Back to Journals » Cancer Management and Research » Volume 11
MARCH5 overexpression contributes to tumor growth and metastasis and associates with poor survival in breast cancer
Authors Tang H ![]() , Peng S, Dong Y, Yang X, Yang P, Yang L, Yang B
, Peng S, Dong Y, Yang X, Yang P, Yang L, Yang B ![]() , Bao G
, Bao G
Received 12 October 2018
Accepted for publication 19 November 2018
Published 24 December 2018 Volume 2019:11 Pages 201—215
DOI https://doi.org/10.2147/CMAR.S190694
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kenan Onel
Haili Tang,1,* Shujia Peng,1,* Yanming Dong,1,* Xiaojun Yang,1 Ping Yang,1 Lin Yang,1 Bing Yang,2 Guoqiang Bao1
1Department of General Surgery, Tangdu Hospital, The Fourth Military Medical University, Xi’an, Shaanxi 710038, China; 2Department of General Surgery, JingYang Country Hospital, XianYang, Shaanxi 713700, China
*These authors contributed equally to this work
Background: Human MARCH5 is a mitochondrial localized E3 ubiquitin-protein ligase that is critical for the regulation of mitochondrial dynamics. A body of evidence has indicated the close links between unbalanced mitochondrial dynamics and cancers. However, the expression, biological functions, and prognostic significance of MARCH5 in breast cancer (BC) have not been determined.
Materials and methods: The mRNA and protein expressions of MARCH5 were evaluated by quantitative real-time PCR and Western blot analysis in BC cell lines and tumor tissues. Clinical prognostic significance of MARCH5 was assessed in 65 patients with BC. The biological functions of MARCH5 were determined by in vitro cell proliferation, apoptosis, cell cycle, migration and invasion assays, and in vivo tumor growth and metastasis assays through knockdown or overexpression of MARCH5 in BC cells. In addition, the underlying mechanisms by which MARCH5 regulated BC cell growth and metastasis were explored.
Results: MARCH5 was substantially upregulated in BC cells mainly due to the downregulation of miR-30a, which contributed to the poor survival of BC patients. MARCH5 promoted the growth and metastasis of BC cells both in vitro and in vivo by inducing G1–S cell cycle arrest and epithelial–mesenchymal transition. Mechanistic investigations revealed that the oncogenic effect of MARCH5 was mainly mediated by increased mitochondrial fission and subsequent ROS production in BC cells.
Conclusion: Our findings demonstrate that MARCH5 plays a critical oncogenic role in BC cells, which provides experimental evidence supporting MARCH5 as a potential therapeutic target in BC therapy.
Keywords: MARCH5, proliferation, invasion, prognosis, BC
Introduction
Breast cancer (BC) is the most common malignancy and the second leading cause of death from cancer among women.1 Although significant progress has been made in early BC treatment, the prognosis of patients with metastasis continues to be poor. Thus, identification of novel molecular mechanisms leading to metastatic growth of GC cells is critical for the development of new therapeutic targets for treatment of this malignancy.
Mitochondria are double-membrane-bound organelle that are vital for ATP production and macromolecule biosynthesis in most eukaryotic organisms. A significant association has been observed between the dysfunction of mitochondrial and many types of cancer.2–4 Investigation of novel oncogenic regulators involved in the dysfunction of mitochondria become imperative and urgent, which will provide insights into the molecular basis of cancer and therapeutic targets for treatment of this disease.
MARCH5, also known as membrane-associated ring finger (C3HC4) 5, is an E3 ubiquitin-protein ligase that is localized in the mitochondrial outer membrane, which is involved in the regulation of mitochondrial morphology through ubiquitination of dynamin-related protein (DMN1L), mitofusin-1 (MFN1), and mitofusin 2 (MFN2) that are pivotal regulators of mitochondrial fission and fusion.5,6 Recently, a body of evidence has indicated the close link between unbalanced mitochondrial fission/fusion and cancer.7,8 The aberrant expressions of DRP1, MFN1, and MFN2 have been demonstrated in human cancers of lung,9 liver,10 and bladder.11 In addition, a few recent studies have implicated the involvement of disruption of mitochondrial fission in BC progression.12–14 However, the expression, biological effects, and prognostic significance of MARCH5, a critical regulator of mitochondrial fission and fusion, in tumorigenesis and development are unknown, especially in BC.
In this study, we will first investigate the expression patterns and prognostic significance of MARCH5 in BC. More importantly, the biological functions and their underlying molecular mechanisms will be explored in depth in this malignancy.
Materials and methods
BC cell lines and tissue sample collection
All human BC cell lines of MDA-MB-231, MDA-MB-468, MDA-MB-435, MCF-7, T47D, and nonmalignant human breast cell lines MCF 10A and MCF 12A were purchased from the ATCC. Cells were cultured in L-15, DMEM, or RPMI-1640 medium supplemented with 10% fetal bovine serum (HyClone).
In addition, 65 primary BC tumor tissue samples were collected during surgical resection at the Tangdu Hospital of the Fourth Military Medical University in Xi’an, China. All samples were histologically confirmed as BC, and written informed consent was obtained from each patient. The study protocol was approved by the Research Ethics Committee of Tangdu Hospital of the Fourth Military Medical University and performed in compliance with the Declaration of Helsinki.
Knockdown and forced expression of target genes
Cells were transfected with siRNA (synthesized by GenePharma, Shanghai, China) against MARCH5 or expression vectors for MARCH5 using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s protocols. The following sequences were targeted for human MARCH5 siRNA: MARCH5#1: 5′-GCACUUGGGAGUAAUUUGA-3′; MARCH5#2: 5′-GCACACGUGUCCGAUUUAU-3′. In addition, synthetic miR-30a precursor (Thermo Fisher Scientific) was used. For generation of MARCH5 overexpression vectors, the coding sequences of MARCH5 were amplified from the cDNA of MCF-7 cells and cloned into the pcDNA™3.1(C) vector (Thermo Fisher Scientific). For generation of shRNA expression vectors targeting MARCH5, the pSilencer™ 3.1-H1 puro vector (Ambion, Austin, TX, USA) was used. Transfection was applied using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instruction.
Real-time PCR analysis
Total RNA was extracted from BC cell lines and tissue and then reverse transcribed into cDNA using random primers. Real-time PCR (RT-PCR) was performed by SYBR Green master mixture (Thermo Fisher Scientific) on a Corbett 6200. For the detection of miR-30a, the U6 snRNA was used as endogenous control. Primer sequences are listed in Table S1.
Western blot analysis
RIPA buffer was used for whole cell extracts. Equal amount of protein was separated on SDS-PAGE and transferred onto polyvinylidene fluoride membranes. After blocking with 5% milk, membranes were immunostained with specific primary antibody and secondary antibody. The reactions were finally detected by an enhanced chemiluminescence system. Primary antibodies and their dilutions are listed in Table S2.
Immunohistochemistry analysis
Immunohistochemistry (IHC) analysis was performed using an IHC detection kit (Thermo Fisher Scientific). All sections were treated with boiling citrate buffer (pH=6) under pressure to unmask epitopes, followed by incubation with primary antibody. Color was developed using DAB for 3 minutes, and then hematoxylin was used for counterstaining. Finally, the expression of MARCH5 was evaluated by assigning the staining intensity and percentage of positive cells.
IHC staining intensities were scored by two investigators. The staining of MARCH5 was evaluated under a light microscope at a magnification of ×100. The staining intensity was graded on a scale of 0–3 (0 for no staining, 1 for weak staining, 2 for moderate staining, and 3 for strong staining). Percentages of the stained area were also scored (0 if 0%, 1 if <25%, 2 if 25%–50%, 3 if 51%–75%, and 4 if >75%). Points for intensity and percentage were calculated to obtain a final semiquantitative IHC score. Median values of the IHC score were chosen as the cutoff point for distinguishing the low and high MARCH5 expression levels.
Cell viability and colony formation assays
MTS assay was used for determination of cell viability (Promega Corporation, Fitchburg, WI, USA, G3581) based on the manufacturer’s instructions. Briefly, BC cells were seeded onto 6-well plates at a density of 1,000 cells/well. Cell viability was determined based on the absorbance of 490 nm at 0, 24, 48, 72, 96, and 120 hours, respectively, after cells were seeded.
For the detection of colony formation, 1,000 BC cells were seeded onto 6-well plates and cultured for approximately 2 weeks. After that, colonies were fixed with 70% ethanol and stained with crystal violet. Finally, the colonies were counted.
