Back to Journals » OncoTargets and Therapy » Volume 11
MALAT1 silencing suppresses prostate cancer progression by upregulating miR-1 and downregulating KRAS
Authors Chang J, Xu W, Du X, Hou J
Received 30 January 2018
Accepted for publication 19 April 2018
Published 15 June 2018 Volume 2018:11 Pages 3461—3473
DOI https://doi.org/10.2147/OTT.S164131
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tohru Yamada
Junkai Chang, Weibo Xu, Xinyi Du, Junqing Hou
Department of Urology, Huaihe Hospital of Henan University, Kaifeng 475000, China
Background: Prostate cancer (PC) is the second leading cause of cancer-related deaths among men. Long noncoding RNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) performed as an oncogene in multiple cancers including PC. However, the molecular mechanisms of MALAT1 implicated in PC progression have not been thoroughly elaborated.
Materials and methods: Reverse transcription-quantitative polymerase chain reaction assay was used to detect the expressions of MALAT1 and microRNA-1 (miR-1). Protein levels of cleaved poly (ADP-ribose) polymerase, cleaved caspase-3, BAX, bcl-2, and KRAS were determined using a western blot assay. Cell proliferation was assessed by colony formation and MTS assays. Cell migration capacity was examined by transwell migration assay (Corning Incorporated, Corning, NY, USA). Apoptosis rate was measured by flow cytometry via double staining of annexin V-FITC and propidium iodide. Luciferase and RNA immunoprecipitation assays were employed to explore the relationship among miR-1, MALAT1, and KRAS.
Results: MALAT1 expression was upregulated and miR-1 expression was downregulated in PC tissues and cell lines. MALAT1 knockdown inhibited cell proliferation and migration, and promoted cell apoptosis in androgen receptor-negative DU145 and PC3 cells. Molecular mechanism explorations disclosed that MALAT1 acted as a molecular sponge of miR-1 in DU145 cells. Moreover, miR-1 downregulation partly abrogated MALAT1 silencing-mediated anti-proliferative, antimigratory, and proapoptotic effects in DU145 and PC3 cells. Further investigation revealed that KRAS was a target of miR-1 in DU145 cells. MALAT1 acted as a competing endogenous RNA of miR-1, resulting in the increase of KRAS expression in DU145 and PC3 cells. Furthermore, miR-1 overexpression hampered proliferation and migration and promoted apoptosis in DU145 and PC3 cells, while these effects were markedly weakened following KRAS upregulation.
Conclusion: MALAT1 knockdown inhibited proliferation and migration and facilitated apoptosis by upregulating miR-1 and downregulating KRAS in androgen receptor-negative PCa cells, providing a new insight into the molecular basis of MALAT1 and a potential biomarker or therapeutic target for suppressing castration-resistant PC.
Keywords: prostate cancer, MALAT1, microRNA-1, KRAS
Introduction
Prostate cancer (PC) is the most common cancer in male and ranks second in cancer-related deaths among men, with an estimated 1,80,890 new cases and 26,120 deaths in 2016 in the USA.1 Although much progress has been made in early detection and clinical management of PC over the past few decades, 30% of PC patients undergo a relapse following radical prostatectomy or adjuvant treatment.2–4 Hence, it is imperative to search for more effective biomarkers to better guide therapeutic decisions for patients with PC.
Noncoding RNAs including long noncoding RNAs (lncRNAs) and microRNAs (miRNAs) have been demonstrated to be critical regulators in the development and progression of cancers.5,6 Moreover, numerous miRNAs and lncRNAs have been reported as tumor suppressors or oncogenes in PC by regulating various biological processes such as proliferation, migration, and apoptosis.7,8 Due to the vital roles in tumorigenesis and pathogenesis of PC, miRNAs and lncRNAs show great potential as predictive, diagnostic, and prognostic biomarkers or therapeutic targets for PC.9,10 lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), also named as nuclear-enriched transcript 2, was generally highly expressed and performed as an oncogene in most of malignant tumors, such as breast cancer, lung cancer, and hepatocellular carcinoma (HCC).11 Previous studies also manifested that increased MALAT1 expression was positively correlated with unfavorable prognosis and advanced clinical stage in pancreatic cancer and PC.12,13 Moreover, MALAT1 was overexpressed in PC tissues and cell lines, and MALAT1 depletion impaired proliferation, migration, and invasion, and induced apoptosis in PC cells.13 However, the underlying molecular mechanisms of MALAT1 involved in PC etiology and pathogenesis have not been thoroughly elaborated.
MicroRNA-1 (miR-1) has been classified to be a tumor suppressor with great potential to block cancer development and therapy resistance in the majority of malignancies, such as bladder cancer, head and neck squamous cell cancer, colorectal cancer, and lung cancer.14 Furthermore, miR-1 expression was downregulated in PC tissues and cells, and ectopic expression of miR-1 inhibited proliferation, migration, invasion, and epithelial–mesenchymal transition in PC cells, and prevented tumorigenesis in mice xenograft model of PC.15–17 Moreover, reduced miR-1 expression was positively correlated with advanced pathogenic stage and poor prognosis in PC.16
Bioinformatics analyses revealed that miR-1 had a chance to interact with MALAT1. Additionally, a previous report disclosed that MALAT1 exerted its oncogenic effect by regulating the expressions of miR-1 and target genes in breast cancer.18 Hence, in the present study, we aimed to further investigate whether the carcinogenic role of MALAT1 was mediated by miR-1 and downstream targets in PC.
Materials and methods
Tissue samples and cell culture
A total of 20 pairs of PC tumor tissues and adjacent normal tissues were obtained from PC patients at our hospital from 2015 to 2016. Our study obtained the approval of Ethics and Scientific Committees of Huaihe Hospital of Henan University. Written informed consent was signed by all tissue donators.
Human PC cell lines (PC3, LNCap, and DU145) were purchased from Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Human normal prostate epithelial cell line (RWPE-1) and PC cell line (C4-2) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). DU145 cells were maintained in minimum essential medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS, Thermo Fisher Scientific), 1× glutamax (Thermo Fisher Scientific), 1× non-essential amino acids (Thermo Fisher Scientific), and sodium pyruvate solution (1 mM, Thermo Fisher Scientific). PC3 cells were cultured in Ham’s F-12 medium (Thermo Fisher Scientific) supplemented with 10% FBS (Thermo Fisher Scientific), L-glutamine (300 mg/L, Sigma-Aldrich, St Louis, MO, USA), and NaHCO3 (1.5 g/L, Sigma-Aldrich). LNCap cells were maintained in RPMI-1640 medium (Thermo Fisher Scientific) containing 10% FBS (Thermo Fisher Scientific), NaHCO3 (1.5 g/L, Sigma-Aldrich), glucose (2.5 g/L, Sigma-Aldrich), and sodium pyruvate (0.11 g/L, Sigma-Aldrich). C4-2 cells were cultured in Dulbecco’s Modified Eagle’s Medium/Ham’s F-12 (DMEM/F-12, Thermo Fisher Scientific) medium containing 10% FBS (Thermo Fisher Scientific), transferrin (5 μg/mL, Sigma-Aldrich), biotin (0.25 μg/mL Sigma-Aldrich), triiodothyronine (13.6 pg/mL, Sigma-Aldrich), adenine (25 μg/mL, Sigma-Aldrich), and insulin (5 μg/mL, Sigma-Aldrich). RWPE-1 cells were maintained in Defined Keratinocyte Serum Free Medium (Thermo Fisher Scientific).
