Back to Archived Journals » Hypoxia » Volume 8
Markers of Hypoxia Correlate with Histologic and Endoscopic Severity of Colitis in Inflammatory Bowel Disease
Authors deZoeten EF ![]() , Battista KD, Colson SB, Lovell MA, Kessler BE, Isfort RW, Fennimore BP, Onyiah JC, Kao DJ
, Battista KD, Colson SB, Lovell MA, Kessler BE, Isfort RW, Fennimore BP, Onyiah JC, Kao DJ ![]() , Yeckes A
, Yeckes A ![]() , Keely S
, Keely S ![]() , Murray M, Hoffenberg EJ
, Murray M, Hoffenberg EJ ![]() , Colgan SP, Gerich ME
, Colgan SP, Gerich ME
Received 11 June 2019
Accepted for publication 16 October 2019
Published 10 February 2020 Volume 2020:8 Pages 1—12
DOI https://doi.org/10.2147/HP.S219049
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Edwin F deZoeten,1,* Kayla D Battista,2,* Steven B Colson,1,* Mark A Lovell,3 Brittelle E Kessler,2 Robert W Isfort,2 Blair P Fennimore,2 Joseph C Onyiah,2 Daniel J Kao,2 Alyson Yeckes,1 Simon Keely,1,4 Monica Murray,2 Edward J Hoffenberg,1 Sean P Colgan,2 Mark E Gerich2
1Department of Pediatrics and the Digestive Health Institute, University of Colorado School of Medicine/Children’s Hospital Colorado, Aurora, CO, USA; 2Department of Medicine and Mucosal Inflammation Program, Division of Gastroenterology and Hepatology, University of Colorado School of Medicine, Aurora, CO, USA; 3Department of Pathology, University of Colorado School of Medicine/Children’s Hospital Colorado, Aurora, CO, USA; 4School of Biomedical Sciences and Pharmacy, University of Newcastle, Newcastle, NSW, Australia
*These authors contributed equally to this work
Correspondence: Edwin F deZoeten
Mucosal Inflammation Program, 12700 E. 19th Ave, Aurora, CO 80045, USA
Tel +1 303-724-7239
Fax +1 303-724-7243
Email [email protected]
Background: Inflammation results in significant shifts in tissue metabolism. Recent studies indicate that inflammation and hypoxia occur concomitantly. We examined whether circulating and tissue markers of hypoxia could serve as surrogate indicators of disease severity in adult and pediatric patients with inflammatory bowel disease (IBD).
Methods: Serum and colonic biopsies were obtained from pediatric subjects with active IBD colitis and adult subjects with active and inactive ulcerative colitis, along with healthy non-colitis controls of all ages. Disease activity was evaluated by endoscopy and histopathology. Levels of serum hypoxia markers (macrophage inflammatory protein-3α [MIP-3α], vascular endothelial growth factor [VEGF], and erythropoietin [EPO]) were measured.
Results: Children with active IBD colitis had higher levels of serum MIP-3α and VEGF compared to non-colitis controls (p< 0.01 and p< 0.05, respectively). In adult subjects with endoscopically active ulcerative colitis, serum MIP-3α and EPO were significantly elevated compared to non-colitis controls (both p< 0.01). In parallel, analysis of colon tissue MIP-3α mRNA and protein in pediatric subjects revealed increased expression in those with IBD colitis compared to controls (p< 0.05 and p< 0.01 for mRNA and protein, respectively). Serum MIP-3α and VEGF significantly increased with histology grade.
Conclusion: Peripheral blood hypoxia markers may be useful indicators of disease activity for pediatric and adult IBD patients.
Keywords: Crohn’s disease, ulcerative colitis, mucosal inflammation, tissue histology
Introduction
The inflammatory bowel diseases (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), affect approximately 0.5% of the North American population and are increasing in incidence.1 At the same time, the financial burden of IBD is substantial and growing. Much of this expense is driven by the cost of newer medical therapies and the cost of hospitalizations for disease flares and/or complications.2 These adverse outcomes appear more likely when therapeutic decisions are based on clinical signs and symptoms as opposed to more objective measures, such as endoscopic evaluation of mucosal inflammation, which is costly as well.3,4 Furthermore, there is an evolving line of evidence, particularly in colitis, indicating that the achievement of histologic endpoints for response to treatment provides significant benefit towards avoiding downstream disease complications.5,6 In this context, non-invasive biomarkers of histologic disease activity could help optimize costly treatment decisions and avoid disease complications.
It is currently thought that IBD results from a combination of environmental factors and maladaptive responses to gastrointestinal flora in a genetically susceptible host that results in chronic mucosal inflammation.7,8 There is an intense interest in understanding metabolism in mucosal inflammation.9 Paramount to all metabolic pathways is the utilization of oxygen, and recent studies have implicated diminished availability and delivery of oxygen with resultant tissue hypoxia as a significant component of the inflammatory response.10 It is thought that inflammation-associated tissue hypoxia occurs through a number of mechanisms.9,11 For example, local oxygen depletion can occur as a result of the influx of large numbers of oxygen-utilizing neutrophils and monocytes.12 Additionally, extensive inflammation can result in damage to the vasculature. As a consequence of localized vasculitis, microthrombi form and abnormal production of vasoactive mediators result in poor tissue perfusion.13,14 Such “inflammatory hypoxia” has been demonstrated to contribute in fundamental ways to the inflammatory process. To date, a study comparing tissue markers of hypoxia with IBD disease activity has not been done. Likewise, it is not known whether circulating markers of hypoxia can serve as surrogates of endoscopic or histologic disease activity.
