Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
MAPK/FoxA2-mediated cigarette smoke-induced squamous metaplasia of bronchial epithelial cells
Authors Du C ![]() , Lu J, Zhou L, Wu B, Zhou F, Gu L, Xu D, Sun Y
, Lu J, Zhou L, Wu B, Zhou F, Gu L, Xu D, Sun Y
Received 5 June 2017
Accepted for publication 10 October 2017
Published 21 November 2017 Volume 2017:12 Pages 3341—3351
DOI https://doi.org/10.2147/COPD.S143279
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Chunling Du,* Jinchang Lu,* Lei Zhou, Bo Wu, Feng Zhou, Liang Gu, Donghui Xu, Yingxin Sun
Department of Respiratory Medicine, Qingpu Branch of Zhongshan Hospital, Fudan University, Shanghai, China
*These authors contributed equally to this work
Objective: To explore the effect of cigarette smoke (CS) on the development of squamous metaplasia in human airway epithelial cells and the role of MAPK- and FoxA2-signaling pathways in the process.
Materials and methods: In an in vitro study, we treated the bronchial epithelial cell line BEAS2B with CS extract, followed by treatment with the ERK inhibitor U0126, the JNK inhibitor SP600125, or the p38 inhibitor SB203580. In vivo, we used a CS-induced rat model. After treatment with CS with or without MAPK inhibitors for 90 days, lung tissues were harvested. p-ERK, p-p38 and p-JNK protein levels in cells and lung tissue were measured using enzyme-linked immunosorbent assays, mRNA- and protein-expression profiles of FoxA2, E-cadherin, CD44, and ZO1 were measured using quantitative real-time polymerase chain reaction and Western blotting, respectively, and morphological changes in bronchial epithelial cells were observed using lung-tissue staining.
Results: In both the in vitro and in vivo studies, phosphorylation of the ERK1/2, JNK, and p38 proteins was significantly increased (P<0.05) and mRNA and protein expression of E-cadherin and FoxA2 significantly decreased (P<0.05) compared with the control group. ERK, JNK, and p38 inhibitors reversed the CS-extract-induced changes in E-cadherin, CD44, and ZO1 mRNA and protein expression (P<0.05), decreased p-ERK, p-p38, and p-JNK protein levels in cells and lung tissue, suppressed bronchial epithelial hyperplasia and local squamous metaplasia, and decreased FoxA2 expression.
Conclusion: MAPK and FoxA2 mediate CS-induced squamous metaplasia. MAPK inhibitors upregulate FoxA2, resulting in a reduction in the degree of squamous metaplasia.
Keywords: MAPK, FoxA2, cigarette smoke, bronchial epithelial cell, squamous metaplasia
Introduction
COPD is characterized by irreversible and progressive airflow limitation and encompasses various degrees of chronic obstructive bronchitis and emphysema. Chronic cigarette smoke (CS) exposure is a key element in the induction of COPD by chronic inflammation and oxidative damage.1 Research indicates that smoking can activate ERK1/2, JNK, p38, ERK5, and AP1 in lung tissue and induce obvious squamous metaplasia and hyperplasia in rat bronchial epithelial cells.2 In addition, the MAPK-signaling pathway is closely associated with smoking-induced abnormal differentiation of bronchial epithelial cells and increased secretion of Muc5AC.3 The MAPK pathway has become an emerging therapeutic target in COPD.4 However, the results of clinical trials conducted to date have not been satisfactory.
FoxA2, a transcription factor that plays a critical role in pulmonary morphogenesis and gene expression, is required for bronchial epithelial cell differentiation. Studies of FoxA2 have mainly focused on its regulation of hepatocyte maturation and differentiation and on its potential as a therapeutic target for type 2 diabetes mellitus.5,6 FoxA2 is considered a suppressor of epithelial–mesenchymal transition (EMT) in human lung cancers,7,8 and long-term CS exposure leads to downregulation of FoxA1 and FoxA2 concomitant with the occurrence of EMT in human bronchial epithelial cells.9 However, associations between MAPK signaling and the molecules regulating differentiation (eg, FoxA2, E-cadherin, CD44, and ZO1) are unclear. In the present study, with E-cadherin, CD44, and ZO1 as epithelial cell markers used in in vitro and in vivo models, we used CS extract (CSE) to stimulate human airway epithelial cells as an in vitro model to evaluate the function of the MAPK-signaling pathway and FoxA2 in bronchial epithelial cell differentiation. Furthermore, we used a rat smoking model to confirm the effects of the MAPK-signaling pathway (ERK1/2, JNK, and p38) and FoxA2 on bronchial epithelial cell differentiation.
Materials and methods
Materials
The bronchial epithelial cell line BEAS2B, an immortalized cell line transformed using an adenovirus 12–SV40 viral vector, was purchased from Bogoo Biotechnology (Shanghai, China) and cultured in Roswell Park Memorial Institute (RPMI) 1640 complete culture medium containing 10% fetal bovine serum (FBS). Healthy 4- to 6-week-old Sprague Dawley rats of specific pathogen-free (SPF) grade with body weights of 200±20 g were purchased from the Department of Laboratory Animal Science of Fudan University and housed in an SPF-grade experimental animal center at Fudan University. The experimental protocol was approved by the ethics committee of Fudan University and followed the Guide for the Care and Use of Laboratory Animals. UO126 (ERK inhibitor), SP600125 (JNK inhibitor), and SB203580 (p38 inhibitor) were purchased from Selleck (S1102, S1460, and S1076; Shanghai, China). The concentration used in cell experiments was 20 μM, in accordance with a previous report,10 and dosages used in animal experiments were 1 mg/kg, 1.5 mg/kg, and 1 mg/kg, respectively.
Preparation of CSE and cell intervention
CSE preparation was modified from Ballweg et al.11 The smoke was obtained by burning four cigarettes (Shanghai Double Happiness (Shanghai, China), tar content 8 mg/cigarette, nicotine in smoke 0.7 mg/cigarette, and carbon monoxide in smoke 10 mg/cigarette); smoke gas was dissolved in 40 mL serum-free culture medium using a negative-pressure pump. pH was adjusted to approximately 7.4 using NaOH. After passage over a 0.22 μm filter to remove bacteria, the solution was used as the 100% CSE solution. CSE was freshly prepared for each experiment, and the working concentration was 1%.
BEAS2B cells were maintained under routine culture conditions. In preparation for experiments, each well of three 24-well culture plates was inoculated with 105 cells from a culture in the logarithmic growth phase. The next day, cell density was approximately 60%. The cell-culture medium was replaced with medium containing 1% freshly prepared CSE with or without MAPK inhibitors. The concentration of all inhibitors was 20 μM. All inhibitors were dissolved in dimethyl sulfoxide (DMSO), so an equal volume of DMSO was added to the control wells. The culture medium was replaced every other day. The experimental design included five groups with four replicated wells in each group per plate. Experimental groups were a control group, CSE-treatment group, CSE + ERK-inhibitor group (CSE + UO126), CSE + JNK-inhibitor group (CSE + SP600125), and CSE + p38-inhibitor group (CSE + SB203580). Cells were cultured in serum-free medium and collected after 1 week of treatment for subsequent evaluation.