Reagents, plasmid construction, and transfection
MiR-1 mimic and its scramble control (miR-NC), miR-1 inhibitor (anti-miR-1) and its scramble control (anti-miR-NC), and small interference RNA targeting MALAT1 (si-MALAT1) and its scramble control (si-NC) were purchased from Gene Pharma Co., Ltd. (Shanghai, China). The sequences of MALAT1 or KRAS were amplified by polymerase chain reaction (PCR) and subcloned into pcDNA3.1 (Thermo Fisher Scientific) to produce MALAT1 or KRAS overexpression plasmid, named as pcDNA-MALAT1 (MALAT1) or pcDNA-KRAS (KRAS). Then, these miRNAs, miRNA inhibitors, siRNAs, or plasmids were transfected into PC cells using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions.
RT-qPCR assay
Total RNAs including miRNAs were extracted from tissues and cells using Trizol reagent (Thermo Fisher Scientific) and quantified with Nanodrop 2000 instrument (Thermo Fisher Scientific). Then, 1 μg RNA was reversely transcribed into first-strand cDNA using M-MLV Reverse Transcriptase and random primers (for the reverse transcription of MALAT1, β-actin) or specific RT primers (for miR-1 or U48 snRNA). Then, the expression analyses of MALAT1, β-actin, miR-1, or U48 snRNA were conducted using SYBR™ Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) and specific quantitative primers. β-Actin was used to normalize the expressions of MALAT1. U48 snRNA acted as an endogenous control of miR-1. RT primer sequences were listed as follows: miR-1 (RT), 5′-GTTGGCTCTGGTGCAGGGTCCGAGGTATTCGCACCAGAGCCAACATACAT-3′; U48 (RT), 5′-GTTGGCTCTGGTGCAGGGTCCGAGGTATTCGCACCAGAGCCAACGGTCAG-3′. Quantitative primer sequences were also listed as follows: miR-1, 5′-CGGCGGTGGAATGTAAAGAAG-3′ (forward) and 5′-GTGCAGGGTCCGAGGT-3′ (reverse); U48, 5′-CGACGAGTGATGATGAC-3′ (forward) and 5′-GTGCAGGGTCCGAGGT-3′ (reverse); MALAT1, 5′-AAAGCAAGGTCTCCCCACAAG-3′ (forward) and 5′-GGTCTGTGCTAGATCAAAAGGCA-3′ (reverse); β-actin, 5′-CTGTCTGGCGGCACCACCAT-3′ (forward) and 5′-GCAACTAAGTCATAGTCCGC-3′ (reverse).
Luciferase assay
Partial sequences of MALAT1 and KRAS 3′-untranslated region (UTR) containing putative miR-1 binding sites were amplified by PCR and constructed into psiCHECK-2 vector (Promega Corporation, Madison, WI, USA) to generate wild-type MALAT1 reporter (MALAT1 [WT]) and wild-type KRAS reporter (KRAS [WT]). GeneArt™ Site-Directed Mutagenesis System (Thermo Fisher Scientific) was used to produce mutant-type MALAT1 reporter (MALAT1 [MUT]) and mutant-type KRAS reporter (KRAS [MUT]). Then, the constructed reporters were, respectively, cotransfected into DU145 cells together with miRNAs (miR-NC, miR-1), miRNA inhibitors (anti-miR-NC, ant-miR-1), or plasmids (pcDNA3.1 vector or pcDNA-MALAT1). At 48 h after transfection, luciferase activities in DU145 cells were determined using a dual-luciferase assay system (Promega) following the protocols of the manufacturer.
Apoptosis rate analysis
Cell apoptosis rates were determined using FITC Annexin V/Dead Cell Apoptosis Kit (Thermo Fisher Scientific) at 48 h upon transfection. Briefly, collected cells were resuspended in 1× annexin-binding buffer, double stained with FITC annexin V and propidium iodide for 15 min at room temperature, and then subjected to a flow cytometry (FACScan; BD Biosciences, Franklin Lakes, NJ, USA) for the detection of apoptotic rates.
RNA immunoprecipitation (RIP) assay
RIP assay was performed in DU145 cells using Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, Billerica, MA, USA) and antibody against IgG or Argonaute2 (Ago2) according to the manufacturer’s instructions. Briefly, DU145 cells were collected at 80%–90% confluency and then lysed by RIP lysis buffer. Next, whole cell extract was treated with RIP buffer supplemented with magnetic beads containing antibodies against IgG (Millipore) or Ago2 (Millipore), followed by the purification of RNA. Then, the expression patterns of MALAT1 and miR-1 in IgG or Ago2 immunoprecipitated complex were detected using RT-qPCR assay.
MTS assay
Cell viability was determined using a CellTiter 96® AQueous One Solution Cell Proliferation Assay kit (Promega) according to the manufacturer’s instructions. Generally, at the indicated time points (0, 24, 48, and 72 h) after transfection, cells (in 100 μL medium) were incubated with 20 μL CellTiter 96 AQueous One Solution reagent for 3 h. Then, cell absorbance was measured at 490 nm.
Colony formation assay
Transfected DU145 and PC3 cells were seeded into 60 mm culture dishes and cultured for 14 days in the complete medium. Then, cells were fixed using methanol for 10 min and stained with 0.1% crystal violet (Sigma-Aldrich) for 15 min. Next, dishes were photographed and colonies with over 50 cells were counted.
Transwell migration assay
Cell migration ability was assessed using transwell plates (Corning Incorporated, Corning, NY, USA) containing 8 μm pore membrane. Transfected cells (5×104) were resuspended in serum-free culture medium and then plated into the upper chamber. Also, a medium containing 20% FBS was introduced into the lower chamber. After 24 h incubation at 37°C, cells on the upper chamber were abandoned. But cells on the lower surface were fixed and stained, followed by the determination of migrated cell numbers in 10 random fields using a microscope.
Western blot assay
At 48 h after transfection, DU145 and PC cells were collected and lysed with RIPA buffer (Beyotime, Shanghai, China) containing protease inhibitor cocktail (MedChem Express, Monmouth Junction, NJ, USA), followed by high-speed centrifugation (12,000 rpm, 15 min, 4°C). Then, whole protein concentrations in cell supernatants were determined using a BCA protein assay kit (Beyotime). Next, equivalent proteins (50 μg/lane) were separated by 10% SDS-polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes (Millipore). After blocking with 5% nonfat milk for 1 h, the membranes were incubated with anti-Cleaved PARP (Cell Signaling Technology, Danvers, MA, USA), anti-cleaved caspase-3 (Cell Signaling Technology), anti-KRAS mAb (Abcam, Cambridge, UK), anti-β-actin mAb (Abcam), anti-Bcl-2pAb (Abcam), and anti-BAXmAb (Abcam) overnight at 4°C. On the next day, the membranes were probed with horseradish peroxidase-conjugated goat-anti-rabbit or goat-anti-mouse secondary antibody (Abcam) for 1 h. Finally, specific protein signals were detected using Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific) and quantified using Quantity One Software Version 4.1.1 (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Statistical analysis
Data were analyzed using SPSS software (IBM, Armonk, NY, USA) and displayed as mean ± SD from at least three independent experiments. Student’s t-test or one-way analysis of variance was employed to perform difference analyses. P-value <0.05 meant the difference was statistically significant.