Murine models of IBD have shown that inflammation involving the colonic epithelium is accompanied by severe oxygen-deprivation and activation of hypoxia-inducible factor (HIF).15 HIF represents a family of transcription factors that regulate genes important in adaptation to low oxygen states.16 Classic targets of HIF stabilization include those involved in inflammation (e.g. MIP-3α), angiogenesis (e.g. VEGF), and erythropoiesis (e.g. EPO).16,17 Prominent induction of HIF target genes has been noted within the intestinal mucosa in murine models of colitis.15,18 Moreover, histopathologic analysis of tissue from human IBD patients has shown stabilization of HIF within the intestinal mucosa of patients with active ulcerative colitis and Crohn’s disease.19 Thus, we hypothesized that both tissue and serum biomarkers of hypoxia should correlate with tissue inflammation and histologic disease activity.
Materials and Methods
Human Subjects
The institutional review board of the University of Colorado approved all human studies. All patients provided written informed consent. All patient’s under the age of 18 provided written assent and their parents or legal guardians provided written informed consent. This study was conducted in accordance with the Declaration of Helsinki. IBD diagnoses were made using standard clinical, radiographic, endoscopic, and histologic criteria. Study data were collected and managed using REDCap electronic data capture tools hosted at the University of Colorado.20
In the pediatric cohort (Table 1), subjects from one to 17 years of age undergoing diagnostic colonoscopy for clinical purposes were recruited. Subjects were excluded if they had known gastrointestinal infection, known causes of hypoxia, history of transplant or neutropenia. Up to 8 mL of blood were collected per patient for serum isolation (4 mL) and for RNA analysis (4 mL). During colonoscopy, up to 8 research biopsies were obtained, 4 from the distal colon and 4 more proximal if there was a shift in severity of tissue inflammation. From each area, one biopsy was taken for tissue histology, two biopsies were snap frozen in liquid nitrogen for protein analysis, and the final biopsy was placed in RNAlater® for RNA isolation. The most recent clinical laboratory data for hemoglobin and hematocrit were recorded.
 |
Table 1 Clinical and Demographic Data for Pediatric Subjects |
The two groups of pediatric study subjects were those found to have active IBD colitis and control patients who were found to have no colitis on biopsy histology. Of the 45 subjects consented, 35 were enrolled in the study and included in the analysis: 18 with active IBD colitis and 17 non-colitis controls. The reasons for non-enrollment were: colitis not deemed consistent with IBD (eosinophilic colitis or mild non-specific colitis) (n=5); colonoscopy not performed (n=2); colonoscopy performed at different site (n=2); and Clostridium difficile infection (n=1). The two groups were similar for gender, ethnicity, age, and body mass index (Table 1). Twelve pediatric subjects had UC, five had CD, and one had IBD unclassified.
Specimens from adult subjects were obtained from the University of Colorado IBD Biorepository (Table 2). Under this protocol, serum and mucosal biopsies were obtained from IBD patients when colonoscopy was performed for either disease activity assessment or colorectal cancer surveillance. Similar specimens were collected from healthy subjects undergoing colonoscopy for colon cancer screening purposes. Endoscopic disease severity was assessed according to the Mayo endoscopic scoring system.21 At the time of endoscopy, serum was extracted from 5 mL of whole blood, snap frozen in liquid nitrogen, and then stored at −80°C. Mucosal biopsies were obtained from the most active site of colitis, either snap frozen in liquid nitrogen or placed in RNAlater®, then stored at −80°C. At the time of specimen collection, clinical disease activity was assessed through calculation of the Mayo score and the simple clinical colitis activity index (SCCAI).21,22 Quality of life was assessed by administration of the inflammatory bowel disease questionnaire (IBDQ).23 Analysis of stool for pathogens was performed prior to endoscopic evaluation in any patient with signs or symptoms of active IBD.
