Back to Journals » OncoTargets and Therapy » Volume 13
Management of Refractory Pediatric Sarcoma: Current Challenges and Future Prospects
Authors Pushpam D, Garg V, Ganguly S ![]() , Biswas B
, Biswas B ![]()
Received 4 February 2020
Accepted for publication 12 May 2020
Published 8 June 2020 Volume 2020:13 Pages 5093—5112
DOI https://doi.org/10.2147/OTT.S193363
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Deepam Pushpam,1,* Vikas Garg,1,* Sandip Ganguly,2 Bivas Biswas2
1Department of Medical Oncology, AIIMS, New Delhi, India; 2Department of Medical Oncology, Tata Medical Center, Kolkata, India
*These authors contributed equally to this work
Correspondence: Bivas Biswas
Department of Medical Oncology, Tata Medical Center, 14 MAR [EW] New Town, Rajarhat, Kolkata 700160, India
Tel +91033 6605 7628
Email [email protected]
Abstract: Paediatric sarcomas are a heterogeneous group of disorders constituting bone sarcoma and various soft tissue sarcomas. Almost one-third of these presents with metastasis at baseline and another one-third recur after initial curative treatment. There is a huge unmet need in this cohort in terms of curative options and/or prolongation of survival. In this review, we have discussed the current treatment options, challenges and future strategies of managing relapsed/refractory paediatric sarcomas. Upfront risk-adapted treatment with multidisciplinary management remains the main strategy to prevent future recurrence or relapse of the disease. In the case of limited local and/or systemic relapse or late relapse, initial multimodality management can be administered. In treatment-refractory cases or where cure is not feasible, the treatment options are limited to novel therapeutics, immunotherapeutic approach, targeted therapies, and metronomic therapies. A better understanding of disease biology, mechanism of treatment refractoriness, identifications of driver mutation, the discovery of novel targeted therapies, cellular vaccine and adapted therapies should be explored in relapsed/refractory cases. Close national and international collaboration for translation research is needed to fulfil the unmet need.
Keywords: paediatric sarcoma, immunotherapy, targeted therapy, relapsed sarcoma, osteosarcoma, disease biology
Introduction
Paediatric sarcoma constitutes a major group of disease that includes osteosarcoma (OGS), Ewing’s sarcoma/primitive neuroectodermal tumor (EWS/PNET), rhabdomyosarcoma (RMS), and heterogeneous group of soft tissue sarcoma (STS), mostly infantile fibrosarcoma, malignant peripheral nerve sheath tumour (MPNST), termed as non-rhabdomyomatous soft tissue sarcoma (NRSTS). With the introduction of multimodality treatment including poly-chemotherapy, surgery and/or radiotherapy, the outcome of localized paediatric sarcoma has improved drastically. However, 20–30% localized sarcoma recurs where cure seldom occurs. On the other hand, a sizeable number of bone1 and soft tissue sarcoma2 presents with metastasis at baseline, and the cure rate is still low with the current armamentarium of systemic anti-cancer therapies. In spite of progress in chemotherapeutic combinations, newer surgical approach and radiation techniques, local as well as distant recurrence is one of the major challenges in the management of paediatric sarcomas. The outcome remains dismal in recurrent or treatment-refractory (primary or secondary) setting with the currently available systemic therapies even with a better understanding of tumorigenesis and discovery of potential therapeutic targets.
In-depth understanding of disease biology, determination of molecular signature, identifications of potentially targetable driver mutation, newer immunotherapeutic approach, risk-adapted treatment strategies, and long-term maintenance therapy remains the optimal approach to treat recurrent and refractory paediatric sarcomas.
In this review, we have discussed the current standard of care in recurrent bone sarcoma & STS in paediatric & young adult patients with emerging therapeutic options, unmet needs and future strategies.
Burden of Problems
Data regarding burden of refractory paediatric sarcoma can be estimated from the patients where cure is not possible as literature regarding the same is limited. There is no consensus regarding optimal treatment in second line as various regimens have been tried and there is no head-to-head comparison amongst the different regimens.
Survival in OGS has plateaued at 70% after the dramatic improvement in the 1980s and 1990s.3 In a large retrospective analysis of 1067 patients, there were 564 (52.85%) recurrences with a median time of 13 months. Post recurrence survival was 18% at 5 years. Approximately 35% had refractory disease.4
For localized EWS, the 5-year overall survival ranges from 65% to 75% whereas in metastatic setting it is less than 30%, except in isolated pulmonary metastasis (approximately 50%).5 The 5-year survival of relapsed EWS is 13%. Patients who relapse in the first 2 years from diagnosis have a 5-year survival of 7% compared to 29% who relapse after 2 years from diagnosis.6 At present, there is no standard protocol to treat relapsed EWS due to paucity of clinical trials.7
In pediatric populations, the most common soft tissue sarcoma is RMS, which accounts for one-half of all soft tissue sarcomas. Relapses occur in around 30% cases of RMS, with most relapses occurring before 2 years after initial diagnosis whereas late relapses (after 5 years of diagnosis) are rare and constitute less than 10% of relapsed cases. Most relapses (80%) occur at distant site and involve lungs, bones or bone marrow. Few patients (20%) relapse at the local site and have a more favourable prognosis.8–11 Prognostic factors associated with recurrent or progressive disease include factors at the time of initial diagnosis, including histological subtype, disease group, stage, age 10 years or younger, size, site, and lack of initial radiation therapy. Distant site of recurrence and time of relapse after diagnosis <18 months are important prognostic factors for survival at the time of relapse.8–10,12
In a retrospective analysis by Intergroup Rhabdomyosarcoma Study Group (IRSG), 605 (25.6%) patients out of 2364 treated on IRS-III, IRS-IV pilot, & IRS-IV experienced disease relapse or progression. Median OS was 9.6 months and 17% of patients survived till 5 years after relapse. Probability of survival after relapse was dependent on histologic subtype, disease group, and stage at initial diagnosis. At the time of relapse distant site recurrence have poor survival compared with local recurrence.8
Of 1398 patients with localized disease at diagnosis treated in International Society of Paediatric Oncology (SIOP) Malignant Mesenchymal Tumour/MMT 84, 89, and 95 studies, 474 (33.9%) had a relapse. Metastatic disease at relapse, prior treatment, initial tumour size >5 cm, and relapse within 18 months of diagnosis were associated with poor outcomes.10 Many other retrospective analyses have found similar prognostic factors for survival.12
NRSTS accounts for approximately 50% of paediatric STS (13). It is a heterogenous group of rare tumours, with common histologies being synovial cell sarcoma, infantile fibrosarcoma and malignant peripheral nerve sheath tumour. Five-year survival from synovial sarcoma has been approximately 77%.13 Survival of infantile fibrosarcoma ranges from 80% to 94%.14,15 Malignant peripheral nerve sheath tumour is relatively chemoresistant. The five-year survival has ranged from 23% to 69%.13
Pathophysiology of Treatment Refractoriness
Pathophysiology of treatment refractoriness is an evolving area of research and there are a lot of knowledge gaps. The studies have focussed on cancer stem cells (CSC), signalling pathways and multidrug resistance. OGS treatment refractoriness has been relatively well studied compared to others.
Gibbs et al16 demonstrated CSC in OGS which has the ability to self-renew, form sarcospheres and were multipotent. These cells had high expression of CD 133, CD 117 and Stro-1.17,18 In vitro studies in OGS cell lines and xenograft mouse model has shown that exposure to chemotherapy transforms a subpopulation of OGS cells to CSC like phenotype.19 These transformed cells have increased the capacity to form sarcospheres in vitro and initiation of tumour in vivo.20 These transformed cells are chemoresistant and are found in refractory OGS.21
It is thought that Wnt-β-catenin signalling pathway plays a key role in the development of OGS and chemotherapy resistance. Increased cytoplasmic expression β-catenin predicts lung metastasis in murine OGS cell line.22 Small interfering RNA (siRNA) mediated silencing of β-catenin leads to chemoresistance to doxorubicin mediated by N-kappaB, inhibition of invasion and motility in human OGS cell line.23 Increased expression of TWIST decreases β-catenin via PI3K pathway leading to cisplatin sensitivity in vitro.24 Knocking down of β-catenin results in methotrexate sensitivity in OGS cell line.25 Apart from Wnt-β-catenin pathway, Hedgehog pathway, Notch, MAP kinase and FGF signalling pathway play a role in maintaining OGS CSC.26
Multidrug resistance in OGS is mediated through different mechanisms. ATP-binding cassette transporters play a key role in drug uptake and transport. Intracellular concentration of the drug is decreased by P-glycoprotein (P-gp), a membrane efflux pump.
P-gp is associated with multidrug resistance in OGS cell lines.27 High P-gp levels have been associated with poor event-free survival and increase the risk of adverse events.28 P-gp has a potential to be used as a biomarker for risk stratification in OGS. Polymorphisms in multidrug resistance-associated protein 2 have been associated with necrosis, methotrexate resistance, myelosuppression and cardiotoxicity.29
Methotrexate is one of the key drugs in OGS and it enters into cells through reduced folate carrier (RFC). Reduced RFC expression is associated with poor histologic response.30,31 Low levels of DNA topoisomerase II β are associated with multidrug resistance in OGS cell line.31 Human glutathione S-transferase P1 (GSTP1) overexpression is associated with poor histological response and prognosis, as it metabolizes chemotherapeutic agents.32,33 There is evidence to suggest that OGS cells develop resistance due to the ability to repair the DNA damaged by cisplatin. Excision repair cross-complementing (ERCC) polymorphisms were associated with event-free survival (EFS) in OGS patients.34–36 In a meta-analysis of 858 OGS patients, ERCC2 polymorphism Lys751Gln was associated with OS and His46His mutation was associated with EFS.36 A key enzyme in the base excision repair pathway (repairs DNA damage) is Apurinic/apyrimidinic exonuclease 1 (APEX 1). Increased expression of APEX 1 is associated with decreased disease-free survival and increased tumour recurrence.37 MicroRNAs modulate OGS drug resistance via DNA damage repair, apoptosis avoidance, suppression of autophagy, activation of cancer stem cells and alteration of OGS associated signal pathways.38
EWS therapy resistance is explained on the basis of CSC, drug-metabolizing enzymes and modulation of signalling pathways. It is thought that EWSCSC arises from primitive mesenchymal stem cells.39 The proposed explanations for chemoresistance of EWS CSC are that they are quiescent, have enhanced ability to repair DNA damage, disrupted apoptotic pathways and high expression of drug-efflux proteins.40 Glutathione S transferases (GST) which belong to the family of Phase II detoxification enzymes are involved in the metabolism of toxic compounds. Low expression of microsomal GST was associated with better prognosis and is correlated with sensitivity to doxorubicin. The same group identified molecular signatures using microarray technology which predicted tumour resistance.41 ERBB4 tyrosine kinase is overexpressed in EWS cell line of chemoresistant and metastatic patients. Overexpression of ERBB4 correlates with poor disease-free survival.42 STAG2 overexpression is associated with metastaticEWS.43 Tumour heterogeneity in tumour cells and microenvironment has also been implicated in chemoresistant EWS.40
Constitutional activation of STAT3 causes resistance to chemotherapy in RMS cell lines.44 In a study that included 31 patients of RMS and 12 patients of NRSTS, biopsy was done at baseline from the primary lesion and from the residual tumour at the end of treatment. The paired samples were analysed for P-gp, multidrug resistance-associated protein 1 (MRP 1) and multidrug resistance 3 (MDR 3). Expression of MRP1, MDR 3 and P-gp was higher than in post-treatment specimens suggesting their role in chemoresistance. Clonal selection of MDR protein-expressing tumour cells and up-regulation of MDR proteins were thought to be the possible explanations for increased expression of MDR proteins in post-treatment specimens.45 Serum and glucocorticoid inducible kinase expression in RMS has been shown to be associated with treatment resistance in RMS cell lines.46 In vitro and in vivo studies in RMS have shown that GST mediates chemotherapy resistance.47
Genomic Profiling, Next-Generation Sequencing and Target-Based Approach
Whole genome sequencing was done in 100 patients of OGS with outcome of relapse, percent tumour necrosis and survival. Intronic and intergenic hotspot regions from 26 genes were identified which were associated with relapse. Mutations in genes belonging to AKR enzyme family, PI3K pathways, cell-cell adhesion processes and variants of SLC22 were associated with tumour necrosis and survival.48 In whole-exome analysis of eight OGS patients, out of which three were nonresponder fifteen genes were identified which are possibly associated with drug resistance, metastasis and can be drug targets.49 In a case, a series of two metastatic and chemo-refractory OGS patients comprehensive molecular profiling was done and molecular targeted therapy was given accordingly. Both the patients did not benefit from the approach.50 Whole exome sequencing, whole transcriptome sequencing, high-density single nucleotide polymorphism array analysis of the tumor and whole exome sequencing of matched germline DNA was done in 59 relapse/refractory paediatric solid tumours to evaluate genome-guided therapy. Out of 59 patients, ten patients were refractory sarcoma [EWS-5, RMS-2, inflammatory myofibroblastic sarcoma-2 and OGS-1]. Actionable mutation with FDA (Unites States Food and Drug Administration) approved drug in adults was seen in four patients while for drugs currently in paediatric trial was seen in five patients. The study showed that multi-dimensional “omics” approach is feasible and can have therapeutic implications.51
The Individualized Therapy for Relapsed Malignancies in Childhood (INFORM) studied 57 patients of relapse paediatric tumours and whole-exome, low-coverage, whole-genome, RNA sequencing, methylation and expression microarray analysis was done. Out of these 57 patients, ten patients of EWS, 5 patients with RMS, 5 patients of OGS and NRSTSwere 3 which were analysable. Eight out of 23 patients had targetable alterations with intermediate or higher prioritization scores.52
In a study of 62 patients with relapsed/refractory paediatric tumours, whole exome sequencing and RNA sequencing were done. Thirteen patients had sarcoma [OGS-7, RMS-4 and EWS-2]. All patients had potentially actionable alterations, which were grouped into targeted therapy, biomarker, risk stratification and diagnostic. Ten patients had actionable alterations for targeted therapy out of which two patients received targeted therapy.53
Comprehensive genomic profiling of 102 advanced/relapse/refractory sarcoma patients were done which included OGS (10), RMS (6) and EWS (3) and actionable mutations were seen in six patients.54 Twenty refractory paediatric sarcoma patients were evaluated for targetable aberrations by array-based expression profiling. All patients had actionable targets. Nine patients received therapy based on actionable alteration and eleven patients did not receive. Median OS and PFS were 8.83 and 6.17 months in the targeted treatment group compared to 4.9 months and 1.7 months in the group which did not receive the targeted therapy, with p values of 0.0014 and 0.0011, respectively. This study showed that patients treated with therapy targeting genetic alterations are likely to benefit.55
In a study of 71 OGS samples from 66 adults and paediatric patients, next-generation sequencing (NGS) was used to identify potentially actionable mutations. Out of these 71 samples, 32 were from metastatic or recurrent lesions. In metastatic/recurrent samples 41.2% had VEGFA whereas the primary site had only 9.7%.56 Amplification of VEGFA has been shown to be a predictor of poor outcomes.57 In the whole cohort, they found genetic changes that can be potentially targeted in 21% and in another 40% they found mutually exclusive VEGFA or PDGFR amplification which can be evaluated as targets. The authors proposed a genetic algorithm classifying approximately 50% of OGS patients eligible for targeted therapy trials.56
Despite encouraging results in identification of targets in refractory paediatric sarcoma, genomic profiling and next-generation sequencing approaches are still experimental.