Results
MALAT1 expression was upregulated and miR-1 expression was downregulated in PC tissues and cells
Firstly, RT-qPCR assay was performed to investigate expression patterns of MALAT1 and miR-1 in PC tissues and cells. The results showed that MALAT1 expression was markedly upregulated (Figure 1A), and miR-1 expression was significantly downregulated (Figure 1C) in PC tissues (n=20) compared with adjacent normal tissues (n=20). Moreover, a remarkable increase of MALAT1 level and a notable decrease of miR-1 level were observed in PC cell lines (PC3, C4-2, LNCap, DU145) when compared with that in a normal human prostate epithelial cell line RWPE-1 (Figure 1B and D). In other words, these results indicated that MALAT1 and miR-1 might be implicated in PC progression.
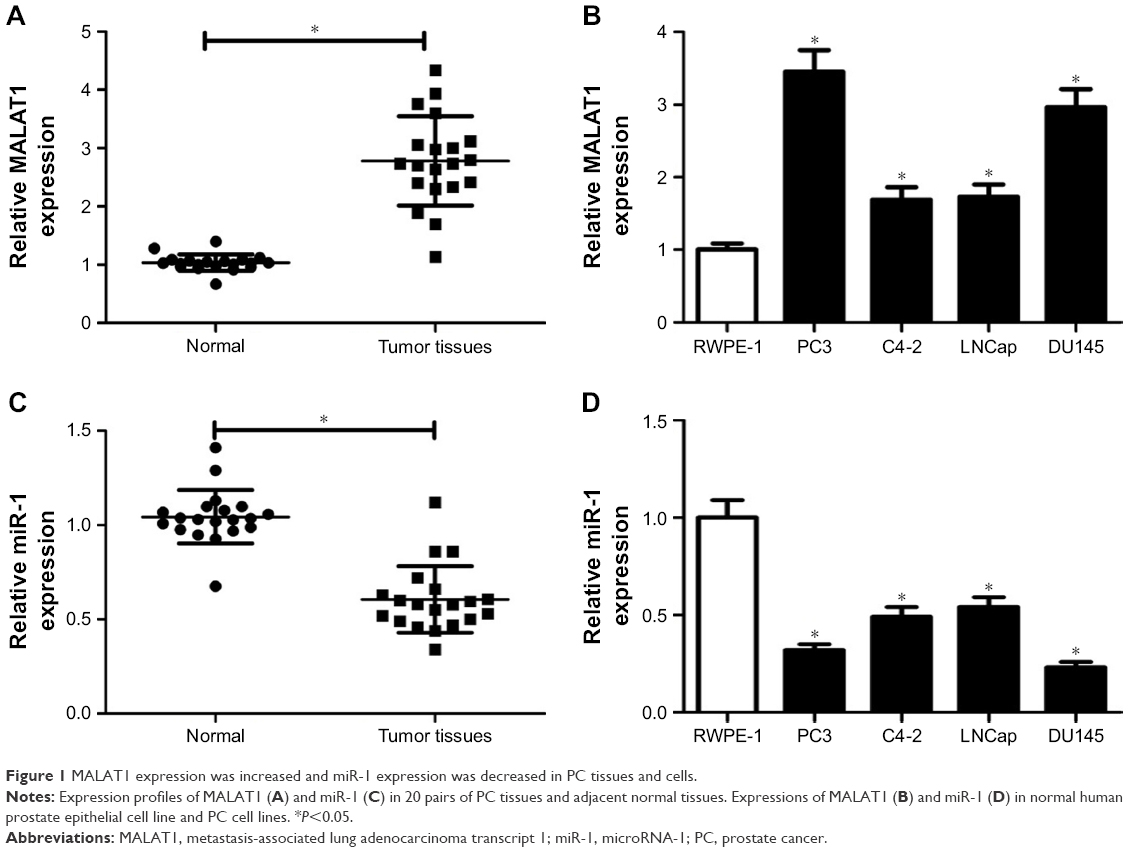 | Figure 1 MALAT1 expression was increased and miR-1 expression was decreased in PC tissues and cells. |
MALAT1 knockdown inhibited proliferation and promoted apoptosis in PC cells
To further investigate the possible biological roles of MALAT1 in PC tumorigenesis, si-MALAT1 and its scramble control (si-NC) were synthesized. Then, knockdown efficiency of si-MALAT1 was measured by RT-qPCR assay in DU145 and PC3 cells. As presented in Figure 2A, compared with scramble control, introduction of si-MALAT1 induced a striking reduction of MALAT1 level in DU145 and PC3 cells, indicating that si-MALAT1 could be used for the following loss-of-function experiments. Next, the effects of MALAT1 downregulation on cell proliferation, migration, and apoptosis were further examined in DU145 and PC3 cells. Colony formation assay showed that MALAT1 knockdown triggered a prominent decrease of colony numbers in DU145 and PC3 cells (Figure 2B). MTS assay revealed that proliferation ability was significantly declined in MALAT1-depleted DU145 and PC3 cells than that in mock cells (Figure 2C). Transwell migration assay further manifested that MALAT1 knockdown inhibited the migration of DU145 and PC3 cells (Figure 2D). Subsequent flow cytometry analysis demonstrated that MALAT1 silencing resulted in a marked elevation of apoptotic rate in DU145 and PC3 cells (Figure 2E). Consistently, western blot assay manifested that MALAT1 deficiency induced the expressions of proapoptosis proteins (cleaved PARP, cleaved caspase-3, and BAX) and inhibited antiapoptosis protein Bcl-2 expression in DU145 and PC3 cells (Figure 2F). All these data suggested that downregulation of MALAT1 impeded proliferation and migration and enhanced apoptosis in PC cells.