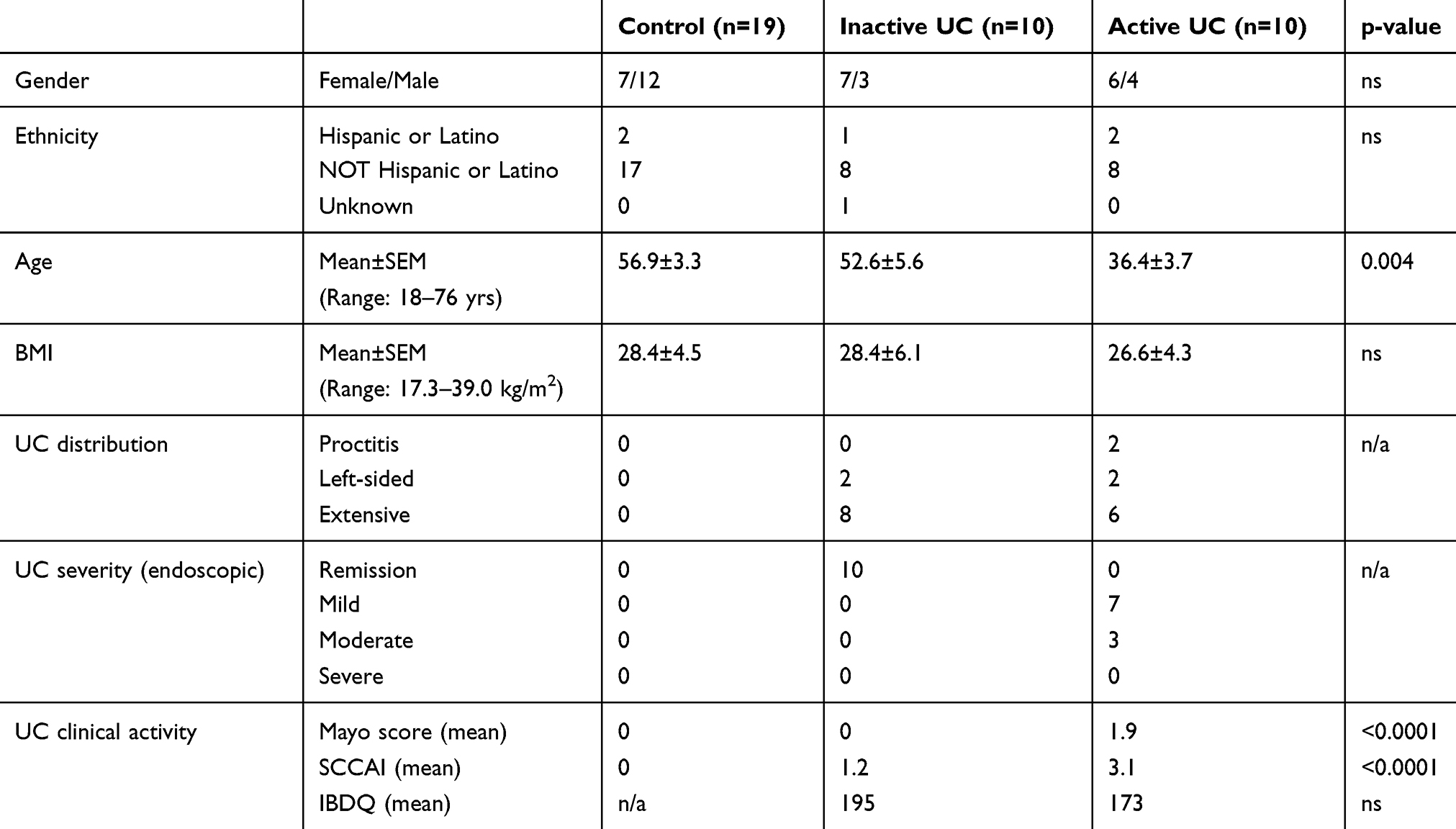 |
Table 2 Clinical and Demographic Data for Adult Subjects |
Ten subjects with endoscopically active UC and ten subjects with inactive UC (in endoscopic remission) were included in the adult IBD cohort (Table 2). Nineteen healthy subjects were included as normal controls. The three groups were similar for gender and ethnicity; however, the active UC subjects were significantly younger than the inactive UC and non-colitis control subjects.
Histopathologic Disease Activity Scoring
A previously reported histology scoring system was used to define severity of colitis (control, mild, moderate or severe).24 Using this system, a single pathologist, blinded to both the severity and disease status, defined severity according to changes within the epithelial compartments (loss of enterocytes and crypt abscess formation) and within the lamina propria (increases in mononuclear cells and neutrophils). A sum of these categories resulted in an overall score for each biopsy of 0 to 3, corresponding to a range of normal to severe colitis. In subjects from whom multiple intestinal biopsies were taken, the biopsy with the highest defined severity was used. When two biopsies had the same severity, the average protein and cDNA levels were calculated. The highest colon biopsy histologic severity was also used for correlations with serum hypoxia markers.
Tissue and Serum Analysis
Serum and tissue protein were analyzed for the presence of HIF-1a and downstream hypoxia markers (MIP-3α, VEGF, and EPO) using an electrochemiluminescence-based ELISA platform (MesoScale Discovery, Gaithersburg, MD) as described previously.25 Briefly, all serum samples (thawed from frozen) were diluted 1:2 in PBS and 50µL samples were analyzed. Tissue biopsies (n=2 biopsies per analysis) were extracted using MesoScale lysis protocol, protein normalized and 100µg total protein was analyzed. Tissue RNA was extracted through homogenization and cell lysis with Trizol® Reagent as described previously.25 Real-time qPCR was performed from cDNA as previously described25 with the following primers: human MIP-3α (CCL20): forward 5ʹ-GCGCAAATCCAAAACAGACT-3ʹ and reverse 5ʹ- CAAGTCCAGTGAGGCACAAA-3ʹ; human EPCAM: forward 5ʹ- GCAGCTCAGGAAGAATGTG-3ʹ and reverse 5ʹ-CAGCCAGCTTTGAGCAAATGAC-3ʹ; human GLUT1: forward 5ʹ-GGGTCAGGCTCCATTAGGATT-3ʹ and reverse 5ʹ- CCCAACTGGTCTCAGGTAAAGAA-3ʹ; human ENO1: forward 5ʹ-ATCGCCAAGGCCGTGAA-3ʹ and reverse 5ʹ-ACGGAGCCAATCTGGTTGAC-3ʹ; human ITF: forward 5ʹ-CCAGGCACTGTTCATCTCAG-3ʹ and reverse 5ʹ- GGAGCATGGGACCTTTATTC-3ʹ; human MDR1: forward 5ʹ- AACGGAAGCCAGAACATTCC-3ʹ and reverse 5ʹ-AGGCTTCCTGTGGCAAAGAG-3ʹ; and human beta actin: forward 5ʹ-GGAGAAAATCTGGCACCACA-3ʹ and reverse 5ʹ- AGAGGCGTACAGGGATAGCA-3ʹ.
Statistical Analysis
Based on power analysis for detecting differences in tissue HIF stabilization, we estimated that 14 patients would be needed in each group in order to detect a significant difference between healthy and IBD at a power of 95% and a 5% level of significance. Thus, we tested 19 adult and 18 pediatric patients with IBD to allow for subgroup severity analysis.
Differences by study group were tested with two-tail t-tests assuming unequal variance or in multiple comparisons they were assessed by non-parametric ANOVA with post-hoc t-tests or by the Kruskal Wallis test (>2 groups) or Mann Whitney U-test (2 groups) and differences across histology grade were assessed by ANOVA with post-hoc t-tests as well by multivariate analysis using the Kruskal Wallis test (>2 groups) or Mann Whitney U-test (2 groups). This study was estimated to have >99% power to detect a between-group difference in VEGF of 139 pg/mL (previously reported by Bousvaros et al26) using a two-sided t-test with α=0.05 and 10 patients per group under the null hypothesis of equal group means. Receiver operating characteristics (ROC) curve analysis27 was used to determine the optimal threshold as measured by sensitivity and specificity. A p-value of <0.05 was considered statistically significant. All data are presented as the mean ± standard error of the mean. Statistical analyses were performed using GraphPad Prism software (La Jolla, CA).