Current Therapeutic Options in Selected Relapsed/Refractory Paediatric Sarcomas
Rhabdomyosarcoma
Treatment at the time of relapse depends on many factors, viz. site of recurrence, previous treatment received, and individual patient preference. There are no clear guidelines for the treatment of relapsed RMS. Treatment of recurrent childhood RMS should include multimodality options including chemotherapy, surgery and radiotherapy whenever feasible as summarized in Table 4. In the case of localized disease where surgery is possible, initial surgical resection should be attempted followed by chemotherapy and radiotherapy. If the initial disease is unresectable, initial chemotherapy followed by local therapy, ie, surgery or radiotherapy should be done. Surgery may also be attempted in patients with fewer lung metastases. Radiotherapy is an important modality in recurrent RMS, especially in cases where a patient has not received prior radiotherapy and if surgical excision is not feasible.58–60 In the case of gross metastatic disease, palliative chemotherapy may be administered based on chemotherapeutic agents received at the time of initial diagnosis.
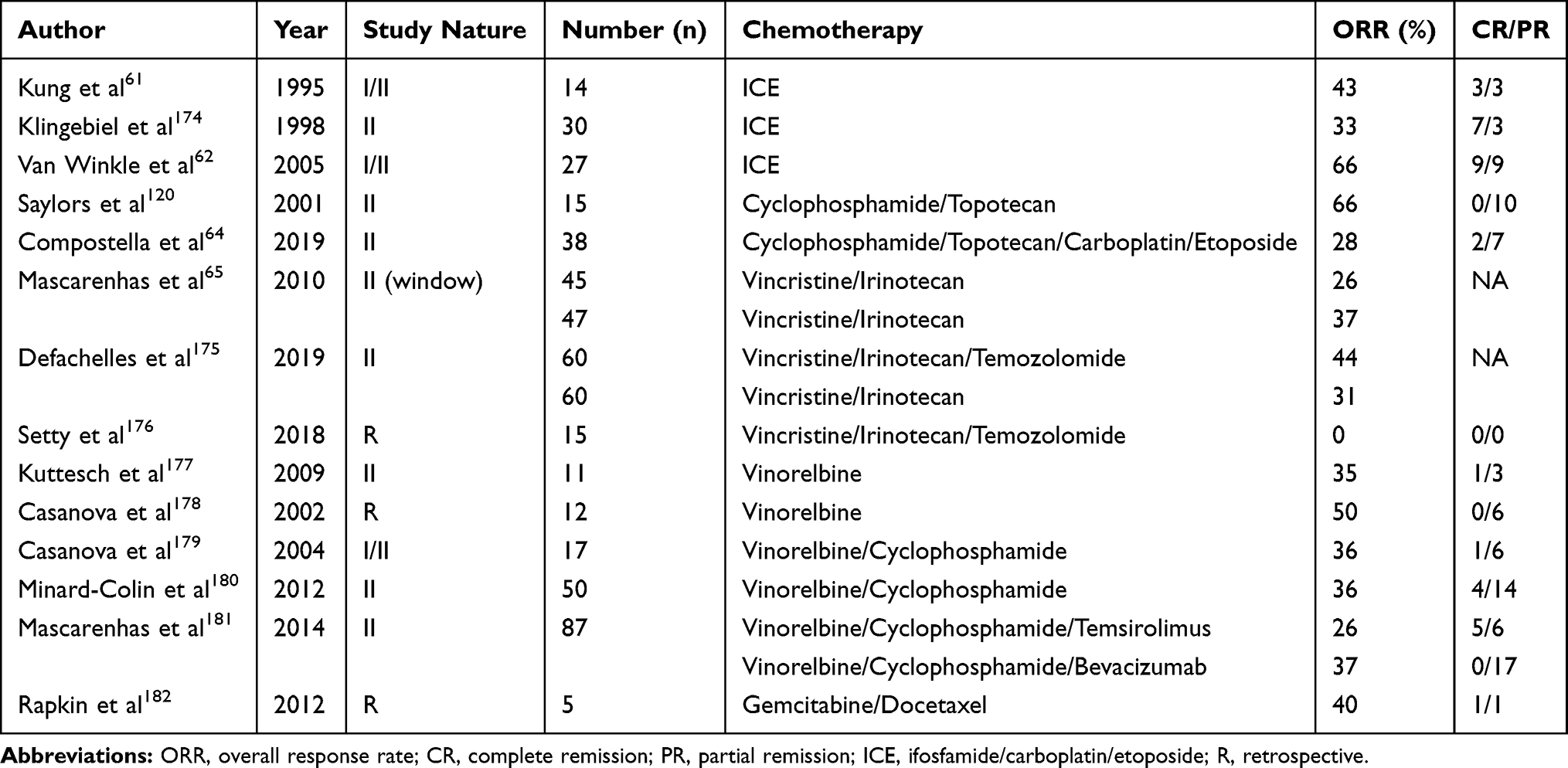 |
Table 1 Various Chemotherapy Options in Relapsed RMS |
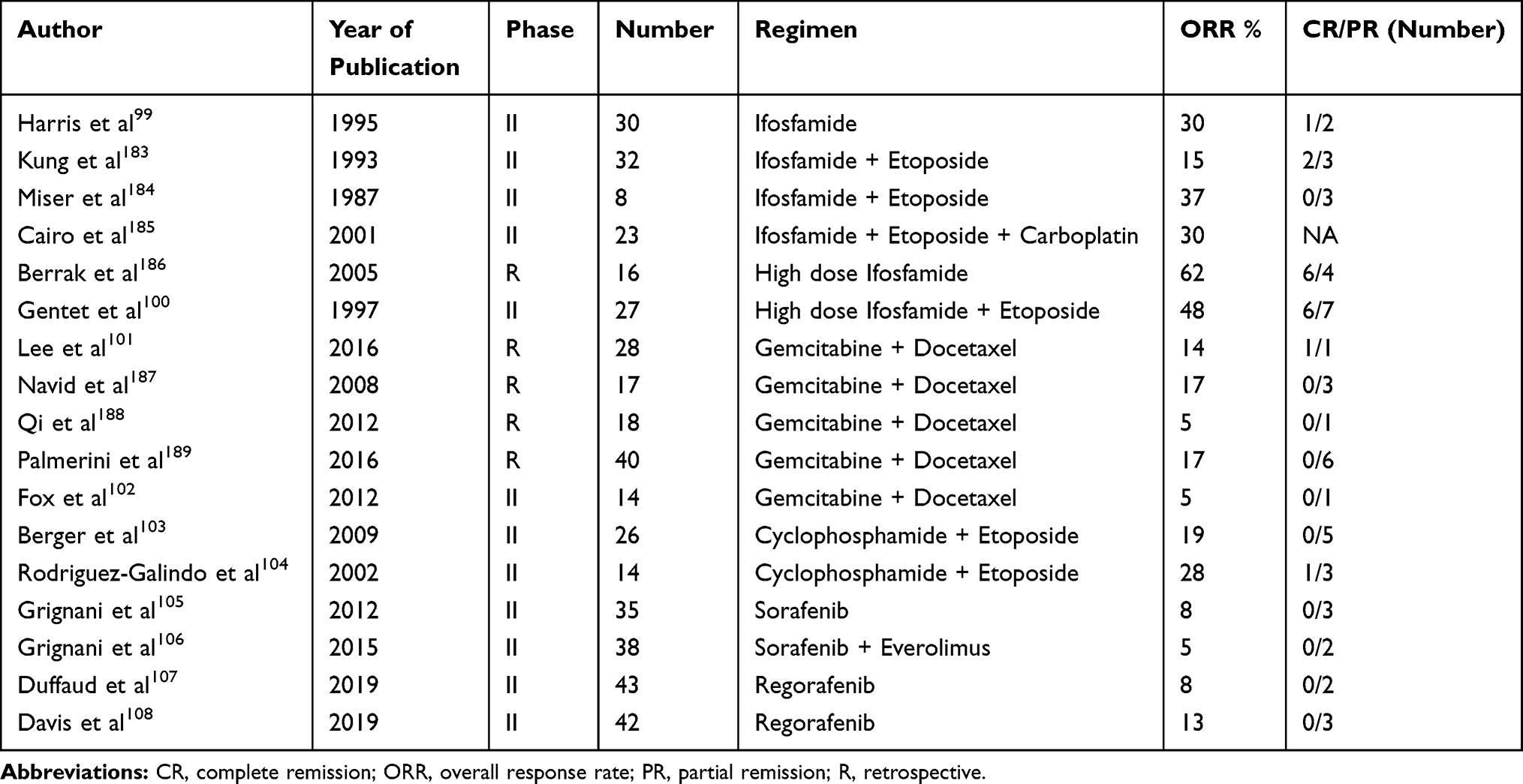 |
Table 2 Studies Using Chemotherapeutic Agents in Relapsed Osteosarcoma |
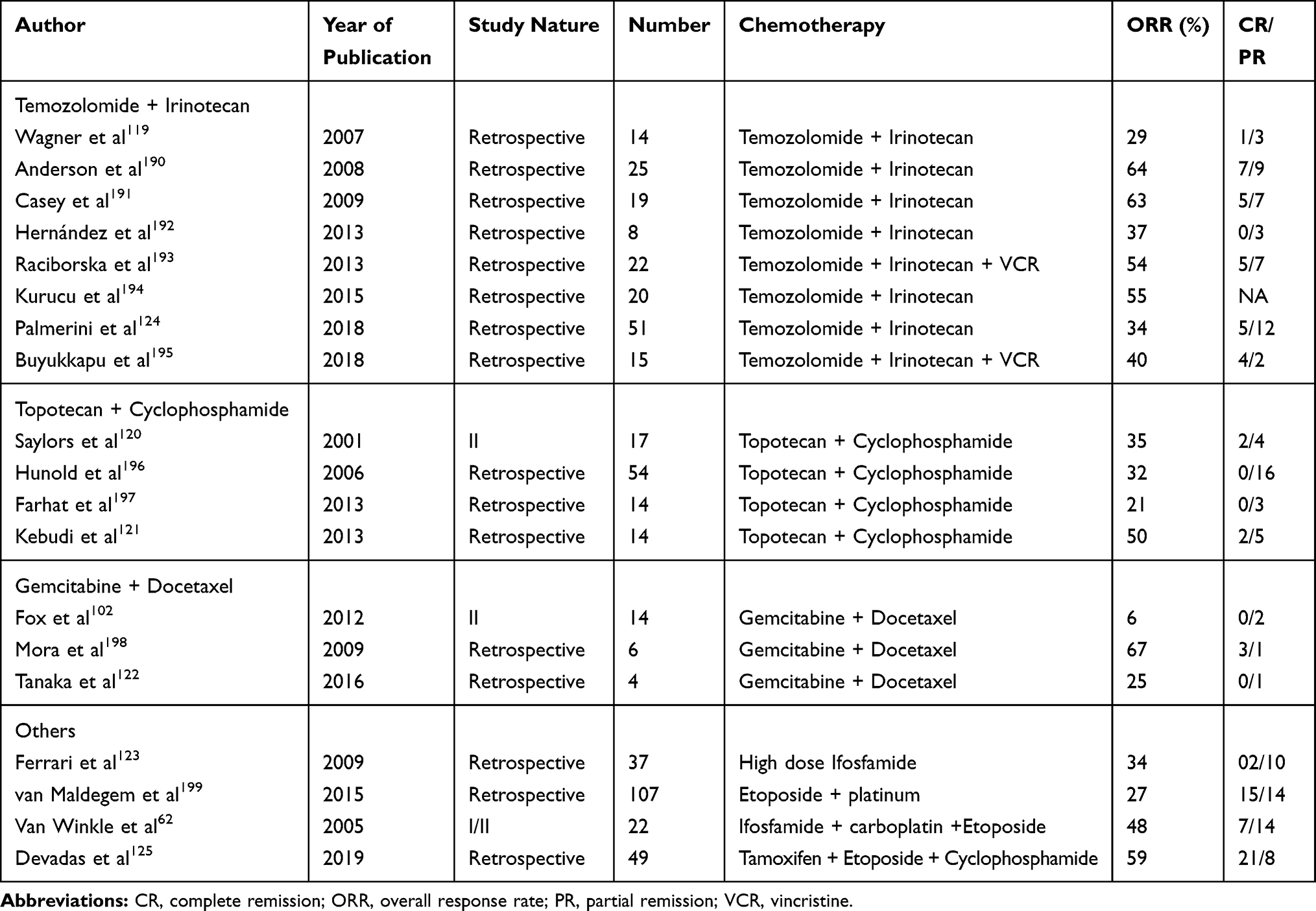 |
Table 3 Chemotherapy Regimens in Relapsed Ewing’s Sarcoma |
 |
Table 4 Summary of Treatment of Relapsed Paediatric Sarcomas |
Chemotherapy has an important role in the treatment of relapsed RMS. However, there is no clear data for the choice of chemotherapy due to lack of comparative trials and heterogeneity of the trial population. Patients should be encouraged to enrol in clinical trials if available. If the patient has previously received two drug VA (vincristine and actinomycin D) therapy, full course of VAC (vincristine, actinomycin D and cyclophosphamide) may be administered at relapse. Patients who have received VAC, options include Ifosfamide, carboplatin, and etoposide (ICE); cyclophosphamide/topotecan; irinotecan, temozolomide, vincristine; gemcitabine, docetaxel; vinorelbine; temsirolimus; and combination of above agents. Various regimens are summarized in Table 1.
