Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 13
Managing Severe Hemophilia A in Children: Pharmacotherapeutic Options
Authors Regling K ![]() , Callaghan MU, Sidonio R Jr
, Callaghan MU, Sidonio R Jr ![]()
Received 15 October 2021
Accepted for publication 27 December 2021
Published 15 February 2022 Volume 2022:13 Pages 27—35
DOI https://doi.org/10.2147/PHMT.S293246
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Roosy Aulakh
Katherine Regling,1,2 Michael U Callaghan,1– 3 Robert Sidonio Jr4
1Pediatric Hematology Oncology, Children’s Hospital of Michigan, Detroit, MI, USA; 2Central Michigan University School of Medicine, Mount Pleasant, MI, USA; 3Agios Pharmaceuticals, Cambridge, MA, USA; 4Emory University and Aflac Cancer and Blood Disorders, Atlanta, GA, USA
Correspondence: Katherine Regling Pediatric Hematology Oncology, Children’s Hospital of Michigan, 3901 Beaubien Blvd, Detroit, MI, 48201, USA Tel +1 313 745 5515 Fax +1 313 745 5237 Email [email protected]
Abstract: Hemophilia A is the most common severe inherited bleeding disorder in males. Initial treatment strategies focused on the use of factor concentrates to prevent joint bleeding and the development of long-term crippling arthropathy. The current standard of care has evolved from regular replacement of factor VIII concentrates which has significantly improved the quality of life for those with severe disease to include and consider novel therapies that augment or bypass the hemostatic pathway (ie, emicizumab, Mim8). Other pipeline therapies that suppress specific natural anticoagulant pathways (ie, antithrombin, TFPI) to reestablish hemostatic balance are under Phase 3 trial investigation. These novel therapeutics have allowed providers more variety in dosing regimens and ease of administration while also maintaining effective bleeding prevention. The possibility of “curative” gene therapy is under exploration, with ongoing clinical trials in adult males.
Keywords: factor VIII, therapeutic management, coagulation, pediatrics
Introduction
Hemophilia A (HA) is an X-linked recessive disorder that results in defective and/or deficient clotting factor (F) VIII and is classified based on percentage of circulating plasma normal FVIII activity.1 Individuals with severe HA (<1% FVIII activity) often display bleeding symptoms early during their first year of life, typically joint and soft tissue bleeding becoming more evident as the child's activity increases. Prior to implementation of factor prophylaxis, the frequent episodes of muscle and joint bleeding lead to debilitating arthropathy and a reduced lifespan. Thus, the primary goal for management of HA has been the prevention of hemarthroses and other life-threatening bleeding episodes.
Prophylaxis with FVIII concentrates was, until recently, the gold standard of treatment for severe HA and has significantly improved the overall bone and joint health in these patients.2,3 In addition, the use of prophylaxis also leads to a marked reduction of intracranial hemorrhage, hematomas, and hospitalizations/emergency room visits, while improving the medical independence of the patient.4
Primary prophylaxis refers to routine, continuous treatment with FVIII concentrates prior to three years of age and any major joint bleed, whereas secondary prophylaxis is the initiation of continuous treatment after three years of age or following more than two joint bleeding episodes.1 Tertiary prophylaxis refers to continuous replacement therapy started after the onset of joint disease.1 The Joint Outcomes Study and the ESPRIT study are two pivotal randomized controlled trials that assessed the value of prophylaxis in comparison to on-demand factor treatment and showed that prophylaxis was more effective when initiated early; resulting in a decreased incidence of joint bleeds and in radiographic evidence of joint damage.2,3 More recently, the Joint Outcome Continuation Study showed that 35% of patients on early prophylaxis (prior to 2.5 years of age) had osteochondral damage on magnetic resonance imaging compared to 77% of those on delayed prophylaxis (after six years of age). Annualized bleeding rates (ABR) were also significantly increased when delaying prophylaxis compared to the early prophylaxis group.5
Today, the most important treatment-related complication is the development of an inhibitor. Approximately, 30% of patients with hemophilia A develop neutralizing antibodies to FVIII, rendering decreased response to FVIII concentrate products and breakthrough bleeding episodes.6 The risk of inhibitor development is highest in those with severe HA and has been associated with certain genetic variants, African American or Latino race and positive family history.7 Inhibitor development occurs within the first 20 exposure days in 79% of those with HA and most, within the first 75 exposure days.8 Conversely, regular prophylaxis has been reported to decrease inhibitor risk by 60% when compared to on-demand treatment.9 Although outside the scope of this review, it is widely recommended that all patients with HA that develop an inhibitor undergo a trial of immune tolerance induction (ITI) to eradicate the inhibitor either in conjunction with or prior to use of bypassing agents (BPAs) or nonfactor replacement therapies.
It is for these reasons, it is recommended that early in life all persons with severe HA should implement prophylaxis with either clotting factor concentrates or more recently, nonfactor replacement strategies. This review discusses current and upcoming prophylactic treatment options for those with severe HA with and without inhibitors including FVIII concentrates and nonfactor therapies.