Results
Epithelial Hypoxia Markers in Colon Tissue
We initially evaluated HIF stabilization in colon biopsies from pediatric subjects. As shown in Figure 1A, tissue HIF-1a protein was increased in colon biopsies from pediatric subjects with active IBD colitis compared to uninflamed controls (p<0.05). We extended these results to screen a panel of epithelial-expressed, hypoxia-inducible genes previously shown to be induced in murine models of colitis,11 including glucose transporter-1 (GLUT1), enolase (ENO1), intestinal trefoil factor (ITF), macrophage inflammatory protein-3α (MIP-3α), and multidrug resistance gene-1 (MDR1) (Supplemental Figure 1). This screen revealed a prominent induction of MIP-3α in pediatric IBD samples compared to controls (8.2 ± 1.97 fold increase, p<0.05) (Figure 1B). Given the potential variability in epithelial content due to ulceration and biopsy technique, we also measured RNA levels of the selective marker epithelial cell adhesion molecule (EpCAM). No differences in EpCAM expression between colitis and control subjects were noted (0.70±0.12-fold change, p=not significant), indicating the amount of epithelial tissue was not significantly different between samples.
In light of the magnitude of MIP-3α mRNA transcript induction, we validated the induction of tissue MIP-3α protein by ELISA. As shown in Figure 1C, MIP-3α protein levels were increased by more than seven-fold in IBD subject tissue compared to uninflamed controls (p<0.01). This prominent increase in MIP-3α, a well-established hypoxia-inducible gene target,28–31 in IBD is consistent with previous studies in murine models of colitis.9
Serum Hypoxia Biomarkers
We next determined whether tissue hypoxia, as measured by elevations in the expression of tissue HIF-inducible proteins, were reflected in circulating serum. To do this, we analyzed serum MIP-3α, VEGF, and EPO protein in pediatric IBD and control subjects. As shown in Figure 2A and B, MIP-3α and VEGF were significantly elevated in subjects with active IBD as compared to controls (p<0.01 and p<0.05, respectively). Serum EPO levels appeared higher in IBD patients compared to controls; however, this increase was not statistically significant (Figure 2C). A possible confounder of serum EPO levels is anemia; although hemoglobin and hematocrit levels were significantly decreased in the IBD cohort, there was not a significant corresponding change in EPO levels (Supplemental Figure 2).
Serum Hypoxia Markers and Histologic Severity of Colitis
Next, we extended our analysis to determine whether severity of colitis, determined histopathologically (Figure 3A), correlated with circulating hypoxia markers within subject populations. As shown in Figure 3B and C, serum MIP-3α and VEGF varied significantly by histologic severity of colitis (p<0.001 and p<0.01, respectively). Serum EPO levels also showed a trend toward increases in the subjects with more severe colitis compared to controls (p=0.077, Figure 3D). MIP-3α showed the most compelling increase in patients with graded tissue histology compared to controls (p<0.001). None of the markers were different between histologically graded mild colitis and non-colitic controls (all p>0.05).
Consistency of Hypoxia Biomarker Findings in Adult IBD Subjects
Expanding upon the results derived from our pediatric IBD cohort, we next tested the universality of these pediatric results in a cohort of adult IBD subjects with active UC. We first validated the finding of HIF-1a stabilization in adults using colonic biopsies. As shown in Figure 4A, tissue HIF-1a protein was increased in colon biopsies from subjects with active UC compared to healthy controls (p<0.05). We then extended the results from our pediatric cohort to determine whether serum hypoxia markers (MIP-3α, VEGF, and EPO) correlated with active colitis in adult IBD patients. As shown in Figure 4B, serum MIP-3α served as an excellent biomarker of active colitis (p<0.01) and was not significantly different between inactive colitis (p>0.05) and healthy control subjects. Serum VEGF trended toward increased levels in active UC (Figure 4C), but the difference was not statistically significant compared to healthy controls (p = 0.13), suggesting that a larger cohort of subjects may be necessary to define VEGF as a relevant biomarker for active disease. Serum EPO levels (Figure 4D) were significantly increased in both inactive (p<0.05) and active colitis (p<0.001) compared to healthy control subjects. Importantly, there were no between-group differences in hematocrit or hemoglobin levels across any of the three adult cohorts (Supplemental Figure 3). Taken together, these results in adult subjects confirm, at least in part, that peripheral blood hypoxia markers can serve as reasonable surrogates for disease activity.
Predictive Quality of Tissue HIF and Serum Targets in Determining Active IBD
Receiver operating characteristics (ROC) curve analysis, a graphical display of true positive and false positive diagnostic rates across a range of cut-off values is valuable in assessment of diagnostics,32 was used to evaluate the performance of these biomarkers in discriminating active IBD from healthy controls. As shown in Figure 5, we pooled results from pediatric and adult IBD samples and analyzed ROC curves by calculation of area under the curve (AUC). These results revealed that tissue HIF-1a (AUC=0.75 with 79.0% sensitivity and 76.0% specificity at 0.9417, Figure 5A), as well as serum MIP-3a (AUC=0.77 with 72.0% sensitivity and 72.7% specificity at 0.8399, Figure 5B), and VEGF (AUC=0.77 with 76.9% sensitivity and 75.8% specificity at 0.8548, Figure 5C) were good predictors of active IBD. Serum EPO did not perform as well in such analysis (AUC=0.68 with 61.5% sensitivity and 72.7% specificity at 1.059, Figure 5D). The combination of serum hypoxia markers (MIP-3a, VEGF, and EPO) did not result in improved ability to discriminate active IBD (AUC=0.77).