Ifosfamide, carboplatin, and etoposide (ICE) has been commonly used regimen in the past, but has significant hematologic toxicity. In a prospective trial of 92 patients with relapsed solid tumours including 14 relapsed RMS, responses were seen in 43% of patients.61 In Children’s Cancer group pooled analysis of three phase I/II trials using ICE in 27 relapsed/refractory RMS patients, the response was observed in two-third of patients.62
Cyclophosphamide/topotecan has been tested in some phase II trials. In one study 10 out of 15 RMS patients responded, however, all patients had partial response.63 In an Italian study, topotecan/cyclophosphamide was combined with carboplatin/etoposide in 38 patients with recurrent or refractory RMS. Response was seen in around one-third of patients but the 5-year OS (17%) and PFS (14%) rates were poor. Rates of haematological toxicity were high.64
Vincristine and irinotecan with/without other drugs are the most commonly used regimens in the current era due to good response rates and manageable toxicity. However, gastrointestinal toxicity and neuropathy remain of concern. A Children’s Oncology Group (COG) prospective, randomized trial (COG-ARST0121) showed no significant difference between responses in two schedules of vincristine/irinotecan.65 In a European Soft Tissue Sarcoma Study Group (EpSSG) study, 120 patients with recurrent or refractory RMS were randomized to vincristine and irinotecan (VI) or vincristine, irinotecan, and temozolomide (VIT). The VIT arm was associated with higher response rates and better survival.66
High dose chemotherapy (HDT) followed by autologous hematopoietic stem cell transplantation (AHSCT) has also been tried for patients with RMS in relapse as well as upfront setting. However, available data did not show a significant benefit with this approach.67,68
PAX-FOXO1 fusion protein is expressed in tumor cells in RMS has been historically difficult to target. Novel approaches are currently being developed designed to target either PAX-FOXO1 or its co-regulators. Experimental approach using nanoparticles as a vehicle to transport siRNA to downregulate PAX-FOXO1 has been demonstrated in vitro studies.69 Cell line studies have shown that targeting BET bromodomain protein BRD4 which is a coregulator of PAX-FOXO1 by JQ1 inhibits PAX-FOXO1 function.70 Similarly, targeting chromatin helix DNA binding protein 4 (CHD4) which acts as a coregulator of PAX-FOXO1is feasible in preclinical models.71 Potential targets in receptor tyrosine kinase are IGF-1R, FGFR4, PDGFR, ALK, MET, VEGFR and ERBB2. NOTCH and Smo are being evaluated as the target in developmental pathways.72
Various trials involving nivolumab (NCT02304458), pembrolizumab (NCT02332668), IGF-1 inhibitors (NCT03041701), Wee1 inhibitor (NCT02095132) and CDK4 inhibitors (NCT03709680) are currently undergoing. The summary of the current ongoing clinical trials involving novel therapeutic approaches to RMS is summarized in Table 5.
Osteosarcoma
OGS is the most common primary bone tumor in the pediatric population.73 Two-third of patients present with localized disease and have good long-term survival. About 20% of cases have metastatic disease at diagnosis and have a poor outcome. However, about one-third of patients with localized OGS and about 80% of metastatic OGS patients relapse even after multimodality treatment. Most relapses (>50%) occur within 2 years of diagnosis and only a few relapses (<5%) after 5 years of initial diagnosis.74 The most common site of relapse is lung followed by bones and local sites. Pulmonary relapse occurs in 60–90% of cases, bone relapse in 10–20%, local relapse in 10–20% and other sites in <10%.75,76 Approach to treatment of relapsed OGS is based on sites of relapse (lungs, bones, local site or multiple), resectability and timing of relapse after the first diagnosis (Table 4).
Long-term survival in relapsed cases may be achieved in up to one-third of cases. Various factors that predict outcome in relapsed osteosarcoma include timing of relapse after initial diagnosis, sites of relapse, presence of initial metastasis, response to neo-adjuvant therapy, achievement of second complete remission (CR2) after relapse and age. Most important among these are timing of relapse and site of relapse.74
Prognosis of locally recurrent OGS without systemic metastasis is poor with long-term survival reported in 10–40% patients in retrospective studies.4,76-82 No benefit was observed with chemotherapy administration after surgical salvage.83 So locally recurrent OGS should be managed by surgical resection whenever possible. The role of chemotherapy and radiotherapy is doubtful after complete surgical resection. Additional chemotherapy may be administered after surgery as followed in unresectable disease.
Osteosarcoma: Lung Metastasis
In the majority of patients with relapsed OGS, lungs are the only site of disease. Surgery (metastasectomy) is the mainstay of management in these cases, with many cases requiring repeated or staged lung resection.83–87 Five-year event-free survival (EFS) for patients after complete surgical resection of pulmonary metastasis ranges from 20% to 45%.84,85 Overall survival with unresectable metastatic disease is less than 5%.86
In the case of isolated lung metastases, the treatment of recurrent OS is primarily surgical. Even patients with subsequent relapses may be cured as long as recurrences are resectable, and repeated thoracotomies may be required.87 Surgical resection of all macroscopic disease should be attempted with thoracoscopy or thoracotomy with palpation of the collapsed lung. There is no comparative trial or clear recommendations for using different approaches viz. thoracoscopy, unilateral or bilateral thoracotomy. Unilateral multiple metastases are usually treated with the addition of chemotherapy but the benefit is less clear. Bilateral thoracotomy with chemotherapy is required for the management of bilateral metastases. Unresectable metastases are treated with chemotherapy with palliative intent. Whether the use of preoperative chemotherapy improves the respectability of bilateral metastases is yet to be defined.88,89 The role of chemotherapy after surgery is controversial.82,90 Limited success has been achieved using radiofrequency ablation and stereotactic radiotherapy as an alternative local treatment option for primary lung or bone metastases.91,92
Osteosarcoma: Bone Metastasis
Patients with bone metastases have a poor prognosis with a 5-year EFS of 11%. However, patients who have late solitary bone relapse have a 5-year EFS of 30%.93,94 Samarium-153 ethylenediamine tetramethylene phosphonate (Sm 153-EDTMP) is a beta-particle–emitting radiopharmaceutical agent, has been studied in patients with locally recurrent or metastatic OGS. Patients with recurrent OGS with bone-only involvement can be managed by surgical resection if feasible. Patients with multiple unresectable bone lesions, 153Sm-EDTMP with or without stem cell support provide good pain relief.95,96
Radium-223 dichloride (Ra 223) is alpha-particle–emitting radiopharmaceutical that is under trials for treating metastatic or recurrent OGS. Preliminary studies suggest that this agent is active in OGS and may have less marrow toxicity and greater efficacy than beta-particle–emitting radiopharmaceuticals such as Sm 153-EDTMP.97,98
Osteosarcoma: Palliative Chemotherapy
Patients with unresectable disease are treated with conventional chemotherapy with palliative intent. The role of chemotherapy for recurrent OS is much less well defined. The choice of chemotherapy depends on the prior disease-free interval and includes ifosfamide or cyclophosphamide, with etoposide and/or carboplatin, gemcitabine and docetaxel, sorafenib or regorafenib and radioactive agents. Various agents and their responses are summarised in Table 2.
Ifosfamide with etoposide/carboplatin has shown some response in retrospective and Phase 2 trials. Responses have been observed in about one-third of cases. Higher responses were observed in high dose ifosfamide.99,100 The combination of gemcitabine and docetaxel has an overall response rate of less than 20%. A 900 mg/m2 dose of gemcitabine was associated with a higher response rate and longer survival than 675 mg/m2 dose.101,102 Cyclophosphamide and etoposide have shown some activity in recurrent OGS in two phase 2 trials.103,104
Targeted inhibition of molecular pathways viz. mTOR, SRC kinases, and vascular endothelial growth factor receptors (VEGFRs) are currently under clinical trials in relapsed or refractory OGS. The Italian Sarcoma Group reported a poor response rate with sorafenib alone or in combination with everolimus. However, disease stabilization was observed in half of the cases at 6 months.105,106 Two phase 2 double-blind, placebo-controlled have evaluated the efficacy of regorafenib in OGS whose disease had progressed after treatment with at least one previous line of chemotherapy for metastatic disease. Responses were observed in 8–13% cases; however, at least stable disease was seen in up to 64% cases at 8 weeks.107,108
In the Italian Sarcoma Group study, carboplatin and etoposide followed by peripheral blood autologous stem cell transplantation, the 3-year OS and DFS rates were 20% and 12%, which is similar to that achieved with the conventional chemotherapeutic approach.109,110 So currently HDT followed by AHSCT is not recommended. Current approaches for treating OGS using novel therapeutics are given in Table 5.
Osteosarcoma: Newer Advances
Immunotherapy targeting the PD1-PDL1 axis has shown only modest activity in relapsed and refractory OGS patients. In the SARC28 study of pembrolizumab, in advanced bone and soft tissue sarcoma patients, only one partial response was observed among 22 patients with OGS, and median progression-free survival was 8 weeks.111 Prospective data from trials involving other immune checkpoint inhibitors are pending. Novel immunotherapeutic agent for the lung metastases includes inhalation of aerosolized granulocyte-macrophage colony-stimulating factor (GM-CSF). GM-CSF stimulates the proliferation and differentiation of hematopoietic progenitor cells and augments the function of neutrophils, monocytes, macrophages, and dendritic cells. However, no significant benefit was observed in 43 patients with pulmonary relapse from OGS in the American Osteosarcoma Study Group (AOST) protocol 0221.112
Osteosarcoma: Ongoing Studies
AOST1421 (NCT02484443) is currently studying combination dinutuximab (anti-GD2 antibody) with sargramostim (GM-CSF) in patients after complete surgical resection of pulmonary metastasis at recurrence.
NCI-COG Paediatric Molecular Analysis for Therapeutic Choice (MATCH) in patients between 1 and 25 years of age is next-generation sequencing-based study in refractory and recurrent solid tumours.Children and adolescents aged 1 to 21 years are eligible for the trial.AOST1321 (NCT02470091) is a single-arm, phase II trial of RANKL inhibitor denosumab, in patients with relapsed or refractory OGS post resection of any measurable disease.
Ewing Sarcoma [EWS]/Primitive Neuroectodermal Tumor [PNET]
EWS is the second most common primary bone tumour after OGS.113 With currently available therapeutic modalities good outcomes have been achieved; however, relapses occur in about 30–40% of localized disease and up to 80% with advanced disease.114 Most of the relapses, about 70–80% occur early (within 2 years of diagnosis). Late (2–5 years) and very late (after 5 years of diagnosis) relapses occur in about 20–30% and 5 −10% patients, respectively.115,116 At relapse, most patients have metastatic disease or combined distant and local disease (75−85%). About 15–25% of relapses occur only at the local site, and these are more common with initially localized disease. Lungs are the most common sites of distant relapse followed by bones.6,114 At the time of relapse, about 52% of patients have symptoms, while remaining patients are diagnosed during follow-up imaging.117
The most important prognostic factors are interval between initial diagnosis and subsequent relapse, and site of relapse. Other prognostic factors for poor survival at relapse include high LDH at relapse, age >15 years, non-pulmonary metastasis and symptomatic relapse.114,117,118
No standard approach is available for the treatment of recurrent EWS. Multimodality treatment including surgery, radiation, and chemotherapy is required and depends on previous therapy, duration from diagnosis to relapse, and site of relapse. Relapsed cases are considered a systemic disease and chemotherapy is recommended in all cases. Survival with only local recurrence is better compared with distant and combined recurrences.118 If operable, the patient should undergo surgery and systemic chemotherapy. Radiotherapy may be used in case of inoperable cases, and if margin positivity after surgery. In cases of metastatic disease, chemotherapy is administered with palliative intent. Accepted approach is summarized in Table 4.