Development of the rat COPD model
CS-induced COPD in rats was established by passive inhalation. This model develops only minimal goblet-cell metaplasia in the smaller airways, although rats do demonstrate metaplasia in the larger airways.12 The rats in the smoking group were exposed to smoke in a homemade fumigation box (0.9×0.6×0.6 m). Each inhalation used 20 cigarettes (Double Happiness) and lasted 30 minutes. Inhalation was conducted twice per day for 3 months.
Forty rats were randomly divided into a blank control group, a CS group, a smoking + U0126 group, a smoking + SB203580 group, and a smoking + SP600125 group. Inhalation was used to establish the COPD animal model in each group, except the blank controls; drug intervention was performed at the same time. Intraperitoneal injection of the ERK1/2 inhibitor U0126 (1 mg/kg), the JNK-specific inhibitor SP600125 (1.5 mg/kg), and the p38-specific inhibitor SB203580 (1 mg/kg) was performed 30 minutes prior to each fumigation. Each dose of inhibitor was dissolved in 0.5 mL normal saline. Drugs were administered once daily for 90 consecutive days. After the model had been successfully established, the rats were killed and lung tissue from one lung of each animal divided into several parts. Two of these were rapidly frozen and stored at −80°C for polymerase chain reaction (PCR) and Western blot, and one was fixed in 4% formalin for subsequent pathological assessment by H&E staining.
Measurement of phosphorylated protein levels of ERK1/2, JNK, and p38 with ELISA
Following CSE treatment of cells in culture, the culture medium was removed and cells washed with PBS. Freshly diluted lysis buffer (200 μL) was then added to each well of the 24-well plate, and the cells were lysed at room temperature for 10 minutes. The lysis buffer was collected, and protein levels of phosphorylated ERK1/2, p38 and JNK were measured using an enzyme-linked immunosorbent assay (ELISA) reagent kit (ab119674; Abcam, Cambridge, UK) as described in the manufacturer’s instructions. In the animal experiments, extraction of lung-tissue protein was performed using a protein-extraction reagent kit for animal tissue (BSP006; Sangon Biotech, Shanghai, China). Total active proteins extracted from tissues were stored at −80°C until future use. For measurement, 50 μL cell-lysis buffer or total protein extract was added, 50 μL corresponding antibody solution then added, and the sample incubated at room temperature for 1 hour. Each well was then washed three times with 200 μL washing buffer. To each well, 100 μL substrate was added, and the sample was incubated at room temperature for 10 minutes. Stop solution was added, and fluorescence detection performed.
Real-time fluorescence quantitative PCR
Tissue blocks were ground into powder in liquid nitrogen, and RNA was extracted from 50 mg of tissue using the Trizol method. The total RNA (4 μL) from each sample was used for reverse transcription using a reverse-transcription reagent kit (PCR005; BioServe, Beltsville, MD, USA). Equal amounts of standard and reverse-transcribed samples (2 μL each) were amplified using a BioServe quantitative detection amplification reagent kit (PCR002; BioServe). mRNA-expression levels of FoxA2, E-cadherin, CD44, and ZO1 were determined using fluorescence quantitative PCR (Hoffman-La Roche Ltd., Basel, Switzerland). Sequences of primers used in the study are listed in Table 1.
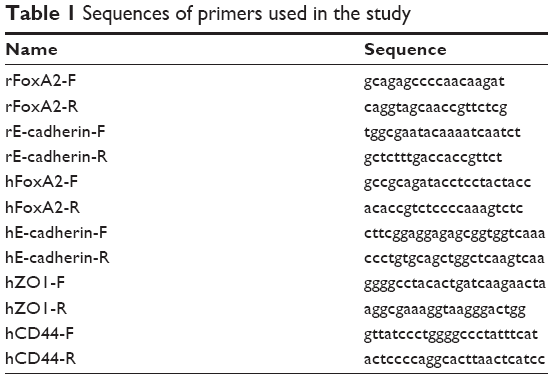 | Table 1 Sequences of primers used in the study |
Measurement of protein levels of FoxA2 and E-cadherin by Western blotting
Protein extracts from cells and tissue were mixed with 5× sample loading buffer, boiled at 100°C for 10 minutes, centrifuged, and cooled to room temperature. An 8% separating gel and 3.9% stacking gel were prepared. Each well was loaded with 20 μL sample. Electrophoresis was performed for 3–5 hours. Proteins were transferred to a nitrocellulose membrane at 300 mA constant current for 1 hour (90 minutes for ZO1). After successful membrane transfer had been confirmed using Ponceau S stain, immunostaining was performed. Primary antibodies used in this study included antiactin mouse monoclonal antibody (sc-47778; Santa Cruz Biotechnology, Dallas, TX, USA), anti-E-cadherin mouse monoclonal antibody (ab1416; Abcam), anti-FoxA2 rabbit polyclonal antibody(ab23630; Abcam), anti-CD44 rabbit polyclonal antibody (ab157107; Abcam), and anti-ZO1 goat polyclonal antibody (ab190085; Abcam). All dilutions were 1:1,000. A horseradish peroxidase-labeled secondary antibody (1:500) was used. Results were determined using enhanced chemiluminescence in a dark room.
Statistical analysis
All data were statistically analyzed using SPSS 17.0 software. Mean values and SD were calculated. Mean values were compared among groups using the F-test. Pairwise comparison was performed using the Q-test. Differences with P<0.05 indicated statistical significance.
Results
Exposure of bronchial epithelial cells to CSE increased phosphorylation levels of ERK1/2, JNK, and p38
To investigate the role of the MAPK pathway in bronchial epithelial cells exposed to CSE, levels of phosphorylated ERK1/2, JNK, and p38 proteins in BEAS2B cells that had been treated with CSE for 48 hours were measured using ELISA. BEAS2B cells were originally established from healthy human bronchial epithelium, and were transformed by the adenovirus 12–SV40 hybrid virus; they are considered airway epithelial cells.13 BEAS2B cells have been shown to maintain typical epithelial morphology and many of the functional characteristics of normal human bronchial epithelial cells.14,15 The results showed that levels of phosphorylated ERK1/2, JNK, and p38 in cells were significantly higher in the CSE group than in the control group (P<0.05), suggesting that activation of the MAPK pathway may be an important mechanism underlying the influence of CSE on bronchial epithelial cell growth (Figure 1).