Discussion
Tissue hypoxia is strongly associated with active inflammation (termed “inflammatory hypoxia”) and its impact in IBD is an area of intense investigation.10 Here, we validated previous findings from murine models of intestinal inflammation and explored the feasibility of using hypoxia-inducible targets as markers of disease activity in human subjects. Our study demonstrates that circulating markers of hypoxia (MIP-3α and VEGF) correlate with histologically defined disease severity in pediatric IBD patients. In adults, the association between serum hypoxia markers and active disease were corroborated when both serum MIP-3α and EPO were found to be significantly increased in active UC. Previous studies have demonstrated a relationship between hypoxia and human IBD.9
A recent study by Cosin-Roger et al33 demonstrated that hypobaric oxygen actually ameliorates intestinal inflammation, seemingly contradicting the large body of work that shows that tissue hypoxia results in worse inflammation; however, this study does not identify a mechanism (microbiome changes or intestinal hypoxia) nor does it assess the effects on intestinal tissue hypoxia of hypobaric oxygen thus making the hypoxic effect difficult to analyze therefore tissue hypoxia remains a likely pro-inflammatory signal in intestinal inflammation. In addition to HIF, several studies have implicated a role for MIP-3α in IBD.34–36 MIP-3α (also termed CCL20) is a CD4 T cell-directed chemokine that traffics lymphocytes and dendritic cells to the epithelium.29 Elevation of MIP-3α has been demonstrated in both peripheral blood monocytes and colonic tissue.34,36
Interestingly, when MIP-3α was studied in colonic biopsies from subjects with IBD, non-IBD colitis, and irritable bowel syndrome, the active IBD patients showed elevations in MIP-3α by mRNA and protein analysis compared to controls, but there were no increases in non-IBD colitis samples.34 This could implicate MIP-3α as a marker novel to IBD induced colitis, which warrants further investigation. These previous MIP-3α studies have mostly involved intestinal epithelial cell lines or adult subjects and have not analyzed serum levels. We found MIP-3α levels in IBD subjects with colitis were universally elevated, including in colon tissue RNA, colon protein, and serum protein. Although this does not provide direct insight into the origins of circulating serum MIP-3α, such findings suggest that colon tissue is at least a significant source of MIP-3α during colitis. In support of this line of reasoning, a recent study demonstrated that MIP-3α levels are increased during active intestinal inflammation and that the primary source appears to be colonic epithelial cells.37 It is notable that while MIP-3α is a well-established hypoxia gene target,28–31 MIP-3α is also regulated by other inflammatory signals, including but not limited to NF-kB.38 It is therefore possible that inflammatory stimuli present within IBD tissue could contribute to the induction of MIP-3α observed in our studies.
EPO, on the other hand, originates primarily from the kidney, where specialized cells that produce EPO are stimulated in response to decreased oxygen tension and predominantly by HIF stabilization.39 EPO has mostly been studied in terms of anemia in parallel to disease40,41 but not as a biomarker for IBD. There are limited reports of elevated serum EPO levels in IBD subjects.42,43 Complicating the picture is the observation that anemia, which is often present in active IBD, can lead to increased production of EPO as an adaptive mechanism to increase red blood cell mass. Notably, correction for the degree of anemia in one study revealed that EPO levels in IBD patients were still elevated above normal.43 In our study, anemia, as measured by hemoglobin and hematocrit, did not correlate to serum EPO levels in our pediatric population, and hemoglobin and hematocrit did not vary by cohort in our adult population. However, we saw a trend toward increased EPO in our severe pediatric IBD samples and significantly increased EPO in our adult active UC patients compared to healthy controls. It is notable that of all the markers measured, we observed the most variability with EPO in this study. We do not know the nature of this variability. Since all study subjects resided in Colorado, we would expect higher baseline hemoglobin, although this can be variable.44 Other potential confounders include different degrees of physiologic adaptation to lower oxygen in those individuals more conditioned to higher altitude.
A number of previous studies found elevations in VEGF in the blood and tissue of IBD subjects; however, this has not been a universal observation.26,45–49 In one study, for example, VEGF was increased in the serum of adult patients with active UC and CD compared to controls, but no differences were seen between subjects with inactive disease and controls.50 We similarly saw no shift in inactive UC patients compared to healthy controls, but did not see the same significant increase in VEGF in serum from patients with active UC. We did, however, find that VEGF was significantly increased in the serum of active pediatric IBD patients, especially with increased disease severity. This finding reflects previous work by Bousvaros et al that showed increased serum VEGF in the pediatric IBD population.26
These hypoxia biomarker studies could also provide insight into new therapeutic options for patients with IBD. For example, studies in murine models of colitis have demonstrated a protective role for HIF stabilizers in different models of intestinal inflammation. The first study used the prolyl hydroxylase (PHD) inhibitor DMOG for the treatment of intestinal inflammation during chemically induced colitis.51 A second study used the HIF stabilizer FG-4497 during TNBS-induced intestinal inflammation. Similar to DMOG, FG-4497 blocks the active site of PHDs.52 In both studies, HIF stabilization was associated with profound improvements of multiple disease parameters, including weight loss, intestinal inflammation, and histologic tissue injury.11,53 Since that time, a significant effort has been made to develop inhibitors of PHDs. The development of these molecules has focused on iron chelators and 2-oxoglutarate mimetics and have undergone human studies in Phase 1–3 clinical trials examining their influence on erythropoietin stimulation in patients with chronic renal anemia (reviewed in (155)). These PHD inhibitors are generally well tolerated and promising results using GSK-1278863 and AKB-6548 produce an increase in circulating erythropoietin. Targeted delivery of such compounds may provide a therapeutic impact while limiting the potential for untoward systemic effects in healthy organs (143, 156). The potential use of local, oral delivery of extended release preparations to the inflamed mucosa could represent a novel therapeutic approach for IBD.54 It is therefore possible to speculate that hypoxia biomarkers represent a protective tissue inflammatory response.