Factor VIII Replacement Products for Hemophilia A
FVIII concentrates were, until the approval of emicizumab, the treatment of choice as they are safe and effective in the prevention of bleeding events. Prophylactic regimens should be individualized based on patient preference and response to therapy with the use of pharmacokinetic (PK) evaluation. A target trough level of 1–2% FVIII activity is sufficient to prevent most breakthrough bleeding, including hemarthroses. However, the Joint Outcome Continuation Study reported that despite full, early prophylaxis only a small proportion of participants had no joint damage through adolescence.5 This suggests that targeting a higher trough level may lead to improved quality of life and overall outcomes.10
Standard half-life (SHL) products have been in use for decades with both plasma-derived and recombinant products available. The human immunodeficiency virus (HIV) and hepatitis pandemics of the 1980s brought concern about the viral safety of plasma-derived FVIII.11 This led to strict development of viral screening protocols for donor selection and viral inactivation methods in product preparation for plasma derived products. Since the adoption of these practices, the safety of plasma-derived FVIII is well established and there has been no transmission of HIV or hepatitis B or C in the US FDA-approved products since the late 1980s.12 The efficacy of plasma-derived FVIII and recombinant FVIII are similar, with an FVIII half-life of approximately 12 hours in adults.13,14 However, there is significant interpatient variability in the dose of factor and terminal half-life. Recent studies have revealed that endogenous von Willebrand factor (VWF) levels, ABO blood group and age play a significant role in the variation of FVIII half-life.15,16 Generally, for each unit of FVIII infused the FVIII activity rises by approximately 2%; thus, most FVIII IV infusion prophylactic regimens start between 25 and 40 units/kg/dose.17,18 With a goal FVIII trough of >1%, the use of SHL products necessitates infusions three to four times per week.
The development of extended half-life (EHL) products has positively influenced the quality of life in those with severe HA with decreased burden of care and the ability to maintain significantly higher trough FVIII levels with routine prophylaxis. These products have improved the half-life of FVIII in circulation to 1.4 to 1.6-fold that of SHL products, by either crystallizable fragment (Fc) fusion or PEGylation technologies.19 The maximum half-life achievable is about 19 hours, because the majority of FVIII (95–97%) is bound to VWF in circulation.20 Unbound FVIII has a six-fold decrease in half-life, whereas unbound VWF appears to have a comparable half-life to FVIII-bound VWF. The clearance of FVIII-VWF complex creates a half-life ceiling and breakthrough may require modifications beyond FVIII.16,19,20 Frequency of infusions may be decreased to two to three times per week, however there is significant variability between individuals due to age, body mass, blood group, von Willebrand factor level, physical activity level and other variables.21,22
As previously mentioned, VWF stabilizes and protects FVIII from degradation and clearance in circulation causing a FVIII half-life ceiling of about 14–19 hours. Ultimately, to extend FVIII half-life further it is dependent upon the uncoupling of FVIII to VWF. A new FVIII replacement product has been developed, BIVV001, that exhibits these properties. BIVV001 is a fusion protein consisting of VWF-D’D3 domain fused to rFVIII via immunoglobulin-G1 Fc domains and 2 XTEN polypeptides (Amunix Pharmaceuticals, Inc.).23 BIVV001 had an increase in FVIII half-life ranging from 25–31 hours and 33–34 hours in mice and monkeys, respectively.23 In early phase trials in men with severe hemophilia A the FVIII half-life with BIVV001 was increased three- to four-fold when compared to rFVIII products.24 The Phase 3 XTEND and XTEND-Kids clinical trials for the use of BIVV001 in severe hemophilia A are underway (NCT04644575, NCT04759131).
A comprehensive list of currently approved SHL and EHL products is available in the World Federation of Hemophilia Online Registry of Clotting Factor Concentrates.25
Nonfactor Therapies for Prophylaxis Hemophilia A
Despite the advances with treatment in EHL FVIII products, the implementation of routine factor prophylaxis still presents with challenges. The need for infusion may decrease to one to two times per week, but this can still be difficult in children with poor venous access, more so in infants. As discussed above, the development of neutralizing antibodies to FVIII is detrimental and occurs most frequently in patients with severe HA. Nonfactor therapeutic options for the treatment of both hemophilia A and B are under development. These new products augment the hemostatic pathway or suppress specific anticoagulant pathways that may help to overcome the current treatment gaps in hemophilia (Figure 1).
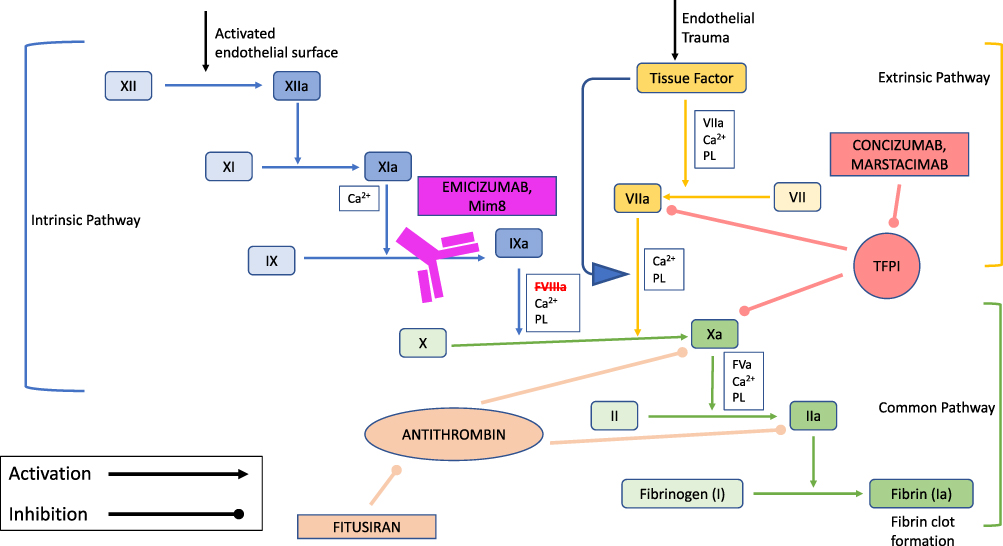 |