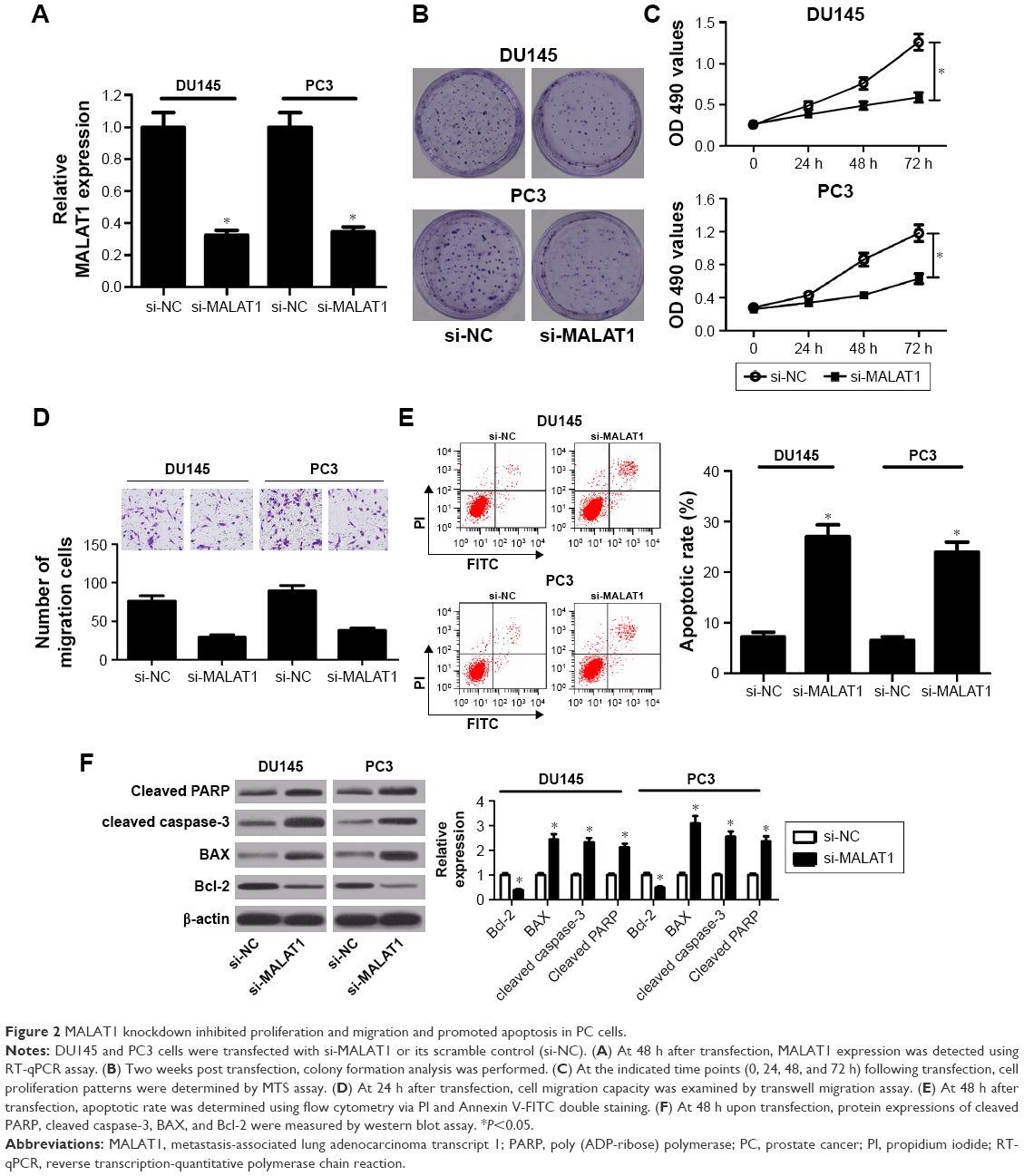 | Figure 2 MALAT1 knockdown inhibited proliferation and migration and promoted apoptosis in PC cells. |
MALAT1 acted as a molecular sponge of miR-1 in PC cells
Abundant evidences propose that lncRNAs could act as miRNA sponges to exert their regulatory effects on target mRNAs.19 Thus, miRcode and starBase online websites were employed to search for miRNAs possessing a chance to interact with MALAT1. Among candidate miRNAs, miR-1 (Figure 3A) was chosen by virtue of its antitumor effect in multiple cancers.20 To further validate the interaction of MALAT1 and miR-1, wild-type MALAT1 reporter containing predicted binding sites of miR-1 and mutant-type MALAT1 reporter with mutant binding sites of miR-1 was constructed. Subsequent luciferase assay revealed that luciferase activity of wild-type MALAT1 reporter was significantly decreased in miR-1-transfected DU145 cells, but was markedly increased in DU145 cells after introduction of anti-miR-1 (Figure 3B). However, there was no change in luciferase activity of mutant-type MALAT1 reporter following miR-1 overexpression or knockdown (Figure 3B), indicating that MALAT1 interacted with miR-1 by putative binding sites. As is known to all, Ago2, a core component of RNA-induced silencing complex (RISC), plays an indispensable role in miRNA-mediated gene silencing. Hence, RIP assay was carried out using Ago2 antibody or IgG control antibody to explore whether MALAT1 and miR-1 were present in the same RISC. Results showed that MALAT1 and miR-1 were substantially enriched in Ago2 immunoprecipitates of DU145 cells compared with control group, hinting the binding probability between MALAT1 and miR-1 (Figure 3C). Then, we further demonstrated that miR-1 expression was inversely associated with MALAT1 expression in 20 cases of PC tissues (Figure 3D). To further determine the influence of MALAT1 on miR-1 expression, MALAT1 overexpression plasmid (MALAT1) or si-MALAT1 was transfected into DU145 cells, followed by the detection of transfection efficiency. As displayed in Figure 3E, introduction of MALAT1 overexpression plasmid significantly elevated MALAT1 expression; conversely, transfection of si-MALAT1 markedly lowered MALAT1 expression in DU145 cells. As expected, miR-1 level was notably downregulated in MALAT1-overexpressed DU145 cells, but was strikingly upregulated in MALAT1-silenced DU145 cells (Figure 3F). In other words, MALAT1 inhibited miR-1 expression by direct interaction in PC cells.
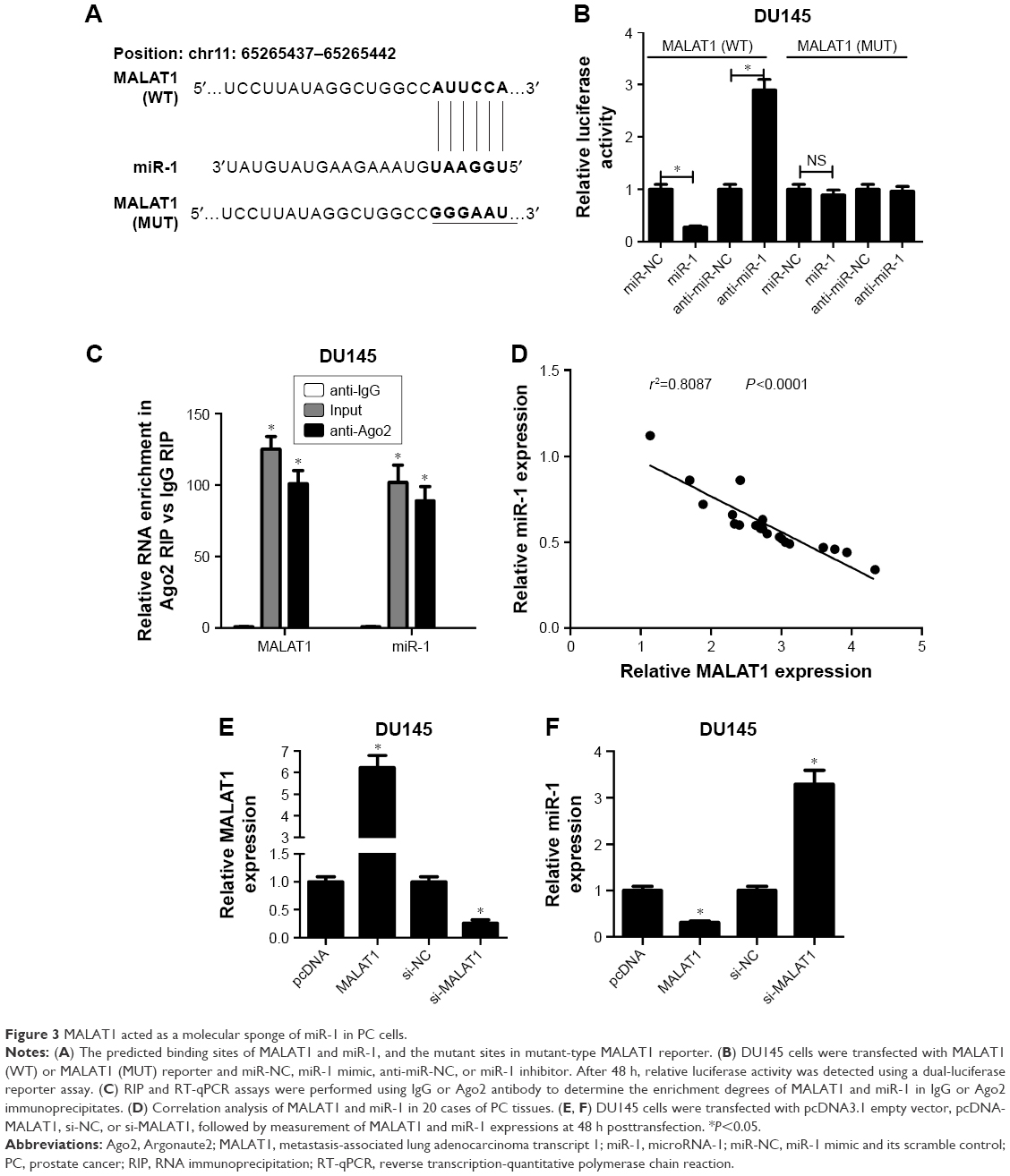 | Figure 3 MALAT1 acted as a molecular sponge of miR-1 in PC cells. |
MiR-1 downregulation partly abrogated MALAT1 silencing-mediated antiproliferative, antimigratory and proapoptotic effects in PC cells
Next, restoration assay was performed to further explore whether miR-1 could influence the carcinogenic roles of MALAT1 in PC cells by transfecting with si-MALAT1 alone, or in combination with anti-miR-1. First, RT-qPCR assay demonstrated that introduction of miR-1 inhibitor weakened the promotive effect of si-MALAT1 on miR-1 expression in DU145 and PC3 cells (Figure 4A). Functional analyses further revealed that miR-1 downregulation relieved the inhibitory effects of MALAT1 knockdown on colony formation capacity (Figure 4B), cell proliferation (Figure 4C), and cell migration (Figure 4D) in DU145 and PC3 cells. Moreover, miR-1 deficiency undermined si-MALAT1-mediated promotive effect on cell apoptosis, presented by decreased apoptotic rate (Figure 4E), reduced protein expressions of cleaved PARP, cleaved caspase-3, and BAX, and increased Bcl-2 protein expression (Figure 4F) in si-MALAT1-transfected DU145 and PC3 cells following miR-1 inhibition. Collectively, these data showed that MALAT1 silencing suppressed cell proliferation and migration, and enhanced cell apoptosis through upregulating miR-1 expression in PC cells.