There are some limitations to this study. HIF regulation of MIP-3α, EPO, and VEGF is well documented, justifying their designation as hypoxia markers; however, it is possible that, in addition to hypoxia, other inflammatory pathways could contribute to our observations here. Individuals with colitis from non-IBD sources, such as infection, may show similar elevation in hypoxia-inducible markers. In the case of children with infectious colitis, colonoscopy is not routinely performed, making this challenging to study. However, future studies that include a subset of non-IBD colitis patients are justified. Likewise, the present studies in adult patients were limited to UC and did not include those with active or inactive CD. At this time, it is unclear if the observed associations are specific to colitis, if they have utility in distinguishing UC from CD, and if their usefulness is modified by disease distribution. Importantly, in this study, stool was not analyzed for fecal calprotectin and sufficient data regarding serum levels of C-reactive protein were not available. Therefore, the test characteristics of hypoxia biomarkers relative to existing, but imperfect, biomarkers of histologic disease activity like fecal calprotectin and CRP remain unclear. Future investigation that compares the performance of hypoxia biomarkers with existing biomarkers should be performed.
In conclusion, the current study supports the hypothesis that metabolic deficits, including hypoxia, are contributing factors to IBD. Our results reveal that serum hypoxia markers strongly correlate with endoscopically active colitis as well as histologically graded colitis. As such, further studies are warranted to determine if peripheral blood hypoxia biomarkers are useful measures of colitis that might have utility in guiding treatment decisions in IBD patients.
 |
Figure 1 Analysis of colon HIF-1a and MIP-3α in pediatric IBD colitis. In panel A, biopsy tissue lysates from pediatric non-colitis controls (n=7) and pediatric subjects with active IBD colitis (n=9) were used to quantify HIF-1a protein by electrochemiluminescent-based ELISA and presented as mean±SEM relative light units/µg total protein where * is p<0.05. In panel B, total RNA was isolated from colonic biopsies derived from pediatric non-colitis controls (n=7) and pediatric subjects with active IBD colitis (n=9). Quantitative PCR was used to analyze MIP-3α mRNA levels. Data are expressed as mean±SEM transcript level derived from the differential threshold cycle number (2e-∆CT) and normalized to healthy controls where * is p<0.05. In panel C, biopsy tissue lysates were used from pediatric non-colitis controls (n=7) and pediatric subjects with active IBD colitis (n=10) to quantify MIP-3α protein by electrochemiluminescent-based ELISA and presented as mean±SEM pg MIP-3α/ug total protein where ** is p<0.01. |
 |
Figure 2 Serum hypoxia marker levels in pediatric IBD colitis. Serum samples from healthy control (n=17) and pediatric subjects with active IBD colitis (n=17) were used to quantify MIP-3α (panel A), VEGF (panel B), or EPO (panel C) protein by electrochemiluminescent-based ELISA and presented as mean±SEM pg/mL where * is p<0.05 and ** is p<0.01. |
 |
Figure 3 Analysis of pediatric serum hypoxia markers in relation to colonic tissue histology. The upper panels (A) depict representative 200x magnification images of hematoxylin and eosin-stained histopathologic tissue used to subgroup patients into normal (score 0), mild (score 1), moderate (score 2), or severe colitis (score 3). In the bottom panels (B–D), serum samples from non-colitis controls (n = 17) and histologic subgroups of pediatric subjects with active IBD colitis were used to quantify (from left to right) MIP-3α, VEGF, and EPO protein by electrochemiluminescent-based ELISA and presented as mean±SEM pg/mL where * is p<0.05, ** is p<0.01 and *** is p<0.001. |
 |
Figure 4 Tissue HIF-1α and serum hypoxia markers in adult ulcerative colitis. Colon biopsies and serum samples from healthy adult controls (n = 19), inactive UC subjects (n = 10) and active UC subjects (n=10) were used to quantify colon HIF-1a (panel A), serum MIP-3α (panel B), serum VEGF (panel C) and serum EPO (panel D) protein by electrochemiluminescent-based ELISA and presented as mean±SEM RLU/µg or pg/mL where ** is p<0.01 and *** is p<0.001. |
 |
Figure 5 ROC curve analysis of tissue HIF-1α and serum hypoxia markers in pediatric and adult IBD. Colon biopsies (panel A) and serum samples (panels B-D) from healthy pediatric and adult controls (n= 26 for panel A and n= 36 for panels B-D) and pediatric and adult IBD subjects (n= 20 for panel A and n= 27 for panels B-D) were used to quantify colon HIF-1a (top panel A), serum MIP-3α (top panel B), serum VEGF (top panel C) and serum EPO (top panel D) protein. Samples were normalized to control values and are presented as mean±SEM where ** is p<0.01 and *** is p<0.001. Bottom panels depict ROC curve analysis for individual hypoxia biomarkers with AUC values. |
Acknowledgment
We would like to acknowledge Kelley Capocelli, MD for providing histology photomicrographs.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Dr Steven B. Colson reports T32 Training grants from NIH, during the conduct of the study. Dr Robert Isfort was a speaker for Takeda Pharmaceuticals, outside the submitted work. Dr Simon Keely reports grants from GossamBio, NHMRC, Cancer institute NSW, and Anatara Life Sciences, personal fees from Aerpio Therapeutics, Gossamer Bio, Anatara Life Sciences, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1):46–54. doi:10.1053/j.gastro.2011.10.001