EWS/PNET: Systemic Therapy
There is no standard second-line regimen following relapse after the first‐line treatment. Several regimens have been tried, mostly retrospective evidence is available with only a few Phase 1/2 trials. No single regimen had been proven to be superior to others. Various regimens (Table 3) with promising results includes temozolomide + irinotecan ± vincristine, cyclophosphamide + topotecan, gemcitabine + docetaxel, high dose ifosfamide, ifosfamide + etoposide ± carboplatin (ICE).62,83,102,119-123 Responses with targeted therapy and immunotherapy have been poor.
Among the various regimens, temozolomide combined with irinotecan has the most encouraging results. Additional vincristine has also been used with this regimen. Among overall 176 combined patients from various studies, responses had been observed in around 40% of patients.119,124 This regimen is well tolerated with lower myelotoxicity and fewer grade 3/4 toxicities (diarrhoea 5–10%, neuropathy 4–12%) compared with other salvage regimens. Irinotecan has been tried by the oral route, and initial studies have shown similar efficacy as the parenteral route. As oral temozolomide is available, this regimen may be considered for oral therapy.119
Topotecan and cyclophosphamide combination has shown some efficacy with 32% response rate in 99 patients from various studies. Grade 3/4 hematological toxicity was observed in more than 50% of patients.120,121 Gemcitabine with docetaxel has 29% response rate in 29 treated patients. Grade 3/4 neutropenia was seen in around 70% of patients.102,122 Ifosfamide, etoposide, and carboplatin (ICE) combination and high dose Ifosfamide had shown good efficacy but the cost of toxicity (100% Grade 3/4 haematological toxicity).62,123 A retrospective study from India has shown encouraging results with oral metronomic therapy using tamoxifen, etoposide, and cyclophosphamide. In 49 relapsed sarcoma patients, the majority of which was EWS, responses were observed in 59% of patients.125 Ongoing clinical trials using novel therapeutics for the management of refractory Ewing’s sarcoma are given in Table no. 5.
The Euro Ewing Consortium is currently conducting a multi-armed phase II/III randomized study (rEECur study) in patients aged 2–50 years with recurrent EWS. (ISRCTN 36453794). In Phase II patients will receive one of four regimens, cyclophosphamide/topotecan, gemcitabine/docetaxel, high dose ifosfamide, or temozolomide/irinotecan. In Phase III two regimens with a higher objective response rate will be tested with progression-free survival as the primary endpoint.
EWS/PNET: Targeted Therapy
Various pathways have been identified in EWS, as targets which may help in disease control. These include insulin growth factor receptor 1 (IGF-1R), vascular endothelial growth factor receptors (VEGFR), MET, EWS-FLI1 fusion, DNA repair protein PARP1, lysine-specific demethylase 1 (LSD-1), and placenta growth factor (PGF).126–133
IGF-1R is expressed on EWS tumour cells and drives the growth of tumour cells. Initial trials with monoclonal antibodies against IGF −1R showed some encouraging results but larger studies failed to provide significant results. Studies have been done in combination with an mTOR inhibitor, but also failed to provide meaningful benefit. Current trials of IGF-1R in combination with conventional chemotherapy are ongoing [NCT02306161].134–137
Targeting angiogenesis has been attempted with modest success using regorafenib, and cabozantinib. In heavily pre-treated adult patients treated with regorafenib, response was observed in 10% and 73% of patients were progression-free at 8 weeks.137 With MET inhibitor cabozantinib responses have been observed in around 28% and 24% of patients were progression-free at 6 months.129
EWS-FLI1 is the principal driver of tumour growth in EWS. EWS-FLI1 fusion protein is a possible target for therapy in relapsed cases; however, it is very difficult to target.131 BRD4 protein is a member of the bromodomain and extra terminal domain (BET) family of proteins, is a transcriptional regulator required for the EWS-FLI1 fusion protein function. Bromodomain inhibitors inhibit gene expression of EWS-FLI1.132 Clinical trials using bromodomain inhibitors are currently ongoing [NCT02419417, NCT03220347].
Poly (ADP-ribose) polymerase (PARP) is a family of proteins involved in various cellular processes including DNA repair and genomic stability. The expression of PARP1 is elevated in EWS. Olaparib did not have an objective clinical response in a phase 2 trial, though patients were not selected based on biomarkers.138 Currently, trials are undergoing which are selecting patients based on actionable mutation olaparib (NCT03233204), talazoparib (NCT02116777) and niraparib (NCT02044120).
Lysine-specific demethylase 1 (LSD-1) regulates histone methylation and influences the epigenetic state of cells, is upregulated in EWS. It is a transcriptional repressor of downstream targets of EWS/FLI1 and inhibits tumour growth.139 Clinical trials are currently ongoing with LSD −1 inhibitors [NCT03514407, NCT03600649]. Other targets that have been identified are NKX2.2, HSP90, FOX01 and ATR.140
EWS/PNET: Immunotherapy
Immunotherapy targeting PD-1/PD-L1 axis has shown disappointing results in relapsed EWS. PDL1 expression has observed in around 20% of cases; however, these have low mutational tumour burden (TMB), and low level of tumour infiltrating lymphocytes (TIL).43,141 No objective responses have been observed in patients treated with the anti-PD-1 antibody pembrolizumab or nivolumab.111,142
A novel strategy in the field of immunotherapy for EWS known as Vigil or FANG has shown promising results. In this tumor cells are obtained by biopsy or resection and transfected with a plasmid containing the rhGMCSF (recombinant human GM-CSF) transgene and the shRNAfurin(short hairpin RNA inactivating furin). This leads to the upregulation of the immune system by inhibition of TGF-beta1 and 2 mediated pathways.139,143 Ghisoli et al144 have reported a 1-year survival of 73% for relapsed EWS patients treated with Vigil compared to 23% for those treated with conventional chemotherapy. A phase III trial is currently undergoing in which relapsed EWS patients are treated with temozolomide/irinotecan combination with and without Vigil [NCT03495921].
EWS/PNET: Role of ASCT
The role of HDT followed by ASCT is doubtful in relapsed EWS due to the lack of any prospective trial in this population. Various retrospective analyses have shown the benefit of HDT followed by ASCT. However, ASCT was done in the patients responding to initial chemotherapy. This limits the role of ASCT in relapsed EWS and it is not routinely recommended.145–147 Temozolomide/Irinotecan-based chemotherapy may be favored due to higher responses and lower toxicity.
Infantile Fibrosarcoma
Infantile fibrosarcoma (IF) also known as congenital fibrosarcoma is a rare soft tissue tumor that presents at birth or develops in the 1st year of life. They represent approximately 5% to 10% of all sarcomas in infants.148–150 It has a reciprocal translocation, t(12;15)(p13;q25), resulting in ETV6/NTRK3 gene fusion in which the ETV6 (TEL) gene from 12p13 fuses with the NTRK3 gene (Trk C) on 15q25.151
The European Paediatric Soft Tissue Sarcoma Study Group evaluated a conservative therapeutic strategy in 50 infants with localized IFS. The initial surgery was performed if it could be done without mutilation. No further therapy was given if negative margins (group I/R0; n=11) or microscopic positive margins (group II/R1; n=8). Non-alkylator-based chemotherapy vincristine-actinomycin (VA) was administered to those with initial inoperable tumours (group III/R2; n=31). Response to chemotherapy was observed in 68% of patients. The 3-year event-free survival was 84% and the overall survival 94.0% at a median follow-up of 4.7 years.152
As discussed, ETV6/NTRK3 gene fusion is found in patients with IF. Larotrectinib is an oral ATP-competitive inhibitor of TRK A, B, and C. In a phase I/II basket trial involving 55 adults and paediatric patients with advanced or metastatic tumours with TRK fusion proteins, seven patients with IF were included. All patients responded to larotrectinib with two patients achieved complete remission and five achieved partial responses.153 Larotrectinib was approved by the FDA for use in adults and children with solid tumours with an NTRK gene fusion without a known acquired resistance mutation, which are either metastatic or where surgical resection is likely to result in severe morbidity, and who have no satisfactory alternative treatments or who have progressed following treatment.
In a patient with IF, upfront surgical resection should be considered. If the tumour is very large and a mutilating surgery is expected, neoadjuvant therapy similar to RMS may be administered. In cases of relapsed and metastatic IF, where the tumour is not surgically amenable, the patient should be started on larotrectinib or enrolled in a clinical trial. Summary of current modalities is mentioned in Table 4, recent novel therapeutic approaches are given in Table 5.
 |
Table 5 Ongoing Studies in Relapsed/Refractory Paediatric Sarcoma |
Non-Chemotherapeutic Approaches in Refractory Pediatric Sarcoma Including Newer Advances
Metronomic Therapy
Metronomic therapy is low-dose, prolonged, continuous administration of drugs that inhibits angiogenesis.154,155 Metronomic therapy was shown to be safe and well tolerated in a study of 16 children with relapsed/refractory patients which included five patients with OGS.156 A randomized controlled trial comparing metronomic therapy with placebo, in extracranial non-hematopoietic paediatric solid tumours, post lines of chemotherapy were done to evaluate the role of metronomic therapy with the proportion of patients without disease progression as an endpoint. There was no significant difference between the two groups. However, in post hoc subgroup analysis patients who received more than three cycles and did not have bone sarcoma benefited with metronomic therapy.157 Metronomic therapy needs to be explored in a palliative setting, in clinical trials designed for this group of patients.
Immune Check-Point Inhibitors
Few STS[1–5%] may have genomic instability in the form of Microsatellite instability [MSI], such as MSI-H, which is the biologic basis of deficient mismatch repair (dMMR).158 They may respond to anti-programmed death receptor 1 [PDl] or anti-programmed death receptor ligand 1 [PDL1] inhibitors. Recently FDA has approved pembrolizumab, a PD1 inhibitor as a treatment option in certain advanced solid tumours including STS that are refractory to standard of care therapy which are MSI-H or dMMR.159 PD-1–expressing TILs and tumour PD-L1 expression were seen in 65% and 58% of tumours in 105 cases of various STS sub-types. Initial immunotherapy trials did show a modest response in advanced STS with few tumour types showed predominantly stable disease.160–163 Second study showed encouraging results in some adult-type STS [dedifferentiated liposarcoma & undifferentiated pleomorphic sarcoma] but none in paediatric sarcoma.111
Ipilimumab a CTLA4 immune checkpoint inhibitor was evaluated in Phase I clinical trial for refractory pediatric sarcoma which included OGS and RMS. No objective responses were seen.161 Similarly, a phase I/II trial in relapsed refractory Ewing’s sarcoma and OGS using nivolumab and combination of nivolumab plus ipilimumab did not show any objective response.163
Immunotherapy probably will not be as effective as in adult oncology because pediatric cancers have less tumor mutational burden, immune system in children is still maturing and regulatory T cells present in the microenvironment of pediatric sarcomas have high immunosuppressive action which hinders immunotherapy.72 Many clinical trials ongoing on refractory STS evaluating the role of immunotherapy and the majority of them include adult patients with STS (Table 5).
Cancer Vaccines
Dendritic cell vaccine is a novel approach that appears to be promising. In metastatic and refractory pediatric sarcoma autologous lymphocyte infusion plus sequential autologous tumor lysate vaccines were administered as adjuvant therapy after completion of conventional therapy. Five-year survival was 63% in patients with EWS and RMS and 74% in patients who did not have any residual disease after conventional therapy.164
CAR-T Therapy
In view of the limitations of immune checkpoint inhibitors in refractory pediatric sarcoma, CAR-T cells have generated a lot of interest. The key challenges lie in the rarity of the disease and finding an appropriate target. To circumvent these problems current research is focussing on developing CAR-T cells against antigens which are present in other tumors as well as refractory pediatric tumors.165 Phase I clinical trials of EGFR as CAR-T cells as target in OS, RMS and EWS (NCT03618381) are going on. GD2 is another target of CAR-T cells that are currently being tried in OGS, EWS and sarcoma (NCT01953900, NCT02107963, NCT03356782). Infantile fibrosarcoma with a high level of tumor-infiltrating lymphocytes with high expression of costimulatory molecules suggests that adoptive T-cell therapy could be an option.166
Promising Immunotherapeutic Approaches in Pediatric Sarcomas
Paediatric sarcomas express fewer neoantigen than adult type and distinguish them distinctly from adult STS in terms of pathogenesis as well as response to immune checkpoint inhibitors.167 Neoantigen expression is vital for immune cell infiltration in the tumor microenvironment and subsequent immune-mediated cell killing. The discovery of neoantigen and development of directed therapy is of paramount importance. Surface targets such as ganglioside GD2 and GD3 are expressed in paediatric sarcomas and also IGF1R in Ewing’s sarcoma. These surface antigens can be targeted with advanced targeted therapy including CAR-T cell therapy.168
However, it is pertinent to note that novel therapeutical approaches that have been successful in other cancers have not yet impacted the management of pediatric sarcoma patients. INFORM study and other studies using the “omics” approach have generated huge genetic, epigenetic, gene expression data along with the identification of new driver mutations, but patients have not benefited in terms of survival from this genetic data acquisition. Immunotherapy with conventional targets has failed in pediatric refractory sarcomas. Drug screens have identified new drugs and are currently under trials, but proof of efficacy is lacking.