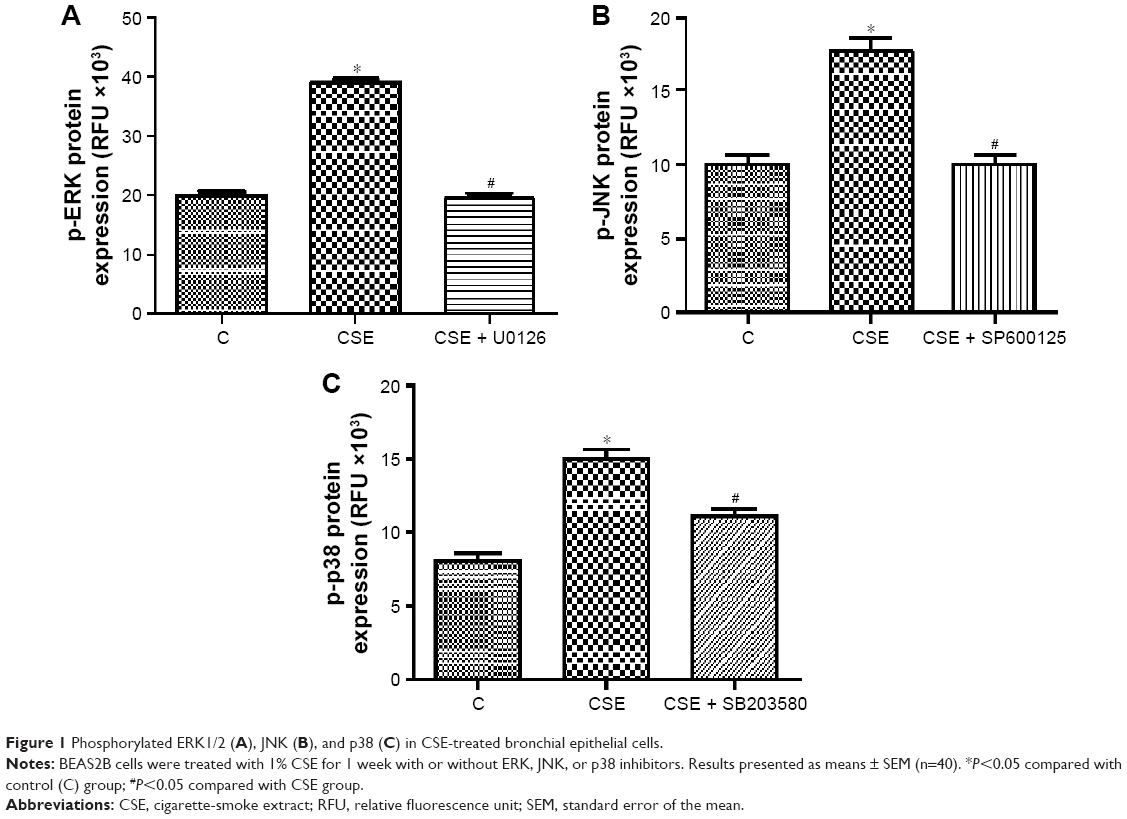 | Figure 1 Phosphorylated ERK1/2 (A), JNK (B), and p38 (C) in CSE-treated bronchial epithelial cells. |
CSE promoted epithelial cell differentiation through MAPK
To investigate the effect of the MAPK-signaling pathway on bronchial epithelial cell differentiation induced by CSE, cells were treated with CSE in the presence or absence of an ERK inhibitor, a JNK inhibitor, or a p38 inhibitor. Cellular RNA was extracted, and mRNA levels of the epithelial cell-differentiation markers E-cadherin, CD44, and ZO1 were determined. mRNA levels of E-cadherin, CD44, and ZO1 were all significantly higher in the CSE-treatment group than in the control group (P<0.05); however, CSE treatment in the presence of ERK, JNK, or p38 inhibitors significantly ameliorated the CSE-induced reduction in the mRNA levels of the three markers (P<0.05) (Figure 2).
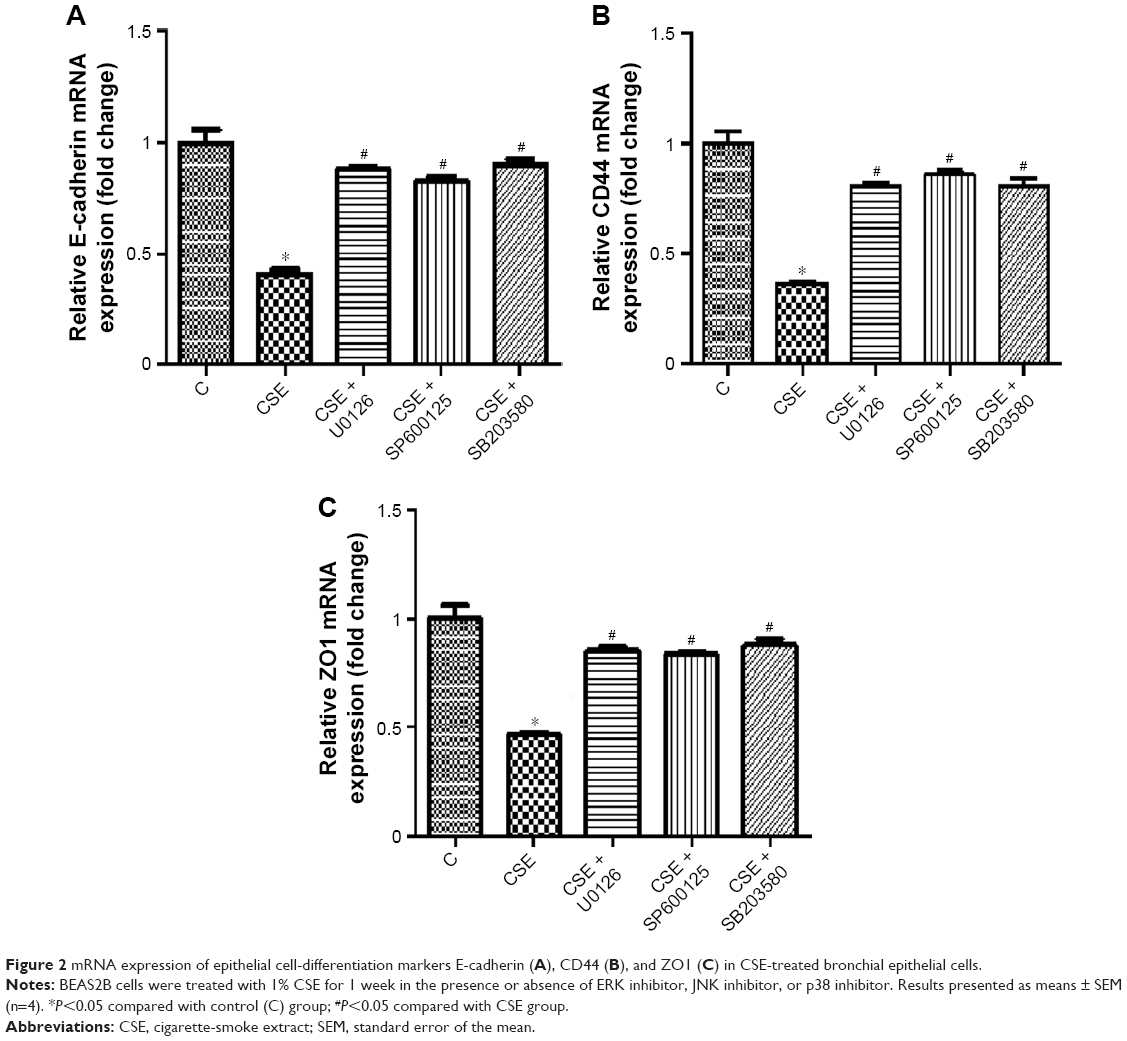 | Figure 2 mRNA expression of epithelial cell-differentiation markers E-cadherin (A), CD44 (B), and ZO1 (C) in CSE-treated bronchial epithelial cells. |
Total protein was extracted from the cells for Western blotting analysis. The results were consistent with the changes observed in mRNA levels. Compared to levels in the control group, protein levels of E-cadherin, CD44, and ZO1 all significantly decreased in the CSE-treatment group, and CSE treatment combined with treatment with ERK, JNK, or p38 inhibitor significantly ameliorated CSE-induced reduction in the protein levels of these three markers (Figure 3). Interestingly, the JNK inhibitor SP600125 significantly increased ZO1 protein, and the effect was stronger than the ERK inhibitor U0126 and the p38 inhibitor SB203580. However, for mRNA expression, the three inhibitors had similar effect on ZO1 expression. As such, these data suggested that MAPK involved in epithelial cell differentiation and the JNK inhibitor perhaps upregulate ZO1 by posttranscription.