Figure 1 Mechanism of action of nonfactor therapies for hemophilia A. Abbreviations: Ca2+, calcium; PL, phospholipid. Notes: Intrinsic pathway factors are depicted in blue (light blue: inactivated, dark blue: activated). Extrinsic pathway factors are depicted in yellow (light yellow: inactivated, dark yellow: activated). Common pathway factors are depicted in green (light green: inactivated, dark green: activated). The monoclonal antibodies (emicizumab and Mim8) are depicted in purple; mimicking FVIII activity through the bridging of factor IXa and factor X. Antithrombin shows its inhibitory effects on factors Xa and IIa; consequently, fitusiran, the siRNA, inhibits the production of antithrombin in the liver allowing for increased thrombin generation. Tissue factor pathway inhibitor (TFPI) demonstrates its inhibitory effect on factor VIIa and factor Xa. The TFPI inhibitors (concizumab and marstacimab) prevent binding of TFPI to factors VIIa and Xa and restore thrombin generation. |
Emicizumab
The most developmentally advanced nonfactor therapy is emicizumab, a humanized bispecific monoclonal antibody, was initially approved in 2017 for patients with hemophilia A with inhibitors.26 Following reporting of the results from the HAVEN 3 and HAVEN 4 clinical trials, approval was granted for adult and pediatric HA patients with and without inhibitors in 2018.27
Emicizumab (Genentech) acts by mimicking FVIII activity through the bridging of factor IXa and factor X, thereby facilitating FIXa-mediated FX activation.28 Phase 1 and 2 clinical studies demonstrated linear pharmacokinetic profiles of four to five weeks with no serious adverse events, including thromboembolic events. The global phase 3 study, HAVEN 1, enrolled 109 HA males with inhibitors; those assigned to the emicizumab prophylaxis group received weekly subcutaneous dosing at 3 mg/kg for four weeks (loading phase), followed by 1.5 mg/kg weekly (maintenance phase) thereafter. Emicizumab prophylaxis led to an 87% reduction in bleeding compared to the no prophylaxis group (ABR of 2.3 vs 23.3 events).26 Thrombotic microangiopathy (TMA) and thrombotic events were reported in a total of five cases and occurred with concurrent use of activated prothrombin complex concentrates. After implementation of a mitigation strategy, no additional thrombotic or TMA events were reported.26 Similar therapeutic results were seen in the HAVEN 2 trial for pediatric patients with inhibitors. A subgroup of 15 patients previously using bypassing agents had a 99% reduction in ABR with the use of emicizumab.29 No thromboembolic events or cases of TMA were reported in the pediatric clinical trial. With the success of emicizumab in inhibitor patients, the HAVEN 3 clinical trials assessed its use in hemophilia A without inhibitors. A total of 152 participants were enrolled and were assigned to receive either 1.5 mg/kg weekly, 3 mg/kg every two weeks or no prophylaxis. Both emicizumab treatment groups had significantly reduced bleeding rates compared to the no prophylaxis group.30 The HAVEN 4 clinical trial evaluated the use of emicizumab dosing every four weeks and showed effective control of bleeding in those with and without inhibitors with similar kinetics compared to the other dosing strategies.31
In all of the HAVEN clinical trials, the most commonly reported adverse events were low-grade injection site reactions, headache and myalgias. The major limitation being the ongoing need for FVIII or bypassing agents for acute bleeding or surgical management. Although emicizumab has low immunogenicity, antidrug antibodies have been reported (about 5% of patients) in both pediatric and adult populations, the majority likely are not clinically significant.32 Since its approval, there remain many clinical questions specific to pediatrics that require ongoing investigation. First and foremost is when to choose emicizumab over routine FVIII prophylaxis in previously untreated patients. There are concerns that lack of FVIII concentrates earlier in life may increase inhibitor risk later on. In addition, It is unclear how the lack of FVIII exposure during infancy will impact long-term outcomes including bone and joint health. A recent survey found that there is widespread practice variation with lack of standardized approach in pediatric patients; patient preference and individual bleeding risk were the major reasons providers considered switching to emicizumab prophylaxis.33 The overall efficacy and safety profile for real-world use continues to evolve and further clinical trials are ongoing.32,34
The principal achievements over regular FVIII prophylaxis include the subcutaneous route for administration, long half-life (~28 days) and efficacy of bleed prevention. Emicizumab is indicated for routine prophylaxis in adults and children of all ages, including newborns, with HA with and without FVIII inhibitors. All patients should undergo a four week loading dose regimen of 3 mg/kg/week, followed by maintenance dosing of either 1.5 mg/kg weekly, 3 mg/kg every two weeks or 6 mg/kg every four weeks. For children, maintenance dosing is most often determined by weight and vial size availability.