Apoptosis and cell cycle assays
Flow cytometry was used for cell cycle and apoptosis analyses. For cell cycle assay, BC cells were fixed in 70% cold ethanol at –20°C for 12 hours and incubated in 1 mL staining solution with RNase (50 U/mL) and propidium iodide (50 µg/mL) for 30 minutes at 4°C. Cell cycle profiles were analyzed by a flow cytometry (Beckman, Fullerton, CA, USA). For apoptosis assay, an Annexin V (FITC-conjugated) apoptosis kit (F-6012, US Everbright Inc , SuZhou, China) was used following the manufacturer’s instructions. Briefly, binding buffer, Annexin V-FITC, and propidium iodide solution were added to BC cells and incubated for 20 minutes at room temperature. Cell apoptotic profiles were analyzed by a flow cytometry (Beckman).
Wound healing and Matrigel invasion assays
Cell migration was assessed by the wound healing assay. Briefly, BC cells were collected and seeded into 6-well plates. When cells grow to 90% confluence, a scratch with a plastic pipette tip was placed in the middle of the wells, followed by washing with PBS. The scratched BC cells were photographed after 0 and 24 hours through a light microscope.
For Matrigel invasion assay, extracellular matrix was coated onto the upper surface of 24-well transwell chamber (BD Companion Plate). Then, 1×105 BC cells were seeded and incubated for 48 hours. Penetrated cells were fixed with 4% paraformaldehyde and stained by crystal violet.
Mitochondrial morphology analysis
MitoTracker Green staining was used for mitochondrial morphology analysis. Briefly, BC cells were seeded into a confocal dish. After an overnight culture, cells were fixed in 4% paraformaldehyde (Sigma-Aldrich Co., St Louis, MO, USA) and incubated with a MitoTracker Green probe (Molecular Probes) for 2 hours. The immunofluorescence images were obtained by a confocal microscope (Olympus Corporation, Tokyo, Japan). Mitochondrial length was determined with the ImageJ software (NIH, Bethesda, MD, USA).
Mouse xenograft model
The expression vector of shMARCH5 was stably transfected into MDA-MB-231 cells for establishment of stable MARCH5 knockdown BC cells. Then, cells were subcutaneously injected into the dorsal flank of Balb/c nude mice (six/group). Tumor volume was monitored every 3 days for 5 weeks. Mice were sacrificed, and the volume and wet weight of tumors were measured. All animal studies were approved by the Institutional Animal Care and Use Committee of the Fourth Military Medical University. Animal experiments were carried out in accordance with the UK Animals (Scientific Procedures) Act, 1986 and associated guidelines.
Tail vein metastatic assay
A total of 1×106 BC cells with different MARCH5 expression levels were injected into the Balb/c nude mice (six/group) through the tail vein. The mice were sacrificed 8 weeks after injection, and the lungs were dissected and stained with H&E.
Statistical analysis
SPSS version 17.0 software was used for statistical analysis. The results are shown as the mean ± SEM, and P-value <0.05 was considered statistically significant. Student’s t-test was used for comparisons between two groups. Pearson correlation analysis was used for correlations between measured variables. The overall survival (OS) and recurrence-free survival were determined by the Kaplan–Meier method.
Results
MARCH5 is upregulated in BC cells and contributes to worse prognosis
First, the expression of MARCH5 was detected in five BC cell lines (MCF-7, T47D, MDA-MB-231, MDA-MB-468, and MDA-MB-435) and two nonmalignant human breast cell lines (MCF-10A and MCF-12A) by quantitative real-time PCR (qRT-PCR) and Western blot analysis. MARCH5 expression was significantly higher in five BC cell lines when compared with nonmalignant breast cell lines (Figure 1A, B). To validate the results from BC cell lines, we determined the expression level of MARCH5 in 18 paired BC tumor tissues and peritumor tissues. In concordance with the results from BC cell lines, a significant upregulation of MARCH5 was also observed in tumor tissues compared with peritumor tissues (Figure 1C, D).
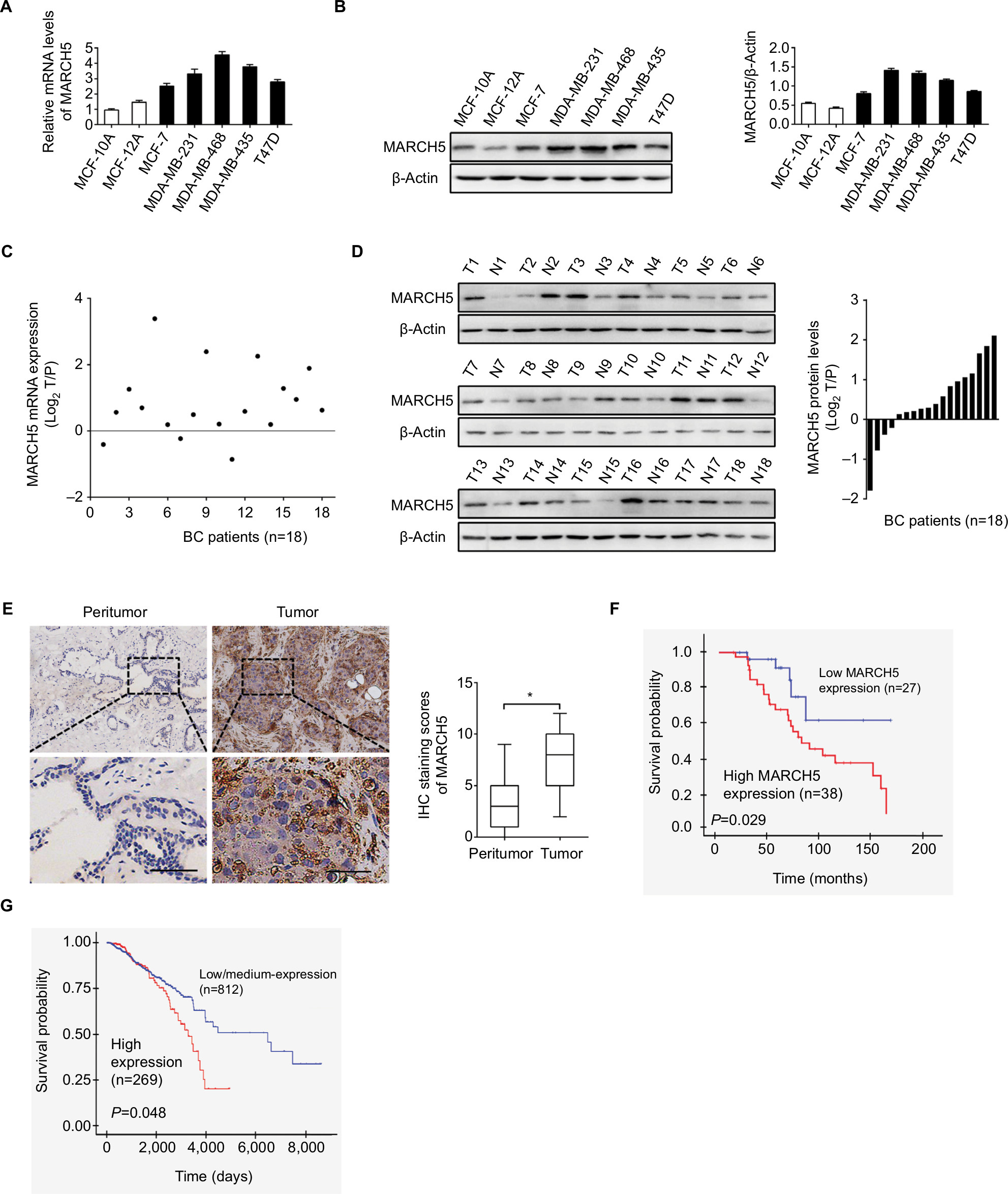 | Figure 1 MARCH5 is upregulated in BC cells and contributes to worse prognosis. Notes: (A and B) Quantitative real-time PCR and Western blot assays for expressions of MARCH5 in five BC cell lines (MDA-MB-231, MDA-MB-468, MDA-MB-435, MCF-7, and T47D) and two nonmalignant human breast cell lines (MCF 10A and MCF 12A). (C and D) Quantitative real-time PCR and Western blot assays for expressions of MARCH5 in 18 paired BC tumor tissues and peritumor tissues (T, tumor; P, peritumor). Scale bars, 50 µm. The relative MARCH5 expression ratio of tumor to peritumor was log2-transformed. (E) Representative immunohistochemical (IHC) staining images (left panel, scale bar, 50 µm) and staining intensities (right panel) of MARCH5 in tumor and peritumor tissues from 65 BC patients. (F) Kaplan–Meier curve for survival analysis of 65 BC patients according to their MARCH5 expression levels. (G) Prognostic significance analysis for MARCH5 in BC using the online web portal UALCAN (http://ualcan.path.uab.edu). *P<0.05. Abbreviation: BC, breast cancer. |
To evaluate the clinical significance of MARCH5 in BC, MARCH5 expression was further evaluated by IHC analysis in another 65 paired tumor and peritumor tissues. As shown in Figure 1E, IHC score of MARCH5 was remarkably increased in tumor tissues of BC when compared with paired peritumor tissues. Kaplan–Meier survival curves showed that BC patients with high expression levels of MARCH5 had significantly poorer survival than those with low MARCH5 (Figure 1F), which was further confirmed by prognostic significance analysis using the online web portal UALCAN15 (http://ualcan.path.uab.edu) (Figure 1G). These findings collectively indicate that MARCH5 is upregulated in BC cells, which predicts a poor prognosis of patients with BC.
