 mutations disrupt COP9 signalosome binding. Am J Physiol Renal Physiol. 2020;318(1):F204–208. doi:10.1152/ajprenal.00497.2019
6. Abou hassan OK, Haidar W, Arabi M, et al. Novel EIF2AK4 mutations in histologically proven pulmonary capillary hemangiomatosis and hereditary pulmonary arterial hypertension. BMC Med Genet. 2019;20(1):176. doi:10.1186/s12881-019-0915-7
7. Li H, Slone J, Fei L, et al. Mitochondrial DNA variants and common diseases: a mathematical model for the diversity of age-related mtDNA mutations. Cells. 2019;8(6):608. doi:10.3390/cells8060608
8. Wallace DC. Mitochondrial genetic medicine. Nat Genet. 2018;50(12):1642–1649. doi:10.1038/s41588-018-0264-z
9. Helm M, Brulé H, Friede D, et al. Search for characteristic structural features of mammalian mitochondrial tRNAs. RNA. 2000;6:1356–1379. doi:10.1017/s1355838200001047
10. Chen X, Zhang Y, Xu B, et al. The mitochondrial calcium uniporter is involved in mitochondrial calcium cycle dysfunction: underlying mechanism of hypertension associated with mitochondrial tRNA Ile A4263G mutation. Int J Biochem Cell Biol. 2016;78:307–314. doi:10.1016/j.biocel.2016.07.018
11. Yang P, Wu P, Liu X, et al. Mitochondrial tRNASer(UCN) 7471delC may be a novel mutation associated with maternally transmitted hypertension. Ir J Med Sci. 2020;189(2):489–496. doi:10.1007/s11845-019-02143-z
12. Yu SS, Du JM, Tang ZD, et al. Molecular characterization of mitochondrial transferRNAGln and transferRNAMet A4401G mutations in a Chinese family with hypertension. Mol Med Rep. 2017;15(4):1832–1836. doi:10.3892/mmr.2017.6216
13. Richter U, McFarland R, Taylor RW, et al. The molecular pathology of pathogenic mitochondrial tRNA variants. FEBS Lett. 2021;595(8):1003–1024. doi:10.1002/1873-3468.14049
14. Liu Y, Li Y, Zhu C, et al. Mitochondrial biogenesis dysfunction and metabolic dysfunction from a novel mitochondrial tRNAMet 4467 C>A mutation in a Han Chinese family with maternally inherited hypertension. Sci Rep. 2017;7(1):3034. doi:10.1038/s41598-017-03303-w
15. Patel KV, Li X, Kondamudi N, et al. Prevalence of apparent treatment-resistant hypertension in the United States according to the 2017 high blood pressure guideline. Mayo Clin Proc. 2019;94(5):776–782. doi:10.1016/j.mayocp.2018.12.033
16. Ding Y, Teng YS, Zhuo GC, et al. The mitochondrial tRNAHis G12192A mutation may modulate the clinical expression of deafness-associated tRNAThr G15927A mutation in a Chinese pedigree. Curr Mol Med. 2019;19(2):136–146. doi:10.2174/1566524019666190308121552
17. Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290(5806):457–465. doi:10.1038/290457a0
18. Ding Y, Xia BH, Zhang CJ, et al. Mitochondrial tRNALeu(UUR) C3275T, tRNAGln T4363C and tRNALys A8343G mutations may be associated with PCOS and metabolic syndrome. Gene. 2018;642:299–306. doi:10.1016/j.gene.2017.11.049
19. Levin L, Zhidkov I, Gurman Y, et al. Functional recurrent mutations in the human mitochondrial phylogeny: dual roles in evolution and disease. Genome Biol Evol. 2013;5(5):
20. Gruber AR, Lorenz R, Bernhart SH, et al. The Vienna RNA websuite. Nucleic Acids Res. 2008;36:W70–4. doi:10.1093/nar/gkn188
21. Cui Y, He DJ. Mitochondrial tRNAIle A4317G mutation may be associated with hearing impairment in a Han Chinese family. Mol Med Rep. 2018;18(6):5159–5165. doi:10.3892/mmr.2018.9519
22. Chomyn A, Lai ST, Shakeley R, et al. Platelet-mediated transformation of mtDNA-less human cells: analysis of phenotypic variability among clones from normal individuals--and complementation behavior of the tRNALys mutation causing myoclonic epilepsy and ragged red fibers. Am J Hum Genet. 1994;54(6):966–974.