Fitusiran
The primary role of antithrombin in the coagulation cascade is to inactivate FXa and thrombin, but also other serine proteases such as FIXa, FXIa and FXIIa; therefore its inhibition leads to a hypercoagulable state. Interestingly, as first observed in murine models of FVIII-deficient mice, suppression of antithrombin improved thrombin generation with subsequent reduction of clinically severe bleeding.35 This led to the development of small interfering ribonucleic acids (siRNAs) to target antithrombin silencing. Sehgal et al showed sustained reduction of antithrombin in mice and nonhuman primates with hemophilia A.36
Fitusiran (Sanofi Genzyme), a siRNA that suppresses antithrombin production in the liver, has been developed and is under investigation for its use in patients with hemophilia A and B with or without inhibitors. Patients with hemophilia had a decrease in antithrombin levels by about 80%; in turn, increasing overall thrombin generation.37 Concomitantly, ABR was significantly reduced in patients without inhibitors receiving fitusiran, with 48% (16/33) subjects experiencing no bleeding during the observation period. For those with inhibitors, ABR was also significantly reduced; 67% (22/33) reported no spontaneous bleeds. All breakthrough bleeding episodes were successfully treated with factor replacement or BPAs.38 The most common treatment related adverse event was injection site reactions. Phase 2 trials were suspended after development of an acute cerebral venous sinus thrombosis (initial incorrect impression was a subarachnoid hemorrhage) in a patient with severe hemophilia A, receiving fitusiran in combination with a FVIII product. After thorough investigation of clinical risk and safety, recommendations were given in relation to reduced dosing of FVIII or BPAs and these phase 2 clinical trials were reopened.39,40 Phase 3 studies are ongoing, with use of prophylactic fitusiran dosing at 80 mg subcutaneous once monthly. Pediatric trials were on hold with the identification of thromboembolic events, however, have since reopened and are actively recruiting males with severe hemophilia A or B (ages 1 to <12 years) (NCT03974113).41
The most commonly reported side effects of fitusiran were injection site reactions and pain, increase in liver transaminases and increase in D-dimer; these adverse events were generally mild and transient in study participants. In the event of breakthrough bleeding, aPCC or rFVIIa have been used successfully, but it is unclear the limitations that would occur in the setting of trauma or surgery. The benefits of fitusiran are once monthly subcutaneous injections, utility in both hemophilia A and B with and without inhibitors and good stability regardless of external temperature fluctuations.37
Tissue Factor Pathway Inhibitors
Tissue factor pathway inhibitor (TFPI) is an important protease inhibitor with three tandem Kunitz-type inhibitor domains (K1, K2, and K3). TFPI is an early inhibitor of the extrinsic coagulation system, blocking of FXa via the K2 domain and the tissue factor-FVIIa complex via the K1 domain.42
Concizumab (Novo Nordisk) was developed as a humanized monoclonal antibody against TFPI by inhibiting the K2 domain, thus preventing binding to FXa. Phase 1 and 2 studies demonstrated that plasma concizumab levels of at least 100 ng/mL were the most effective at reducing the frequency of bleeding episodes and was well tolerated with low rates of adverse events.43,44 To reach a goal plasma concentration of 100 ng/mL, Phase 2 clinical trials implemented a starting daily dose of 0.15 mg/kg given subcutaneously which was found to be suboptimal, as few (13%) experienced zero bleeds. Thus, the ABR for each patient was calculated based on their final dose levels and determined that 0.25 mg/kg per dose provided optimum efficacy while maintaining safety.44 During Phase 3 clinical trials, nonfatal thrombotic events occurred in three patients leading to a brief suspension, however the trial has since resumed with new safety mitigation measures and treatment guidelines.45 Mild injection site reactions were the most common reported side effect for concizumab. However, a total of six patients developed low-titer antidrug antibodies with no observed clinic effect.44 Its benefits include subcutaneous daily dosing with good stability in an easy to use device.43
Marstacimab (Pfizer) is a human monoclonal immunoglobulin G isotype, subclass 1 (IgG1) that targets the K2 domain of TFPI. Phase 1/2 clinical trials in hemophilia A or B with or without inhibitors, assessed four dosing regimens. Four serious adverse events occurred, which were determined to be unrelated to treatment. The most common reaction was injection site reactions, and no thrombotic events occurred. Three patients developed non-neutralizing antidrug antibodies with no effect on safety or pharmacokinetics.46 Phase 3 clinical trials (NCT03938792) are in recruitment. Men with hemophilia A or B, greater than 12 years of age, will receive a 300 mg subcutaneous loading dose, followed by 150 mg subcutaneous dosing weekly (with potential for dose escalation to 300 mg weekly).47
Mim8
Kjellev et al analyzed over 30,000 bispecific antibodies compatible for anti-FIXa and anti-FX to develop a novel, next-generation FVIII mimetic human bispecific antibody, Mim8. In vitro analysis used congenital HA plasma for thrombin generation assay and whole blood from healthy volunteers for thromboelastography. Mim8 was able to normalize both thrombin generation and blood clot formation, with approximately 15 times greater potency than emicizumab.48 Animal models have been underway to assess potency and pharmacokinetics with Mim8. HA mice showed reduction of bleeding with severe tail-vein transection and demonstrated a half-life of 14 days in the cynomolgus monkey.49
These in vitro and in vivo characterizations are encouraging for the clinical development of Mim8 as a potential next-generation FVIII-mimetic prophylactic treatment for individuals with hemophilia A. Currently, Phase 1 and Phase 2 clinical trials are underway (NCT04204408).
Future Directions
Gene Therapy
Therapeutic options for HA over the past 40 years have been vast, but only recently focused on simple replacement. Despite the expansive landscape of available options, there remain gaps in efficacy, safety and perceived ease of care. Gene therapy is a realistic therapeutic option for hemophilia, as even a small increase in blood factor levels would significantly improve the bleeding phenotype in severely affected patients. Initial gene therapy work was first successful in hemophilia B, but has now expanded to HA and offers the possibility of a decade long suspension of need for prophylaxis or cure and ability to live life with fewer restrictions.
Gene therapy in hemophilia has been achieved by using in vivo gene transfer to the liver with adeno-associated viral (AAV) vectors. Early trials were obstructed by the size and poor expression of FVIII, but have been addressed with removal of the FVIII B-domain which is not required for cofactor activity.50 These advances have led to many clinical trials for hemophilia A; current Phase 3 trials are listed in Table 1.