Knockdown of MARCH5 suppressed BC cell growth by inducing G1–S cell cycle arrest
To investigate the biological functions of MARCH in BC cells, MARCH5 was knocked down by siRNA in MDA-MB-231 and MDA-MB-468 cells, which have relative high MARCH5 expression (shown in Figure 1A, B). As shown in Figure S1A, B, a significant decrease in MARCH5 at both mRNA and protein levels was observed in MDA-MB-231 and MDA-MB-468 cells transfected with siMARCH5 when compared with scramble siRNA groups. MARCH5 knockdown significantly suppressed BC cell growth, as evidenced by MTS cell viability and colony formation assays (Figure 2A, B). Considering that growth promotive effect could be caused by induction of cell cycle progression or inhibition of apoptosis, or both. To further explore the mechanism by which MARCH5 promotes the growth of GC cells, we determined whether MARCH5 regulated cell cycle distribution and apoptosis of GC cells by flow cytometry. A significant increase of cells in G1 phase and decrease of cells in S phase were found in BC cells when MARCH5 was knocked down (Figure 2C). Consistently, fewer proliferating cells were detected by EdU (5-ethynyl-2′-deoxyuridine) incorporation assay in BC cells with MARCH5 knocked down compared with their control cells (Figure 2D). However, no significant changes of cell apoptosis were observed in BC cells when MARCH5 was knocked down, as evidenced by both the flow cytometry (Figure 2E) and Western blot analysis (Figure 2F, G). Collectively, these results suggest that MARCH5 promoted the growth of BC cells mainly through induction of G1–S cell cycle transition.
 | Figure 2 Knockdown of MARCH5 suppressed BC cell growth by inducing G1–S cell cycle arrest. Notes: (A) MTS cell viability analysis in MDA-MB-231 and MDA-MB-468 cells with transfection of siMARCH5 or siCtrl as indicated (siMARCH5, siRNA against MARCH5; siCtrl, control siRNA). (B) Colony formation assay in BC cells with treatment as indicated. (C) Flow cytometry-based cell cycle analysis in MDA-MB-231 and MDA-MB-468 cells with treatment as indicated. (D) EdU incorporation assay for determination of cell proliferation in BC cells with treatment as indicated. Scale bars, 50 µm. (E) Annexin V/PI staining for cell apoptosis analysis in BC cells with treatment as indicated. (F) Western blot analysis for cytochrome c (cyt c) levels in cytoplasm and mitochondria of BC cells with treatment as indicated. β-actin and COX IV were loading controls of cytoplasm (Cyto) and mitochondria (Mito). (G) Western blot analysis for levels of cleaved caspase 9 and caspase 3 in BC cells with treatment as indicated. *P<0.05. Abbreviations: BC, breast cancer; EdU, 5-ethynyl-2′-deoxyuridine; PI, propidium iodide; ns, not significant. |
Knockdown of MARCH5 inhibited the migration and invasion abilities of BC cells through inhibition of epithelial–mesenchymal transition (EMT)
We also investigated the role of MARCH5 in the migration and invasion of BC cells by scratch wound healing and Matrigel invasion assays. As shown in Figure 3A, B, knockdown of MARCH5 significantly reduced both the wound closure and invasion of BC cells. EMT is a process by which epithelial cancer cells acquire more migratory mesenchymal phenotype characterized by reduced cell–cell contact and increased cell motility.16,17 We thus explored whether EMT was involved in the suppression of BC migration and invasion when MARCH5 was knocked down. qRT-PCR and Western blot analysis indicated that knockdown of MARCH5 significantly increased the levels of epithelial markers of E-cadherin and ZO-1 but reduced the levels of mesenchymal markers of N-cadherin and vimentin in BC cells (Figure 3C, D). Except for EMT, increased matrix metalloproteinase (MMP) expression also have been implicated in tumor metastasis.18 To further determine whether MMPs were also involved in MARCH5-mediated BC cell invasion, the expression levels of four most common MMPs were analyzed by qRT-PCR and Western blot analysis in BC cells with MARCH5 knockdown. As shown in Figure 3E, F, no significant change of the expressions of MMPs was observed in BC cells when MARCH5 was knocked down, suggesting that MMP production were not involved in MARCH5-mediated invasion of BC cells. These findings collectively suggest that MARCH5 promotes the migration and invasion abilities of BC cells through induction of EMT transition.