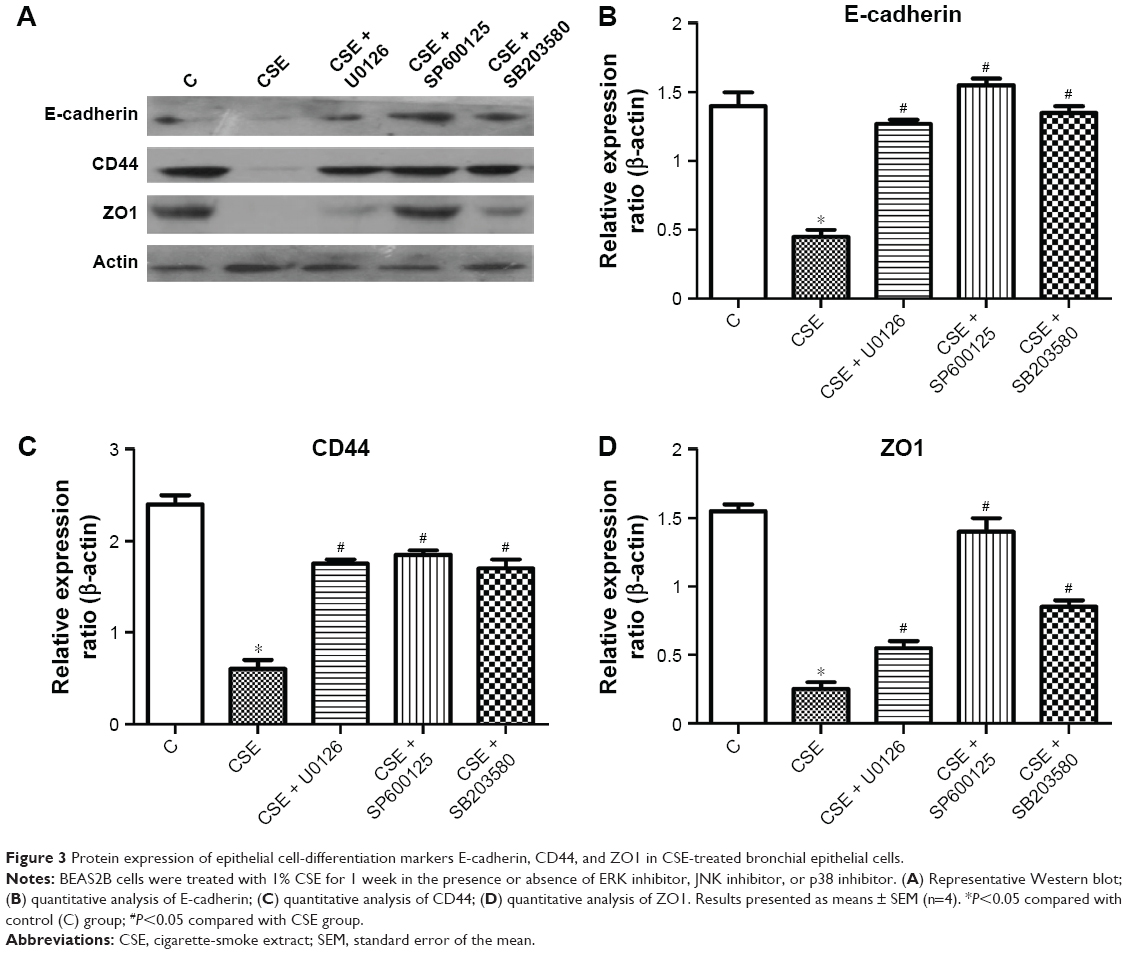 | Figure 3 Protein expression of epithelial cell-differentiation markers E-cadherin, CD44, and ZO1 in CSE-treated bronchial epithelial cells. |
MAPK inhibitors reversed pathological changes of squamous metaplasia
To evaluate the effect of the MAPK pathway further, we established a rat smoking model and determined the action of MAPK inhibitors in this model. Pathological assessment of the experimental animals showed that the tracheae, bronchi, and alveolar structures of rats in the control group were intact; the bronchiolar epithelial cells of these animals did not show degeneration, and no exudate was found in the alveolar cavity (see Figure 4). The bronchial epithelia of the animals in the established smoking-model group displayed mild hyperplasia, the number of lymphoid follicles in the bronchial epithelial layer increased, local squamous metaplasia occurred, and considerable lymphocyte infiltration of the bronchial wall was observed. These results suggested that the smoking model had been successfully established. Treatment of the animals with an ERK1/2-, JNK-, or p38-specific inhibitor during exposure to CS prevented the development of airway inflammation and squamous metaplasia.
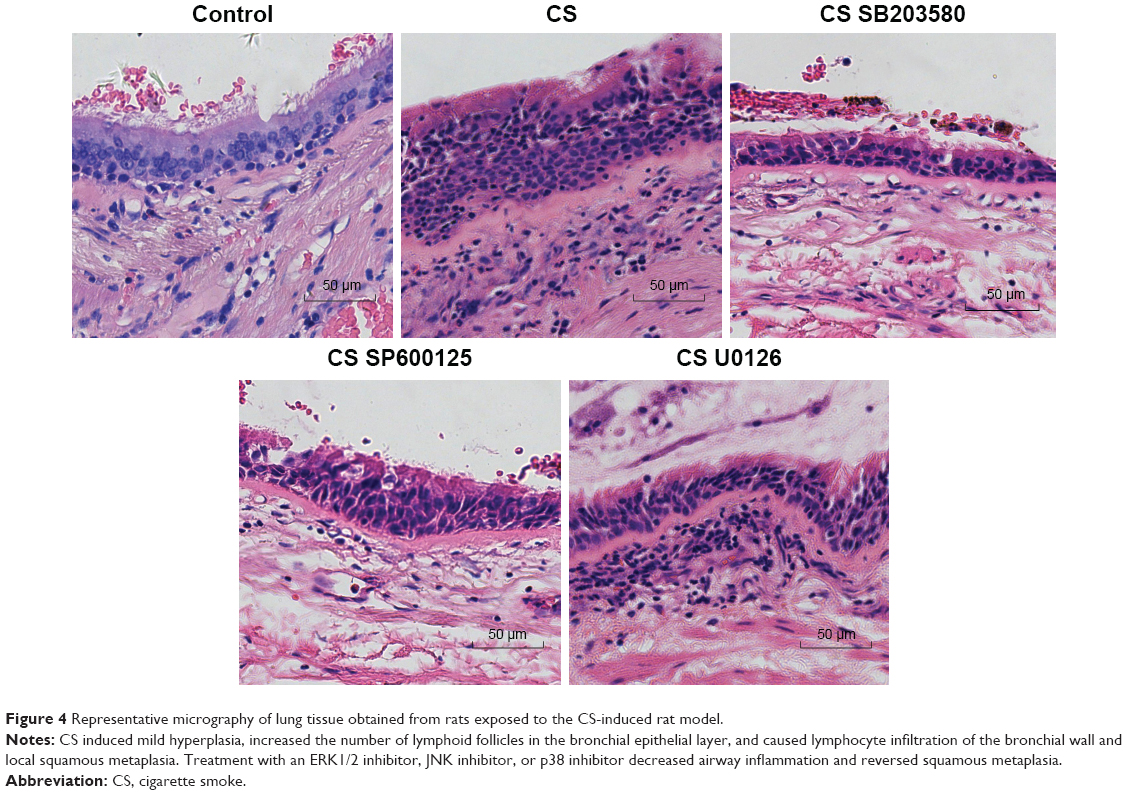 | Figure 4 Representative micrography of lung tissue obtained from rats exposed to the CS-induced rat model. |
To confirm that changes occurred in the MAPK pathway in the rat smoking model, we measured phosphorylation levels of ERK1/2, JNK, and p38 (Figure 5). Phosphorylation levels of all three proteins increased significantly in the lung tissue of rats exposed to CS in the smoking model (P<0.05 vs control). However, relative phosphorylation levels of ERK1/2, JNK, and p38 in animals in the established smoking-model group did not differ significantly from one another.
