")
Back to Archived Journals » Vaccine: Development and Therapy » Volume 6
Management of visceral leishmaniasis with therapeutic vaccines
Authors Rawat K, Yadav N, Joshi S, Ratnapriya S, Sahasrabuddhe A, Dube A
Received 15 April 2016
Accepted for publication 29 June 2016
Published 28 September 2016 Volume 2016:6 Pages 33—45
DOI https://doi.org/10.2147/VDT.S110654
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Don Diamond
Keerti Rawat,1 Narendra K Yadav,1 Sumit Joshi,1 Sneha Ratnapriya,1 Amogh A Sahasrabuddhe,2 Anuradha Dube1
1Division of Parasitology, 2Division of Molecular and Structural Biology, Council of Scientific and Industrial Research-Central Drug Research Institute, Lucknow, India
Abstract: Visceral leishmaniasis (VL), a phlebotomine-borne neglected tropical disease, is caused by parasites of the Leishmania donovani complex. While L. donovani infection is restricted to the Indian subcontinent and East Africa, where transmission is anthroponotic, Leishmania infantum occurs in Europe, North Africa, and parts of Latin America, where it is zoonotic in nature with dogs as reservoir hosts. Though the incidence of VL caused by L. infantum has been on the decline, L. donovani continues to cause epidemics periodically. By and large, a small proportion of L. donovani infection manifests as clinical disease but majority of the infected individuals remain asymptomatic and contribute to the perpetuation of the VL transmission cycle via the sand fly vector. This is one of the major stumbling blocks to World Health Organization initiatives to eliminate this deadly disease by 2020. These parasites reside within the host macrophages and impair the immune system of the infected individual, which ultimately results in marked immunosuppression. In the absence of any safe and effective vector control measure, attempts have been made to design therapeutic vaccine(s) that can exclusively target infected macrophages. So far, two vaccines – a glycoprotein complex from L. donovani promastigote, fucose–mannose ligand with saponin, commercialized as Leishmune®, as well as a polyprotein vaccine formulation, Leish-111f + monophosphoryl lipid A plus squalene emulsion in combination with glucantime, have been successfully evaluated for their immunotherapeutic potential against canine VL. However, encouraging results obtained from several experimental trials so far against human VL are still to be translated clinically. This review provides an overview on the various strategies tried and tested for developing therapeutic vaccines against this dreaded disease.
Keywords: immunosuppression, Leishmune, Leish-111f, adjuvant, antileishmanial
Introduction
The Leishmania donovani complex (comprising L. donovani and L. infantum chagasi), an obligate intracellular protozoan parasite, is responsible for a fatal progressive systemic disease – visceral leishmaniasis (VL), commonly referred to as “kala azar” or “black fever”.1 This disease can be of either zoonotic (from animal-to-human, or vice versa) or anthroponotic type (human-to-human cycle) based on their transmission characteristics.1 Approximately 90% of the annual 100,000 VL cases were recorded from the Asian (Bangladesh and India), African (Ethiopia, Sudan, and South Sudan), and South American (Brazil) continents, where it affects the underprivileged communities residing in remote areas with limited access to primary health care facilities,1 although the actual number of VL cases is much higher due to underreporting.2–4 L. donovani, the causal agent in the Indian subcontinent and East Africa, is transmitted through Phlebotomus argentipes and Phlebotomus orientalis, respectively, in a predominantly anthroponotic life cycle.5 However, L. infantum, transmitted through Lutzomyia spp., is mainly responsible for VL in the Mediterranean region and the American subcontinent. The parasites remain either as extracellular and flagellated promastigotes within the sandfly or as intracellular and nonflagellated amastigotes within the mammalian host. This digenetic lifestyle ensures better survival of the parasite within vector and host.6
Leishmania parasites mainly affect mononuclear phagocytic cells of the reticuloendothelial organs, such as liver, spleen, and bone marrow. However, they are usually disseminated to other visceral organs (gut, lung, etc), as well as the skin, particularly seen in L. infantum-affected areas.7 Persistent fever, hepatosplenomegaly, pancytopenia, and hypergammaglobulinemia are the characteristic features of human VL.1 The case fatality rate of this disease is 100% because the patient dies in the absence of treatment. Available chemotherapeutics have several demerits, and their usage is also limited due to the fact that only ailing individuals with clinical symptoms are benefited.
Human VL occurs in a more heterogeneous form with varied chronicity. Only few infected individuals develop clinical symptoms, and on this basis, they are the ones who are subjected to antileishmanial chemotherapy. However, the majority of the endemic population remains with subclinical infection or as asymptomatic carriers (~6–10 times more than the number of VL patients) and their role in disease transmission is yet to be confirmed.8 Moreover, the immunocompromised state of VL patients makes them susceptible to other secondary infections. Emergence of parasite resistance following drug treatment and increased reporting of disease relapse is another major problem. Furthermore, because the treatment end point is not defined, as patients often fail to report back 6 months posttreatment, it is difficult to ascertain the complete cure from the disease.