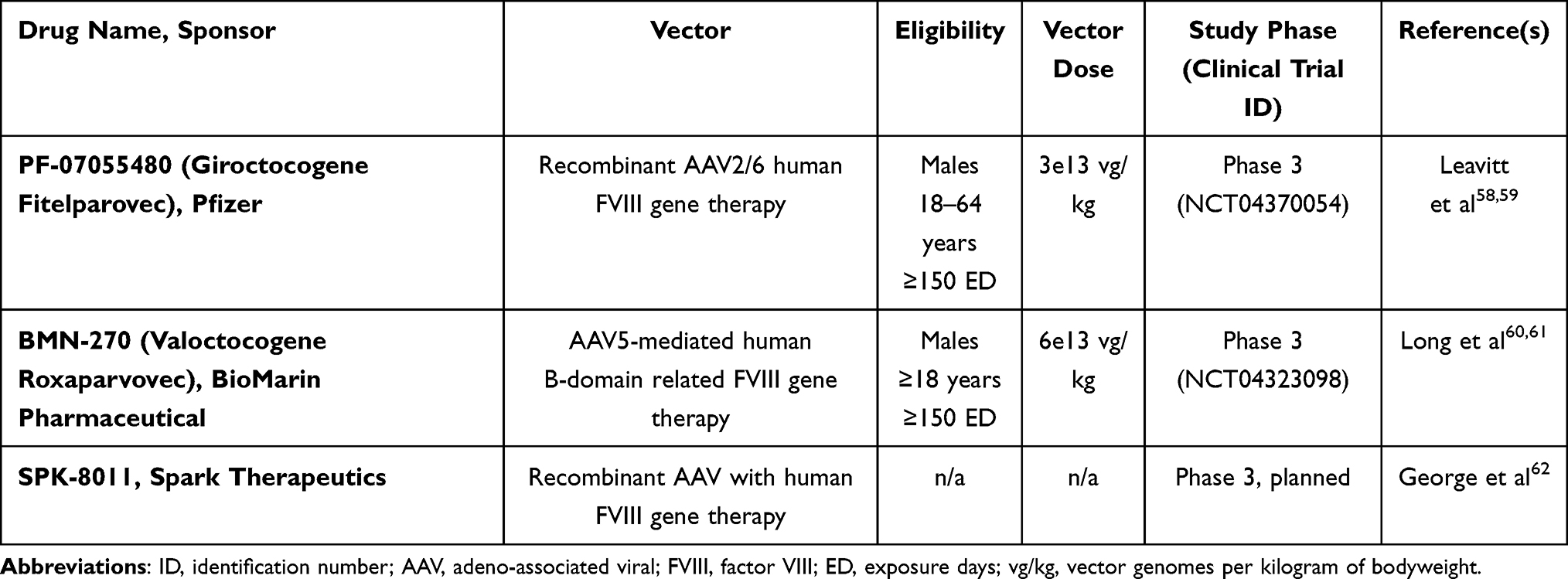 |
Table 1 Current Gene Therapy Clinical Trials for Hemophilia A in Phase 3 |
Safety concerns with AAV vector technology include acute liver toxicity with loss or reduced gene expression and preexisting neutralizing anti-AAV antibodies which may limit gene transfer.51 Specifically, these concerns heighten for young pediatric patients given small liver size and likely loss of transgene expression over time with liver growth and dilution. The exaggerated humoral response that occurs with initial vector infusion prevents the ability for repeated infusions later in life, thus making current technologies for gene therapy an unrealistic option in most children.52 There is of course concern for the costs associated with gene therapy treatment. However, with time, the decreased need for regular use of factor and nonfactor products and decreased episodes of breakthrough bleeding would undoubtedly prevent long-term sequelae of hemophilia and in turn benefit the health-care system as a whole.
Phase 1 investigation of gene therapy by transplantation of hematopoietic stem cells (HSCT) transduced ex vivo with lentivirus vectors is underway. The Atlanta trial will use autologous CD34+ HSCT transduced ex vivo with CD68-ET3 lentiviral vector following a conditioning regimen of busulfan and anti-thymocyte globulin (NCT04418414).53 Similarly, the Wisconsin trial is using a reduced intensity conditioning regimen with melphalan and fludarabine, followed by an infusion of autologous CD34+ peripheral blood stem cells, transduced with a lentiviral vector (Pleighlet, MUT6) encoding the B domain deleted form of human FVIII (NCT03818763).54
Genetically Modified Human Cell Therapy
Another concept under investigation is the use of genetically modified human cells. In order for this to be successful, the cell must be protected from host immune cells to prevent the foreign body response and pericapsular fibrotic overgrowth that can negatively impact long-term efficacy of these therapies.55 Certain biomaterials (ie hydrogels) can act as a physical barrier of protection from the host immune system.
Sigilon has created a platform that consists of genetically modified allogeneic human cells that are able to produce a therapeutic protein of interest (ie FVIII), that is encapsulated in a two-compartment sphere to support the function of cells while protecting it from the host immune response. SIG-001 is a buffered suspension of 1.5 mm alginate spheres encapsulating hFVIII-expressing human cells that has been shown to correct the bleeding phenotype and produced durable hFVIII levels in HA mice.55 The first human clinical Phase 1/2 trials began in early 2020 and will include those with severe and moderately-severe HA who have had over 150 exposure days to FVIII products.56 The clinical trial (NCT04541628) has since been suspended due to one patient developing inhibitors to SIG-001.57
Conclusion
With nearly a half century of investigation, our ability to provide safe and efficient care for children with hemophilia has grown immensely. The care of pediatric patients with hemophilia requires a thoughtful approach of exactly when to start prophylaxis and with what product. There are many factors that the medical team needs to consider including age, previous exposure days and venous access. Today, both FVIII concentrate products and emicizumab are suitable options for primary prophylaxis in pediatric hemophilia A. The use of other nonfactor therapies is under investigation, however their applicability in young children still needs to be studied. Continued understanding of the complexities of the coagulation system has led to advancements in therapy, specifically in nonfactor therapies, for hemophilia with prospects for significant mitigation of bleeding in the near future and possibility of cure in the distant future. Additional clinical studies and long-term outcome data are needed to establish ideal use and ensure safety for these patients.