Future Direction
Systemic combination chemotherapy has reached a plateau in terms of cure rate & survival after huge success over the last 3 decades and has complemented by advanced surgical techniques, improved quality of life with limb salvage therapy and better prosthesis with newer engineering and improved radiation technique sparing short-&long-term treatment-related toxicities. Now, the focus is on further improving the survival, lowering long-term treatment-related toxicities, better salvage therapy in case of recurrent disease and to find the cure with research & development of newer therapeutics for metastatic disease. Newer targets in refractory sarcomas are shown in Figure 1.
 |
Figure 1 Newer targets in refractory pediatric sarcomas. Abbreviations: BET, bromodomain extra terminal motif; CD Cyclin D, CDK 4/6 cyclin-dependent kinase 4/6; CTLA4, cytotoxic T-lymphocyte-associated protein 4; GD2, disialoganglioside 2; IGF1R, insulin-like growth factor receptor 1; LSD1, lysine-specific histone demethylase 1; MET, mesenchymal–epithelial transition; NTRK, neurotropic tropomyosin receptor kinase; PARP, poly ADP ribose polymerase; PD1, programmed death receptor 1; PDL1, programmed death receptor Ligand 1; RTK, receptor tyrosine kinase; VEGFR, vascular endothelial growth factor receptor. |
Various immunotherapeutic approaches – both as single agents and in combination are in clinical studies or published with the mixed outcome and some are promising.169 Chemo-immunotherapy combination therapies can be one of the potential strategies in those with a high burden or upfront metastatic disease after the huge success of the same approach in non-small cell lung cancer.170 Cellular vaccine171 & adaptive cell therapy,172 defining kinome and development of small molecule targeted therapies [NCT02601950] are emerging & theoretically promising treatment strategies in paediatric sarcoma especially translocation associated sarcomas.
Maintenance immunotherapeutic strategy after completion of definitive initial treatment may improve the long-term outcome just like PACIFIC173 study demonstrated huge success of Durvalumab [an anti-PDL1 antibody] in stage 3 NSCLC after definitive chemoradiation and should be investigated in paediatric sarcomas. A strategy that decreases the chance of recurrence in paediatric sarcomas should be a better strategy rather than treating them on recurrence.
In the case of treatment refractoriness, a newer immunotherapeutic strategy like CAR-T cells can be an emerging & promising therapy that can cure even treatment-resistant cases, just like in haematological malignancies. International and national collaboration of multi-pronged treatment strategy, personalized & precision medicine and a better understanding of tumour genome, identification of treatment resistance mechanism, exploitation of host immunity remains the future treatment strategy in the management of relapsed & refractory paediatric sarcomas.
Conclusions
Paediatric sarcomas are heterogeneous group of disorders and nearly one-third of them presents with metastasis at presentation and another one-third relapsed after initial curative therapies. Not much of therapeutic developments happened in relapsed/refractory paediatric sarcomas and current treatment options are very much limited in terms of cure or long-term control. Multimodality treatment with chemotherapy, surgery and/or radiotherapy can be curative options in some limited relapsed cases or when relapse occurs after a long remission. But, in the majority of remaining cases, treatment refractoriness remains the main challenge and possesses a huge unmet need. Current newer strategies including targeted therapies, salvage chemotherapy, ASCT, immunotherapeutic approach, adoptive cellular therapies only produced modest & temporary responses, so far. Better understanding of disease biology, mechanism of treatment refractoriness, well-designed clinical trials, combination chemo-immunotherapeutic strategy, better national and international collaboration, translational research remains the key to success and fulfils the unmet need.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Biswas B, Rastogi S, Khan SA, et al. Hypoalbuminaemia is an independent predictor of poor outcome in metastatic Ewing’s sarcoma family of tumours: a single institutional experience of 150 cases treated with uniform chemotherapy protocol. Clin Oncol. 2014;26(11):722–729. doi:10.1016/j.clon.2014.05.006
2. Iqbal N, Shukla NK, Deo SVS, et al. Prognostic factors affecting survival in metastatic soft tissue sarcoma: an analysis of 110 patients. Clin Transl Oncol. 2016;18(3):310–316. doi:10.1007/s12094-015-1369-9
3. Anderson ME. Update on survival in osteosarcoma. Orthop Clin North Am. 2016;47(1):283–292. doi:10.1016/j.ocl.2015.08.022
4. Gelderblom H, Jinks RC, Sydes M, et al. Survival after recurrent osteosarcoma: data from 3 European Osteosarcoma Intergroup (EOI) randomized controlled trials. Eur J Cancer. 2011;47(6):895–902.
5. Gaspar N, Hawkins DS, Dirksen U, et al. Ewing sarcoma: current management and future approaches through collaboration. J Clin Oncol. 2015;33(27):3036–3046. doi:10.1200/JCO.2014.59.5256
6. Stahl M, Ranft A, Paulussen M, et al. Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr Blood Cancer. 2011;57(4):549–553. doi:10.1002/pbc.23040
7. Grünewald TGP, Cidre-Aranaz F, Surdez D, et al. Ewing sarcoma. Nat Rev Dis Primer. 2018;4:1.
8. Pappo AS, Anderson JR, Crist WM, et al. Survival after relapse in children and adolescents with rhabdomyosarcoma: a report from the intergroup rhabdomyosarcoma study Group. J Clin Oncol. 1999;17(11):3487–3493. doi:10.1200/JCO.1999.17.11.3487
9. Dantonello TM, Int-Veen C, Winkler P, et al. Initial patient characteristics can predict pattern and risk of relapse in localized rhabdomyosarcoma. J Clin Oncol. 2008;26(3):406–413. doi:10.1200/JCO.2007.12.2382
10. Chisholm JC, Marandet J, Rey A, et al. Prognostic factors after relapse in nonmetastatic rhabdomyosarcoma: a nomogram to better define patients who can be salvaged with further therapy. J Clin Oncol. 2011;29(10):1319–1325. doi:10.1200/JCO.2010.32.1984
11. Sung L, Anderson JR, Donaldson SS, et al. Late events occurring five years or more after successful therapy for childhood rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Eur J CanceR. 1990. 2004;40(12):1878–1885.
12. Mazzoleni S, Bisogno G, Garaventa A, et al. Outcomes and prognostic factors after recurrence in children and adolescents with nonmetastatic rhabdomyosarcoma. Cancer. 2005;104(1):183–190. doi:10.1002/cncr.21138
13. Dasgupta R, Rodeberg D. Non-rhabdomyosarcoma. Semin Pediatr Surg. 2016;25(5):284–289. doi:10.1053/j.sempedsurg.2016.09.012
14. Parida L, Fernandez-Pineda I, Uffman JK, et al. Clinical management of infantile fibrosarcoma: a retrospective single-institution review. Pediatr Surg Int. 2013;29(7):703–708. doi:10.1007/s00383-013-3326-4
15. Loh ML, Ahn P, Perez-Atayde AR, Gebhardt MC, Shamberger RC, Grier HE. Treatment of infantile fibrosarcoma with chemotherapy and surgery: results from the Dana-Farber Cancer Institute and Children’s Hospital, Boston. J Pediatr Hematol Oncol. 2002;24(9):722–726.
16. Gibbs CP, Kukekov VG, Reith JD, et al. Stem-like cells in bone sarcomas: implications for tumorigenesis. Neoplasia. 2005;7(11):967–976. doi:10.1593/neo.05394
17. Tirino V, Desiderio V, d’Aquino R, et al. Detection and characterization of CD133+ cancer stem cells in human solid tumours. PLoS One. 2008;3(10):e3469.
18. Adhikari AS, Agarwal N, Wood BM, et al. CD117 and Stro-1 identify osteosarcoma tumor-initiating cells associated with metastasis and drug resistance. Cancer Res. 2010;70(11):4602–4612. doi:10.1158/0008-5472.CAN-09-3463
19. Martins-Neves SR, Paiva-Oliveira DI, Wijers-Koster PM, et al. Chemotherapy induces stemness in osteosarcoma cells through activation of Wnt/β-catenin signaling. Cancer Lett. 2016;370(2):286–295. doi:10.1016/j.canlet.2015.11.013
20. Tang Q-L, Liang Y, Xie X-B, et al. Enrichment of osteosarcoma stem cells by chemotherapy. Chin J Cancer. 2011;30(6):426–432. doi:10.5732/cjc.011.10127
21. Brown HK, Tellez-Gabriel M, Heymann D. Cancer stem cells in osteosarcoma. Cancer Lett. 2017;386:189–195. doi:10.1016/j.canlet.2016.11.019
22. Iwaya K, Ogawa H, Kuroda M, Izumi M, Ishida T, Mukai K. Cytoplasmic and/or nuclear staining of beta-catenin is associated with lung metastasis. Clin Exp Metastasis. 2003;20(6):525–529. doi:10.1023/A:1025821229013
23. Zhang F, Chen A, Chen J, Yu T, Guo F. siRNA-mediated silencing of beta-catenin suppresses invasion and chemosensitivity to doxorubicin in MG-63 osteosarcoma cells. Asian Pac J Cancer. 2011;12(1):239–245.
24. Wu J, Liao Q, He H, Zhong D, Yin K. TWIST interacts with β-catenin signaling on osteosarcoma cell survival against cisplatin. Mol Carcinog. 2014;53(6):440–446. doi:10.1002/mc.21991
25. Ma Y, Ren Y, Han EQ, et al. Inhibition of the Wnt-β-catenin and Notch signaling pathways sensitizes osteosarcoma cells to chemotherapy. Biochem Biophys Res Commun. 2013;431(2):274–279. doi:10.1016/j.bbrc.2012.12.118
26. Basu-Roy U, Basilico C, Mansukhani A. Perspectives on cancer stem cells in osteosarcoma. Cancer Lett. 2013;338(1):158–167. doi:10.1016/j.canlet.2012.05.028
27. Rajkumar T, Yamuna M. Multiple pathways are involved in drug resistance to doxorubicin in an osteosarcoma cell line. Anticancer Drugs. 2008;19(3):257–265. doi:10.1097/CAD.0b013e3282f435b6
28. Baldini N, Scotlandi K, Barbanti-Bròdano G, et al. Expression of P-glycoprotein in high-grade osteosarcomas in relation to clinical outcome. N Engl J Med. 1995;333(21):1380–1385. doi:10.1056/NEJM199511233332103
29. Windsor RE, Strauss SJ, Kallis C, Wood NE, Whelan JS. Germline genetic polymorphisms may influence chemotherapy response and disease outcome in osteosarcoma: a pilot study. Cancer. 2012;118(7):1856–1867. doi:10.1002/cncr.26472
30. Ifergan I, Meller I, Issakov J, Assaraf YG. Reduced folate carrier protein expression in osteosarcoma: implications for the prediction of tumor chemosensitivity. Cancer. 2003;98(9):1958–1966. doi:10.1002/cncr.11741
31. Patiño-García A, Zalacaín M, Marrodán L, San-Julián M, Sierrasesúmaga L. Methotrexate in pediatric osteosarcoma: response and toxicity in relation to genetic polymorphisms and dihydrofolate reductase and reduced folate carrier 1 expression. J Pediatr. 2009;154(5):688–693. doi:10.1016/j.jpeds.2008.11.030
32. Uozaki H, Horiuchi H, Ishida T, Iijima T, Imamura T, Machinami R. Overexpression of resistance-related proteins (metallothioneins, glutathione-S-transferase π, heat shock protein 27, and lung resistance-related protein) in osteosarcoma. Cancer. 1997;79(12):2336–2344. doi:10.1002/(SICI)1097-0142(19970615)79:12<2336::AID-CNCR7>3.0.CO;2-J
33. Wei L, Song X-R, Wang X-W, Li M, Zuo W-S. Expression of MDR1 and GST-pi in osteosarcoma and soft tissue sarcoma and their correlation with chemotherapy resistance. Zhonghua Zhong Liu Za Zhi. 2006;28(6):445–448.