Occasionally, approximately 10%–50% of VL patients, during or after the treatment, develop post kala azar dermal leishmaniasis (PKDL), which is characterized by maculopapular or nodular skin rashes.9 It is commonly seen in East Africa and the Indian subcontinent and requires prolonged and expensive treatment.9 These individuals play a crucial role in VL transmission because they can act as a potential source of kala azar.10
Due to the geographical overlap between VL- and HIV-affected areas, there is increase in the number of Leishmania/HIV-coinfected individuals, showing atypical presentations of VL.11 Because both are opportunistic infections, they impose a similar detrimental effect on the immune system, resulting in increased frequency of treatment failure and relapse cases. However, antiretroviral therapy significantly decreases disease progression and relapses, with increased survival of the coinfected patients.11 Increased incidence of HIV cases resulted in the spread of this coinfection in ~35 countries of the world, including India.12 In Leishmania/HIV-coinfected patients, characteristic features of VL such as splenomegaly may be absent;13 however, there is involvement of atypical organs such as lungs or the gastrointestinal system.14
Immunopathology of VL
Humans
Active VL patients demonstrate depressed cell-mediated immune response, with a negative leishmanin skin test.15 Moreover, their peripheral blood mononuclear cells (PBMCs) neither proliferate nor produce interferon (IFN)-γ in the presence of Leishmania antigen.16,17 Conversely, stimulation of PBMCs of cured VL patients with parasite antigen enhances the production of IFN-γ, thus showing that immunosuppression is not due to the antigen-specific T-cell response but several other elements might be involved in the undesirable clinical outcomes.18 Various studies on human VL suggested that the immune response did not clearly skew toward T-helper cell type 2 (Th2 type) as increased synthesis of cytokines and chemokines of the Th1 type was also noticed.19 Clinical studies strongly depict that there is an elevated level of serum interleukin (IL)-10 in active VL patients, confirmed by increased messenger RNA (mRNA) transcripts in the spleen, bone marrow, and lymph nodes.18 In addition, regulatory T (T-reg)-cells, found to produce IL-10 in the bone marrow of VL patients, could suppress antiparasitic immunity.20 The mRNA expression of transforming growth factor beta (TGF-β) was also found to be elevated in splenic aspirates of active VL patients.21 In addition to this, a typical VL patient showed an elevated level of antibodies in the plasma, leading to the formation of immune complexes.22
PKDL, which generally appears on the skin, either due to the ineffective treatment of VL or the suppression of immunity due to persistent parasites, differs immunologically from VL in several ways.9 Increased levels of IL-4, IL-10, and IFN-γ were observed in PKDL lesions, with increased infiltration of cluster of differentiation 3 (CD3+) T-cell.23 Other studies reveal that there was a stronger parasite-specific T-cell response evident at the onset of PKDL.24 Furthermore, PKDL lesions showed T-reg cell aggregation to be positively associated with parasite burden. Although most PKDL cases showed IL-10-mediated immunosuppression, there must be involvement of other deregulated inflammatory responses.25
Animal models
Rodent
Mice models, especially BALB/c, as well as Syrian hamsters are widely used for experimental VL studies and each has its own limitations.26 Susceptible mouse strains initially allow parasite replication in the liver, with a resolution of disease in the later stages of infection; therefore, they serve as a more suitable model of acute infection rather than of the progressive chronic VL.27 Although not representing the ideal pathological signs, the parasites cause splenomegaly, with atrophy of the lymphoid follicles, which progresses for a longer duration in the spleen and is not seen in the liver.28 Similar to the human immune response, which is characterized by a mixed Th1/Th2 type, the spleen of mice showed increased levels of tumor necrosis factor alpha (TNF-α) as well as IL-10. Secretion of IL-10 induced by TNF-α promoted disease progression by rendering the macrophages unresponsive to activation signals and also inhibit the priming of T-cell responses by acting on dendritic cells.29 Hence, BALB/c mice work as better models of self-healing or subclinical VL infection.26
Another rodent model, the Syrian golden hamster, closely mimics the clinicopathologic features of active human and canine VL, which gradually develops into progressive fatal disease, ultimately causing the death of the host.26 There is an elevated level of Th1 cytokines and significant amounts of IL-10 and TGF-β mRNA transcripts in Leishmania-infected hamsters, with reduced expression of inducible nitric oxide synthase. This promotes the multiplication and survival of the parasite, similar to the situation in humans.30 However, in contrast to humans, hamsters show the development of severe ascites and glomerulonephritis.31 In spite of being a highly relevant VL model, the hamster is not frequently used due to the unavailability of commercial reagents for immunological and molecular studies.26,27
Dog
Dog, being a natural host of L. infantum, serves as a pertinent model for VL.32 Canine VL is a multisystemic disease associated with poor body condition score, lymphadenomegaly, and typical granulomatous inflammatory reactions.32 Infected dogs show a balance between Th1 and Th2 cytokines, with increased production of IL-10 and INF-γ.33 However, there are measurable levels of IL-4 mRNA transcripts, which are related to the parasitic load as well as clinical symptoms.33,34 Moreover, there is a reduction in CD4+ and CD8+ populations during infection, which are restored after drug therapy.35 Contrarily, there is significant production of IL-2 and TNF-α by the PBMCs of asymptomatic dogs in comparison to symptomatic and uninfected ones.36 Dogs, although the most appropriate model for canine VL, are used in limited number for studies due to their higher costs and ethical concerns.27
Thus, in a nutshell, hamster is a better model than mice to study progressive VL as the latter varies in immunopathological features relative to human cases. However, the nonavailability of immunological markers makes its use difficult for the evaluation of vaccine candidates, although some recent studies have indicated that the immune status after vaccination could be analyzed by other techniques such as real-time polymerase chain reaction.30,37,38 Because dogs act as a key natural reservoir of the visceralizing form of Leishmania, they constitute a model of choice for studying zoonotic VL.26 Continuous efforts are being made to develop strategies that could mimic natural transmission by either using lower infectious doses of parasites or bioactive saliva or using natural reservoir hosts, which ultimately will lead to better understanding of the disease dynamics. This will ultimately help in generation of more meaningful data regarding the immune response that parallels human disease, and thus contribute immensely to the development of improved experimental models to evaluate possible vaccine candidates.
Feasibility of a suitable vaccine against VL
The only control measure for VL presently available is chemotherapy, which includes pentavalent antimonials (meglumine antimoniate and sodium stibogluconate [SSG]), amphotericin B and its liposomal formulation, paromomycin, and miltefosine either alone or in combination.1 The World Health Organization, in 2005, has initiated elimination campaigns in VL-endemic regions through early case detection, followed by complete treatment with a target to reduce the number of diseased individuals. This drive has successfully saved many lives through the administration of antileishmanial drugs. Although these chemotherapeutics are effective, they have several demerits, such as lesser availability, high cost of treatment (drugs and hospitalization), low efficacy, adverse effects such as toxicity, and requirement of longer regimens with the invasive route of administration (parenteral).39 Moreover, with the emergence of drug-unresponsive cases, it is difficult to curb this disease completely with chemotherapy alone.40 Furthermore, the presence of asymptomatic individuals results in disease transmission, especially in hyperendemic pockets of the globe, defeating the goal of the current VL control program.41 Hence, there is an urgent need for alternative treatment strategies that become truly accessible in order to sustain the accomplishment of the current kala azar elimination program.41 Over the past few decades, researchers have been trying to develop a vaccine as a cost-effective treatment strategy against VL because this is thought to be critical for the complete eradication of this dreadful disease.41 It was evident that recovered VL patients develop strong immunity, making them resistant to subsequent clinical infections, which indicates that prevention of VL through either prophylactic or therapeutic vaccination is feasible.42 Identification of appropriate vaccine candidates through a proper understanding of the immunobiology and pathogenesis of the disease is required for the success of the vaccine development program.43 Additionally, selection of either a suitable adjuvant or immunomodulator or an effective delivery system can further boost the immune responses and generate long-lasting immunity.44 Although a little emphasis was given to the development of therapeutic vaccines, it would be more useful for individuals with active infection so as to modulate their immune responses toward a cure.