Abbreviations
AAV, adeno-associated viral; ABR, annualized bleeding rate; BPA, bypassing agent; EHL, extended half-life; F, clotting factor; Fc, crystallizable fragment; HSCT, hematopoietic stem cell transplant; HA, hemophilia A; HIV, human immunodeficiency virus; ITI, immune tolerance induction; PK, pharmacokinetic; siRNA, small interfering ribonucleic acid; SHL, standard half-life; TFPI, tissue factor pathway inhibitor; TMA, thrombotic microangiopathy; VWF, von Willebrand factor.
Author contributions
KR wrote and edited the manuscript. RFS and MUC reviewed, edited and contributed to the manuscript.
Disclosure
KR received a 2020 HTRS/Novo Nordisk Clinical Fellowship Award in Hemophilia and Rare Bleeding Disorders from the Hemostasis and Thrombosis Research Society (HTRS), which was supported by an educational grant from Novo Nordisk Inc. MUC has received honoraria from Sanofi, Genentech/Roche, Kedrion, Catalyst Biosciences, Hema Biologics, Takeda, Pfizer, Bayer, Novo Nordisk, Octapharma, Spark, BioMarin, Uniqure, Global Blood Therapeutics, Chiesi. MUC is employed by Agios Pharmaceuticals. RS has received honoraria from Sobi/Sanofi, Genentech/Roche, Grifols, Sigilon, Hema Biologics, Takeda, Pfizer, Bayer, Catalyst, Guardian Therapeutics, Novo Nordisk, Octapharma. RS has investigator initiated grant funding from Takeda (ATHN 9 and SAFE study), Genentech/Octapharma (Emi PUPs and Nuwiq ITI) and Octapharma (MOTIVATE study). The authors report no other conflicts of interest in this work.
References
1. Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935–1939. doi:10.1111/jth.12672
2. Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535–544. doi:10.1056/NEJMoa067659
3. Gringeri A, Lundin B, Von Mackensen S, et al. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT study). J Thromb Haemost. 2011;9(4):700–710. doi:10.1111/j.1538-7836.2011.04214.x
4. Panicker J, Warrier I, Thomas R, Lusher JM. The overall effectiveness of prophylaxis in severe haemophilia. Haemophilia. 2003;9(3):272–278. doi:10.1046/j.1365-2516.2003.00757.x
5. Warren BB, Thornhill D, Stein J, et al. Young adult outcomes of childhood prophylaxis for severe hemophilia A: results of the joint outcome continuation study. Blood Adv. 2020;4(11):2451–2459. doi:10.1182/bloodadvances.2019001311
6. Gouw SC, Van Den Berg HM, Fischer K, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. Blood. 2013;121(20):4046–4055. doi:10.1182/blood-2012-09-457036
7. Ragni MV, Ojeifo O, Feng J, et al. Risk factors for inhibitor formation in haemophilia: a prevalent case-control study. Haemophilia. 2009;15(5):1074–1082. doi:10.1111/j.1365-2516.2009.02058.x
8. Van Den Berg HM, Fischer K, Carcao M, et al. Timing of inhibitor development in more than 1000 previously untreated patients with severe hemophilia A. Blood. 2019;134(3):317–320. doi:10.1182/blood.2019000658
9. Gouw SC, Van Der Bom JG, Van Den Berg HM. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood. 2007;109(11):4648–4654. doi:10.1182/blood-2006-11-056291
10. den Uijl IEM, Mauser Bunschoten EP, Roosendaal G, et al. Clinical severity of haemophilia A: does the classification of the 1950s still stand? Haemophilia. 2011;17(6):849–853. doi:10.1111/j.1365-2516.2011.02539.x
11. Mannucci PM. Back to the future: a recent history of haemophilia treatment. Haemophilia. 2008;14(SUPPL. 3):10–18. doi:10.1111/j.1365-2516.2008.01708.x
12. Tabor E. The epidemiology of virus transmission by plasma derivatives: clinical studies verifying the lack of transmission of hepatitis B and C viruses and HIV type 1. Transfusion. 1999;39(December):1160–1168. doi:10.1046/j.1537-2995.1999.39111160.x
13. Björkman S, Blanchette VS, FischeR K, et al. Comparative pharmacokinetics of plasma- and albumin-free recombinant factor VIII in children and adults: the influence of blood sampling schedule on observed age-related differences and implications for dose tailoring. J Thromb Haemost. 2010;8(4):730–736. doi:10.1111/j.1538-7836.2010.03757.x
14. Morfini M, Marchesini E, Paladino E, Santoro C, Zanon E, Iorio A. Pharmacokinetics of plasma-derived vs. recombinant FVIII concentrates: a comparative study. Haemophilia. 2015;21(2):204–209. doi:10.1111/hae.12550
15. Versloot O, Iserman E, Chelle P, et al. Terminal half-life of FVIII and FIX according to age, blood group and concentrate type: data from the WAPPS database. J Thromb Haemost. 2021;19(8):1896–1906. doi:10.1111/jth.15395
16. Turecek PL, Johnsen JM, Pipe SW, O’Donnell JS. Biological mechanisms underlying inter-individual variation in factor VIII clearance in haemophilia. Haemophilia. 2020;26(4):575–583. doi:10.1111/hae.14078