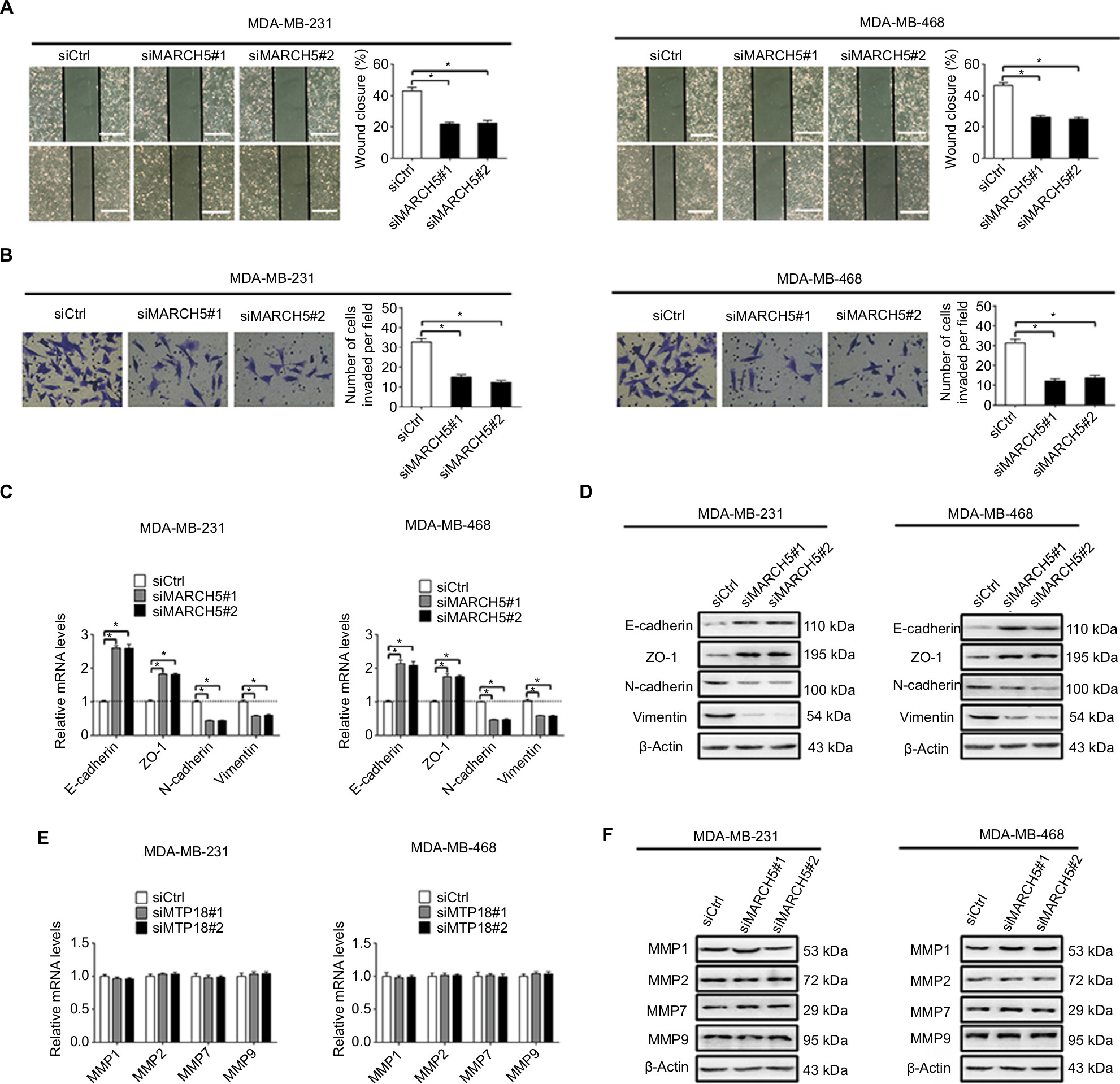 | Figure 3 Knockdown of MARCH5 inhibited the migration and invasion abilities of BC cells through inhibition of EMT. Notes: (A and B) Cell migration and invasion were evaluated by the wound healing migration and Matrigel invasion assays in MDA-MB-231 and MDA-MB-468 cells after transfection with siMARCH5 or siCtrl (siMARCH5, siRNA against MARCH5; siCtrl, control siRNA). Scale bars, 50 µm. (C and D) The expressions of EMT markers were evaluated by qRT-PCR and Western blot analysis in BC cells treated as indicated. (E and F) The mRNA levels of four matrix metalloproteinases (MMPs) were evaluated by qRT-PCR analysis in BC cells treated as indicated. *P<0.05. Abbreviations: BC, breast cancer; EMT, epithelial–mesenchymal transition; qRT-PCR, quantitative real-time PCR. |
Knockdown of MARCH5 suppressed BC growth and metastasis in vivo
To study the role of MARCH5 in BC growth in vivo, subcutaneous xenograft models were established by injecting MDA-MB-231 cells with stable MARCH5 knockdown in 4-week-old male nude mice. As shown in Figure 4A, tumors developed from MDA-MB-231 cells with stable MARCH5 knockdown (shMARCH5) grew slower than those in controls (shCtrl). Consistently, the volume and net wet weight of tumors from shMARCH5 group were also significantly decreased compared with those in control group (Figure 4B). In addition, IHC staining showed significantly lower levels of MARCH5 in tumors obtained from the nude mice injected with shMARCH5-transfected cells than in the controls, suggesting that the tumor growth inhibition effect was exerted by MARCH5 knockdown (Figure 4C). As expected, Ki-67 staining showed significantly fewer proliferating cells in shMARCH5 xenografts when compared with control (Figure 4D). Moreover, in vivo metastatic assay via tail vein injection of GC cells further showed significantly decreased formation of metastatic nodules in the lungs in shMARCH5 group than those in the shCtrl group (Figure 4E).
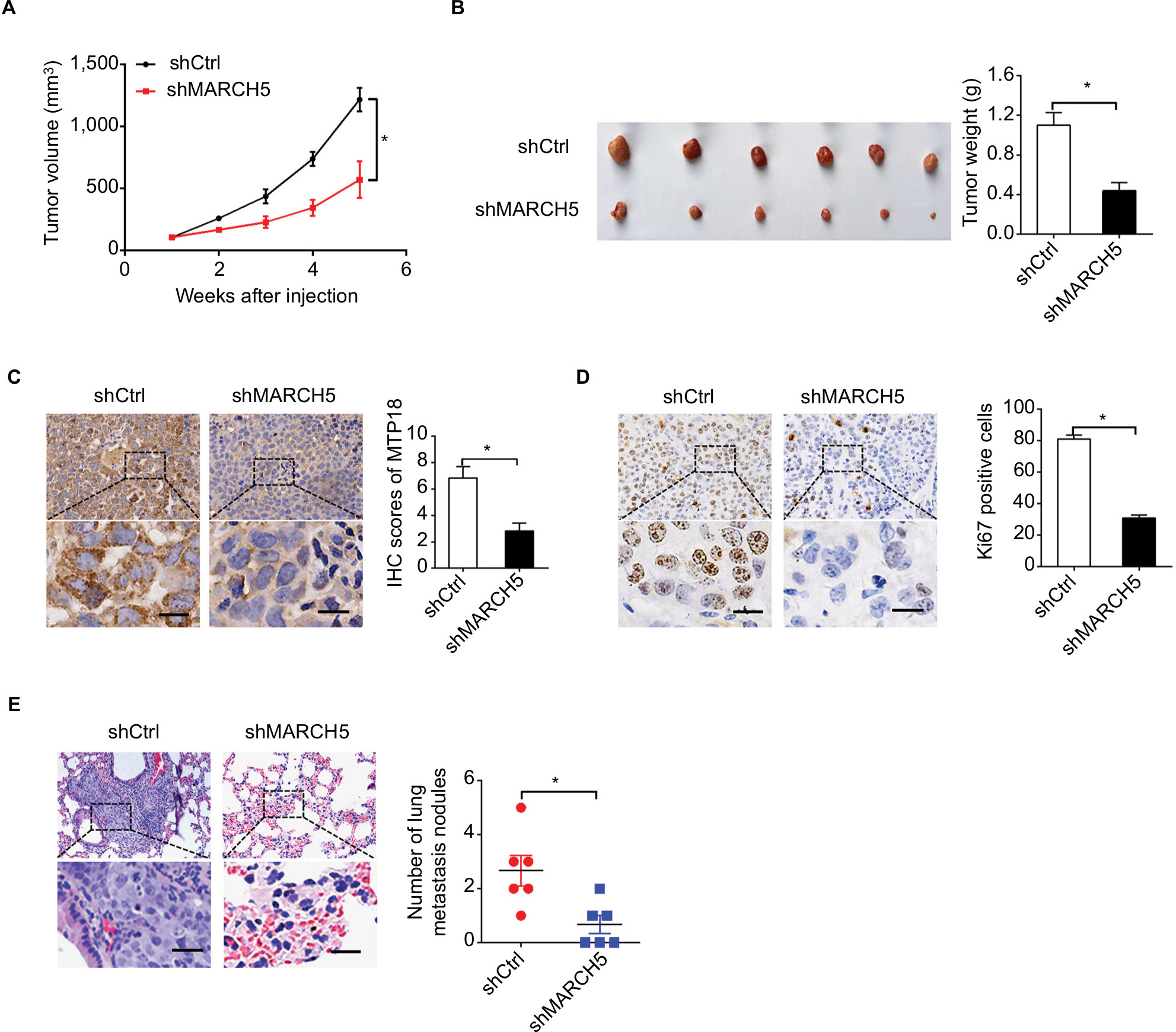 | Figure 4 Knockdown of MARCH5 suppressed breast cancer growth and metastasis in vivo. Notes: (A) Growth of subcutaneous xenografts established from MDA-MB-231 cells with stable MARCH5 knockdown or control cells. shMARCH5, shRNA expression vector against MARCH5; shCtrl, control shRNA. (B) The weight of tumors dissected from mice was compared between the shMARCH5 and shCtrl groups. (C and D) The expression levels of MARCH5 and Ki-67 were detected by IHC analysis in the two groups of subcutaneous xenografts tumors. Scale bars, 10 µm. (E) Representative images and numbers of lung metastatic foci in each group are shown. Scale bars, 10 µm. *P<0.05. |
Forced expression of MARCH5 promoted growth and metastasis of BC cells
To further confirm the promotive effects of MARCH5 on the growth and metastasis of BC cell, MARCH5 was upregulated by transfecting the MARCH5 expression vector or empty vector into MCF-7 and T47D cells, which have relative low MARCH5 expression (shown in Figure 1A, B). Upregulation of MARCH5 was evidenced by qRT-PCR and Western blot analysis (Figure S2A, B). As shown in Figure 5A, B, upregulation of MARCH5 significantly enhanced cell viability and clonogenicity of MCF-7 and T47D cells. As expected, MARCH5 upregulation also induced G1–S cell cycle progression and EdU incorporation (Figure 5C–E), which further supported the tumor growth promotive effect of MARCH5 by causing cell cycle progression in BC. Moreover, wound healing and Matrigel invasion assays indicated that MARCH5 upregulation significantly increased both migration and invasion abilities of MCF-7 and T47D cells (Figure 5F, G). Collectively, these results suggest a crucial oncogenic role for MARCH5 in the regulation of BC growth and metastasis.