23. Ding Y, Zhang S, Guo Q, et al. Mitochondrial Diabetes is Associated with the ND4 G11696A Mutation. Biomolecules. 2023;13(6):907. doi:10.3390/biom13060907
24. Lan C, Huang X, Liao X, et al. PUS1 May Be a Potential Prognostic Biomarker and Therapeutic Target for Hepatocellular Carcinoma. Pharmgenomics Pers Med. 2023;16:337–355. doi:10.2147/PGPM.S405621
25. Ding Y, Yu J, Guo Q, et al. Molecular characterization of two Chinese pedigrees with maternally inherited hypertension. J Gene Med. 2021;23(4):e3328. doi:10.1002/jgm.3328
26. Yarham JW, Al-Dosary M, Blakely EL, et al. A comparative analysis approach to determining the pathogenicity of mitochondrial tRNA mutations. Hum Mutat. 2011;32(11):1319–1325. doi:10.1002/humu.21575
27. Bibb MJ, Van Etten RA, Wright CT, et al. Sequence and gene organization of mouse mitochondrial DNA. Cell. 1981;26(2 Pt 2):167–180. doi:10.1016/0092-8674(81)90300-7
28. Gadaleta G, Pepe G, De Candia G, et al. The complete nucleotide sequence of the Rattus norvegicus mitochondrial genome: cryptic signals revealed by comparative analysis between vertebrates. J Mol Evol. 1989;28(6):497–516. doi:10.1007/BF02602930
29. Roe A, Ma DP, Wilson RK, et al. The complete nucleotide sequence of the Xenopus laevis mitochondrial genome. J Biol Chem. 1985;260(17):9759–9774.
30. Wong LC, Chen T, Wang J, et al. Interpretation of mitochondrial tRNA variants. Genet Med. 2020;22(5):917–926. doi:10.1038/s41436-019-0746-0
31. Lee SR, Han J. Mitochondrial mutations in cardiac disorders. Adv Exp Med Biol. 2017;982:81–111. doi:10.1007/978-3-319-55330-6_5
32. Aibara S, Singh V, Modelska A, et al. Structural basis of mitochondrial translation. Elife. 2020;9:e58362. doi:10.7554/eLife.58362
33. Nakano M, Imamura H, Sasaoka N, et al. ATP Maintenance via Two Types of ATP Regulators Mitigates Pathological Phenotypes in Mouse Models of Parkinson’s Disease. EBioMedicine. 2017;22:225–241. doi:10.1016/j.ebiom.2017.07.024
34. Lertsuwan J, Svasti J, Satayavivad J. 8-Chloroadenosine Induces ER Stress and Apoptotic Cell Death in Cholangiocarcinoma Cells. Anticancer Res. 2023;43(12):5425–5436. doi:10.21873/anticanres.16746
35. Kong QP, Bandelt HJ, Sun C, et al. Updating the East Asian mtDNA phylogeny: a prerequisite for the identification of pathogenic mutations. Hum Mol Genet. 2006;15(13):
36. Suzuki T, Nagao A, Suzuki T. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu Rev Genet. 2011;45:299–329. doi:10.1146/annurev-genet-110410-132531
37. Sprinzl M, Vassilenko KS. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 2005;33:D139–40. doi:10.1093/nar/gki012
38. Das AS, Alfonzo JD, Accornero F. The importance of RNA modifications: from cells to muscle physiology. Wiley Interdiscip Rev RNA. 2022;13(4):e1700. doi:10.1002/wrna.1700
39. Ji Y, Liang M, Zhang J, et al. Mitochondrial haplotypes may modulate the phenotypic manifestation of the LHON-associated ND1 G3460A mutation in Chinese families. J Hum Genet. 2014;59(3):134–140. doi:10.1038/jhg.2013.134
40. Ding Y, Li Y, You J, et al. Mitochondrial tRNA(Glu) A14693G variant may modulate the phenotypic manifestation of deafness-associated 12S rRNA A1555G mutation in a Han Chinese family. J Genet Genomics. 2009;36(4):241–250. doi:10.1016/S1673-8527(08)60111-3