17. Blanchette VS. Prophylaxis in the haemophilia population. Haemophilia. 2010;16(SUPPL. 5):181–188. doi:10.1111/j.1365-2516.2010.02318.x
18. Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl6):1–158. doi:10.1111/hae.14046
19. Peters R, Harris T. Advances and innovations in haemophilia treatment. Nat Rev Drug Discov. 2018;17(7):493–508. doi:10.1038/nrd.2018.70
20. Pipe SW, Montgomery RR, Pratt KP, Lenting PJ, Lillicrap D. Life in the shadow of a dominant partner: the FVIII-VWF association and its clinical implications for hemophilia A. Blood. 2016;128(16):2007–2016. doi:10.1182/blood-2016-04-713289
21. Ragni MV. New and emerging agents for the treatment of hemophilia: focus on extended half-life recombinant clotting proteins. Drugs. 2015;75(14):1587–1600. doi:10.1007/s40265-015-0451-5
22. Ragni MV, Croteau SE, Morfini M, Cnossen MH, Iorio A. Pharmacokinetics and the transition to extended half-life factor concentrates: communication from the SSC of the ISTH. J Thromb Haemost. 2018;16(7):1437–1441. doi:10.1111/jth.14153
23. Chhabra ES, Liu T, Kulman J, et al. BIVV001, a new class of factor VIII replacement for hemophilia A that is independent of von Willebrand factor in primates and mice. Blood. 2020;135(17):1484–1496. doi:10.1182/BLOOD.2019001292
24. Konkle BA, Shapiro AD, Quon DV, et al. BIVV001 fusion protein as Factor VIII replacement therapy for Hemophilia A. N Engl J Med. 2020;383(11):1018–1027. doi:10.1056/nejmoa2002699
25. World Federation of Hemophilia. Online registry of clotting factor concentrates. World Federation of Hemophilia website; 2018.
26. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377(9):809–818. doi:10.1056/NEJMoa1703068
27. U.S. Food & Drug Administration. FDA approves emicizumab-kxwh for hemophilia A with or without factor VIII inhibitors; 2018.
28. Lenting PJ, Denis CV, Christophe OD. Emicizumab, a bispecific antibody recognizing coagulation factors IX and X: how does it actually compare to factor VIII? Blood. 2017;130(23):2463–2468. doi:10.1182/blood-2017-08-801662
29. Young G, Liesner R, Chang T, et al. A multicenter, open-label phase 3 study of emicizumab prophylaxis in children with hemophilia A with inhibitors. Blood. 2019;134(24):2127–2138. doi:10.1182/blood.2019001869
30. Mahlangu J, Oldenburg J, Paz-Priel I, et al. Emicizumab prophylaxis in patients who have hemophilia a without inhibitors. N Engl J Med. 2018;379(9):811–822. doi:10.1056/NEJMoa1803550
31. Pipe SW, Shima M, Lehle M, et al. Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open-label, non-randomised phase 3 study. Lancet Haematol. 2019;6(6):e295–e305. doi:10.1016/S2352-3026(19)30054-7
32. Jimenez-Yuste V, Peyvandi F, Klamroth R, et al. Final analysis of the STASEY trial: a single-arm, multicenter, open-label, Phase III clinical trial evaluation the safety and tolerability of emicizumab prophylaxis in persons with Hemophilia A (PwHA) with Factor (F) VIII Inhibitors. Res Pr Thromb Haemost. 2021;5(Suppl1):17–21.
33. Lucas TL, Gupta S, Davis JA, Corrales-Medina FF. Emicizumab-Kxwh for previously untreated patients with haemophilia: the conversation begins. Blood. 2019;134(Supplement_1):2409. doi:10.1182/blood-2019-127270
34. ATHN 7: hemophilia natural history study; 2018. Available from: https://clinicaltrials.gov/ct2/show/NCT03619863.
35. Bolliger D, Szlam F, Suzuki N, Matsushita T, Tanaka KA. Heterozygous antithrombin deficiency improves in vivo haemostasis in factor VIII-deficient mice. Thromb Haemost. 2010;103(6):1233–1238. doi:10.1160/TH09-10-0732
36. Sehgal A, Barros S, Ivanciu L, et al. An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat Med. 2015;21(5):492–497. doi:10.1038/nm.3847
37. Pasi KJ, Lissitchkov T, Mamonov V, et al. Targeting of antithrombin in hemophilia A or B with investigational siRNA therapeutic fitusiran—Results of the phase 1 inhibitor cohort. J Thromb Haemost. 2021;19(6):1436–1446. doi:10.1111/jth.15270
38. Pasi KJ, Georgiev P, Mant T, et al. Fitusiran, an investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia: updated results from a Phase 1 and Phase 1/2 extension study in patients with inhibitors. Blood. 2016;128(22):1397. doi:10.1182/blood.v128.22.1397.1397
39. Alnylam suspends fitusiran dosing due to thrombotic event in Phase 2 open-label extension study. World Federation of Hemophilia; 2017. Available from: https://news.wfh.org/alnylam-suspends-fitusiran-dosing-due-thrombotic-event-phase-2-open-label-extension-study/.
40. Update: FDA lifts suspension of fitusiran trial. World Federation of Hemophilia; 2017. Available from: https://news.wfh.org/update-fda-lifts-suspension-fitusiran-trial/.