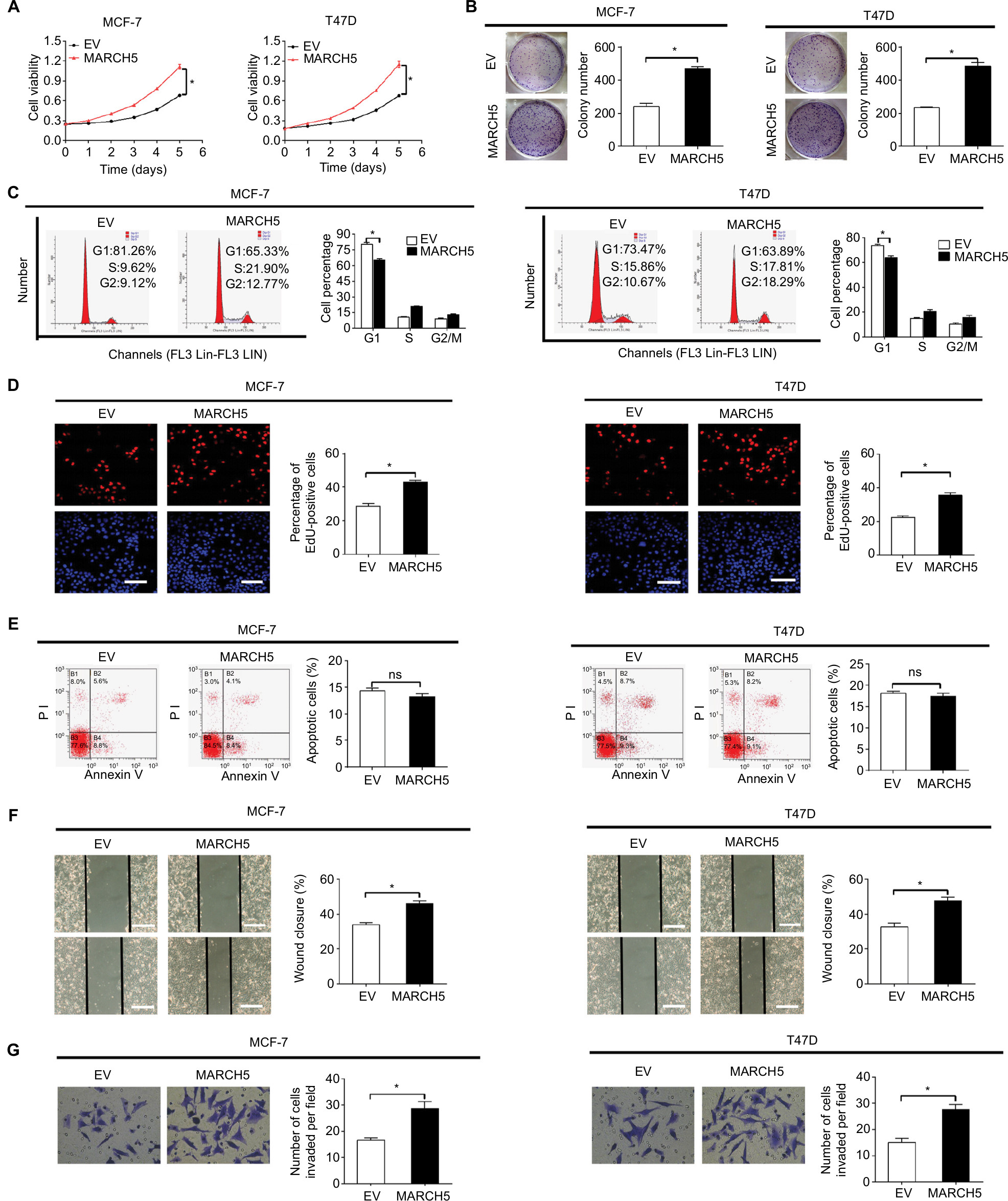 | Figure 5 Forced expression of MARCH5 promoted growth and metastasis of BC cells. Notes: (A and B) MTS cell viability and colony formation were analyzed in MCF-7 and T47D cells after transfection with MARCH5 (MARCH5) expression vector or empty vector (EV). (C) Flow cytometry-based cell cycle analysis in BC cells with treatment as indicated. (D) Cell proliferation was analyzed by the EdU incorporation assay in BC cells with treatment as indicated. Scale bar, 20 µm. (E) Cell apoptosis was analyzed by the flow cytometry in BC cells treated as indicated. (F and G) Wound healing migration and transwell Matrigel invasion assays in BC cells treated as indicated. Scale bars, 50 µm. *P<0.05. Abbreviations: BC, breast cancer; EdU, 5-ethynyl-2′-deoxyuridine; EV, empty vector; ns, not significant. |
Overexpression of MARCH5 is mainly mediated by downregulation of miR-30a
MicroRNAs are crucial components in the network of gene expression regulation. A previous study has reported MARCH5 as a novel target of miR-30a.19 Considering that miR-30a has been proved as a frequently downregulated tumor suppressor in various types of cancer,20 including BC.21 We hypothesized that miR-30a may be involved in the upregulation of MARCH5 in BC cells. To test this possibility, synthetic precursor of miR-30a was transfected into MCF-7 and T47D cells. Forced expression of miR-30a significantly downregulated the expression of MARCH5 at both mRNA and protein levels in BC cells (Figure 6A, B). To provide further supports, we examined the levels miR-30a in 18 BC tissue samples. We found a significant negative correlation between the levels of miR-30a and MARCH5 (r=–0.545, P=0.019) (Figure 6C). Moreover, forced expression of miR-30a significantly attenuated both the tumor growth and metastasis of BC cells promoted by MARCH5 (Figure 6D–G).
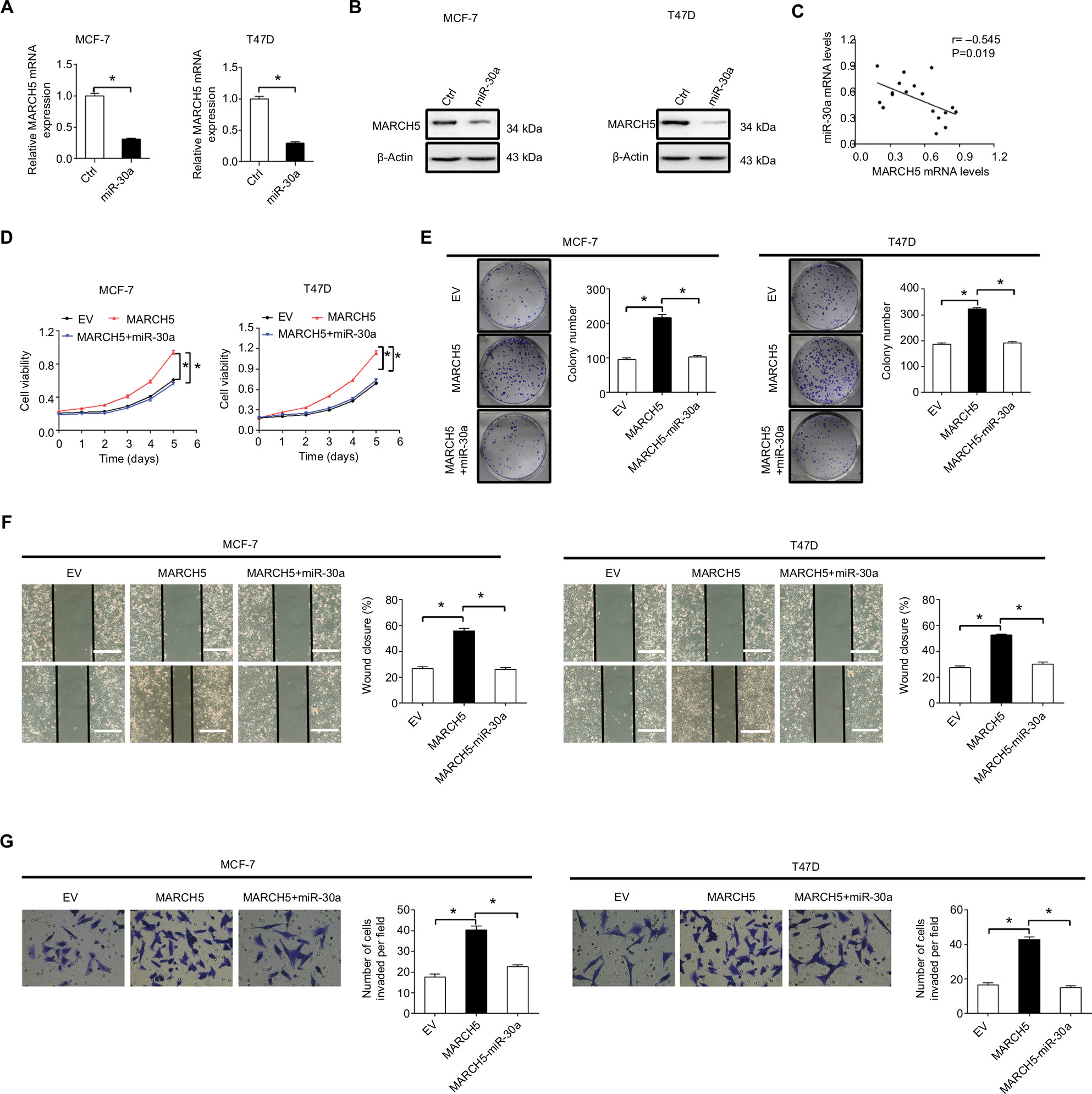 | Figure 6 Overexpression of MARCH5 is mainly mediated by downregulation of miR-30a. Notes: (A and B) MARCH5 expression was evaluated by quantitative real-time PCR and Western blot in MCF-7 and T47D cells after transfection with the synthetic miR-30a precursor. (C) The correlation between mRNA levels of MARCH5 and miR-30a was determined in 18 tumor tissues of BC. (D and E) MTS cell viability and colony formation were analyzed in MCF-7 and T47D cells treated as indicated. (F and G) Wound healing migration and transwell Matrigel invasion assays in MCF-7 and T47D cells treated as indicated. Scale bars, 50 µm. *P<0.05. Abbreviations: BC, breast cancer; EV, empty vector. |
MARCH5 promotes tumor growth and metastasis through elevation of mitochondrial fission-mediated ROS production
Previous studies have demonstrated MARCH5 as a critical regulator of mitochondrial fission and fusion.5 To explore the molecular basis for the oncogenic properties of MARCH5 in BC, we first assessed the morphological changes of mitochondria in BC cells after MARCH5 was knocked down or upregulated. As shown in Figure 7A, MARCH5 knockdown promoted extensive elongation of mitochondria in MDA-MB-231 cells, while MARCH5 overexpression resulted in a remarkable fragmentation of mitochondria in MCF-7 cells, as evidenced by a MitoTracker Green staining.