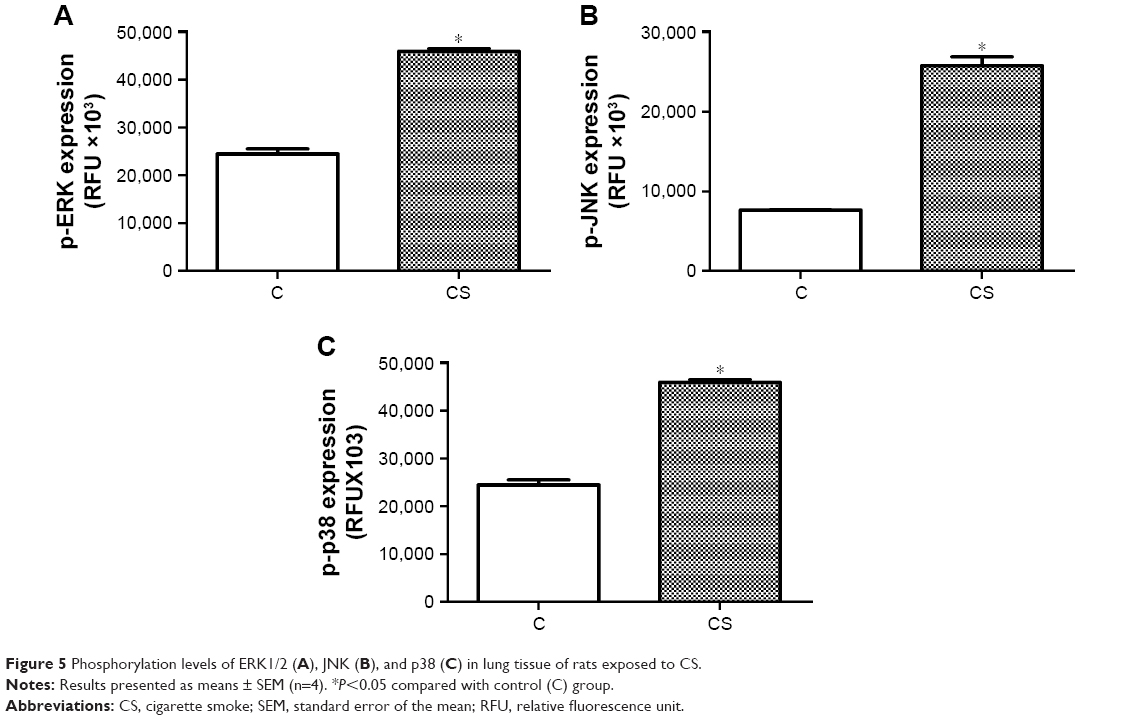 | Figure 5 Phosphorylation levels of ERK1/2 (A), JNK (B), and p38 (C) in lung tissue of rats exposed to CS. |
To confirm further that treatment with MAPK inhibitors reduces squamous epithelial metaplasia, we measured E-cadherin expression at the mRNA and protein levels (Figure 6). ERK1/2, JNK, and p38 inhibitors reversed CS-induced downregulation of E-cadherin expression in rat lung tissue at both protein and mRNA levels; there were no significant differences in the effectiveness of the ERK1/2, JNK, and p38 inhibitors.
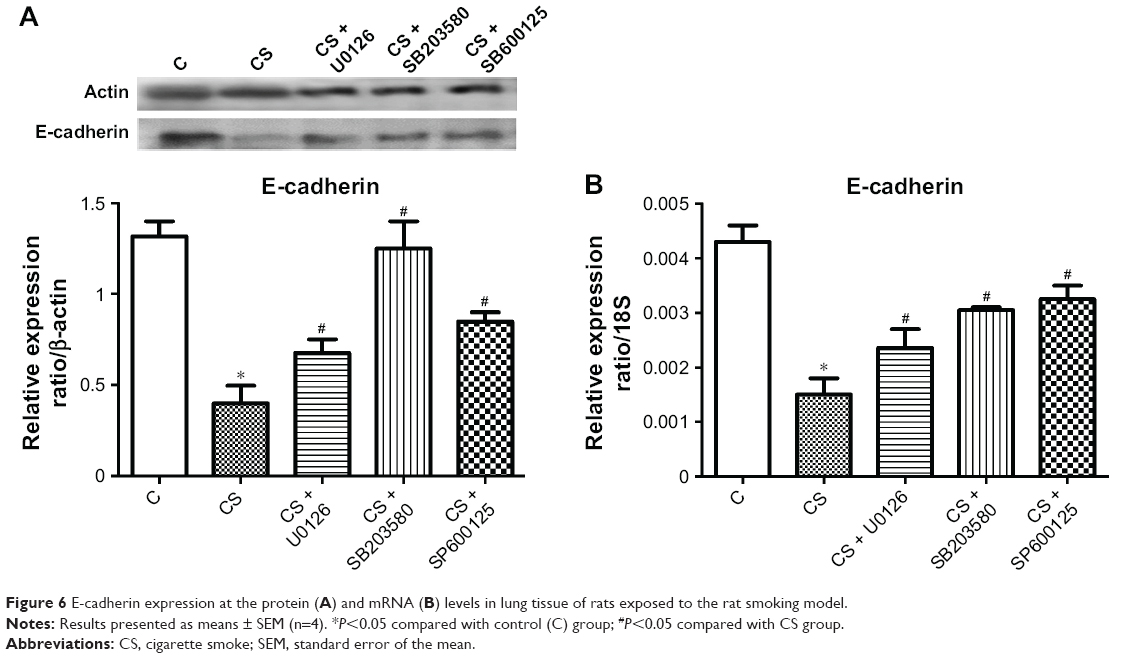 | Figure 6 E-cadherin expression at the protein (A) and mRNA (B) levels in lung tissue of rats exposed to the rat smoking model. |
MAPK inhibitors reversed squamous epithelial metaplasia by increasing FoxA2 expression
To investigate the effects of MAPK-pathway activation on specific downstream transcription factors, we measured FoxA2 levels in the BEAS2B cell line and in the rat model. After CSE treatment, FoxA2 mRNA levels in BEAS2B cells were significantly decreased compared with the blank control (P<0.05) (Figure 7). After treatment of cells with the ERK1/2 inhibitor UO126, the JNK inhibitor SP600125, or the p38 inhibitor SB203580, FoxA2 mRNA levels increased. Protein-expression levels also increased significantly compared with cells treated with CSE alone (P<0.05).