Current status of prophylactic vaccines
Prophylactic vaccines were thought of as obvious priority, for which several molecules were identified and evaluated as vaccine targets, as reviewed by Joshi et al.45 However, only a few of them reached the stage of clinical trials and were commercialized. LeishTec® and Leishmune® were developed and registered in Brazil, while CaniLeish® was developed in Europe, against canine VL. LeishTec® (recombinant A2 antigen plus saponin) conferred only 40% protection, whereas Leishmune® (fucose–mannose ligand [FML]-QuilA) and CaniLeish® (LiESP/QA-21) offered significant protection in naturally infected dogs.46 Apart from these trials, another vaccine candidate LEISH-F1, a polyprotein expressed in bacteria, has made it into Phase I and Phase II clinical trials.47 Further, when used in conjunction with chemotherapy, it appeared to have shortened the time to cure.48,49 With the encouraging results from LEISH-F1, a new construct – LEISH-F2 – was designed, lacking a histidine tag due to regulatory issues, which also progressed to clinical trials (Phase I and Phase II).50 Additionally, LEISH-F3, composed of nucleoside hydrolase from L. donovani and sterol 24-c-methyltransferase from L. infantum, formulated with a Toll-like receptor 4 (TLR4)-based adjuvant, glucopyranosyl lipid A, was found to be safe and immunogenic.51 Beyond these trials, an ongoing research project named Multivalent Vaccine for Human Visceral Leishmaniasis (MeLeVaClin) is also looking for newer vaccine strategies against VL in preclinical models.52
Therapeutic vaccines against VL
Therapeutic vaccines are thought to be essential, particularly in the case of persistent chronic infections, because in such cases, either intracellular parasites evade the host immune system and establish themselves in a more secured way or the available control intervention is ineffective.53 In the case of VL, the majority of inhabitants in disease-affected areas are either healthy endemic individuals or asymptomatic ones, who serve as the reservoir of the parasite or may become symptomatic in future.41 Hence, there is need for a therapeutic vaccine that can be effectively used to stimulate the patients’ own immune defense system in these endemic populations, which would be a major asset in controlling the progression of disease.54 These therapeutic vaccines have been reported to be extremely successful against several chronic diseases,55 such as HIV infection,56 tuberculosis,57 Chagas disease,58 human papilloma virus infection,59 as well as cancer,60,61 indicating that the era of successful therapeutic vaccination has arrived.
The therapeutic strategy involves the use of biological molecules (whole or their components) in combination with either adjuvants or drugs to modulate the immune responses of Leishmania-infected individuals toward the protective type.62 Therefore, this strategy, which restores or induces an effective immune response without any side effects, could be a promising alternative to conventional chemotherapeutics.62,63 This review focuses on the various therapeutic approaches against VL, as summarized in Table 1.
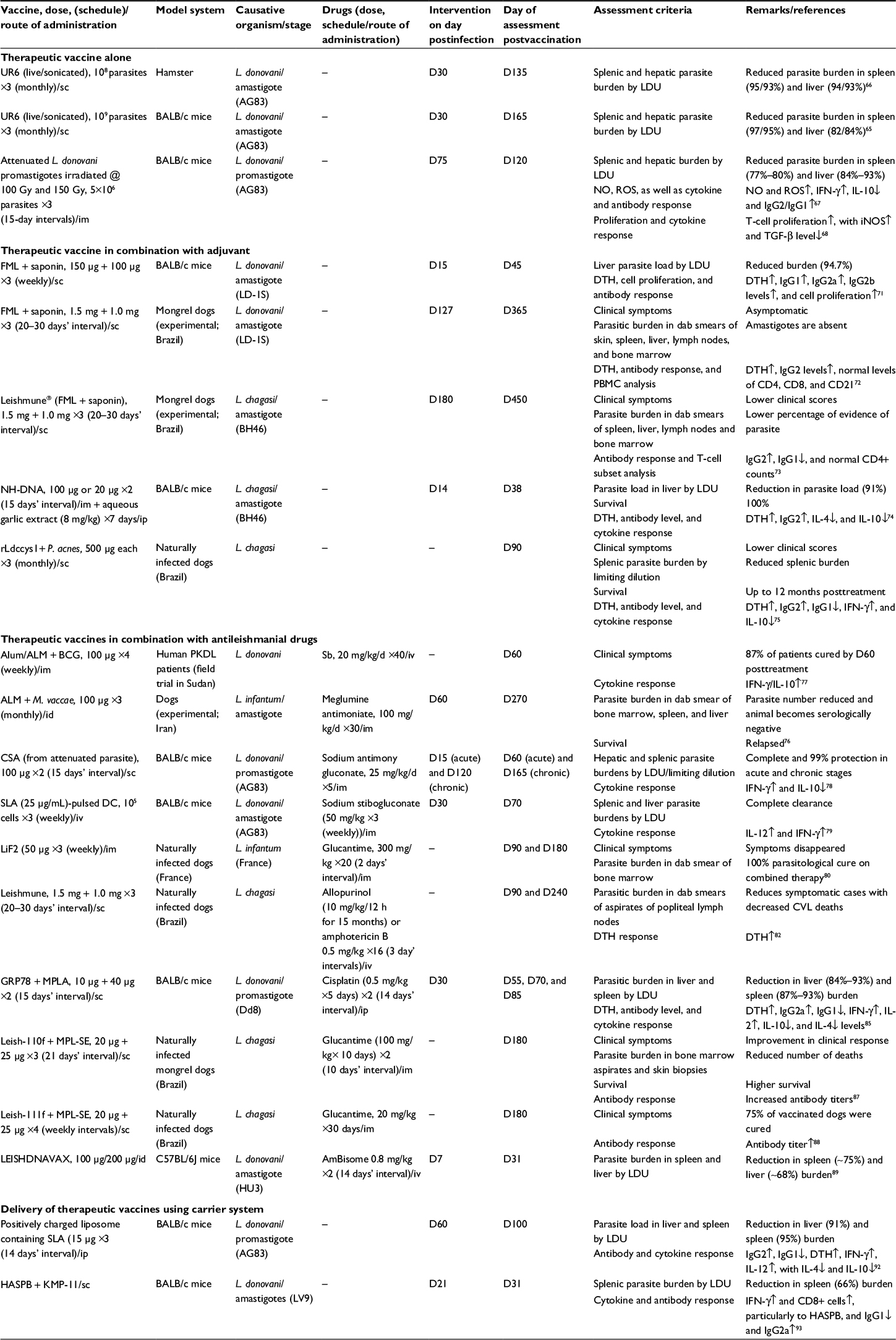 | Table 1 A summary of therapeutic vaccines evaluated against VL Notes: Numbers separated by / indicate reduction in parasite burden by live/sonicated parasites. ↑, Increase; ↓, decrease. Abbreviations: ALM, autoclaved Leishmania major; BCG, Bacillus Calmette–Guérin; CD, cluster of differentiation; CSA, complete soluble antigen; CVL, canine visceral leishmaniasis; DTH, delayed type hypersensitivity; DC, dentritic cell; FML, fucose–mannose ligand; GRP78, glucose-regulated protein 78; HASPB, hydrophilic acylated surface protein B; id, intradermal; IFN, interferon; IgG, immunoglobulin G; IL, interleukin; im, intramuscular; iNOS, inducible nitric oxide synthase; ip, intraperitoneal; iv, intravenous; KMP-11, kinetoplastid membrane protein-11; L. chagasi, Leishmania chagasi; L. donovani, Leishmania donovani; LDU, Leishman Donovan Units; LiF2, L. infantum-derived fraction 2, 94–67 kDa; MPLA, monophosphoryl lipid A; MPL-SE, monophosphoryl lipid A plus squalene emulsion; NH, nucleoside hydrolase; NO, nitric oxide; PBMC, peripheral blood mononuclear cell; PKDL, post kala azar dermal leishmaniasis; rLdccys1, recombinant cysteine proteinase from L. (L.) infantum chagasi; ROS, reactive oxygen species; sc, subcutaneous; SLA, soluble Leishmania antigen; TGF-β, transforming growth factor beta; Sb, sodium stibogluconate. |
Use of therapeutic vaccines alone
Considering the fact that strong immunity was developed in individuals who had recovered from cutaneous leishmaniasis (CL), leishmanization (LZ), a process of vaccination using live parasites, was effectively practiced in Western and Southwestern Asia.64 Subsequently, a large number of prophylactic vaccination trials were conducted using these parasites with a higher success rate.45 Similarly, these live parasites were assessed for their immunotherapeutic potential also. Mukhopadhyay et al65,66 evaluated the immunotherapeutic effects of live attenuated UR6 leishmanial parasite, which fails to develop the disease in infected hamster and BALB/c mice. It was observed that this parasite alone confers significant protection by reduction of parasite load in the spleen and the liver of these animal models, respectively. Later on, Datta et al67 assessed the therapeutic efficacy of attenuated (gamma-irradiated) L. donovani parasite in infected mice and reported the clearance of the parasites in the 100 Gy and 150 Gy treated infected mice, facilitated by the restoration of T-cell response and increased generation of nitric oxide and reactive oxygen species. Further studies on these attenuated parasites demonstrated that PDK1, PI3K, and p38MAPK signaling pathways are involved in the increased level of nitric oxide and decreased expression of TGF-β responsible for the predominant Th1-type response.68
Use of therapeutic vaccines in combination with adjuvant
Immune modulation via immunomodulatory agents plays a beneficial role in the regulation of chronic diseases that show either suboptimal or excessive immune response.27 These elements are required for inducing a very strong and long-lasting immune response (humoral as well as cellular), which makes them promising adjuvants for the control of VL. So far, for the immunoprophylaxis and immunotherapy of VL, plant-based immunomodulators (such as saponin, garlic extract, and so on) and nonvirulent bacteria-based adjuvants (eg, Bacillus Calmette–Guérin [BCG], Propionibacterium acnes, and so on) have been widely used.