34. Biason P, Hattinger CM, Innocenti F, et al. Nucleotide excision repair gene variants and association with survival in osteosarcoma patients treated with neoadjuvant chemotherapy. Pharmacogenomics J. 2012;Dec;12(6):476–483. doi:10.1038/tpj.2011.33
35. Caronia D, Patiño-García A, Milne RL, et al. Common variations in ERCC2 are associated with response to cisplatin chemotherapy and clinical outcome in osteosarcoma patients. Pharmacogenomics J. 2009;9(5):347–353. doi:10.1038/tpj.2009.19
36. Li J, Liu S, Wang W, et al. ERCC polymorphisms and prognosis of patients with osteosarcoma. Tumour Biol. 2014;35(10):10129–10136. doi:10.1007/s13277-014-2322-1
37. Yang J, Yang D, Cogdell D, et al. APEX1 gene amplification and its protein overexpression in osteosarcoma: correlation with recurrence, metastasis, and survival. Technol Cancer Res Treat. 2010;9(2):161–169. doi:10.1177/153303461000900205
38. Chen R, Wang G, Zheng Y, Hua Y, Cai Z. Drug resistance‐related microRNAs in osteosarcoma: translating basic evidence into therapeutic strategies. J Cell Mol Med. 2019;23(4):2280–2292. doi:10.1111/jcmm.14064
39. Lin PP, Wang Y, Lozano G. Mesenchymal stem cells and the origin of Ewing’s sarcoma. Sarcoma. 2011;2011.
40. Ahmed AA, Zia H, Wagner L. Therapy resistance mechanisms in Ewing’s sarcoma family tumors. Cancer Chemother Pharmacol. 2014;73(4):657–663. doi:10.1007/s00280-014-2392-1
41. Scotlandi K, Remondini D, Castellani G, et al. Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J Clin Oncol. 2009;27(13):2209–2216. doi:10.1200/JCO.2008.19.2542
42. Mendoza-Naranjo A, El-Naggar A, Wai DH, et al. ERBB4 confers metastatic capacity in Ewing sarcoma. EMBO Mol Med. 2013;5(7):1087–1102. doi:10.1002/emmm.201202343
43. Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326–1341. doi:10.1158/2159-8290.CD-13-1037
44. Wu X, Xiao H, Wang R, Liu L, Li C, Lin J. Persistent GP130/STAT3 signaling contributes to the resistance of doxorubicin, cisplatin, and MEK inhibitor in human rhabdomyosarcoma cells. Curr Cancer Drug Targets. 2016;16(7):631–638. doi:10.2174/1568009615666150916093110
45. Citti A, Boldrini R, Inserra A, et al. Expression of multidrug resistance-associated proteins in paediatric soft tissue sarcomas before and after chemotherapy. Int J Oncol. 2012;41(1):117–124. doi:10.3892/ijo.2012.1433
46. Schmid E, Stagno MJ, Yan J, et al. Serum and glucocorticoid inducible kinase 1-sensitive survival, proliferation and migration of rhabdomyosarcoma cells. Cell Physiol Biochem. 2017;43(3):1301–1308. doi:10.1159/000481842
47. Seitz G, Bonin M, Fuchs J, et al. Inhibition of glutathione-S-transferase as a treatment strategy for multidrug resistance in childhood rhabdomyosarcoma. Int J Oncol. 2010;36(2):491–500.
48. Bhuvaneshwar K, Harris M, Gusev Y, et al. Genome sequencing analysis of blood cells identifies germline haplotypes strongly associated with drug resistance in osteosarcoma patients. BMC Cancer. 2019;19:1. doi:10.1186/s12885-019-5474-y
49. Chiappetta C, Mancini M, Lessi F, et al. Whole-exome analysis in osteosarcoma to identify a personalized therapy. Oncotarget. 2017;8:46. doi:10.18632/oncotarget.19010
50. Subbiah V, Wagner MJ, McGuire MF, et al. Personalized comprehensive molecular profiling of high risk osteosarcoma: implications and limitations for precision medicine. Oncotarget. 2015;6(38):38. doi:10.18632/oncotarget.5841
51. Chang W, Brohl AS, Patidar R, et al. MultiDimensional clinOmics for precision therapy of children and adolescent young adults with relapsed and refractory cancer: a report from the center for cancer research. Clin Cancer Res. 2016;22(15):3810–3820. doi:10.1158/1078-0432.CCR-15-2717
52. Worst BC, van Tilburg CM, Balasubramanian GP, et al. Next-generation personalised medicine for high-risk paediatric cancer patients – the INFORM pilot study. Eur J Cancer. 2016;65:91–101. doi:10.1016/j.ejca.2016.06.009
53. Khater F, Vairy S, Langlois S, et al. Molecular profiling of hard-to-treat childhood and adolescent cancers. JAMA Netw Open. 2019;2(4):e192906. doi:10.1001/jamanetworkopen.2019.2906
54. Groisberg R, Hong DS, Holla V, et al. Clinical genomic profiling to identify actionable alterations for investigational therapies in patients with diverse sarcomas. Oncotarget. 2017;8:24. doi:10.18632/oncotarget.16845
55. Weidenbusch B, Richter GHS, Kesper MS, et al. Transcriptome based individualized therapy of refractory pediatric sarcomas: feasibility, tolerability and efficacy. Oncotarget. 2018;9(29):29. doi:10.18632/oncotarget.25087
56. Suehara Y, Alex D, Bowman A, et al. Clinical genomic sequencing of pediatric and adult osteosarcoma reveals distinct molecular subsets with potentially targetable alterations. Clin Cancer Res. 2019;25(21):6346–6356. doi:10.1158/1078-0432.CCR-18-4032
57. Yang J, Yang D, Sun Y, et al. Genetic amplification of the vascular endothelial growth factor (VEGF) pathway genes, including VEGFA, in human osteosarcoma. Cancer. 2011;117(21):4925–4938. doi:10.1002/cncr.26116
58. Hayes-Jordan A, Doherty DK, West SD, et al. Outcome after surgical resection of recurrent rhabdomyosarcoma. J Pediatr Surg. 2006;41(4):633–638. doi:10.1016/j.jpedsurg.2005.12.002
59. De Corti F, Bisogno G, Dall’Igna P, et al. Does surgery have a role in the treatment of local relapses of non-metastatic rhabdomyosarcoma? Pediatr Blood Cancer. 2011;57(7):1261–1265. doi:10.1002/pbc.23225
60. Angelini L, Bisogno G, Alaggio R, et al. Prognostic factors in children undergoing salvage surgery for bladder/prostate rhabdomyosarcoma. J Pediatr Urol. 2016;12(4):
61. Kung FH, Desai SJ, Dickerman JD, et al. Ifosfamide/carboplatin/etoposide (ICE) for recurrent malignant solid tumors of childhood: a Pediatric Oncology Group Phase I/II study. J Pediatr Hematol Oncol. 1995;17(3):265–269. doi:10.1097/00043426-199508000-00009
62. Van Winkle P, Angiolillo A, Krailo M, et al. Ifosfamide, carboplatin, and etoposide (ICE) reinduction chemotherapy in a large cohort of children and adolescents with recurrent/refractory sarcoma: the Children’s Cancer Group (CCG) experience. Pediatr Blood Cancer. 2005;44(4):338–347. doi:10.1002/pbc.20227
63. Rl S, Kc S, J S J, et al. Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a pediatric oncology group Phase II Study. J Clin Oncol. 2001.
64. Compostella A, Affinita MC, Casanova M, et al. Topotecan/carboplatin regimen for refractory/recurrent rhabdomyosarcoma in children: report from the AIEOP Soft Tissue Sarcoma Committee. Tumori. 2019;105(2):138–143. doi:10.1177/0300891618792479
65. Mascarenhas L, Lyden ER, Breitfeld PP, et al. Randomized phase II window trial of two schedules of irinotecan with vincristine in patients with first relapse or progression of rhabdomyosarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2010;28(30):4658–4663. doi:10.1200/JCO.2010.29.7390
66. Abstracts S. Pediatr blood cancer. 2019;66(S4):e27989.
67. Weigel BJ, Breitfeld PP, Hawkins D, Crist WM, Baker KS. Role of high-dose chemotherapy with hematopoietic stem cell rescue in the treatment of metastatic or recurrent rhabdomyosarcoma. J Pediatr Hematol Oncol. 2001;23(5):272–276. doi:10.1097/00043426-200106000-00007
68. Peinemann F, Kröger N, Bartel C, et al. High-dose chemotherapy followed by autologous stem cell transplantation for metastatic rhabdomyosarcoma–a systematic review. PLoS One. 2011;6(2):e17127. doi:10.1371/journal.pone.0017127
69. Rengaswamy V, Zimmer D, Süss R, Rössler J. RGD liposome-protamine-siRNA (LPR) nanoparticles targeting PAX3-FOXO1 for alveolar rhabdomyosarcoma therapy. J Controlled Release. 2016;235:319–327. doi:10.1016/j.jconrel.2016.05.063
70. Gryder BE, Yohe ME, Chou H-C, et al. PAX3–FOXO1 establishes myogenic super enhancers and confers bet bromodomain vulnerability. Cancer Discov. 2017;7(8):884–899. doi:10.1158/2159-8290.CD-16-1297
71. Böhm M, Wachtel M, Marques JG, et al. Helicase CHD4 is an epigenetic coregulator of PAX3-FOXO1 in alveolar rhabdomyosarcoma. J Clin Invest. 2016;126(11):4237–4249. doi:10.1172/JCI85057
72. Chen C, Dorado Garcia H, Scheer M, Henssen AG. Current and future treatment strategies for rhabdomyosarcoma. Front Oncol. 2019;20;9(20):1903–1904. doi:10.1016/0006-2952(75)90415-3
73. Arndt CA, Crist WM. Common musculoskeletal tumors of childhood and adolescence. N Engl J Med. 1999;341(5):342–352. doi:10.1056/NEJM199907293410507
74. Link MP, Goorin AM, Miser AW, et al. The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med. 1986;314(25):1600–1606. doi:10.1056/NEJM198606193142502
75. Gelderblom H, Jinks RC, Sydes M, et al. Survival after recurrent osteosarcoma: data from 3 European Osteosarcoma Intergroup (EOI) randomized controlled trials. Eur J Cancer. 2011;47(6):895–902.
76. Kempf-Bielack B, Bielack SS, Jürgens H, et al. Osteosarcoma relapse after combined modality therapy: an analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). J Clin Oncol. 2005;23(3):559–568. doi:10.1200/JCO.2005.04.063
77. Ferrari S, Briccoli A, Mercuri M, et al. Late relapse in osteosarcoma. J Pediatr Hematol Oncol. 2006;28(7):418–422. doi:10.1097/01.mph.0000212944.82361.1d
78. Bacci G, Forni C, Longhi A, et al. Local recurrence and local control of non-metastatic osteosarcoma of the extremities: a 27-year experience in a single institution. J Surg Oncol. 2007;96(2):118–123. doi:10.1002/jso.20628
79. Bacci G, Ferrari S, Lari S, et al. Osteosarcoma of the limb. Amputation or limb salvage in patients treated by neoadjuvant chemotherapy. J Bone Joint Surg Br. 2002;84(1):88–92.
80. Grimer RJ, Sommerville S, Warnock D, et al. Management and outcome after local recurrence of osteosarcoma. Eur J Cancer. 2005;41(4):578–583. doi:10.1016/j.ejca.2004.11.012
81. Nathan SS, Gorlick R, Bukata S, et al. Treatment algorithm for locally recurrent osteosarcoma based on local disease-free interval and the presence of lung metastasis. Cancer. 2006;107(7):1607–1616. doi:10.1002/cncr.22197
82. Bacci G, Briccoli A, Longhi A, et al. Treatment and outcome of recurrent osteosarcoma: experience at Rizzoli in 235 patients initially treated with neoadjuvant chemotherapy. Acta Oncol Stockh Swed. 2005;44(7):748–755. doi:10.1080/02841860500327503
83. Palmerini E, Torricelli E, Cascinu S, et al. Is there a role for chemotherapy after local relapse in high-grade osteosarcoma? Pediatr Blood Cancer. 2019;66(8):e27792. doi:10.1002/pbc.27792
84. Harting MT, Blakely ML. Management of osteosarcoma pulmonary metastases. Semin Pediatr Surg. 2006;15(1):25–29. doi:10.1053/j.sempedsurg.2005.11.005
85. Chou AJ, Merola PR, Wexler LH, et al. Treatment of osteosarcoma at first recurrence after contemporary therapy: the memorial sloan-kettering cancer center experience. Cancer. 2005;104(10):2214–2221. doi:10.1002/cncr.21417
86. Tabone MD, Kalifa C, Rodary C, Raquin M, Valteau-Couanet D, Lemerle J. Osteosarcoma recurrences in pediatric patients previously treated with intensive chemotherapy.. J Clin Oncol. 1994;12(12):2614–2620. doi:10.1200/JCO.1994.12.12.2614
87. Bielack SS, Kempf-Bielack B, Branscheid D, et al. Second and subsequent recurrences of osteosarcoma: presentation, treatment, and outcomes of 249 consecutive cooperative osteosarcoma study group patients. J Clin Oncol. 2009;27(4):557–565. doi:10.1200/JCO.2008.16.2305
88. Briccoli A, Rocca M, Salone M, Guzzardella GA, Balladelli A, Bacci G. High grade osteosarcoma of the extremities metastatic to the lung: long-term results in 323 patients treated combining surgery and chemotherapy, 1985–2005. Surg Oncol. 2010;19(4):193–199. doi:10.1016/j.suronc.2009.05.002
89. Duffaud F, Digue L, Mercier C, et al. Recurrences following primary osteosarcoma in adolescents and adults previously treated with chemotherapy. Eur J Cancer. 2003;39(14):2050–2057. doi:10.1016/S0959-8049(03)00435-0
90. Hawkins DS, Arndt CAS. Pattern of disease recurrence and prognostic factors in patients with osteosarcoma treated with contemporary chemotherapy. Cancer. 2003;98(11):2447–2456. doi:10.1002/cncr.11799
91. de Baere T, Tselikas L, Gravel G, et al. Interventional radiology: role in the treatment of sarcomas. Eur J Cance. 2018;94:148–155. doi:10.1016/j.ejca.2018.02.017
92. Yu W, Liu Z, Tang L, Lin F, Yao Y, Shen Z. Efficacy and safety of stereotactic radiosurgery for pulmonary metastases from osteosarcoma: experience in 73 patients. Sci Rep. 2017;7(1):17480. doi:10.1038/s41598-017-14521-7
93. Bacci G, Longhi A, Bertoni F, et al. Bone metastases in osteosarcoma patients treated with neoadjuvant or adjuvant chemotherapy: the Rizzoli experience in 52 patients. Acta Orthop. 2006;77(6):938–943. doi:10.1080/17453670610013268
94. Franke M, Hardes J, Helmke K, et al. Solitary skeletal osteosarcoma recurrence. Findings from the Cooperative Osteosarcoma Study Group. Pediatr Blood Cancer. 2011;56(5):771–776.