41. Uusimaa J, Finnilä S, Remes AM, et al. Molecular epidemiology of childhood mitochondrial encephalomyopathies in a Finnish population: sequence analysis of entire mtDNA of 17 children reveals heteroplasmic mutations in tRNAArg, tRNAGlu, and tRNALeu(UUR) genes. Pediatrics. 2004;114(2):443–450. doi:10.1542/peds.114.2.443
42. Suzuki T. The expanding world of tRNA modifications and their disease relevance. Nat Rev Mol Cell Biol. 2021;22(6):375–392. doi:10.1038/s41580-021-00342-0
43. Ding Y, Zhang S, Guo Q, et al. Mitochondrial Diabetes is Associated with tRNALeu(UUR) A3243G and ND6 T14502C Mutations. Diabetes Metab Syndr Obes. 2022;15:1687–1701. doi:10.2147/DMSO.S363978
44. Ikeda M, Ide T, Fujino T, et al. Overexpression of TFAM or twinkle increases mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress. PLoS One. 2015;10(3):e0119687. doi:10.1371/journal.pone.0119687
45. Huang J, Tan L, Shen R, et al. Decreased Peripheral Mitochondrial DNA Copy Number is Associated with the Risk of Heart Failure and Long-term Outcomes. Medicine (Baltimore). 2016;95(15):e3323. doi:10.1097/MD.0000000000003323
46. Chen YB, Aon MA, Hsu YT, et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011;195(2):263–276. doi:10.1083/jcb.201108059
47. Gong S, Peng Y, Jiang P, et al. A deafness-associated tRNAHis mutation alters the mitochondrial function, ROS production and membrane potential. Nucleic Acids Res. 2014;42(12):8039–8048. doi:10.1093/nar/gku466
48. Schaar CE, Dues DJ, Spielbauer KK, et al. Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 2015;11(2):e1004972. doi:10.1371/journal.pgen.1004972
49. Zhang Y, Chang P, Liu Z. ADD1 Single Nucleotide Polymorphisms Are Associated With Essential Hypertension Among Han and Mongolian Population in Inner Mongolia Area. Front Genet. 2022;13:931803. doi:10.3389/fgene.2022.931803
50. Li D, Shao NY, Moonen JR, et al. ALDH1A3 Coordinates Metabolism With Gene Regulation in Pulmonary Arterial Hypertension. Circulation. 2021;143(21):2074–2090. doi:10.1161/CIRCULATIONAHA.120.048845
51. Ray A, Stelloh C, Liu Y, et al. Histone Modifications and Their Contributions to Hypertension. Hypertension. 2023. doi:10.1161/HYPERTENSIONAHA.123.21755
52. Zhao L, Jia YN, Liu QSJ, et al. Association between Mitochondrial DNA Methylation and Hypertension Risk: a Cross-sectional Study in Chinese Northern Population. Biomed Environ Sci. 2023;36(10):972–978. doi:10.3967/bes2023.122
53. Tashi QZ, Tsering SB, Zhou NN, et al. A Study on the Molecular Mechanism of High Altitude Heart Disease in Children. Pharmgenomics Pers Med. 2022;15:721–731. doi:10.2147/PGPM.S356206
54. Vasan RS, Beiser A, Seshadri S, et al. Residual lifetime risk for developing hypertension in middle-aged women and men: the Framingham Heart Study. JAMA. 2002;287(8):1003–1010. doi:10.1001/jama.287.8.1003
55. Gao F, Liang T, Lu YW, et al. A defect in mitochondrial protein translation influences mitonuclear communication in the heart. Nat Commun. 2023;14(1):1595. doi:10.1038/s41467-023-37291-5
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.