A saponin formulation of FML, a glycoprotein complex that is expressed throughout the life cycle of L. donovani parasite, was found to be safe, protective, and immunogenic in experimental rodent (mouse and hamster) models and against canine VL, with significant prophylactic efficacy, as reviewed by Palatnik-de-Sousa.69 This formulation became the first commercial Leishmune veterinary vaccine, licensed after a series of canine VL field studies.70 The vaccine was further evaluated for its immunotherapeutic potential in mice and resulted in the reduction of parasitic burden in liver with significant lymphoproliferative response.71 Later, it was successfully evaluated therapeutically against both experimentally (L. donovani) and naturally (L. chagasi) infected dogs.72 Santos et al73 also investigated the therapeutic efficacy of Leishmune with increased concentrations of saponin in infected, seropositive, and symptomatic dogs with experimental canine VL, where they found a reduction in clinical outcome and modulation of the response toward the Th1 type. In another study, a 36 kDa glycoprotein – nucleoside hydrolase NH36 – in the FML complex was observed to be protective in mice as the native recombinant protein or as a bivalent DNA vaccine.74 When used along with garlic extract, it showed immunoprotection in L. chagasi-infected BALB/c mice, with a significant reduction in liver parasitic load and 100% survival.74
Likewise, Ferreira et al75 explored the therapeutic potential of recombinant cysteine proteinase from L. infantum chagasi (rLdccys1), in conjunction with P. acnes as adjuvant, which was earlier found to be protective in both canine and murine models. Dogs that underwent the treatment showed reduced parasitic load in spleen without increase in clinical symptoms, accompanied by enhanced levels of IFN-γ and immunoglobulin G2 (IgG2) antibodies.
Therapeutic vaccines in combination with antileishmanial drugs
Therapeutic vaccines, whole parasite (live attenuated/killed), soluble antigen, or recombinant antigen, when used either alone or with antileishmanial drugs, may be more effective in controlling a variety of VL forms. Assessment of the immunotherapeutic potential of autoclaved L. major (ALM) antigen mixed with Mycobacterium vaccae (as adjuvant), with or without meglumine antimoniate, against experimental canine VL revealed a reduction in the parasitic count, particularly in dogs treated with ALM with adjuvant and administered along with drug.76 The ALM vaccine was also evaluated therapeutically in PKDL patients in combination with antileishmanial drug in order to reduce the incidence of relapse. The patients receiving a weekly dose of alum-ALM + BCG with SSG showed significant healing, probably due to elevated IFN-γ production, thus modulating the patient’s immune system toward Th1 type.77 In another study, assessment of complete soluble antigen of attenuated parasites in combination with suboptimal dose of sodium antimony gluconate in Leishmania-infected BALB/c mice (both antimony sensitive and resistant) resulted in parasitic clearance, probably due to enhanced T-cell response.78
Using another approach, Ghosh et al79 assessed the therapeutic efficacy of soluble Leishmania antigen (SLA) entrapped in dendritic cell (DC), in combination with drugs (SSG) against murine VL. The results showed complete clearance of parasite load from both liver and spleen, suggesting that the antileishmanial potential of SLA was enhanced by DC-based immunotherapy or vice versa as they are inefficient in curing established infection when administered alone.
The understanding of VL pathogenesis suggested the possible reasons for the failure of whole-parasite vaccines (killed or live attenuated) as either they do not provide life-long immunity or their immunogenicity may get altered due to attenuation. Moreover, the complexity of the parasite makes it difficult to select suitable vaccine candidates.45 Various crude fractions of the parasite were, therefore, tested for their prophylactic potential so as to draw some conclusive results,45 and subsequently some of these fractions were also evaluated for their therapeutic potential. L. infantum-derived Fraction 2 (LiF2) antigen, either alone or in conjunction with chemotherapy, was evaluated in naturally infected dogs, wherein complete parasitological healing was observed in dogs treated with combination therapy but the absence of clinical outcomes was not correlated with it.80 Furthermore, over the past few years, the soluble and other fractions of parasites were evaluated through various proteomic tools, resulting in the identification of various proteins that were tested as suitable vaccine candidates against VL.81 Leishmune enriched with saponin was used in combination with allopurinol or amphotericin B against naturally infected dogs, which resulted in the control of disease, rendering most dogs asymptomatic. This suggested that combining chemotherapy with vaccine helps in disease elimination in dogs.82
Immunization with glucose-regulated protein GRP78 antigen (a 78 kDa Ca2+-binding chaperone molecule) of Leishmania, in combination with various adjuvants, imparts optimum protection with monophosphoryl lipid A (MPLA).83 Encouraged with these results, the therapeutic potential of recombinant GRP78 with MPLA was further assessed in L. donovani-infected mice in combination with cisplatin, which is a platinum-based anticancer drug that has also shown antileishmanial activity.84 The treatment resulted in marked parasitic inhibition, with increased delayed type hypersensitivity response and elevated IFN-γ production.85
Later on, it was thought that a multicomponent/polyprotein vaccine would elicit an enhanced protective response as it overcomes the genetic variability of the mammalian immune system.86 Moreover, it would have lower manufacturing cost and more straightforward quality control testing.47 Therefore, various polyprotein vaccines were developed and evaluated for their therapeutic potential. Leish-111f, in formulation with monophosphoryl lipid A plus squalene emulsion (MPL-SE) (Leish-111f + MPL-SE), failed to prevent naturally infected canine VL cases,43 although it was found to be protective in mouse models of CL and VL. However, an immunotherapeutic evaluation of Leish-111f + MPL-SE in combination with N-methyl meglumine antimoniate (Glucantime) was studied by Miret et al87 in natural L. chagasi-infected dogs, wherein evaluation of immunological (both humoral and cellular) responses revealed improvement of the clinical picture in dogs, both in the chemotherapy-alone and immunochemotherapy cohorts, with reduced death. In another study, two separate trials (open and blinded) to evaluate the therapeutic efficacy of Leish-111f + MPL-SE with or without glucantime in naturally infected canine VL cases, were performed by Trigo et al.88 In the open trial, there was 75% efficacy in canine VL cases receiving a combination of Leish-111f + MPL-SE + glucantime, when monitored up to 36 months posttherapy. This efficacy was subsequently confirmed in a blinded trial. Results of these trials suggested that well-characterized polyprotein vaccines, as an adjunct, can proficiently boost the current chemotherapy efficiency against canine VL in lower doses. In addition to these approaches, Seifert et al89 studied the immunotherapeutic role of novel T-cell epitope-based DNA vaccine (LEISHDNAVAX) along with AmBisome in experimental VL, whereby they found significant reduction in splenic parasite burden, with no difference in the numbers of CD3+ cells.