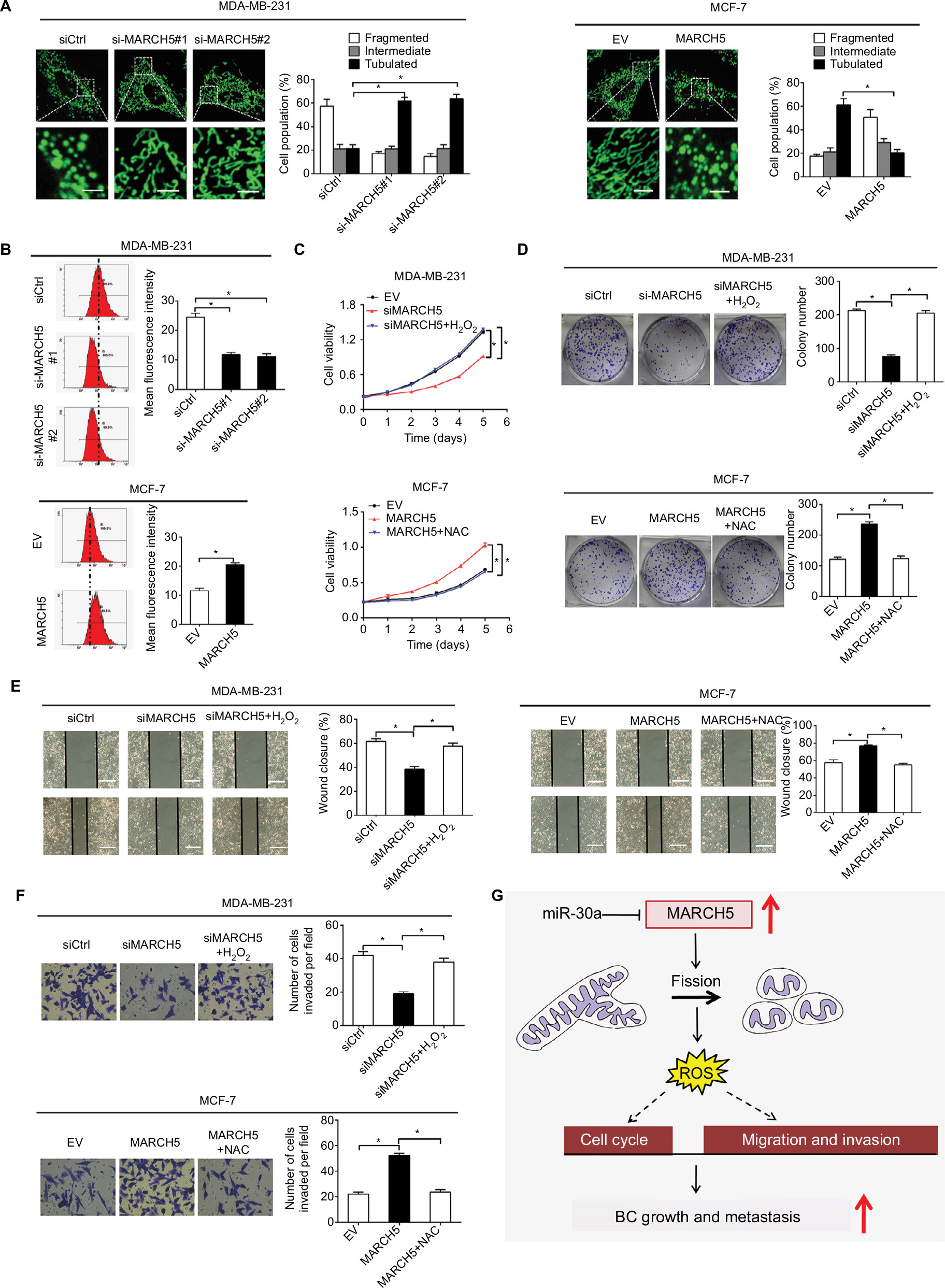 | Figure 7 MARCH5 promotes tumor growth and metastasis through elevation of mitochondrial fission-mediated ROS production. Notes: (A) Left panel: Representative mitochondrial morphology of MDA-MB-231 cells with MARCH5 knockdown and MCF-7 cells with MARCH5 overexpression. Scale bars, 1 µm. Right panel: The proportions of BC cells (30 cells for each sample) with fragmented, intermediate, and tubulated were quantified. (B) The intracellular ROS levels were determined by flow cytometry in BC cells treated as indicated. (C and D) MTS cell viability and colony formation abilities were analyzed in BC cells treated with H2O2 (90 µM, 12 hours) or NAC (30 µM, 12 hours) as indicated. (E and F) Wound healing and transwell Matrigel invasion assays in BC cells treated with H2O2 (90 µM, 12 hours) or NAC (30 µM, 12 hours) as indicated. Scale bars, 50 µm. (G) Schematic depicting the oncogenic role of MARCH5 upregulation in tumor growth and metastasis of BC and their underlying mechanism. *P<0.05. Abbreviations: BC, breast cancer; EV, empty vector. |
Mitochondria is a major source of ROS, which play an important role in the regulation of tumor growth and metastasis by activating several oncogenic signaling pathways.22 Thus, we speculated that mitochondrial fission-regulated ROS production may be involved in the regulation of tumor growth and metastasis by MARCH5 in BC. As shown in Figure 7B, ROS production was significantly reduced when MARCH5 was knockdown in MDA-MB-231 cells, while significantly elevated upon overexpression of MARCH5 in MCF-7 cells. To test whether elevated production of ROS is involved in the oncogenic properties of MARCH5, BC cells were treated with H2O2 or NAC (a ROS scavenger).23 Our results showed that H2O2 treatment significantly increased the growth and metastasis of MDA-MB-231 cells suppressed by MARCH5 knockdown (Figure 7C–F). In contrast, NAC treatment significantly decreased the growth and metastasis of MCF-7 cells induced by MARCH5 overexpression. Collectively, these results suggest that the oncogenic effects of MARCH5 in BC cells are mainly mediated by increased mitochondrial fission and subsequently ROS production.