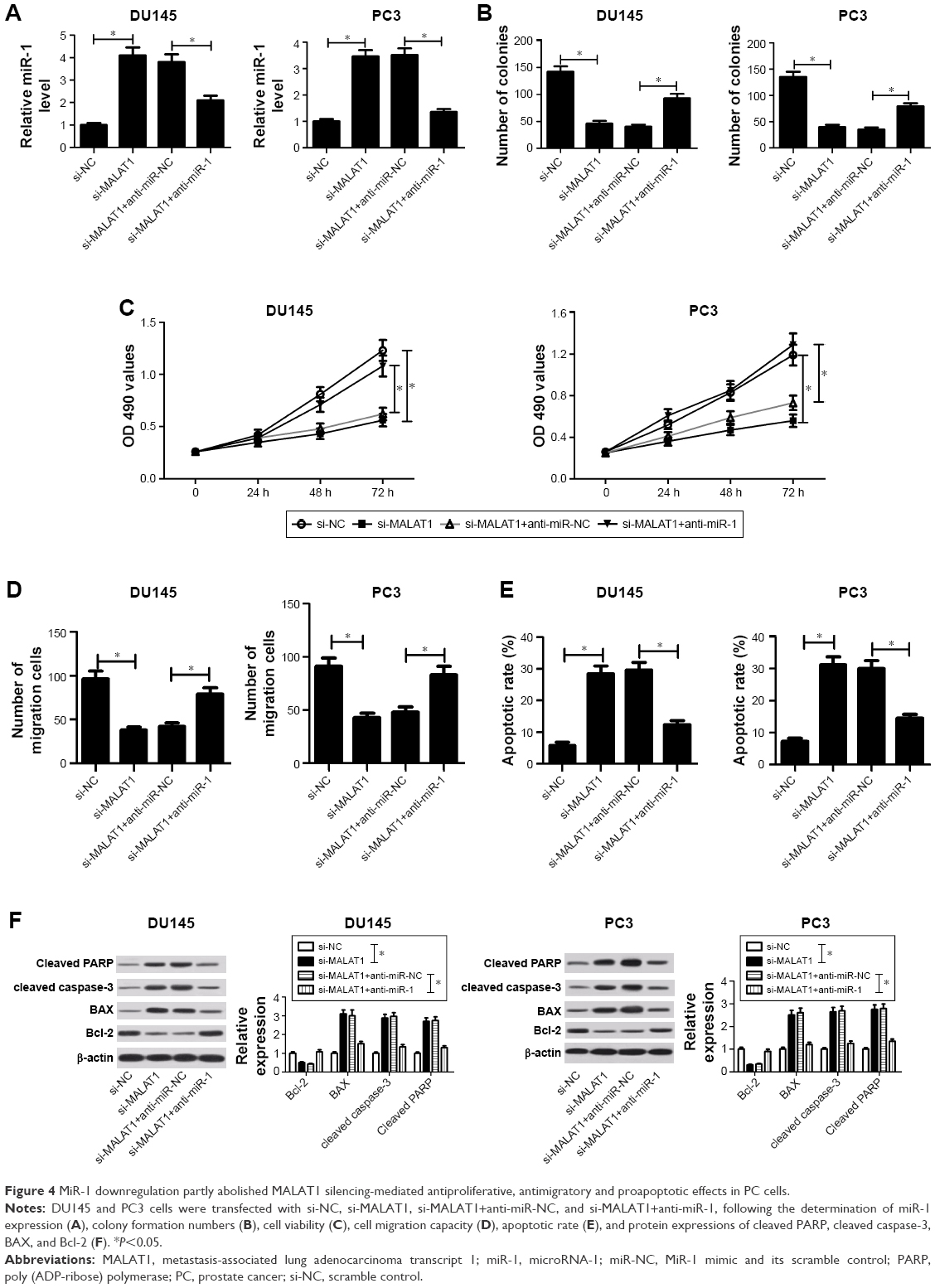 | Figure 4 MiR-1 downregulation partly abolished MALAT1 silencing-mediated antiproliferative, antimigratory and proapoptotic effects in PC cells. |
MALAT1 acted as a competing endogenous RNA (ceRNA) of miR-1 to sequester miR-1 away from KRAS in PC cells
Next, Targetscan software was used to identify potential target genes of miR-1. Among candidate genes, KRAS (Figure 5A) was selected due to its oncogenic effect in human cancers.21 To further test whether KRAS was a direct target of miR-1, WT-KRAS reporter containing predicted miR-1 binding sites and MUT-KRAS reporter with mutant miR-1 binding sites were generated. Then, luciferase assay manifested that the luciferase activity of WT-KRAS reporter was significantly repressed in miR-1-overexpressing DU145 cells, but was markedly raised in miR-1-lacking DU145 cells (Figure 5B). However, the change of miR-1 expression made no difference on the luciferase activity of MUT-KRAS reporter, indicating that miR-1 could interact with KRAS 3′-UTR by putative binding sites. Moreover, ectopic expression of miR-1 resulted in a striking drop of KRAS expression, and miR-1 silencing triggered an evident increase of KRAS expression in DU145 cells (Figure 5C). Taken together, these data stated that KRAS was a target of miR-1. Next, we further demonstrated that MALAT1 overexpression abolished the miR-1-mediated inhibitory effect on the luciferase activity of WT-KRAS reporter in DU145 cells (Figure 5D). Also, MALAT1 upregulation promoted KRAS expression, while this effect was substantially weakened in DU145 cells after transfection with miR-1 (Figure 5E). In summary, these data suggested that MALAT1 acted as a ceRNA of miR-1 to sequester miR-1 from its target gene KRAS, inducing the upregulation of KRAS expression in PC cells.