The higher efficacy of combined therapy, as evident from experimental and human studies, is possibly due to the consequence of drug-induced reduction in the parasite load, aided by the stimulation of sensitized T-lymphocytes by specific mitogenic antigens, ultimately leading to the production of lymphokines, which in turn further induced the activation of macrophages for killing the residual parasites in the hosts.80
Delivery of therapeutic vaccines using carrier systems
Due to severe suppression of cell-mediated immune response in active VL cases, there is a strong need to have immunostimulatory formulations containing materials such as additives, which can generate a strong cell-mediated immune response to the already delivered insufficiently immunogenic antigens to provide optimum therapeutic efficacy against this disease. Of the various vaccine-delivery vehicles being developed to date, liposomes, nondegradable nanoparticles, viral vectors, and virus-like particles have been shown to generate cellular immunity, thus becoming important carrier systems.
Liposomes are phospholipid vesicles that have been evaluated both as adjuvant and as vehicles for antigens. Versatility and plasticity are the key features of liposome-based vaccine delivery systems.90 In particular, cationic liposomes offer long-acting depot effect at the site of injection with strong Th1- and Th17-mediated immunostimulatory effect. Although many studies on the liposomized delivery of prophylactic antigens against VL have been carried out, very few reports are available for therapeutic vaccines.91 In one such study, SLA, entrapped in positively charged liposomes, was used against Leishmania-infected BALB/c mice, wherein it showed 90% parasite clearance from liver and spleen, both in conjunction with production of Th1 cytokines (IFN-γ and IL-12).92 These studies showed that liposomized SLA-based immunotherapy significantly reduced parasite burden through the switching of immune responses toward Th1 type.
Recently, Maroof et al93 developed an adenovirus-based vaccine (Ad5-KH), which was assessed for its therapeutic efficacy in L. donovani-infected BALB/c mice. Adenovirus offers a frequent viral-vector platform for several vaccines against Alzheimer’s disease, influenza, tetanus, and HIV infection.44 Results showed moderate inhibition of splenic parasitic burden, with increased humoral and cellular (CD8+ T-cell) responses following a single vaccination. These observations indicated the utility of the adenoviral vector, which can deliver leishmanial antigens into Leishmania-infected animals in a better protective manner.93
Future perspectives and conclusion
Because VL is a complex heterogeneous disease, with its pathology well associated with various innate and adaptive immune mechanisms, it warrants designing a unified strategy aimed at targeting different mediators and pathways in a comprehensive manner. This necessitates the complete understanding of the actual immunological mechanisms involved in the control of disease, which can ultimately facilitate the development of effective therapeutic vaccines and help in achieving VL elimination.94 Of all the therapeutic vaccination protocols evaluated so far with encouraging results in experimental human VL, none has been translated clinically.In order to have successful therapeutic vaccines against human VL, the following points, as depicted in Figure 1, need to be addressed:
- An understanding of VL disease pathology, along with the underlying basic mechanisms of immunosuppression, is required for searching for novel immunotherapeutic vaccine targets against specific affected regions of the host in established infection. This would necessitate application of genomics, proteomics, and computational modeling to identify/design potential proteins/epitopes as vaccine targets. Immunogens with ability to potentiate Th1-type cellular immune response would be the most ideal ones.45,95
- For poorly immunogenic molecular vaccines, adjuvants and specific formulation(s) or vector system(s) with enhanced immunogenicity have to be developed for inducing strong immune responses.
- There is a need for an impeccable experimental model to minimize the variations in experimental results, such as classical cytokine measurement and parasite burden, which can be extrapolated to human settings (patients).
- Development of a standardized operating procedure, including pharmacokinetic considerations such as assessment of the dose and duration of therapy, for evaluation and comparison of the immunotherapeutic benefits of all emerging strategies is needed. This would ultimately reduce the huge variations in readouts from different immunological assays.
- Identification of immunological biomarkers for assessment of the efficacy of the vaccines is required.
- There is the need to address several issues regarding the mechanism of action as well as the safety and efficacy considerations of therapeutic vaccines.
- Last but not the least, more studies are required on the management of cases of asymptomatic, PKDL, as well as HIV–VL coinfections through therapeutic vaccines as they serve as silent reservoirs in endemic areas and, as such, will hamper the sustainability of the success of ongoing VL elimination programs. Lessons needs to be learnt from the experience of smallpox and polio eradication for complete elimination of VL.
 | Figure 1 Roadmap for development of therapeutic vaccines against VL. Abbreviations: PKDL, post kala azar dermal leishmaniasis; VL, visceral leishmaniasis. |
The prospects for therapeutic vaccines against infectious diseases are better today than ever before. So far, ten therapeutic vaccines are in the offing for humans, particularly against cancer96 (cancer of the brain, cervix, skin, lung, breast, head and neck, and pancreas), celiac disease,97 and recurrent vulvovaginal candidiasis.98 Furthermore, some of the chronic viral infections, importantly infections with hepatitis B virus, human papilloma virus, HIV, and so on, conceptually may present a suitable target for active specific immunotherapy. These immunotherapeutic interventions represent an effective strategy in controlling the chronic infections in days to come.99 Thus, the continuous concerted efforts in academic research will eventually lead to the development of efficient therapeutic vaccine(s) against VL.