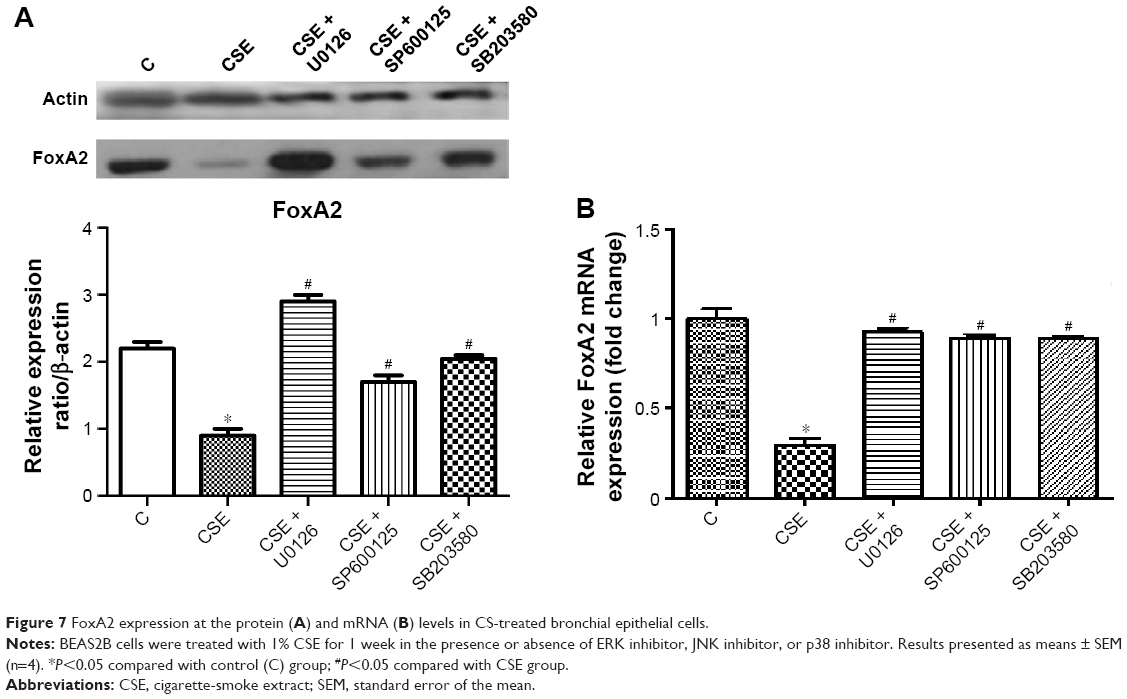 | Figure 7 FoxA2 expression at the protein (A) and mRNA (B) levels in CS-treated bronchial epithelial cells. |
Similar results were obtained in the rat smoking model (Figure 8). The relative level of FoxA2 protein expression in the lung tissue of the rats in the blank control group was 1.405, whereas that of the rats in the smoking group was 0.234. Compared to the blank control group, the level significantly decreased (P<0.05). FoxA2 protein levels in the smoking + U0126 group, the smoking + SB203580 group, and the smoking + SP600125 group were 0.656, 1.135, and 0.865, respectively. Compared to those in the smoking group, the levels all significantly improved (P<0.05). These results suggest that the MAPK pathway may regulate squamous metaplasia through FoxA2 (Figure 9) and that agents that affect the FoxA2 pathway might reverse squamous metaplasia and improve therapy for COPD.
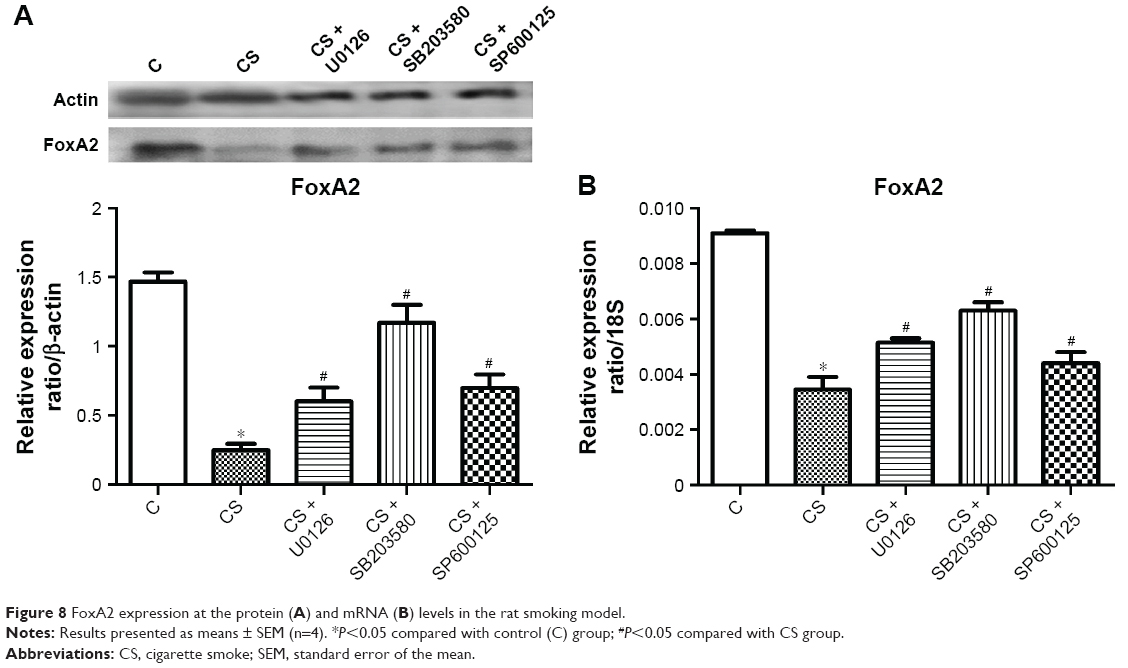 | Figure 8 FoxA2 expression at the protein (A) and mRNA (B) levels in the rat smoking model. |
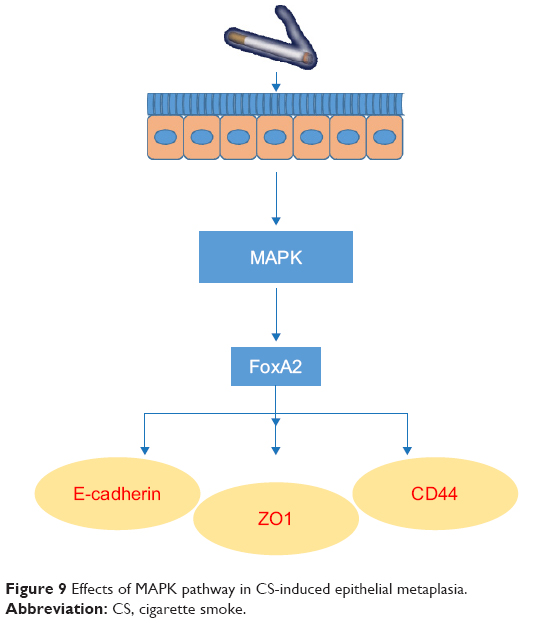 | Figure 9 Effects of MAPK pathway in CS-induced epithelial metaplasia. |
Discussion
Using normal human bronchial epithelial cells as an in vitro model, the present study showed that mRNA and protein levels of FoxA2, E-cadherin, CD44, and ZO1 in bronchial epithelial cells all decreased significantly after CSE treatment, indicating abnormal differentiation of bronchial epithelial cells. The results obtained in this study using an animal model of smoking showed that exposure to CS induced mild hyperplasia of the bronchial epithelia, an increase in the number of lymphoid follicles in the bronchial epithelial layer, and focal alveolar inflammatory changes, as well as lymphocyte infiltration of the bronchial wall and local squamous metaplasia. Levels of p-ERK, p-p38 and p-JNK proteins in bronchial cells and lung tissue increased significantly in the smoking group, indicating that the MAPK-signaling pathway is involved in smoking-induced abnormal epithelial cell differentiation. After treatment with MAPK inhibitors, inflammation and squamous metaplasia were significantly ameliorated and changes in FoxA2 and E-cadherin reversed. Overall, the results of the present study show that ERK, p38, and JNK participate in the induction of airway inflammation and abnormal airway epithelial cell metaplasia by smoking and that these effects are negatively regulated by FoxA2.