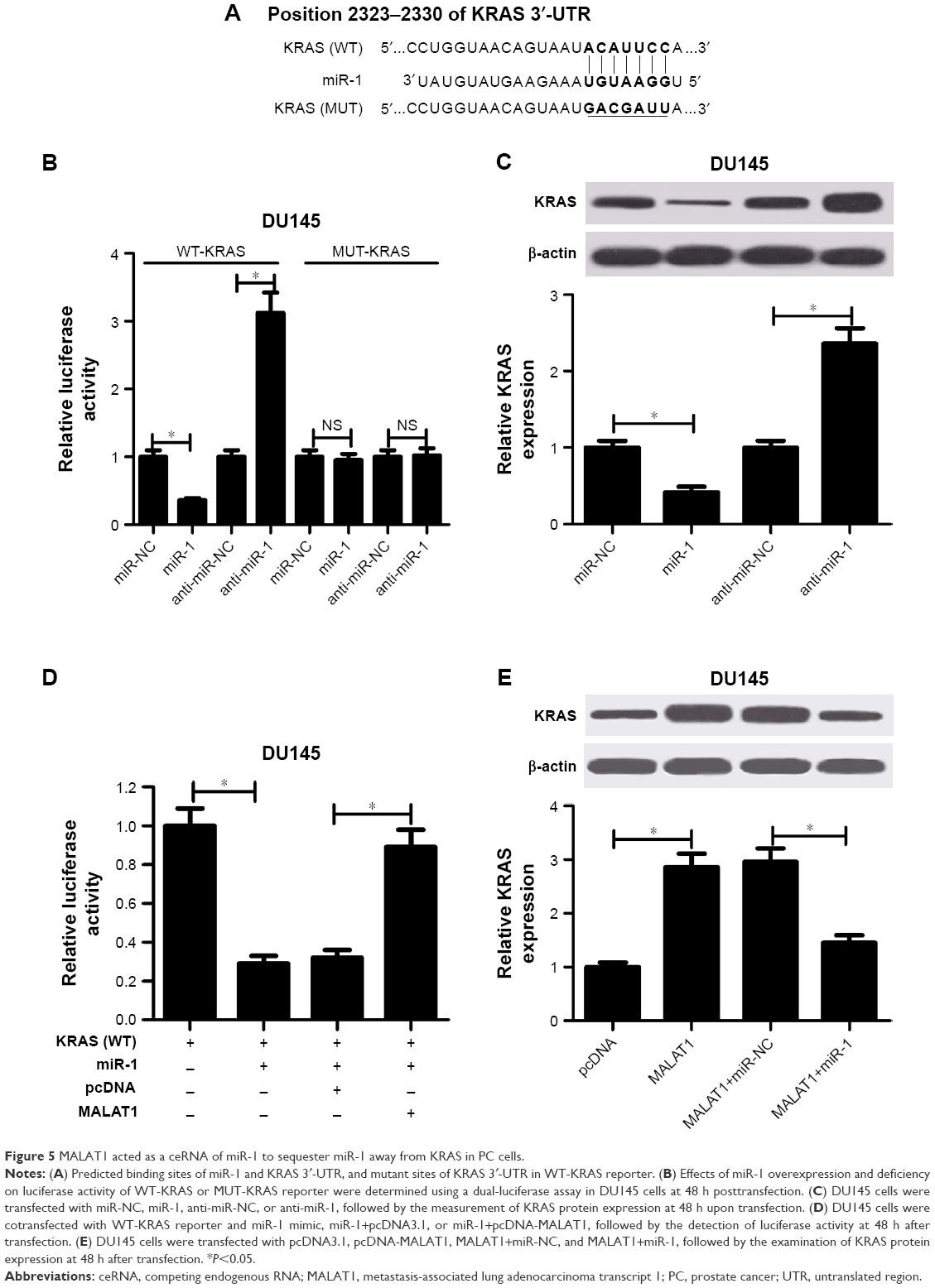 | Figure 5 MALAT1 acted as a ceRNA of miR-1 to sequester miR-1 away from KRAS in PC cells. |
KRAS overexpression weakened miR-1-mediated antiproliferative, antimigratory and proapoptotic effects in PC cells
Next, we further explored the effect of miR-1 on PC cell proliferation, migration, and apoptosis. Western blot assay demonstrated that the introduction of miR-1 mimic markedly inhibited KRAS protein expression, while the effects were notably attenuated by KRAS overexpression in DU145 and PC3 cells (Figure 6A). Functional analyses manifested that enforced expression of miR-1 triggered diminished colony formation capacity (Figure 6B), reduced cell viability (Figure 6C), weakened migration ability (Figure 6D), increased apoptotic rate (Figure 6E), as well as decreased protein expressions of cleaved PARP, cleaved caspase-3, and Bcl-2 (Figure 6F) and enhanced BAX protein expression (Figure 6F) in DU145 and PC3 cells. In other words, miR-1 overexpression hindered cell proliferation and migration and induced cell apoptosis in PC cells. Furthermore, KRAS overexpression resulted in the increase of clone number (Figure 6B), cell viability (Figure 6C), cell migration ability (Figure 6D), and protein expressions of cleaved PARP, cleaved caspase-3, and Bcl-2 (Figure 6F) together with reduced apoptotic rate (Figure 6E) and decreased BAX protein expression (Figure 6F) in miR-1-transfected PC cells. In other words, restoration of KRAS expression markedly abated miR-1-mediated antipro (Figure 6B and C), antimigratory (Figure 6D), and proapoptotic effects (Figure 6E and F) in PC cells. All these results made us draw a conclusion that miR-1 upregulation blocked proliferation and migration and facilitated apoptosis in PC cells by inhibiting KRAS expression.