2. Park KT, Bass D. Inflammatory bowel disease-attributable costs and cost-effective strategies in the United States: a review. Inflamm Bowel Dis. 2011;17(7):1603–1609. doi:10.1002/ibd.21488
3. Froslie KF, Jahnsen J, Moum BA, Vatn MH. Mucosal healing in inflammatory bowel disease: results from a Norwegian population-based cohort. Gastroenterology. 2007;133(2):412–422. doi:10.1053/j.gastro.2007.05.051
4. Laharie D, Filippi J, Roblin X, et al. Impact of mucosal healing on long-term outcomes in ulcerative colitis treated with infliximab: a multicenter experience. Aliment Pharmacol Ther. 2013;37(10):998–1004. doi:10.1111/apt.2013.37.issue-10
5. Travis SP, Higgins PD, Orchard T, et al. Review article: defining remission in ulcerative colitis. Aliment Pharmacol Ther. 2011;34(2):113–124. doi:10.1111/j.1365-2036.2011.04701.x
6. Zenlea T, Yee EU, Rosenberg L, et al. Histology grade is independently associated with relapse risk in patients with ulcerative colitis in clinical remission: a prospective study. Am J Gastroenterol. 2016;111(5):685–690. doi:10.1038/ajg.2016.50
7. Abraham C, Cho JH. Inflammatory bowel disease. N Eng J Med. 2009;361:2066–2076. doi:10.1056/NEJMra0804647
8. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474(7351):307–317. doi:10.1038/nature10209
9. Glover LE, Colgan SP. Hypoxia and metabolic factors that influence inflammatory bowel disease pathogenesis. Gastroenterology. 2011;140(6):1748–1755. doi:10.1053/j.gastro.2011.01.056
10. Colgan SP, Campbell EL, Kominsky DJ. Hypoxia and mucosal inflammation. Ann Rev Pathol. 2016;11:77–100. doi:10.1146/annurev-pathol-012615-044231
11. Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. 2010;7(5):281–287. doi:10.1038/nrgastro.2010.39
12. Kominsky DJ, Campbell EL, Colgan SP. Metabolic shifts in immunity and inflammation. J Immunol. 2010;184(8):4062–4068. doi:10.4049/jimmunol.0903002
13. Hatoum OA, Binion DG, Otterson MF, Gutterman DD. Acquired microvascular dysfunction in inflammatory bowel disease: loss of nitric oxide-mediated vasodilation. Gastroenterology. 2003;125(1):58–69. doi:10.1016/S0016-5085(03)00699-1
14. Hatoum OA, Heidemann J, Binion DG. The intestinal microvasculature as a therapeutic target in inflammatory bowel disease. Ann N Y Acad Sci. 2006;1072:78–97. doi:10.1196/annals.1326.003
15. Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114(8):1098–1106. doi:10.1172/JCI200421086
16. Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda). 2009;24:97–106. doi:10.1152/physiol.00045.2008
17. Ratcliffe PJ. HIF-1 and HIF-2: working alone or together in hypoxia? J Clin Invest. 2007;117(4):862–865. doi:10.1172/JCI31750
18. Shah YM, Ito S, Morimura K, et al. Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology. 2008;134(7):2036–2048. doi:10.1053/j.gastro.2008.03.009
19. Giatromanolaki A, Sivridis E, Maltezos E, et al. Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. J Clin Pathol. 2003;56(3):209–213. doi:10.1136/jcp.56.3.209
20. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap) – a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381. doi:10.1016/j.jbi.2008.08.010
21. Schroeder KW, Tremaine WJ, Ilstrup DM. Coated oral 5-aminosalicylic acid therapy for mildly to moderately active ulcerative colitis. A randomized study. N Engl J Med. 1987;317(26):1625–1629. doi:10.1056/NEJM198712243172603
22. Walmsley RS, Ayres RC, Pounder RE, Allan RN. A simple clinical colitis activity index. Gut. 1998;43(1):29–32. doi:10.1136/gut.43.1.29
23. Guyatt G, Mitchell A, Irvine EJ, et al. A new measure of health status for clinical trials in inflammatory bowel disease. Gastroenterology. 1989;96(3):804–810. doi:10.1016/S0016-5085(89)80080-0
24. Saverymuttu SH, Camilleri M, Rees H, Lavender JP, Hodgson HJ, Chadwick VS. Indium 111- granulocyte scanning in the assessment of disease extent and disease activity in inflammatory bowel disease. A comparison with colonoscopy, histology, and fecal indium 111-granulocyte excretion. Gastroenterology. 1986;90(5 Pt 1):1121–1128. doi:10.1016/0016-5085(86)90376-8
25. MacManus CF, Campbell EL, Keely S, Burgess A, Kominsky DJ, Colgan SP. Anti-inflammatory actions of adrenomedullin through fine tuning of HIF stabilization. FASEB J. 2011;25(6):1856–1864. doi:10.1096/fj.10-170316
26. Bousvaros A, Leichtner A, Zurakowski D, et al. Elevated serum vascular endothelial growth factor in children and young adults with Crohn’s disease. Dig Dis Sci. 1999;44(2):424–430. doi:10.1023/A:1026635308127
27. Grund B, Sabin C. Analysis of biomarker data: logs, odds ratios, and receiver operating characteristic curves. Curr. 2010;5(6):473–479.
28. Bosco MC, Delfino S, Ferlito F, et al. Hypoxic synovial environment and expression of macrophage inflammatory protein 3gamma/CCL20 in juvenile idiopathic arthritis. Arthritis Rheum. 2008;58(6):1833–1838. doi:10.1002/art.23516
29. Bosco MC, Puppo M, Santangelo C, et al. Hypoxia modifies the transcriptome of primary human monocytes: modulation of novel immune-related genes and identification of CC-chemokine ligand 20 as a new hypoxia-inducible gene. J Immunol. 2006;177(3):1941–1955. doi:10.4049/jimmunol.177.3.1941
30. Martin-Rendon E, Hale SJ, Ryan D, et al. Transcriptional profiling of human cord blood CD133+ and cultured bone marrow mesenchymal stem cells in response to hypoxia. Stem Cells. 2007;25(4):1003–1012. doi:10.1634/stemcells.2006-0398
31. Ye LY, Chen W, Bai XL, et al. Hypoxia-induced epithelial-to-mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res. 2016;76(4):818–830. doi:10.1158/0008-5472.CAN-15-0977
32. Matchar DB, Orlando LA. The relationship between test and outcome. In: Price CP, editor. Evidence-Based Laboratory Medicine; Principles, Practice and Outcomes. Washington DC: AACC Press; 2007:53–66.