As an important signaling system that mediates cell responses, the MAPK pathway is present in a variety of cells. This pathway is involved in numerous physiological processes, including cell growth, development, differentiation, apoptosis, and survival.16 The abnormal activation of many kinases in the MAPK pathway is closely associated with many diseases. Smoking is one of the most important risk factors for COPD, and squamous metaplasia is increased in the bronchial epithelium of smokers with COPD.17 Studies have shown that smoking activates ERK1/2, JNK, and p38 in lung tissue, induces significant squamous metaplasia and hyperplasia of bronchial epithelial cells,2 and causes airway inflammation and high levels of mucus secretion in rats,3 but few studies have established the protective effects of MAPK inhibitors, and there is little evidence on the interaction between FoxA2 and MAPK. Our results showed that p-ERK, p-p38, and p-JNK protein levels in the bronchial and lung tissue of rats in the smoking model group increased significantly. Furthermore, the bronchial epithelia of these animals showed mild hyperplasia, lymphoid follicles in the bronchial epithelial layer increased, focal alveolar inflammatory changes occurred, a large amount of lymphocyte infiltration occurred in the bronchial wall, and local squamous metaplasia occurred. Administration of ERK-, p38-, and JNK-specific inhibitors reduced squamous metaplasia, indicating that ERK, p38, and JNK all participate in the development of squamous metaplasia caused by smoking. These results are consistent with previous reports on ERK, p38, and JNK.2,3 Indeed, specific p38 inhibitors have been tested in clinical trials as treatment for COPD, but these trials were discontinued, due to their side effects and ineffective action. To date, no clinical trials of ERK or JNK inhibitors as treatment for COPD have been undertaken, presumably because of toxicity issues.18
In our study, we used the three epithelial cell markers ZO1, E-cadherin, and CD44 to identify squamous metaplasia. These three markers are also considered markers of EMT. A previous publication suggested that ZO1 showed downregulation and E-cadherin exhibited upregulation in nasal squamous metaplasia.19 Our results revealed downregulation in both ZO1 and E-cadherin expression, perhaps due to the different situation between nasal and airway epithelia. CD44 is considered a novel biomarker of non-small-cell lung tumors, squamous metaplasia of the lung, and activated type II pneumocytes.20 Therefore, these three markers can be used to indicate the process of squamous metaplasia. Of course, other epithelial cell markers, such as cytokeratins and involucrin might also be considered. Cytokeratin expression may include CK5/6, CK13, CK7, CK8, and CK18, and varies with epithelial cell type and stage of differentiation, whereas involucrin is a marker of terminal squamous differentiation.21,22 The presence of these markers can distinguish squamous metaplasia from other types of epithelial metaplasia.
The Fox protein family consists of transcription factors that display a winged-helix structure in the DNA-binding region. Seventeen subfamilies of Fox proteins have been identified. Fox proteins are involved in many biological processes, including embryonic development, cell-cycle regulation, sugar and lipid metabolism, biological aging, and immunoregulation. FoxA proteins are mainly expressed in endodermal tissue, and they influence embryonic development, cell differentiation, and cell functions.23 The FoxA family includes FoxA1, FoxA2, and FoxA3. Only FoxA1 and FoxA2 are expressed in the lung.24 FoxA2 is a transcription factor that plays critical roles in pulmonary morphogenesis and gene expression;25,26 it is also necessary for airway epithelial cell differentiation, and plays important roles in cell division and the cell cycle. Nicotine can induce the differentiation of normal human bronchial epithelial cells into neuroendocrine-like cells,27 and FoxA2 is expressed in all types of neuroendocrine lung tumors.28 For these reasons, FoxA2 is considered a very strong tumor suppressor of lung cancer.29 Our study first suggested that CSE induced a reduction in FoxA2 levels, indicating that FoxA2 has an important regulatory function in the abnormal bronchial differentiation induced by smoking. CSE + MAPK inhibitors rescued downregulated FoxA2 protein and mRNA levels, but these were still significantly lower than control group. These results paralleled pathological changes in hyperplasia and metaplasia. Meanwhile, we showed that expression levels of E-cadherin, CD44, and ZO1 correlated positively with FoxA2 expression. However, whether the expression of these markers is regulated by FoxA2 is still unknown. A recent study suggested that CS exposure leads to downregulation of FoxA1 and FoxA2 concomitant with the occurrence of EMT and in vitro transformation of human bronchial epithelial cells.9 E-cadherin localizes to the lateral surfaces of epithelial cells, and plays an important role in the maintenance of airway-epithelia integrity. These reports are consistent with our findings in studies of squamous metaplasia showing that exposure to CS downregulates FoxA2 expression and that this downregulation is positively correlated with E-cadherin expression. In addition to indicating that E-cadherin expression may be regulated by FoxA2, our results also suggest that CD44 and ZO1 may be regulated by FoxA2.
In the present study, downregulation of FoxA2 expression was accompanied by downregulation of E-cadherin expression in airway epithelial cells and in lung tissue of animals exposed to CS, further indicating that smoking results in abnormal differentiation of airway epithelial cells. ERK-, p38-, and JNK-specific inhibitors were all shown to upregulate the expression of FoxA2 significantly, as well as that of E-cadherin, providing further evidence that the ERK-, p38-, and JNK-signaling pathways negatively regulate FoxA2 expression. FoxA2, like the downstream molecules of the MAPK pathway that mediate the effects of MAPK inhibitors, may thus be an effective diagnostic marker for COPD. Due to its broad inhibition of kinases, FoxA2 may be a good alternative target for potential agents that might inhibit the development of COPD.