Acknowledgment
This manuscript bears CSIR-CDRI communication number 9309.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. 2010. Control of the leishmaniases: Report of a meeting of the WHO Expert Committee on the Control of Leishmaniasis, Geneva, 22–26 March 2010. WHO Technical Report Series, no. 949. WHO, Geneva, Switzerland. Available from: http://apps.who.int/iris/bitstream/10665/44412/1/WHO_TRS_949_eng.pdf. Accessed July 26, 2016. | ||
Singh VP, Ranjan A, Topno RK, et al. Estimation of under-reporting of visceral leishmaniasis cases in Bihar, India. Am J Trop Med Hyg. 2010;82(1):9–11. | ||
Singh SP, Reddy DC, Rai M, Sundar S. Serious underreporting of visceral leishmaniasis through passive case reporting in Bihar, India. Trop Med Int Health. 2006;11(6):899–905. | ||
Alvar J, Velez ID, Bern C, et al; WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. PLoS One. 2012;7(5):e35671. | ||
Ready PD. Epidemiology of visceral leishmaniasis. Clin Epidemiol. 2014;6:147–154. | ||
Rodrigues V, Cordeiro-da-Silva A, Laforge M, Silvestre R, Estaquier J. Regulation of immunity during visceral Leishmania infection. Parasit Vectors. 2016;9(1):118. | ||
Diro E, Lynen L, Ritmeijer K, Boelaert M, Hailu A, van Griensven J. Visceral leishmaniasis and HIV coinfection in East Africa. PLoS Negl Trop Dis. 2014;8(6):e2869. | ||
Singh OP, Hasker E, Sacks D, Boelaert M, Sundar S. Asymptomatic Leishmania infection: a new challenge for Leishmania control. Clin Infect Dis. 2014;58(10):1424–1429. | ||
Zijlstra EE, Musa AM, Khalil EA, el-Hassan IM, el-Hassan AM. Post-kala-azar dermal leishmaniasis. Lancet Infect Dis. 2003;3(2):87–98. | ||
Desjeux P, Ghosh RS, Dhalaria P, Strub-Wourgaft N, Zijlstra EE. Report of the post kala-azar dermal leishmaniasis (PKDL) consortium meeting, New Delhi, India, 27–29 June 2012. Parasit Vectors. 2013;6:196. | ||
Lindoso Jé A, Cota Gá F, da Cruz AM, et al. Visceral leishmaniasis and HIV coinfection in Latin America. PLoS Negl Trop Dis. 2014;8(9):e3136. | ||
Cruz I, Nieto J, Moreno J, Canavate C, Desjeux P, Alvar J. Leishmania/HIV co-infections in the second decade. Indian J Med Res. 2006;123(3):357–388. | ||
Mondain-Miton V, Toussaint-Gari M, Hofman P, et al. Atypical leishmaniasis in a patient infected with human immunodeficiency virus. Clin Infect Dis. 1995;21(3):663–665. | ||
Monge-Maillo B, Norman FF, Cruz I, Alvar J, Lopez-Velez R. Visceral leishmaniasis and HIV coinfection in the Mediterranean region. PLoS Negl Trop Dis. 2014;8(8):e3021. | ||
Gidwani K, Rai M, Chakravarty J, Boelaert M, Sundar S. Evaluation of leishmanin skin test in Indian visceral leishmaniasis. Am J Trop Med Hyg. 2009;80(4):566–567. | ||
Sacks DL, Lal SL, Shrivastava SN, Blackwell J, Neva FA. An analysis of T cell responsiveness in Indian kala-azar. J Immunol. 1987;138(3):908–913. | ||
Caldas A, Favali C, Aquino D, et al. Balance of IL-10 and interferon-gamma plasma levels in human visceral leishmaniasis: implications in the pathogenesis. BMC Infect Dis. 2005;5:113. | ||
Singh OP, Gidwani K, Kumar R, et al. Reassessment of immune correlates in human visceral leishmaniasis as defined by cytokine release in whole blood. Clin Vaccine Immunol. 2012;19(6):961–966. | ||
Faleiro RJ, Kumar R, Hafner LM, Engwerda CR. Immune regulation during chronic visceral leishmaniasis. PLoS Negl Trop Dis. 2014;8(7):e2914. | ||
Rai AK, Thakur CP, Singh A, et al. Regulatory T cells suppress T cell activation at the pathologic site of human visceral leishmaniasis. PLoS One. 2012;7(2):e31551. | ||
Nylen S, Maurya R, Eidsmo L, Manandhar KD, Sundar S, Sacks D. Splenic accumulation of IL-10 mRNA in T cells distinct from CD4+CD25+ (Foxp3) regulatory T cells in human visceral leishmaniasis. J Exp Med. 2007;204(4):805–817. | ||
Buxbaum LU, Scott P. Interleukin 10- and Fcgamma receptor-deficient mice resolve Leishmania mexicana lesions. Infect Immun. 2005;73(4):2101–2108. | ||
Ismail A, El Hassan AM, Kemp K, et al. Immunopathology of post kala-azar dermal leishmaniasis (PKDL): T-cell phenotypes and cytokine profile. J Pathol. 1999;189(4):615–622. | ||
Gasim S, Elhassan AM, Kharazmi A, Khalil EA, Ismail A, Theander TG. The development of post-kala-azar dermal leishmaniasis (PKDL) is associated with acquisition of Leishmania reactivity by peripheral blood mononuclear cells (PBMC). Clin Exp Immunol. 2000;119(3):523–529. | ||
Katara GK, Ansari NA, Verma S, Ramesh V, Salotra P. Foxp3 and IL-10 expression correlates with parasite burden in lesional tissues of post kala azar dermal leishmaniasis (PKDL) patients. PLoS Negl Trop Dis. 2011;5(5):e1171. | ||
Garg R, Dube A. Animal models for vaccine studies for visceral leishmaniasis. Indian J Med Res. 2006;123(3):439–454. | ||
Kumar R, Nylen S. Immunobiology of visceral leishmaniasis. Front Immunol. 2012;3:251. | ||
Engwerda CR, Ato M, Kaye PM. Macrophages, pathology and parasite persistence in experimental visceral leishmaniasis. Trends Parasitol. 2004;20(11):524–530. | ||
Nylen S, Gautam S. Immunological perspectives of leishmaniasis. J Glob Infect Dis. 2010;2(2):135–146. | ||
Melby PC, Chandrasekar B, Zhao W, Coe JE. The hamster as a model of human visceral leishmaniasis: progressive disease and impaired generation of nitric oxide in the face of a prominent Th1-like cytokine response. J Immunol. 2001;166(3):1912–1920. | ||
Sartori A, Roque-Barreira MC, Coe J, Campos-Neto A. Immune complex glomerulonephritis in experimental kala-azar. II: detection and characterization of parasite antigens and antibodies eluted from kidneys of Leishmania donovani-infected hamsters. Clin Exp Immunol. 1992;87(3):386–392. | ||
Alvar J, Canavate C, Molina R, Moreno J, Nieto J. Canine leishmaniasis. Adv Parasitol. 2004;57:1–88. | ||
Lage RS, Oliveira GC, Busek SU, et al. Analysis of the cytokine profile in spleen cells from dogs naturally infected by Leishmania chagasi. Vet Immunol Immunopathol. 2007;115(1–2):135–145. | ||