95. Anderson PM, Wiseman GA, Dispenzieri A, et al. High-dose samarium-153 ethylene diamine tetramethylene phosphonate: low toxicity of skeletal irradiation in patients with osteosarcoma and bone metastases. J Clin Oncol. 2002;20(1):189–196. doi:10.1200/JCO.2002.20.1.189
96. Loeb DM, Garrett-Mayer E, Hobbs RF, et al. Dose-finding study of 153Sm-EDTMP in patients with poor-prognosis osteosarcoma. Cancer. 2009;115(11):2514–2522. doi:10.1002/cncr.24286
97. Subbiah V, Anderson P, Rohren E. Alpha emitter radium 223 in high-risk osteosarcoma: first clinical evidence of response and blood-brain barrier penetration. JAMA Oncol. 2015;1(2):253–255. doi:10.1001/jamaoncol.2014.289
98. Anderson PM, Subbiah V, Rohren E. Bone-seeking radiopharmaceuticals as targeted agents of osteosarcoma: samarium-153-EDTMP and radium-223. Adv Exp Med Biol. 2014;804:291–304.
99. Harris MB, Cantor AB, Goorin AM, et al. Treatment of osteosarcoma with ifosfamide: comparison of response in pediatric patients with recurrent disease versus patients previously untreated: a Pediatric Oncology Group study. Med Pediatr Oncol. 1995;24(2):87–92. doi:10.1002/mpo.2950240205
100. Gentet JC, Brunat-Mentigny M, Demaille MC, et al. Ifosfamide and etoposide in childhood osteosarcoma. A phase II study of the French Society of Paediatric Oncology. Eur J Cancer. 1997;33(2):232–237.
101. Lee JA, Jeon D-G, Cho WH, et al. Higher gemcitabine dose was associated with better outcome of osteosarcoma patients receiving gemcitabine-docetaxel chemotherapy. Pediatr Blood Cancer. 2016;63(9):1552–1556. doi:10.1002/pbc.26058
102. Fox E, Patel S, Wathen JK, et al. Phase II study of sequential gemcitabine followed by docetaxel for recurrent Ewing sarcoma, osteosarcoma, or unresectable or locally recurrent chondrosarcoma: results of Sarcoma Alliance for Research Through Collaboration Study 003. Oncologist. 2012;17(3):321. doi:10.1634/theoncologist.2010-0265
103. Berger M, Massimo B, Grignani G, et al. Phase 2 trial of two courses of cyclophosphamide and etoposide for relapsed high-risk osteosarcoma patients. Cancer. 2009;115(13):2980–2987. doi:10.1002/cncr.24368
104. Rodríguez-Galindo C, Daw NC, Kaste SC, et al. Treatment of refractory osteosarcoma with fractionated cyclophosphamide and etoposide. J Pediatr Hematol Oncol. 2002;24(4):250–255. doi:10.1097/00043426-200205000-00006
105. Grignani G, Palmerini E, Dileo P, et al. A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: an Italian Sarcoma Group study. Ann Oncol. 2012;23(2):508–516. doi:10.1093/annonc/mdr151
106. Grignani G, Palmerini E, Ferraresi V, et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: a non-randomised phase 2 clinical trial. Lancet Oncol. 2015;16(1):98–107. doi:10.1016/S1470-2045(14)71136-2
107. Duffaud F, Mir O, Boudou-Rouquette P, et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: a non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 2019;20(1):120–133. doi:10.1016/S1470-2045(18)30742-3
108. Davis LE, Bolejack V, Ryan CW, et al. Randomized double-blind phase II study of regorafenib in patients with metastatic osteosarcoma. J Clin Oncol. 2019;37(16):1424–1431. doi:10.1200/JCO.18.02374
109. Lashkari A, Chow WA, Valdes F, et al. Tandem high-dose chemotherapy followed by autologous transplantation in patients with locally advanced or metastatic sarcoma. Anticancer Res. 2009;29(8):3281–3288.
110. Fagioli F, Aglietta M, Tienghi A, et al. High-dose chemotherapy in the treatment of relapsed osteosarcoma: an Italian sarcoma group study. J Clin Oncol. 2002;20(8):2150–2156.
111. Tawbi HA, Burgess M, Bolejack V, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017;18(11):1493–1501. doi:10.1016/S1470-2045(17)30624-1
112. Arndt CAS, Koshkina NV, Inwards CY, et al. Inhaled granulocyte-macrophage colony stimulating factor for first pulmonary recurrence of osteosarcoma: effects on disease-free survival and immunomodulation. a report from the Children’s Oncology Group. Clin Cancer Res. 2010;16(15):4024–4030.
113. US Department of Health and Human Services; National Cancer Institute. Cancer Incidence and Survival Among Children and Adolescents: United States SEER Program 1975–1995. American Psychological Association; 1999.
114. Leavey PJ, Mascarenhas L, Marina N, et al. Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children’s Oncology Group. Pediatr Blood Cancer. 2008;51(3):334–338. doi:10.1002/pbc.21618
115. Bacci G, Longhi A, Ferrari S, et al. Pattern of relapse in 290 patients with nonmetastatic Ewing’s sarcoma family tumors treated at a single institution with adjuvant and neoadjuvant chemotherapy between 1972 and 1999. Eur J Surg Oncol. 2006;32(9):974–979. doi:10.1016/j.ejso.2006.01.023
116. Wasilewski-Masker K, Liu Q, Yasui Y, et al. Late Recurrence in Pediatric Cancer: A Report From the Childhood Cancer Survivor Study. JNCI J Natl Cancer Inst. 2009;101(24):1709–1720. doi:10.1093/jnci/djp417
117. Heinemann M, Ranft A, Langer T, et al. Recurrence of Ewing sarcoma: is detection by imaging follow-up protocol associated with survival advantage? Pediatr Blood Cancer. 2018;65(7):e27011. doi:10.1002/pbc.27011
118. Rodriguez-Galindo C, Billups CA, Kun LE, et al. Survival after recurrence of Ewing tumors: the St Jude Children’s Research Hospital experience, 1979-1999. Cancer. 2002;94(2):561–569. doi:10.1002/cncr.10192
119. Wagner LM, McAllister N, Goldsby RE, et al. Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Pediatr Blood Cancer. 2007;48(2):132–139. doi:10.1002/pbc.20697
120. Saylors RL, Stine KC, Sullivan J, et al. Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. J Clin Oncol. 2001;19(15):3463–3469. doi:10.1200/JCO.2001.19.15.3463
121. Kebudi R, Cakir FB, Gorgun O, Agaoglu FY, Darendeliler E. A modified protocol with vincristine, topotecan, and cyclophosphamide for recurrent/progressive Ewing sarcoma family tumors. Pediatr Hematol Oncol. 2013;30(3):170–177. doi:10.3109/08880018.2013.767868
122. Tanaka K, Joyama S, Chuman H, et al. Feasibility and efficacy of gemcitabine and docetaxel combination chemotherapy for bone and soft tissue sarcomas: multi-institutional retrospective analysis of 134 patients. World J Surg Oncol. 2016;14:306. doi:10.1186/s12957-016-1059-2
123. Ferrari S, Del Prever AB, Palmerini E, et al. Response to high-dose ifosfamide in patients with advanced/recurrent Ewing sarcoma. Pediatr Blood Cancer. 2009;52(5):581–584. doi:10.1002/pbc.21917
124. Palmerini E, Jones RL, Setola E, et al. Irinotecan and temozolomide in recurrent Ewing sarcoma: an analysis in 51 adult and pediatric patients. Acta Oncol Stockh Swed. 2018;57(7):958–964. doi:10.1080/0284186X.2018.1449250
125. Devadas SK, Banavali S. Retrospective analysis of outcomes of patients with relapsed, refractory and metastatic sarcomas who have received metronomic chemotherapy. Gulf J Oncolog. 2019;1(30):22–28.
126. Kolb EA, Gorlick R. Development of IGF-IR inhibitors in pediatric sarcomas. Curr Oncol Rep. 2009;11(4):307–313. doi:10.1007/s11912-009-0043-1
127. Richter GHS, Fasan A, Hauer K, et al. G-Protein coupled receptor 64 promotes invasiveness and metastasis in Ewing sarcomas through PGF and MMP1. J Pathol. 2013;230(1):70–81. doi:10.1002/path.4170
128. Attia S, Bolejack V, Ganjoo KN, et al. A phase II trial of regorafenib (REGO) in patients (pts) with advanced Ewing sarcoma and related tumors (EWS) of soft tissue and bone: SARC024 trial results. J Clin Oncol. 2017;35(15_suppl):11005. doi:10.1200/JCO.2017.35.15_suppl.11005
129. Cabozantinib in Patients With Advanced Osteosarcomas and Ewing sarcomas: a French Sarcoma Group (FSG)/US National Cancer Institute phase II collab … | oncologyPRO [Internet]. Available from: https://oncologypro.esmo.org/Meeting-Resources/ESMO-2018-Congress/Cabozantinib-in-Patients-With-Advanced-Osteosarcomas-and-Ewing-sarcomas-a-French-Sarcoma-Group-FSG-US-National-Cancer-Institute-phase-II-collaborative-study.
130. van Maldegem AM, JVMG B, Peterse EFP, Hogendoorn PCW, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171–180. doi:10.1016/j.ejca.2015.09.009
131. Zöllner SK, Selvanathan SP, Graham GT, et al. Inhibition of the oncogenic fusion protein EWS-FLI1 causes G2-M cell cycle arrest and enhanced vincristine sensitivity in Ewing’s sarcoma. Sci Signal. 2017;10:499. doi:10.1126/scisignal.aam8429
132. Gollavilli PN, Pawar A, Wilder-Romans K, et al. EWS/ETS-driven Ewing sarcoma requires BET bromodomain proteins. Cancer Res. 2018;78(16):4760–4773. doi:10.1158/0008-5472.CAN-18-0484
133. Theisen ER, Pishas KI, Saund RS, Lessnick SL. Therapeutic opportunities in Ewing sarcoma: EWS-FLI inhibition via LSD1 targeting. Oncotarget. 2016;7(14):17616–17630. doi:10.18632/oncotarget.7124
134. Pappo AS, Patel SR, Crowley J, et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol. 2011;29(34):4541–4547. doi:10.1200/JCO.2010.34.0000
135. Malempati S, Weigel B, Ingle AM, et al. Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(3):256–262. doi:10.1200/JCO.2011.37.4355
136. Tap WD, Demetri G, Barnette P, et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol. 2012;30(15):1849–1856. doi:10.1200/JCO.2011.37.2359
137. Wagner LM, Fouladi M, Ahmed A, et al. Phase II Study of cixutumumab in combination with temsirolimus in pediatric patients and young adults with recurrent or refractory sarcoma: a report from the children’s oncology group. Pediatr Blood Cancer. 2015;62(3):440–444. doi:10.1002/pbc.25334
138. Choy E, Butrynski JE, Harmon DC, et al. Phase II study of olaparib in patients with refractory Ewing sarcoma following failure of standard chemotherapy. BMC Cancer. 2014;5(14):813. doi:10.1186/1471-2407-14-813
139. Ghisoli M, Barve M, Schneider R, et al. Pilot trial of FANG immunotherapy in Ewing’s sarcoma. Mol Ther. 2015;23(6):1103–1109. doi:10.1038/mt.2015.43
140. Gargallo P, Juan A, Yáñez Y, et al. Precision medicine in Ewing sarcoma: a translational point of view. Clin Transl Oncol. 2020. doi:10.1007/s12094-020-02298-7
141. Machado I, López-Guerrero JA, Scotlandi K, Picci P, Llombart-Bosch A. Immunohistochemical analysis and prognostic significance of PD-L1, PD-1, and CD8+ tumor-infiltrating lymphocytes in Ewing’s sarcoma family of tumors (ESFT). Virchows Arch. 2018;472(5):815–824. doi:10.1007/s00428-018-2316-2
142. Van Mater D, Wagner L. Management of recurrent Ewing sarcoma: challenges and approaches. OncoTargets Ther. 2019;27(12):2279–2288. doi:10.2147/OTT.S170585
143. Rao DD, Jay C, Wang Z, et al. Preclinical Justification of pbi-shRNA EWS/FLI1 Lipoplex (LPX) Treatment for Ewing’s Sarcoma. Mol Ther. 2016;24(8):1412–1422. doi:10.1038/mt.2016.93
144. Ghisoli M, Barve M, Mennel R, et al. Three-year follow up of GMCSF/bi-shRNA(furin) DNA-transfected Autologous Tumor Immunotherapy (Vigil) in metastatic advanced ewing’s sarcoma. Mol Ther. 2016;24(8):1478–1483. doi:10.1038/mt.2016.86
145. Rasper M, Jabar S, Ranft A, Jürgens H, Amler S, Dirksen U. The value of high-dose chemotherapy in patients with first relapsed Ewing sarcoma. Pediatr Blood Cancer. 2014;61(8):1382–1386. doi:10.1002/pbc.25042
146. Barker LM, Pendergrass TW, Sanders JE, Hawkins DS. Survival after recurrence of Ewing’s sarcoma family of tumors. J Clin Oncol. 2005;23(19):4354–4362. doi:10.1200/JCO.2005.05.105
147. McTiernan A, Driver D, Michelagnoli MP, Kilby AM, Whelan JS. High dose chemotherapy with bone marrow or peripheral stem cell rescue is an effective treatment option for patients with relapsed or progressive Ewing’s sarcoma family of tumours. Ann Oncol. 2006;17(8):1301–1305. doi:10.1093/annonc/mdl108
148. Hayani A, Mahoney DH, Hawkins HK, Steuber CP, Hurwitz R, Fernbach DJ. Soft-tissue sarcomas other than rhabdomyosarcoma in children. Med Pediatr Oncol. 1992;20(2):114–118. doi:10.1002/mpo.2950200205
149. Orbach D, Rey A, Oberlin O, et al. Soft tissue sarcoma or malignant mesenchymal tumors in the first year of life: experience of the International Society of Pediatric Oncology (SIOP) Malignant Mesenchymal Tumor Committee. J Clin Oncol. 2005;23(19):4363–4371.