In summary, smoking was shown to cause squamous metaplasia of epithelial cells. Along with those changes, ERK, p38, and JNK expression was upregulated and FoxA2 expression decreased. Interventions using ERK, p38, and JNK inhibitors decreased squamous metaplasia after exposure of cells or animals to CS, and FoxA2 expression increased. The results thus showed that MAPK and FoxA2 mediated the development of squamous metaplasia in smoking rats. However, the mechanism by which MAPK-signaling pathways directly or indirectly regulate FoxA2, E-cadherin, CD44, and ZO1 and whether there are intermediary regulatory pathways linking FoxA2 and E-cadherin, CD44, and ZO1 are still unclear. Therefore, further studies are required.
Acknowledgment
This research was supported by the Shanghai Municipal Commission of Health and Family Planning (20124277).
Disclosure
The authors report no conflicts of interest in this work.
References
Di Stefano A, Caramori G, Ricciardolo FL, Capelli A, Adcock IM, Donner CF. Cellular and molecular mechanisms in chronic obstructive pulmonary disease: an overview. Clin Exp Allergy. 2004;34(8): 1156–1167. | ||
Zhong CY, Zhou YM, Douglas GC, Witschi H, Pinkerton KE. MAPK/AP-1 signal pathway in tobacco smoke-induced cell proliferation and squamous metaplasia in the lungs of rats. Carcinogenesis. 2005;26(12):2187–2195. | ||
Xiao J, Wang K, Feng YL, Chen XR, Xu D, Zhang MK. Role of extracellular signal-regulated kinase 1/2 in cigarette smoke-induced mucus hypersecretion in a rat model. Chin Med J (Engl). 2011;124(20): 3327–3333. | ||
Mercer BA, D’Armiento JM. Emerging role of MAP kinase pathways as therapeutic targets in COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(2):137–150. | ||
Kaestner KH. The hepatocyte nuclear factor 3 (HNF3 or FOXA) family in metabolism. Trends Endocrinol Metab. 2000;11(7):281–285. | ||
Kaestner KH. The FoxA factors in organogenesis and differentiation. Curr Opin Genet Dev. 2010;20(5):527–532. | ||
Tang Y, Shu G, Yuan X, Jing N, Song J. FOXA2 functions as a suppressor of tumor metastasis by inhibition of epithelial-to-mesenchymal transition in human lung cancers. Cell Res. 2011;21(2):316–326. | ||
Zhang Z, Yang XF, Huang KQ, et al. The clinicopathological significances and biological functions of parafibromin expression in head and neck squamous cell carcinomas. Tumour Biol. 2015;36(12): 9487–9497. | ||
Bersaas A, Arnoldussen YJ, Sjøberg M, Haugen A, Mollerup S. Epithelial-mesenchymal transition and FOXA genes during tobacco smoke carcinogen induced transformation of human bronchial epithelial cells. Toxicol In Vitro. 2016;35:55–65. | ||
Shen N, Gong T, Wang JD, et al. Cigarette smoke-induced pulmonary inflammatory responses are mediated by EGR-1/GGPPS/MAPK signaling. Am J Pathol. 2011;178(1):110–118. | ||
Ballweg K, Mutze K, Königshoff M, Eickelberg O, Meiners S. Cigarette smoke extract affects mitochondrial function in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2014;307(11):L895–L907. | ||
Wright JL, Cosio M, Churg A. Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2008;295(1): L1–L15. | ||
Ke Y, Reddel RR, Gerwin BI, et al. Human bronchial epithelial cells with integrated SV40 virus T antigen genes retain the ability to undergo squamous differentiation. Differentiation. 1988;38(1):60–66. | ||
Albright CD, Jones RT, Hudson EA, Fontana JA, Trump BF, Resau JH. Transformed human bronchial epithelial cells (BEAS-2B) alter the growth and morphology of normal human bronchial epithelial cells in vitro. Cell Biol Toxicol. 1990;6(4):379–398. | ||
Atsuta J, Sterbinsky SA, Plitt J, Schwiebert LM, Bochner BS, Schleimer RP. Phenotyping and cytokine regulation of the BEAS-2B human bronchial epithelial cell: demonstration of inducible expression of the adhesion molecules VCAM-1 and ICAM-1. Am J Respir Cell Mol Biol. 1997;17(5):571–582. | ||
Yang SH, Sharrocks AD, Whitmarsh AJ. MAP kinase signalling cascades and transcriptional regulation. Gene. 2013;513(1):1–13. | ||
Rigden HM, Alias A, Havelock T, et al. Squamous metaplasia is increased in the bronchial epithelium of smokers with chronic obstructive pulmonary disease. PLoS One. 2016;11(5):e0156009. | ||
Barnes PJ. Kinases as novel therapeutic targets in asthma and chronic obstructive pulmonary disease. Pharmacol Rev. 2016;68(3):788–815. | ||
Jang YJ, Kim HG, Koo TW, Chung PS. Localization of ZO-1 and E-cadherin in the nasal polyp epithelium. Eur Arch Otorhinolaryngol. 2002;259(9):465–469. | ||
Penno MB, August JT, Baylin SB, et al. Expression of CD44 in human lung tumors. Cancer Res. 1994;54(5):1381–1387. | ||
Gray AC, McLeod JD, Clothier RH. A review of in vitro modelling approaches to the identification and modulation of squamous metaplasia in the human tracheobronchial epithelium. Altern Lab Anim. 2007;35(5):493–504. | ||
Merrifield J, O’Donnell R, Davies DE, Djukanovic R, Wilson SJ. A panel of antibodies for identifying squamous metaplasia in endobronchial biopsies from smokers. Biotech Histochem. 2011;86(5):340–344. | ||
Hromas R, Costa R. The hepatocyte nuclear factor-3/forkhead transcription regulatory family in development, inflammation, and neoplasia. Crit Rev Oncol Hematol. 1995;20(1–2):129–140. | ||
Monaghan AP, Kaestner KH, Grau E, Schutz G. Postimplantation expression patterns indicate a role for the mouse forkhead/HNF-3 α, β and γ genes in determination of the definitive endoderm, chordamesoderm and neuroectoderm. Development. 1993;119(3):567–578. | ||
Wan H, Dingle S, Xu Y, et al. Compensatory roles of Foxa1 and FoxA2 during lung morphogenesis. J Biolo Chem. 2005;280(14):13809–13816. | ||
Wan H, Kaestner KH, Ang SL, et al. FoxA2 regulates alveolarization and goblet cell hyperplasia. Development. 2004;131(4):953–964. | ||
Martinez-Garcia E, Irigoyen M, Anso E, Martinez-Irujo JJ, Rouzaut A. Recurrent exposure to nicotine differentiates human bronchial epithelial cells via epidermal growth factor receptor activation. Toxicol Appl Pharmacol. 2008;228(3):334–342. | ||
Khoor A, Stahlman MT, Johnson JM, Olson SJ, Whitsett JA. Forkhead box A2 transcription factor is expressed in all types of neuroendocrine lung tumors. Hum Pathol. 2004;35(5):560–564. | ||
Halmos B, Basseres DS, Monti S, et al. A transcriptional profiling study of CCAAT/enhancer binding protein targets identifies hepatocyte nuclear factor 3β as a novel tumor suppressor in lung cancer. Cancer Res. 2004;64(12):4137–4147. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.