Discussion
In this study, we observed that MARCH5, a critical regulator of mitochondria dynamics, is commonly upregulated in BC cell lines and tumor tissues mainly due to the downregulation of miR-30a. In addition, our results revealed that high expression levels of MARCH5 predict poorer overall survival and recurrence-free survival of patients with BC. Consistently, abnormal expressions of other mitochondrial dynamic regulators have also been demonstrated in different types of cancer. For example, Huang et al have shown that mitochondrial DRP1 is upregulated and MFN1 is downregulated in hepatocellular carcinoma (HCC), both of which contribute to poor survival of patients with HCC.10 In addition, Rehman et al reported that DRP1 is upregulated and MFN2 is downregulated in lung cancer cells.9 Moreover, in BC cells, Zhao et al found that mitochondria were more fragmented in metastatic BC cells with higher levels of DRP1 and lower levels of MFN1 compared with nonmetastatic BC cells.12 These observations support the interpretation that dysfunction of mitochondrial dynamics plays a critical role in the progression of most tumor types.
Deregulation of miR-30a has been reported in various malignances, including lung, thyroid, gastric, and colon cancers.20,24–26 Moreover, a frequently downregulated miR-30a has also been observed in BC cells.21 This study demonstrated that MARCH5 is a novel target of miR-30a in BC and downregulated miR-30a was involved in the overexpression of MARCH5 in BC, suggesting that miR-30a is a critical tumor suppressor microRNA in BC. However, considering that a wide range of mechanisms are used by eukaryotes for the regulation of gene expression, we still cannot rule out the possibility that other molecular mechanisms may be involved in the overexpression of MARCH5 in BC, which still needs further clarification.
Overexpression of MARCH5 in BC suggested that MARCH5 could function as a potential oncogene. We therefore investigated the biological functions of MARCH5 by gain- and loss-of-function assays in human BC cell lines. Knockdown of MARCH5 significantly inhibited cell growth and metastasis in BC cell lines MDA-MB-231 and MDA-MB-468. Conversely, forced expression of MARCH5 significantly enhanced cell growth and metastasis in BC cell lines MCF-7 and T47D. Consistently, a previous study in ovarian cancer also demonstrated that MARCH5 promoted the migration and invasion of SKOV3 cells both in vitro and in vivo.19 MARCH5 has also been shown to act as a prosurvival factor under stress conditions in HeLa cells.27 Moreover, our study further indicated that MARCH5 promoted BC cell growth and metastasis mainly through induction of G1–S cell cycle and EMT, but not through suppression of apoptosis and induction of MMP production. These results together support a pro-oncogenic role for MARCH5 in tumor progression.
The close link between mitochondrial fragmentation and cancer has been well established in many different types of cancer, including BC. Zhao et al showed that MFN silencing-mediated mitochondrial fragmentation significantly enhanced metastatic abilities of BC cells.12 Our results showed that MARCH5 overexpression induced a remarkable fragmentation of mitochondria, indicating the involvement of mitochondrial fragmentation in BC progression. Mitochondria is the main source of ROS, which is critical for tumor progression.22 A previous study in HCC has demonstrated a promoting effect of mitochondrial fragmentation on ROS production.10 Our study showed a similar effect of MARCH5-mediated mitochondrial fragmentation on the production of ROS in BC cells. Moreover, we demonstrated that elevated ROS production was involved in the promotion of BC growth and metastasis by MARCH5.
In summary, our results show that MARCH5 is frequently upregulated in BC, and upregulation of MARCH5 plays a critical oncogenic role in breast carcinogenesis by promotion of both BC growth and metastasis (Figure 7G), which provides experimental evidence supporting MARCH5 as a potential therapeutic target in BC therapy.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (grant no. 81272201).
Disclosure
The authors report no conflicts of interest in this work.
References
Wallace MC, Preen D, Jeffrey GP, Adams LA. The evolving epidemiology of hepatocellular carcinoma: a global perspective. Expert Rev Gastroenterol Hepatol. 2015;9(6):765–779. | ||
Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–698. | ||
Boland ML, Chourasia AH, Macleod KF. Mitochondrial dysfunction in cancer. Front Oncol. 2013;3:292. | ||