33. Cosin-Roger J, Simmen S, Melhem H, et al. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat Commun. 2017;8(1):98. doi:10.1038/s41467-017-00213-3
34. Kaser A, Ludwiczek O, Holzmann S, et al. Increased expression of CCL20 in human inflammatory bowel disease. J Clin Immunol. 2004;24(1):74–85. doi:10.1023/B:JOCI.0000018066.46279.6b
35. Kwon JH, Keates S, Bassani L, Mayer LF, Keates AC. Colonic epithelial cells are a major site of macrophage inflammatory protein 3alpha (MIP-3alpha) production in normal colon and inflammatory bowel disease. Gut. 2002;51(6):818–826. doi:10.1136/gut.51.6.818
36. Lee HJ, Choi SC, Lee MH, et al. Increased expression of MIP-3alpha/CCL20 in peripheral blood mononuclear cells from patients with ulcerative colitis and its down-regulation by sulfasalazine and glucocorticoid treatment. Inflamm Bowel Dis. 2005;11(12):1070–1079. doi:10.1097/01.MIB.0000187576.26043.ac
37. Skovdahl HK, Granlund A, Ostvik AE, et al. Expression of CCL20 and its corresponding receptor CCR6 is enhanced in active inflammatory bowel disease, and TLR3 mediates CCL20 expression in colonic epithelial cells. PLoS ONE. 2015;10(11):e0141710. doi:10.1371/journal.pone.0141710
38. Fujiie S, Hieshima K, Izawa D, et al. Proinflammatory cytokines induce liver and activation- regulated chemokine/macrophage inflammatory protein-3alpha/CCL20 in mucosal epithelial cells through NF-kappaB [correction of NK-kappaB]. Int Immunol. 2001;13(10):1255–1263. doi:10.1093/intimm/13.10.1255
39. Maxwell PH, Eckardt KU. HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nat Rev Nephrol. 2016;12(3):157–168. doi:10.1038/nrneph.2015.193
40. Katsanos KH, Tatsioni A, Natsi D, Sigounas D, Christodoulou DK, Tsianos EV. Recombinant human erythropoietin in patients with inflammatory bowel disease and refractory anemia: a 15-year single center experience. J Crohns Colitis. 2012;6(1):56–61. doi:10.1016/j.crohns.2011.07.004
41. Liu S, Ren J, Hong Z, et al. Efficacy of erythropoietin combined with enteral nutrition for the treatment of anemia in Crohn’s disease: a prospective cohort study. Nutr Clin Pract. 2013;28(1):120–127. doi:10.1177/0884533612462744
42. Kapsoritakis AN, Psychos AK, Sfiridaki A, Zintzaras E, Potamianos SP. Finger clubbing and erythropoietin serum levels in active IBD. Inflamm Bowel Dis. 2006;12(6):535–536. doi:10.1097/00054725-200606000-00014
43. Tsitsika A, Stamoulakatou A, Kafritsa Y, et al. Erythropoietin levels in children and adolescents with inflammatory bowel disease. J Pediatr Hematol Oncol. 2005;27(2):93–96. doi:10.1097/01.mph.0000153441.34407.d9
44. Brothers MD, Doan BK, Zupan MF, Wile AL, Wilber RL, Byrnes WC. Hematological and physiological adaptations following 46 weeks of moderate altitude residence. High Alt Med Biol. 2010;11(3):199–208.s. doi:10.1089/ham.2009.1090
45. Ferrante M, Pierik M, Henckaerts L, et al. The role of vascular endothelial growth factor (VEGF) in inflammatory bowel disease. Inflamm Bowel Dis. 2006;12(9):870–878. doi:10.1097/01.mib.0000235095.01608.10
46. Magro F, Araujo F, Pereira P, Meireles E, Diniz-Ribeiro M, Velosom FT. Soluble selectins, sICAM, sVCAM, and angiogenic proteins in different activity groups of patients with inflammatory bowel disease. Dig Dis Sci. 2004;49(7–8):1265–1274. doi:10.1023/B:DDAS.0000037822.55717.31
47. Pousa ID, Mate J, Salcedo-Mora X, Abreu MT, Moreno-Otero R, Gisbert JP. Role of vascular endothelial growth factor and angiopoietin systems in serum of Crohn’s disease patients. Inflamm Bowel Dis. 2008;14(1):61–67. doi:10.1002/ibd.20269
48. Scaldaferri F, Vetrano S, Sans M, et al. VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology. 2009;136(2):585–595. doi:10.1053/j.gastro.2008.09.064
49. Griga T, Voigt E, Gretzer B, Brasch F, May B. Increased production of vascular endothelial growth factor by intestinal mucosa of patients with inflammatory bowel disease. Hepatogastroenterology. 1999;46(26):920–923.
50. Griga T, Tromm A, Spranger J, May B. Increased serum levels of vascular endothelial growth factor in patients with inflammatory bowel disease. Scand J Gastroenterol. 1998;33(5):504–508. doi:10.1080/00365529850172070
51. Cummins EP, Seeballuck F, Keely SJ, et al. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. 2008;134(1):156–165. doi:10.1053/j.gastro.2007.10.012
52. Fraisl P, Aragones J, Carmeliet P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat Rev Drug Discov. 2009;8(2):139–152. doi:10.1038/nrd2761
53. Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med. 2007;85(12):1295–1300. doi:10.1007/s00109-007-0277-z
54. Tambuwala MM, Manresa MC, Cummins EP, Aversa V, Coulter IS, Taylor CT. Targeted delivery of the hydroxylase inhibitor DMOG provides enhanced efficacy with reduced systemic exposure in a murine model of colitis. J Control Release. 2015;217:221–227. doi:10.1016/j.jconrel.2015.09.022
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.