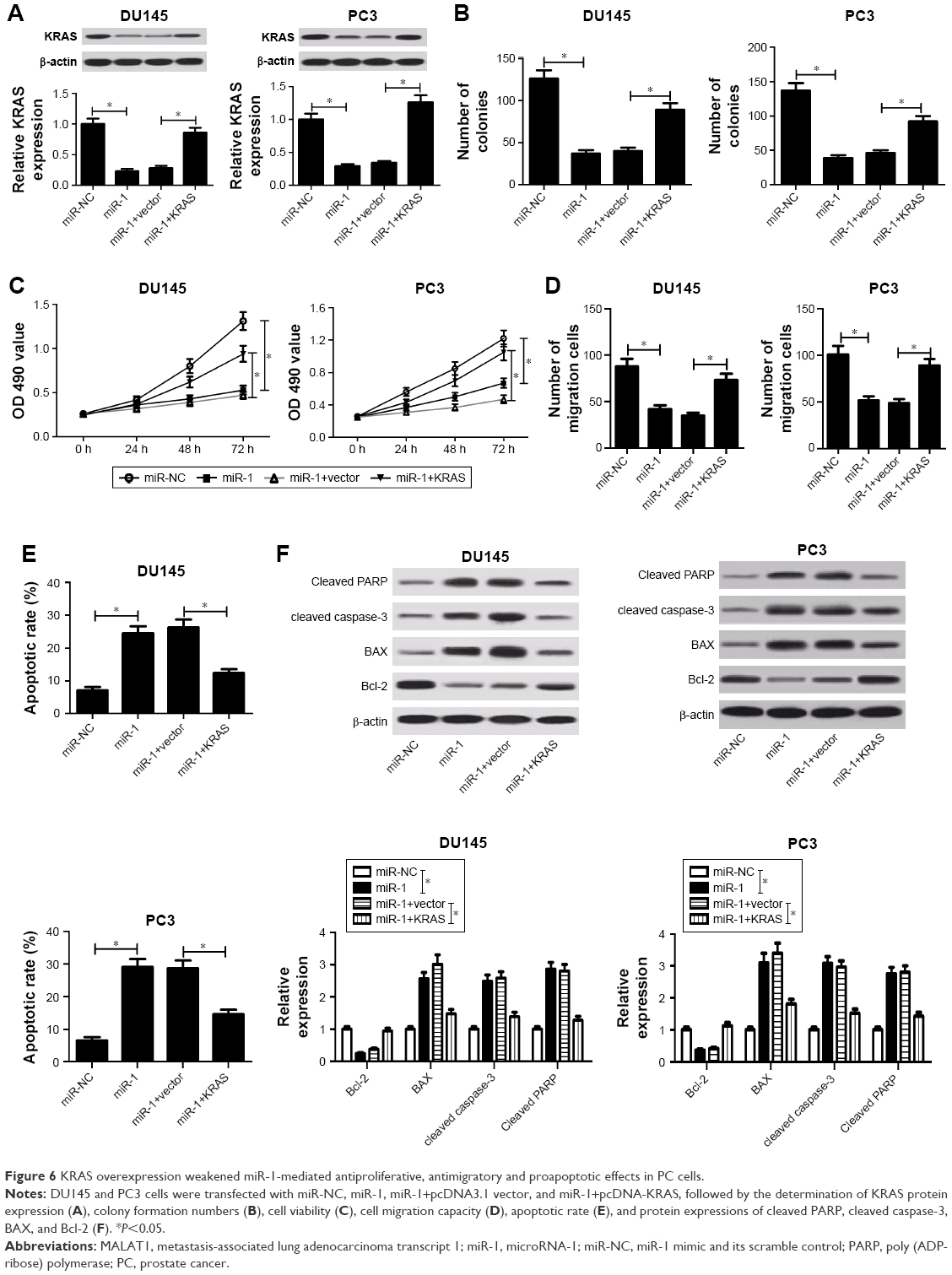 | Figure 6 KRAS overexpression weakened miR-1-mediated antiproliferative, antimigratory and proapoptotic effects in PC cells. |
Discussion
PC is a serious threat for male health, accounting for ~20% in newly diagnosed cancers.1 With the advent of prostate-specific antigen (PSA) as a diagnostic biomarker of PC, a notable reduction in PC-induced death was observed over the past few decades.22 However, increased PSA is not enough to distinguish PC and other benign or malignant prostatic diseases such as benign prostatic hyperplasia and prostatitis.23 Therefore, extensive efforts have been made to search for more effective molecular targets or biomarkers for the prediction, diagnosis, therapy, and prevention of PC.2,24 By virtue of the critical roles of lncRNAs and miRNAs in the initiation and progression of PC, some researchers proposed that lncRNAs and miRNAs might act as potential biomarkers or therapeutic targets of PC.25,26
A growing body of evidence proved that MALAT1 was closely related to various pathophysiological processes in different diseases, ranging from diabetes complications to cancers.27 For instance, decreased MALAT1 suppressed cell metastasis and proliferation by regulating ROCK1/ROCK2 via sponging miR-144-3p in osteosarcoma.28 Lai et al also showed that MALAT1 expression was abnormally upregulated in HCC cell lines and tumor tissues, and MALAT1 downregulation suppressed proliferation, migration, and invasion, and facilitated apoptosis in HCC cells.29
MiR-1 has been shown to be frequently downregulated in multiple cancers, and miR-1 exerted an antitumor effect by regulating the expressions of oncogenes and/or transcriptional factors.20 For example, miR-1 overexpression inhibited cell proliferation and invasion, and induced cell apoptosis and cell cycle arrest in renal cell cancer.30 Liao et al elucidated that miR-1 suppressed proliferation and promoted apoptosis in esophageal cancer cells by inhibiting nonreceptor tyrosine kinase c-Src.31
In the present study, we demonstrated that MALAT1 expression was upregulated and miR-1 expression was downregulated in PC tumor tissues and cell lines, which was in accordance with previous studies.9,13,15,16 Subsequent loss-of-function experiments demonstrated that siRNA-mediated MALAT1 silencing hampered cell proliferation and migration, and facilitated cell apoptosis in PC cells. In accordance with our results, downregulation of MALAT1 repressed tumorigenesis through preventing cell growth, invasion, and migration in castration-resistant prostate cancer.13
To further explore the molecular basis of MALAT1 in PC progression, miRcode and starBase online websites were used to identify potential miRNAs that could interact with MALAT1. Among candidate miRNAs, miR-1 was selected due to its antitumor effect in multiple cancers.20 Subsequent luciferase and RIP assays further demonstrated that MALAT1 could bind to miR-1. Additionally, we further revealed that MALAT1 upregulation suppressed miR-1 expression, and MALAT1 downregulation facilitated miR-1 expression in PC cells. Subsequently, restoration experiments were performed to explore whether the oncogenic effect of MALAT1 was mediated by downregulating miR-1 expression. Results showed that miR-1 downregulation partly abrogated MALAT1 silencing-mediated antiproliferative, antimigratory and proapoptotic effects in PC cells. All these data indicated that MALAT1 knockdown inhibited proliferation and migration, and accelerated apoptosis via increasing miR-1 expression. In agreement with our findings, Chou et al demonstrated that MALAT1 facilitated cell migration and invasion by acting as a molecular sponge of miR-1 and promoting cell division cycle 42 (cdc42) expression in breast cancer.18 Jin et al disclosed that MALAT1 silencing inhibited cell proliferation and migration and induced apoptosis by regulating miR-1/slug axis in triple-negative breast cancer.32
It is widely accepted that miRNAs can exert their functions by regulating the expression of target genes. Hence, Targetscan software was used to search for potential target genes of miR-1. Among candidate genes, KRAS (a small GTPase transductor) was selected by virtue of its oncogenic effect in human cancers.21,33 For instance, Bryant et al demonstrated that KRAS facilitated growth and progression of pancreatic ductal adenocarcinoma by regulating glucose and glutamine metabolism and autophagy.34 Barbie et al also pointed out that KRAS exerted its oncogenic effect by regulating downstream NF-kB survival signals such as p105, c-Rel, and BCL-XL.35 Moreover, Wang et al showed that knockdown of KRAS and UBE2L3 fusion suppressed cell proliferation, invasion, and xenograft growth in PC.36 Following luciferase and western blot assays further manifested that KRAS was a target of miR-1, and miR-1 inhibited KRAS expression by direct interaction.
Accumulating evidence supports the hypothesis that lncRNAs act as ceRNAs to absorb miRNAs like sponges, resulting in the upregulation of target gene expressions.37 Thus, the influence of MALAT1 on the interaction between miR-1 and KRAS was further investigated in PC cells. Results showed that MALAT1 overexpression abated the miR-1-mediated suppressive effect on the luciferase activity of wild-type KRAS reporter. Moreover, enforced expression of MALAT1 facilitated KRAS expression, while this effect was abrogated by miR-1 upregulation in PC cells. In other words, these results declared that MALAT1 performed as a ceRNA of miR-1 to sequester miR-1 away from KRAS, triggering the increase of KRAS expression in PC cells.