Guerra F, Guaragnella N, Arbini AA, Bucci C, Giannattasio S, Moro L. Mitochondrial dysfunction: a novel potential driver of epithelial-to-mesenchymal transition in cancer. Front Oncol. 2017;7:295. | ||
Nagashima S, Tokuyama T, Yonashiro R, Inatome R, Yanagi S. Roles of mitochondrial ubiquitin ligase MITOL/MARCH5 in mitochondrial dynamics and diseases. J Biochem. 2014;155(5):273–279. | ||
Xu S, Cherok E, Das S, et al. Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol Biol Cell. 2016;27(2):349–359. | ||
Srinivasan S, Guha M, Kashina A, Avadhani NG. Mitochondrial dysfunction and mitochondrial dynamics – the cancer connection. Biochim Biophys Acta Bioenerg. 2017;1858(8):602–614. | ||
Simula L, Nazio F, Campello S. The mitochondrial dynamics in cancer and immune-surveillance. Semin Cancer Biol. 2017;47:29–42. | ||
Rehman J, Zhang HJ, Toth PT, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012;26(5):2175–2186. | ||
Huang Q, Zhan L, Cao H, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy. 2016;12(6):999–1014. | ||
Jin B, Fu G, Pan H, et al. Anti-tumour efficacy of mitofusin-2 in urinary bladder carcinoma. Med Oncol. 2011;28(Suppl 1):S373–S380. | ||
Zhao J, Zhang J, Yu M, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013;32(40):4814–4824. | ||
von Eyss B, Jaenicke LA, Kortlever RM, et al. A MYC-driven change in mitochondrial dynamics limits YAP/TAZ function in mammary epithelial cells and breast cancer. Cancer Cell. 2015;28(6):743–757. | ||
Han XJ, Yang ZJ, Jiang LP, et al. Mitochondrial dynamics regulates hypoxia-induced migration and antineoplastic activity of cisplatin in breast cancer cells. Int J Oncol. 2015;46(2):691–700. | ||
Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649–658. | ||
Przybyla L, Muncie JM, Weaver VM. Mechanical control of epithelial-to-mesenchymal transitions in development and cancer. Annu Rev Cell Dev Biol. 2016;32:527–554. | ||
Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 2015;25(11):675–686. | ||
Kessenbrock K, Wang CY, Werb Z. Matrix metalloproteinases in stem cell regulation and cancer. Matrix Biol. 2015;44–46:184–190. | ||
Hu J, Meng Y, Zhang Z, et al. MARCH5 RNA promotes autophagy, migration, and invasion of ovarian cancer cells. Autophagy. 2017;13(2):333–344. | ||
Yang X, Chen Y, Chen L. The versatile role of microRNA-30a in human cancer. Cell Physiol Biochem. 2017;41(4):1616–1632. | ||
Fu J, Xu X, Kang L, et al. miR-30a suppresses breast cancer cell proliferation and migration by targeting Eya2. Biochem Biophys Res Commun. 2014;445(2):314–319. | ||
Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer. 2014;14(11):709–721. | ||
Downs I, Liu J, Aw TY, Adegboyega PA, Ajuebor MN. The ROS scavenger, NAC, regulates hepatic Vα14iNKT cells signaling during Fas mAb-dependent fulminant liver failure. PLoS One. 2012;7(6): e38051. | ||
Kumarswamy R, Mudduluru G, Ceppi P, et al. MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snai1 and is downregulated in non-small cell lung cancer. Int J Cancer. 2012;130(9):2044–2053. | ||
Li X, Zhang Y, Zhang Y, Ding J, Wu K, Fan D. Survival prediction of gastric cancer by a seven-microRNA signature. Gut. 2010;59(5):579–585. | ||
Zhang Q, Tang Q, Qin D, et al. Role of microRNA 30a targeting insulin receptor substrate 2 in colorectal tumorigenesis. Mol Cell Biol. 2015;35(6):988–1000. | ||
Park YY, Nguyen OT, Kang H, Cho H. MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis. 2014;5:e1172. |
Supplementary materials
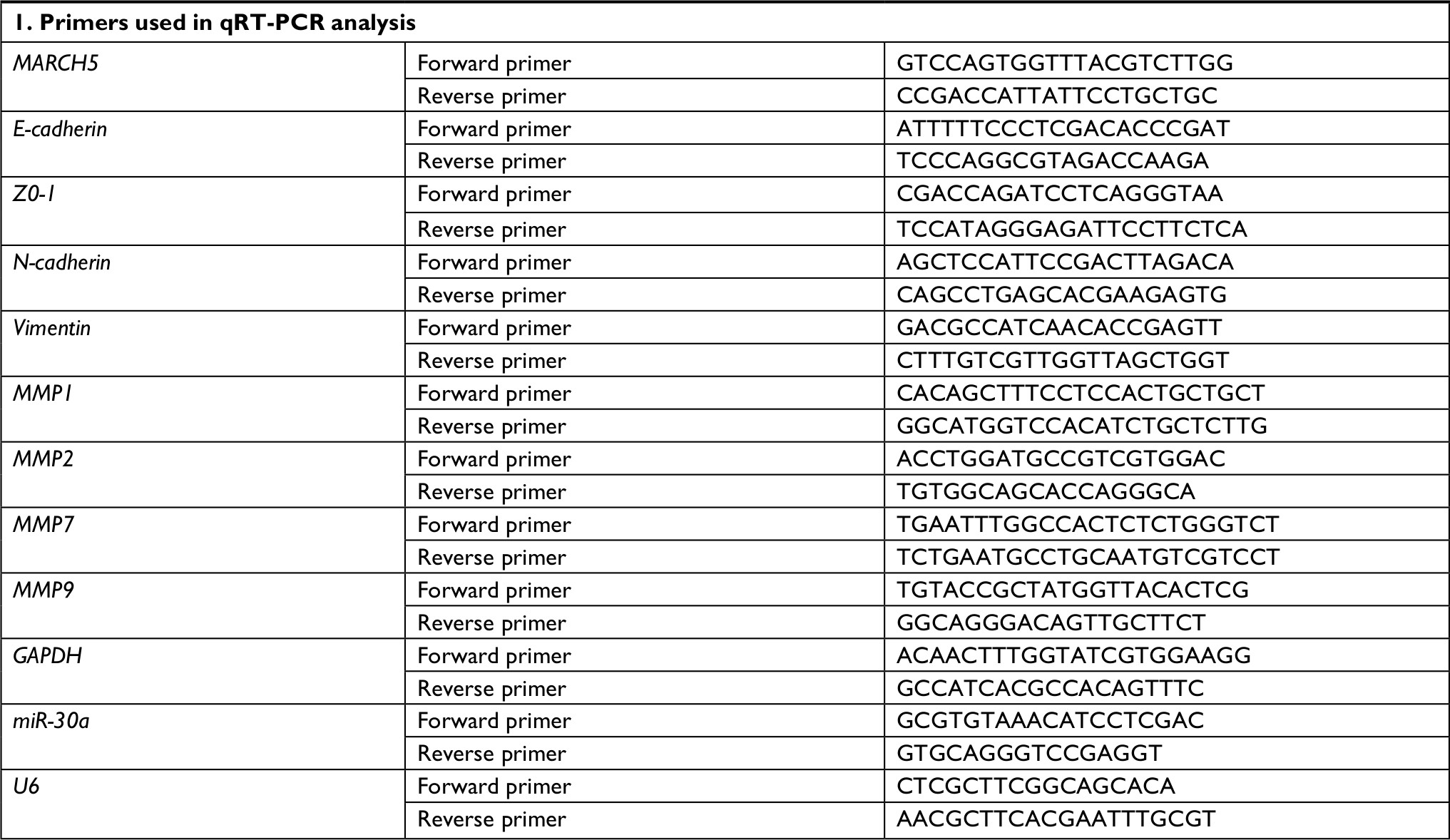 | Table S1 Sequence of primers used for quantitative real-time PCR (qRT-PCR) analysis |
 | Table S2 Primary antibodies used for Western blot and immunohistochemistry analysis |
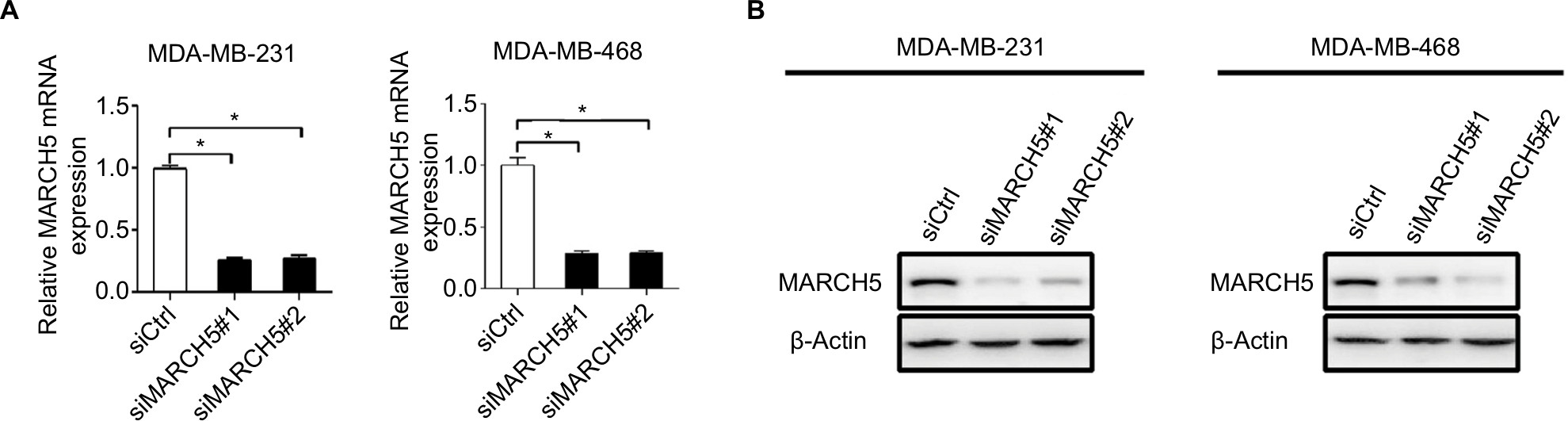 | Figure S1 Knockdown of MARCH5 in MDA-MB-231 and MDA-MB-468 cells was confirmed by quantitative real-time PCR (qRT-PCR) (A) and Western blot (B) analysis at mRNA and protein levels after transfection with siRNAs against MARCH5 (siMARCH5) or control siRNA (siCtrl). Note: Data were presented as the mean ± SEM from three independent repeats, *P<0.05. |
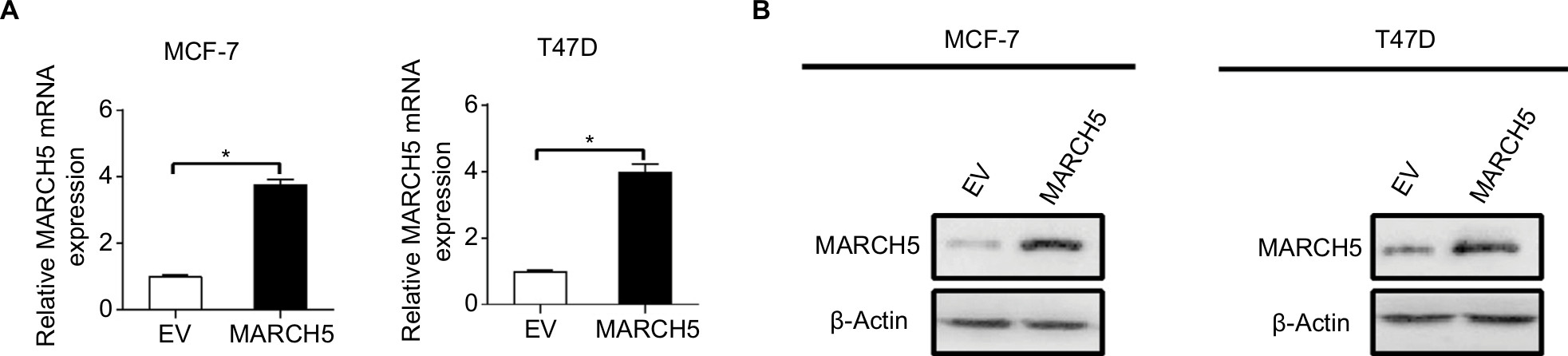 | Figure S2 Forced expression of MARCH5 in MCF-7 and T47D cells was confirmed by quantitative real-time PCR (qRT-PCR) (A) and Western blot (B) analysis at mRNA and protein levels after transfection with expression vector encoding MARCH5 (MARCH5) or empty vector (EV). Note: Data were presented as the mean ± SEM from three independent repeats, *P<0.05. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.