41. Taylor NP. Sanofi plans 2021 restart for pediatric hemophilia trial hit by fallout from thrombotic events. Fierce Biotech; 2021. Available from: https://www.fiercebiotech.com/biotech/sanofi-plans-2021-restart-for-pediatric-hemophilia-trial-hit-by-fallout-from-thrombotic.
42. Kato H. Tissue factor pathway inhibitor; its structure, function and clinical significance. Pol J Pharmacol. 1996;48(1):67–72.
43. Eichler H, Angchaisuksiri P, Kavakli K, et al. Concizumab restores thrombin generation potential in patients with haemophilia: pharmacokinetic/pharmacodynamic modelling results of concizumab phase 1/1b data. Haemophilia. 2019;25(1):60–66. doi:10.1111/hae.13627
44. Shapiro AD, Angchaisuksiri P, Astermark J, et al. Subcutaneous concizumab prophylaxis in hemophilia A and hemophilia A/B with inhibitors: phase 2 trial results. Blood. 2019;134(22):1973–1982. doi:10.1182/blood.2019001542
45. Ray F. Novo nordisk resumes Phase 3 trials of concizumab in Hemophilia A and B. Hemophilia News Today; 2020.
46. Mahlangu J, Lamas J, Morales J, et al. A Phase 1b/2 study of the safety, tolerability, pharmacokinetics, pharmacodynamics, and efficacy of PF-06741086, an anti-TFPI monoclonal antibody, in patients with Severe Hemophilia A or B. Res Pr Thromb Haemost. 2019;3:85–86.
47. Pfizer doses first participant in phase 3 study evaluating anti-TFPI investigational therapy, marstacimab, for people with severe hemophilia A and B with or without inhibitors. Dow Jones Institutional News. Available from: https://cmich.idm.oclc.org/login?url=https://www.proquest.com/wire-feeds/press-release-pfizer-doses-first-participant/docview/2463381584/se-2?accountid=10181.
48. Kjellev SL, Østergaard H, Greisen PJ, et al. Mim8 - A next-generation FVIII mimetic bi-specific antibody - potently restores the hemostatic capacity in hemophilia a settings in vitro and in vivo. Blood. 2019;134(Supplement_1):96. doi:10.1182/blood-2019-122817
49. Østergaard H, Lund J, Greisen P, et al. FVIIIa-mimetic bispecific antibody (Mim8) ameliorates bleeding upon severe vascular challenge in hemophilia A mice. Blood. 2021;138:1258–1268. doi:10.1182/blood.2020010331
50. McIntosh J, Lenting PJ, Rosales C, et al. Therapeutic levels of FVIII following a single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood. 2013;121(17):3335–3344. doi:10.1182/blood-2012-10-462200
51. Nathwani AC. Gene therapy for hemophilia. Hematol Am Soc Hematol Educ Progr. 2019;1:1–8. doi:10.1002/pbc.26865
52. Gollomp KL, Doshi BS, Arruda VR. Gene therapy for hemophilia: progress to date and challenges moving forward. Transfus Apher Sci. 2019;58(5):602–612. doi:10.1016/j.transci.2019.08.012
53. Hematopoietic Stem Cell Transplantation Gene Therapy for Treatment of Severe Hemophilia A. https://clinicaltrials.gov/ct2/show/NCT04418414.
54. Gene therapy trial for platelet derived factor VIII production in Hemophilia A. 2021. Available from: https://www.fiercebiotech.com/biotech/sanofi-plans-2021-restart-for-pediatric-hemophilia-trial-hit-by-fallout-from-thrombotic.
55. Carmona G, Barney L, Sewell J, et al. Correcting rare blood disorders using coagulation factors produced in vivo by shielded living therapeutics products. Blood. 2019;134(Supplement 1):2065. doi:10.1182/blood-2019-127050
56. Shapiro A, Konkle B, Croteau S, et al. First-in-human Phase 1/2 clinical trial of SIG-001, an innovative shielded cell therapy platform, for hemophilia A. Blood. 2020;136(Supplement 1):8. doi:10.1182/blood.2020006611
57. Al Idrus A. FDA puts Sigilon’s hemophilia cell therapy trial on hold thanks to safety concerns. Fierce Biotech; 2021.
58. Leavitt A, Konkle B, Stine K, et al. Updated follow-up of the alta study, a Phase 1/2 study of giroctocogene fitelparvovec (SB-525) gene therapy in adults with severe Hemophilia A. Blood. 2020;136(Supplement1):12. doi:10.1182/blood-2020-137648
59. Study to evaluate the efficacy and safety of PF-07055480/giroctocogene fitelparvovec gene therapy in moderately severe to severe Hemophilia A adults. Available from: https://clinicaltrials.gov/ct2/results?cond=&term=NCT04370054&cntry=&state=&city=&dist=.
60. Long BR, Veron P, Kuranda K, et al. Early phase clinical immunogenicity of valoctocogene roxaparvovec, an AAV5-mediated gene therapy for Hemophilia A. Mol Ther. 2021;29(2):597–610. doi:10.1016/j.ymthe.2020.12.008
61. Study to Evaluate the Efficacy and Safety of Valoctogene Roxaparvovec, With Prophylactic Steroids in Hemophilia A (GENEr8-3). https://clinicaltrials.gov/ct2/show/NCT04323098.
62. George LA, Monahan PE, Eyster ME, et al. Phase I/II Trial of SPK-8011: stable and durable FVIII expression after AAV gene transfer for Hemophilia A. Res Pr Thromb Haemost. 2021;5(Suppl 1):17–21.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.