Quinnell RJ, Courtenay O, Shaw MA, et al. Tissue cytokine responses in canine visceral leishmaniasis. J Infect Dis. 2001;183(9):1421–1424. | ||
Bourdoiseau G, Bonnefont C, Hoareau E, Boehringer C, Stolle T, Chabanne L. Specific IgG1 and IgG2 antibody and lymphocyte subset levels in naturally Leishmania infantum-infected treated and untreated dogs. Vet Immunol Immunopathol. 1997;59(1–2):21–30. | ||
Baneth G, Koutinas AF, Solano-Gallego L, Bourdeau P, Ferrer L. Canine leishmaniosis – new concepts and insights on an expanding zoonosis: part one. Trends Parasitol. 2008;24(7):324–330. | ||
Samant M, Gupta R, Kumari S, et al. Immunization with the DNA-encoding N-terminal domain of proteophosphoglycan of Leishmania donovani generates Th1-type immunoprotective response against experimental visceral leishmaniasis. J Immunol. 2009;183(1):470–479. | ||
Basu R, Bhaumik S, Basu JM, Naskar K, De T, Roy S. Kinetoplastid membrane protein-11 DNA vaccination induces complete protection against both pentavalent antimonial-sensitive and -resistant strains of Leishmania donovani that correlates with inducible nitric oxide synthase activity and IL-4 generation: evidence for mixed Th1- and Th2-like responses in visceral leishmaniasis. J Immunol. 2005;174(11):7160–7171. | ||
Savoia D. Recent updates and perspectives on leishmaniasis. J Infect Dev Ctries. 2015;9(6):588–596. | ||
Sundar S, Singh A, Singh OP. Strategies to overcome antileishmanial drugs unresponsiveness. Re Dai Yi Xue Za Zhi. 2014;2014:646932. | ||
Engwerda CR, Matlashewski G. Development of Leishmania vaccines in the era of visceral leishmaniasis elimination. Trans R Soc Trop Med Hyg. 2015;109(7):423–424. | ||
Chappuis F, Sundar S, Hailu A, et al. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat Rev Microbiol. 2007;5(11):873–882. | ||
Kumar R, Engwerda C. Vaccines to prevent leishmaniasis. Clin Transl Immunol. 2014;3:e13. | ||
Mohan T, Verma P, Rao DN. Novel adjuvants & delivery vehicles for vaccines development: a road ahead. Indian J Med Res. 2013;138(5):779–795. | ||
Joshi S, Rawat K, Yadav NK, Kumar V, Siddiqi MI, Dube A. Visceral leishmaniasis: advancements in vaccine development via classical and molecular approaches. Front Immunol. 2014;5:380. | ||
Gradoni L. Canine Leishmania vaccines: still a long way to go. Vet Parasitol. 2015;208(1–2):94–100. | ||
Skeiky YA, Coler RN, Brannon M, et al. Protective efficacy of a tandemly linked, multi-subunit recombinant leishmanial vaccine (Leish-111f) formulated in MPL adjuvant. Vaccine. 2002;20(27–28):3292–3303. | ||
Nascimento E, Fernandes DF, Vieira EP, et al. A clinical trial to evaluate the safety and immunogenicity of the LEISH-F1+MPL-SE vaccine when used in combination with meglumine antimoniate for the treatment of cutaneous leishmaniasis. Vaccine. 2010;28(40):6581–6587. | ||
Llanos-Cuentas A, Calderon W, Cruz M, et al. A clinical trial to evaluate the safety and immunogenicity of the LEISH-F1+MPL-SE vaccine when used in combination with sodium stibogluconate for the treatment of mucosal leishmaniasis. Vaccine. 2010;28(46):7427–7435. | ||
Bertholet S, Goto Y, Carter L, et al. Optimized subunit vaccine protects against experimental leishmaniasis. Vaccine. 2009;27(50):7036–7045. | ||
Coler RN, Duthie MS, Hofmeyer KA, et al. From mouse to man: safety, immunogenicity and efficacy of a candidate leishmaniasis vaccine LEISH-F3+GLA-SE. Clin Transl Immunol. 2015;4(4):e35. | ||
Jain K, Jain NK. Vaccines for visceral leishmaniasis: a review. J Immunol Methods. 2015;422:1–12. | ||
Sela M, Hilleman MR. Therapeutic vaccines: realities of today and hopes for tomorrow. Proc Natl Acad Sci U S A. 2004;101(suppl 2):14559. | ||
Gupta G, Oghumu S, Satoskar AR. Mechanisms of immune evasion in leishmaniasis. Adv Appl Microbiol. 2013;82:155–184. | ||
Bachmann MF, Dyer MR. Therapeutic vaccination for chronic diseases: a new class of drugs in sight. Nat Rev Drug Discov. 2004;3(1):81–88. | ||
Mylvaganam GH, Silvestri G, Amara RR. HIV therapeutic vaccines: moving towards a functional cure. Curr Opin Immunol. 2015;35:1–8. | ||
Groschel MI, Prabowo SA, Cardona PJ, Stanford JL, van der Werf TS. Therapeutic vaccines for tuberculosis – a systematic review. Vaccine. 2014;32(26):3162–3168. | ||
Beaumier CM, Gillespie PM, Strych U, Hayward T, Hotez PJ, Bottazzi ME. Status of vaccine research and development of vaccines for Chagas disease. Vaccine. 2016;34(26):2996–3000. | ||
Kim TJ, Jin HT, Hur SY, et al. Clearance of persistent HPV infection and cervical lesion by therapeutic DNA vaccine in CIN3 patients. Nat Commun. 2014;5:5317. | ||
Melero I, Gaudernack G, Gerritsen W, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11(9):509–524. | ||
Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Invest. 2015;125(9):3401–3412. | ||
Roatt BM, Aguiar-Soares RD, Coura-Vital W, et al. Immunotherapy and immunochemotherapy in visceral leishmaniasis: promising treatments for this neglected disease. Front Immunol. 2014;5:272. | ||
Okwor I, Uzonna JE. Immunotherapy as a strategy for treatment of leishmaniasis: a review of the literature. Immunotherapy. 2009;1(5):765–776. | ||
Mutiso JM, Macharia JC, Kiio MN, Ichagichu JM, Rikoi H, Gicheru MM. Development of Leishmania vaccines: predicting the future from past and present experience. J Biomed Res. 2013;27(2):85–102. | ||
Mukhopadhyay S, Bhattacharyya S, Majhi R, et al. Use of an attenuated leishmanial parasite as an immunoprophylactic and immunotherapeutic agent against murine visceral leishmaniasis. Clin Diagn Lab Immunol. 2000;7(2):233–240. | ||
Mukhopadhyay S, Sen P, Bhattacharyya S, Majumdar S, Roy S. Immunoprophylaxis and immunotherapy against experimental visceral leishmaniasis. Vaccine. 1999;17(3):291–300. | ||
Datta S, Manna M, Khanra S, et al. Therapeutic immunization with radio-attenuated Leishmania parasites through i.m. route revealed protection against the experimental murine visceral leishmaniasis. Parasitol Res. 2012;111(1):361–369. | ||