150. Coffin CM, Dehner LP. Soft tissue tumors in first year of life: a report of 190 cases. Pediatr Pathol. 1990;10(4):509–526. doi:10.3109/15513819009067140
151. Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. 1998;18(2):184–187. doi:10.1038/ng0298-184
152. Orbach D, Rey A, Cecchetto G, et al. Infantile fibrosarcoma: management based on the European experience. J Clin Oncol. 2010;28(2):318–323. doi:10.1200/JCO.2009.21.9972
153. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378(8):731–739. doi:10.1056/NEJMoa1714448
154. André N, Carré M, Pasquier E. Metronomics: towards personalized chemotherapy? Nat Rev Clin Oncol. 2014;11(7):413–431. doi:10.1038/nrclinonc.2014.89
155. Porkholm M, Toiviainen-Salo S, Seuri R, et al. Metronomic therapy can increase quality of life during paediatric palliative cancer care, but careful patient selection is essential. Acta Paediatr. 2016;105(8):946–951. doi:10.1111/apa.13338
156. André N, Abed S, Orbach D, et al. Pilot study of a pediatric metronomic 4-drug regimen. Oncotarget. 2011;2(12):960–965. doi:10.18632/oncotarget.358
157. Pramanik R, Agarwala S, Gupta YK, et al. Metronomic chemotherapy vs best supportive care in progressive pediatric solid malignant tumors: a randomized clinical trial. JAMA. 2017;3(9):1222.
158. Bonneville R, Krook MA, Kautto EA, et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol. 2017;2017.
159. Marabelle A, Le DT, Ascierto PA, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the Phase II KEYNOTE-158 Study. J Clin Oncol. 2020;38(1):1–10. doi:10.1200/JCO.19.02105
160. Kim JR, Moon YJ, Kwon KS, et al. Tumor infiltrating PD1-positive lymphocytes and the expression of PD-L1 predict poor prognosis of soft tissue sarcomas. PLoS One. 2013;8(12):e82870. doi:10.1371/journal.pone.0082870
161. Merchant MS, Wright M, Baird K, et al. Phase I clinical trial of ipilimumab in pediatric patients with advanced solid tumors. Clin Cancer Res. 2016;22(6):1364–1370. doi:10.1158/1078-0432.CCR-15-0491
162. Groisberg R, Hong DS, Behrang A, et al. Characteristics and outcomes of patients with advanced sarcoma enrolled in early phase immunotherapy trials. J Immunother Cancer. 2017;5(1):100. doi:10.1186/s40425-017-0301-y
163. Davis KL, Fox E, Reid JM, et al. ADVL1412: initial results of a phase I/II study of nivolumab and ipilimumab in pediatric patients with relapsed/refractory solid tumors—A COG study. J Clin Oncol. 2017;35(15_suppl):10526. doi:10.1200/JCO.2017.35.15_suppl.10526
164. Merchant MS, Bernstein D, Amoako M, et al. Adjuvant immunotherapy to improve outcome in high-risk pediatric sarcomas. Clin Cancer Res. 2016;22(13):3182–3191. doi:10.1158/1078-0432.CCR-15-2550
165. Folkert IW, Devalaraja S, Linette GP, Weber K, Haldar M. Primary bone tumors: challenges and opportunities for CAR‐T therapies. J Bone Miner Res. 2019;34(10):1780–1788. doi:10.1002/jbmr.3852
166. Zhu H, Gu S, Yin M, et al. Analysis of infantile fibrosarcoma reveals extensive T-cell responses within tumors: implications for immunotherapy. Pediatr Blood Cancer. 2018;65(2):e26813. doi:10.1002/pbc.26813
167. Chang T-C, Carter RA, Li Y, et al. The neoepitope landscape in pediatric cancers. Genome Med. 2017;9(1):78. doi:10.1186/s13073-017-0468-3
168. Pilbeam K, Wang H, Taras E, et al. Targeting pediatric sarcoma with a bispecific ligand immunotoxin targeting urokinase and epidermal growth factor receptors. Oncotarget. 2017;9(15):11938–11947. doi:10.18632/oncotarget.21187
169. Sp D, Mr M, Ba VT, et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, Phase 2 Trials. Lancet Oncol. 2018.
170. Gandhi L, Rodríguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378(22):2078–2092. doi:10.1056/NEJMoa1801005
171. Geiger JD, Hutchinson RJ, Hohenkirk LF, et al. Vaccination of pediatric solid tumor patients with tumor lysate-pulsed dendritic cells can expand specific T cells and mediate tumor regression. Cancer Res. 2001;61(23):8513–8519.
172. Ahmed N, Brawley VS, Hegde M, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 2015;33(15):1688–1696. doi:10.1200/JCO.2014.58.0225
173. Antonia SJ, Villegas A, Daniel D, et al. Overall survival with durvalumab after chemoradiotherapy in Stage III NSCLC. N Engl J Med. 2018;13;379(24):2342–2350. doi:10.1056/NEJMoa1809697
174. Klingebiel T, Pertl U, Hess CF, et al. Treatment of children with relapsed soft tissue sarcoma: report of the German CESS/CWS REZ 91 trial. Med Pediatr Oncol. 1998;30(5):269–275. doi:10.1002/(SICI)1096-911X(199805)30:5<269::AID-MPO2>3.0.CO;2-D
175. Defachelles AS, Bogart E, Casanova M, et al. Randomized phase 2 trial of the combination of vincristine and irinotecan with or without temozolomide, in children and adults with refractory or relapsed rhabdomyosarcoma (RMS). J Clin Oncol. 2019;37(15_suppl):10000. doi:10.1200/JCO.2019.37.15_suppl.10000
176. Setty BA, Stanek JR, Mascarenhas L, et al. VIncristine, irinotecan, and temozolomide in children and adolescents with relapsed rhabdomyosarcoma. Pediatr Blood Cancer. 2018;65(1):e26728. doi:10.1002/pbc.26728
177. Kuttesch JF, Krailo MD, Madden T, Johansen M, Bleyer A. Children’s Oncology Group. Phase II evaluation of intravenous vinorelbine (Navelbine) in recurrent or refractory pediatric malignancies: a Children’s Oncology Group study. Pediatr Blood Cancer. 2009;53(4):590–593. doi:10.1002/pbc.22133
178. Casanova M, Ferrari A, Spreafico F, et al. Vinorelbine in previously treated advanced childhood sarcomas: evidence of activity in rhabdomyosarcoma. Cancer. 2002;94(12):3263–3268. doi:10.1002/cncr.10600
179. Casanova M, Ferrari A, Bisogno G, et al. Vinorelbine and low-dose cyclophosphamide in the treatment of pediatric sarcomas: pilot study for the upcoming European Rhabdomyosarcoma Protocol. Cancer. 2004;101(7):1664–1671. doi:10.1002/cncr.20544
180. Minard-Colin V, Ichante J-L, Nguyen L, et al. Phase II study of vinorelbine and continuous low doses cyclophosphamide in children and young adults with a relapsed or refractory malignant solid tumour: good tolerance profile and efficacy in rhabdomyosarcoma–a report from the Société Française des Cancers et leucémies de l’Enfant et de l’adolescent (SFCE). Eur J Cancer Oxf Engl 1990. 2012;48(15):2409–2416.
181. Mascarenhas L, Meyer WH, Lyden E, et al. Randomized phase II trial of bevacizumab and temsirolimus in combination with vinorelbine (V) and cyclophosphamide (C) for first relapse/disease progression of rhabdomyosarcoma (RMS): A report from the Children’s Oncology Group (COG). J Clin Oncol. 2014;32(15_suppl):10003. doi:10.1200/jco.2014.32.15_suppl.10003
182. Rapkin L, Qayed M, Brill P, et al. Gemcitabine and docetaxel (GEMDOX) for the treatment of relapsed and refractory pediatric sarcomas. Pediatr Blood Cancer. 2012;59(5):854–858. doi:10.1002/pbc.24101
183. Kung FH, Pratt CB, Vega RA, et al. Ifosfamide/etoposide combination in the treatment of recurrent malignant solid tumors of childhood. A Pediatric Oncology Group Phase II study. Cancer. 1993;71(5):1898–1903. doi:10.1002/1097-0142(19930301)71:5<1898::AID-CNCR2820710529>3.0.CO;2-Q
184. Miser JS, Kinsella TJ, Triche TJ, et al. Ifosfamide with mesna uroprotection and etoposide: an effective regimen in the treatment of recurrent sarcomas and other tumors of children and young adults. J Clin Oncol off J Am Soc Clin Oncol. 1987;5(8):1191–1198. doi:10.1200/JCO.1987.5.8.1191
185. Cairo MS, Shen V, Krailo MD, et al. Prospective randomized trial between two doses of granulocyte colony-stimulating factor after ifosfamide, carboplatin, and etoposide in children with recurrent or refractory solid tumors: a children’s cancer group report. J Pediatr Hematol Oncol. 2001;23(1):30–38. doi:10.1097/00043426-200101000-00008
186. Berrak SG, Pearson M, Berberoğlu S, Ilhan IE, Jaffe N. High-dose ifosfamide in relapsed pediatric osteosarcoma: therapeutic effects and renal toxicity. Pediatr Blood Cancer. 2005;44(3):215–219. doi:10.1002/pbc.20228
187. Navid F, Willert JR, McCarville MB, et al. Combination of gemcitabine and docetaxel in the treatment of children and young adults with refractory bone sarcoma. Cancer. 2008;113(2):419–425. doi:10.1002/cncr.23586
188. Qi W-X, He A-N, Tang L-N, Shen Z, Lin F, Yao Y. Efficacy and safety of gemcitabine-docetaxel combination therapy for recurrent or refractory high-grade osteosarcoma in China: a retrospective study of 18 patients. Jpn J Clin Oncol. 2012;42(5):427–431. doi:10.1093/jjco/hys030
189. Palmerini E, Jones RL, Marchesi E, et al. Gemcitabine and docetaxel in relapsed and unresectable high-grade osteosarcoma and spindle cell sarcoma of bone. BMC Cancer. 2016;16:280. doi:10.1186/s12885-016-2312-3
190. Anderson P, Kopp L, Anderson N, et al. Novel bone cancer drugs: investigational agents and control paradigms for primary bone sarcomas (Ewing’s sarcoma and osteosarcoma). Expert Opin Investig Drugs. 2008;17(11):1703–1715. doi:10.1517/13543784.17.11.1703
191. Casey DA, Wexler LH, Merchant MS, et al. Irinotecan and temozolomide for Ewing sarcoma: the Memorial Sloan-Kettering experience. Pediatr Blood Cancer. 2009;53(6):1029–1034. doi:10.1002/pbc.22206
192. Hernández-Marqués C, Lassaletta-Atienza A, Ruiz Hernández A, et al. [Irinotecan plus temozolomide in refractory or relapsed pediatric solid tumors]. An Pediatr Barc Spain. 2013;79(2):68–74. Spanish.
193. Raciborska A, Bilska K, Drabko K, et al. Vincristine, irinotecan, and temozolomide in patients with relapsed and refractory Ewing sarcoma. Pediatr Blood Cancer. 2013;60(10):1621–1625. doi:10.1002/pbc.24621
194. Kurucu N, Sari N, Ilhan IE. Irinotecan and temozolomide treatment for relapsed Ewing sarcoma: a single-center experience and review of the literature. Pediatr Hematol Oncol. 2015;32(1):50–59. doi:10.3109/08880018.2014.954070
195. Büyükkapu Bay S, Kebudi R, Görgün O, Zülfikar B, Darendeliler E, Çakır FB. Vincristine, irinotecan, and temozolomide treatment for refractory/relapsed pediatric solid tumors: A single center experience. J Oncol Pharm Pract off Publ Int Soc Oncol Pharm Pract. 2019;25(6):1343–1348. doi:10.1177/1078155218790798
196. Hunold A, Weddeling N, Paulussen M, Ranft A, Liebscher C, Jürgens H. Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer. 2006;47(6):795–800. doi:10.1002/pbc.20719
197. Farhat R, Raad R, Khoury NJ, et al. Cyclophosphamide and topotecan as first-line salvage therapy in patients with relapsed ewing sarcoma at a single institution. J Pediatr Hematol Oncol. 2013;35(5):356–360. doi:10.1097/MPH.0b013e318270a343
198. Mora J, Cruz CO, Parareda A, de Torres C. Treatment of relapsed/refractory pediatric sarcomas with gemcitabine and docetaxel. J Pediatr Hematol Oncol. 2009;31(10):723–729. doi:10.1097/MPH.0b013e3181b2598c
199. van Maldegem AM, Benson C, Rutkowski P, et al. Etoposide and carbo-or cisplatin combination therapy in refractory or relapsed Ewing sarcoma: a large retrospective study. Pediatr Blood Cancer. 2015;62(1):40–44. doi:10.1002/pbc.25230
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.