Datta S, Roy S, Manna M. Therapy with radio-attenuated vaccine in experimental murine visceral leishmaniasis showed enhanced T cell and inducible nitric oxide synthase levels, suppressed tumor growth factor-beta production with higher expression of some signaling molecules. Braz J Infect Dis. 2015;19(1):36–42. | ||
Palatnik-de-Sousa CB. Vaccines for leishmaniasis in the fore coming 25 years. Vaccine. 2008;26(14):1709–1724. | ||
Palatnik-de-Sousa CB. Vaccines for canine leishmaniasis. Front Immunol. 2012;3:69. | ||
Santos WR, Aguiar IA, Paraguai de Souza E, de Lima VM, Palatnik M, Palatnik-de-Sousa CB. Immunotherapy against murine experimental visceral leishmaniasis with the FML-vaccine. Vaccine. 2003;21(32):4668–4676. | ||
Borja-Cabrera GP, Cruz Mendes A, Paraguai de Souza E, et al. Effective immunotherapy against canine visceral leishmaniasis with the FML-vaccine. Vaccine. 2004;22(17–18):2234–2243. | ||
Santos FN, Borja-Cabrera GP, Miyashiro LM, et al. Immunotherapy against experimental canine visceral leishmaniasis with the saponin enriched-Leishmune vaccine. Vaccine. 2007;25(33):6176–6190. | ||
Gamboa-Leon R, Paraguai de Souza E, Borja-Cabrera GP, et al. Immunotherapy against visceral leishmaniasis with the nucleoside hydrolase-DNA vaccine of Leishmania donovani. Vaccine. 2006;24(22):4863–4873. | ||
Ferreira JH, Silva Ldos S, Longo-Maugeri IM, Katz S, Barbieri CL. Use of a recombinant cysteine proteinase from Leishmania (Leishmania) infantum chagasi for the immunotherapy of canine visceral leishmaniasis. PLoS Negl Trop Dis. 2014;8(3):e2729. | ||
Jamshidi S, Avizeh R, Mohebali M, Bokaie S. Immunotherapy using autoclaved L. major antigens and M. vaccae with meglumine antimoniate, for the treatment of experimental canine visceral leishmaniasis. Iran J Parasitol. 2011;6(3):26–34. | ||
Musa AM, Khalil EA, Mahgoub FA, et al. Immunochemotherapy of persistent post-kala-azar dermal leishmaniasis: a novel approach to treatment. Trans R Soc Trop Med Hyg. 2008;102(1):58–63. | ||
Bhaumik SK, Naskar K, De T. Complete protection against experimental visceral leishmaniasis with complete soluble antigen from attenuated Leishmania donovani promastigotes involves Th1-immunity and down-regulation of IL-10. Eur J Immunol. 2009;39(8):2146–2160. | ||
Ghosh M, Pal C, Ray M, Maitra S, Mandal L, Bandyopadhyay S. Dendritic cell-based immunotherapy combined with antimony-based chemotherapy cures established murine visceral leishmaniasis. J Immunol. 2003;170(11):5625–5629. | ||
Neogy AB, Vouldoukis I, da Costa JM, Monjour L. Exploitation of parasite-derived antigen in therapeutic success against canine visceral leishmaniosis. Veterinary Group of Lupino. Vet Parasitol. 1994;54(4):367–373. | ||
Coler RN, Reed SG. Second-generation vaccines against leishmaniasis. Trends Parasitol. 2005;21(5):244–249. | ||
Borja-Cabrera GP, Santos FN, Santos FB, et al. Immunotherapy with the saponin enriched-Leishmune vaccine versus immunochemotherapy in dogs with natural canine visceral leishmaniasis. Vaccine. 2010;28(3):597–603. | ||
Nagill R, Kaur S. Enhanced efficacy and immunogenicity of 78kDa antigen formulated in various adjuvants against murine visceral leishmaniasis. Vaccine. 2010;28(23):4002–4012. | ||
Kaur S, Sachdeva H, Dhuria S, Sharma M, Kaur T. Antileishmanial effect of cisplatin against murine visceral leishmaniasis. Parasitol Int. 2010;59(1):62–69. | ||
Joshi J, Kaur S. To investigate the therapeutic potential of immunochemotherapy with cisplatin + 78 kDa + MPL-A against Leishmania donovani in BALB/c mice. Parasite Immunol. 2014;36(1):3–12. | ||
Goto Y, Bhatia A, Raman VS, et al. KSAC, the first defined polyprotein vaccine candidate for visceral leishmaniasis. Clin Vaccine Immunol. 2011;18(7):1118–1124. | ||
Miret J, Nascimento E, Sampaio W, et al. Evaluation of an immunochemotherapeutic protocol constituted of N-methyl meglumine antimoniate (Glucantime) and the recombinant Leish-110f + MPL-SE vaccine to treat canine visceral leishmaniasis. Vaccine. 2008;26(12):1585–1594. | ||
Trigo J, Abbehusen M, Netto EM, et al. Treatment of canine visceral leishmaniasis by the vaccine Leish-111f+MPL-SE. Vaccine. 2010;28(19):3333–3340. | ||
Seifert K, Juhls C, Salguero FJ, Croft SL. Sequential chemoimmunotherapy of experimental visceral leishmaniasis using a single low dose of liposomal amphotericin B and a novel DNA vaccine candidate. Antimicrob Agents Chemother. 2015;59(9):5819–5823. | ||
Schwendener RA. Liposomes as vaccine delivery systems: a review of the recent advances. Ther Adv Vaccines. 2014;2(6):159–182. | ||
Das A, Ali N. Vaccine development against Leishmania donovani. Front Immunol. 2012;3:99. | ||
Bhowmick S, Ravindran R, Ali N. Leishmanial antigens in liposomes promote protective immunity and provide immunotherapy against visceral leishmaniasis via polarized Th1 response. Vaccine. 2007;25:6544–6556. | ||
Maroof A, Brown N, Smith B, et al. Therapeutic vaccination with recombinant adenovirus reduces splenic parasite burden in experimental visceral leishmaniasis. J Infect Dis. 2012;205(5):853–863. | ||
Khamesipour A. Therapeutic vaccines for leishmaniasis. Expert Opin Biol Ther. 2014;14(11):1641–1649. | ||
Seyed N, Taheri T, Rafati S. Post-genomics and vaccine improvement for Leishmania. Front Microbiol. 2016;7:467. | ||
Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang XY. Therapeutic cancer vaccines: past, present and future. Adv Cancer Res. 2013;119:421–475. | ||
Bakshi A, Stephen S, Borum ML, Doman DB. Emerging therapeutic options for celiac disease: potential alternatives to a gluten-free diet. Gastroenterol Hepatol. 2012;8(9):582–588. | ||
De Bernardis F, Arancia S, Sandini S, Graziani S, Norelli S. Studies of immune responses in candida vaginitis. Pathogens. 2015;4(4):697–707. | ||
Moingeon P, Almond J, de Wilde M. Therapeutic vaccines against infectious diseases. Curr Opin Microbiol. 2003;6(5):462–471. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.