Afterward, the effects of miR-1 and KRAS on cell proliferation, migration, and apoptosis were further explored in PC cells. Results delineated that miR-1 overexpression hindered proliferation and migration as well as promoted apoptosis, while miR-1-mediated antiproliferative, antimigratory and proapoptotic effects were markedly attenuated by KRAS upregulation in PC cells, indicating that miR-1 suppressed PC progression by regulating KRAS. In line with our data, Chen et al manifested that KRAS was a target of miR-1, and miR-1 exerted its antitumor effect by repressing KRAS expression in nasopharyngeal cancer.38 Also, a previous document discovered that restoration of miR-1 expression hindered proliferation and motility, and increased apoptosis at least partly by targeting KRAS and MALAT1 in breast cancer.39 In other words, our study highlighted a novel MALAT1/miR-1/KRAS regulatory pathway in PC development.
Conclusion
Our study revealed that MALAT1 facilitated cell proliferation and migration, and inhibited cell apoptosis by downregulating miR-1 and upregulating KRAS in androgen receptor-negative PC cells, providing a new insight into the molecular basis of MALAT1 in PC progression and elucidating a promising biomarker or therapeutic target for suppressing castration-resistant PC. Nevertheless, more in vivo experiments and clinical researches are needed to further validate our findings.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Cuzick J, Thorat MA, Andriole G, et al. Prevention and early detection of prostate cancer. Lancet Oncol. 2014;15(11):484–492. | ||
Glaser Z, Gordetsky JB, Porter KK, Varambally S, Rais-Bahrami S. Prostate cancer imaging and biomarkers guiding safe selection of active surveillance. Front Oncol. 2017;7:256. | ||
Miyake H, Fujisawa M. Prognostic prediction following radical prostatectomy for prostate cancer using conventional as well as molecular biological approaches. Int J Urol. 2013;20(3):301–311. | ||
Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. Non-coding RNAs: regulators of disease. J Pathol. 2010;220(2):126–139. | ||
Liz J, Esteller M. lncRNAs and microRNAs with a role in cancer development. Biochim Biophys Acta. 2016;1859(1):169–176. | ||
Smolle MA, Bauernhofer T, Pummer K, Calin GA, Pichler M. Current insights into long non-coding RNAs (LncRNAs) in prostate cancer. Int J Mol Sci. 2017;18(2). | ||
Kanwal R, Plaga AR, Liu X, Shukla GC, Gupta S. MicroRNAs in prostate cancer: functional role as biomarkers. Cancer Lett. 2017;407:9–20. | ||
Mouraviev V, Lee B, Patel V, et al. Clinical prospects of long noncoding RNAs as novel biomarkers and therapeutic targets in prostate cancer. Prostate Cancer Prostatic Dis. 2016;19(1):14–20. | ||
Cannistraci A, Di Pace AL, Da Maria R, Bonci D. MicroRNA as new tools for prostate cancer risk assessment and therapeutic intervention: results from clinical data set and patients’ samples. Biomed Res Int. 2014;2014:146170. | ||
Gutschner T, Hämmerle M, Diederichs S. MALAT1 – a paradigm for long noncoding RNA function in cancer. J Mol Med (Berl). 2013;91(7):791–801. | ||
Pang EJ, Yang R, Fu XB, Liu YF. Overexpression of long non-coding RNA MALAT1 is correlated with clinical progression and unfavorable prognosis in pancreatic cancer. Tumour Biol. 2015;36(4):2403–2407. | ||
Ren S, Liu Y, Xu W, et al. Long Noncoding RNA MALAT-1 is a new potential therapeutic target for castration resistant prostate cancer. J Urol. 2013;190(6):2278–2287. | ||
Weiss M, Brandenburg LO, Burchardt M, Stope MB. MicroRNA-1 properties in cancer regulatory networks and tumor biology. Crit Rev Oncol Hematol. 2016;104:71–77. | ||
Kojima S, Chiyomaru T, Kawakami K, et al. Tumour suppressors miR-1 and miR-133a target the oncogenic function of purine nucleoside phosphorylase (PNP) in prostate cancer. Br J Cancer. 2012;106(2):405–413. | ||
Hudson RS, Yi M, Esposito D, et al. MicroRNA-1 is a candidate tumor suppressor and prognostic marker in human prostate cancer. Nucleic Acids Res. 2012;40(8):3689–3703. | ||
Liu Y, Yin J, Abou-Kheir W, et al. MiR-1 and miR-200 inhibit EMT via Slug-dependent and tumorigenesis via Slug-independent mechanisms. Oncogene. 2013;32(3):296–306. | ||
Chou J, Wang B, Zheng T, et al. MALAT1 induced migration and invasion of human breast cancer cells by competitively binding miR-1 with cdc42. Biochem Biophys Res Commun. 2016;472(1):262–269. | ||
Davies WD, Perlmutter DM. Analyzing MiRNA-LncRNA interactions. Methods Mol Biol. 2016;1402(1):271–286. | ||
Han C, Shen JK, Hornicek FJ, Kan Q, Duan Z. Regulation of microRNA-1 (miR-1) expression in human cancer. Biochim Biophys Acta. 2017;1860:227–232. | ||
Jančík S, Drábek J, Radzioch D, Hajdúch M. Clinical relevance of KRAS in human cancers. J Biomed Biotechnol. 2010;2010(16):150960. | ||
Stamey TA, Caldwell M, Mcneal JE, Nolley R, Hemenez M, Downs J. The prostate specific antigen era in the United States is over for prostate cancer: what happened in the last 20 years? J Urol. 2004;172(4 Pt 1):1297–1301. | ||
Loeb S, Gashti SN, Catalona WJ. Exclusion of inflammation in the differential diagnosis of an elevated prostate-specific antigen (PSA). Urol Oncol. 2009;27(1):64–66. | ||
Wallace T, Torre T, Grob M, et al. Current approaches, challenges and future directions for monitoring treatment response in prostate cancer. J Cancer. 2014;5(1):3–24. | ||
Massillo C, Dalton GN, Farre PL, De Luca P, De Siervi A. Implications of microRNA dysregulation in the development of prostate cancer. Reproduction. 2017;154(4):R81–R97. | ||
Liu D, Xu B, Chen S, et al. Long non-coding RNAs and prostate cancer. J Nanosci Nanotechnol. 2013;13(5):3186–3194. | ||
Wu Y, Huang C, Meng X, Li J. Long Noncoding RNA MALAT1: insights into its biogenesis and implications in human disease. Curr Pharm Des. 2015;21(34):5017–5028. | ||
Wang Y, Zhang Y, Yang T, et al. Long non-coding RNA MALAT1 for promoting metastasis and proliferation by acting as a ceRNA of miR-144-3p in osteosarcoma cells. Oncotarget. 2017;8(35):59417–59434. | ||
Lai M, Yang Z, Zhou L, et al. Long non-coding RNA MALAT-1 overexpression predicts tumor recurrence of hepatocellular carcinoma after liver transplantation. Med Oncol. 2012;29(3):1810–1816. | ||
Kawakami K, Enokida H, Chiyomaru T, et al. The functional significance of miR-1 and miR-133a in renal cell carcinoma. Eur J Cancer. 2012;48(6):827–836. | ||
Liao Z, Wang X, Liang H, et al. miR-1 suppresses the proliferation and promotes the apoptosis of esophageal carcinoma cells by targeting Src. Cancer Med. 2017;6(12):2957–2965. | ||
Jin C, Yan B, Lu Q, Lin Y, Ma L. Reciprocal regulation of Hsa-miR-1 and long noncoding RNA MALAT1 promotes triple-negative breast cancer development. Tumor Biol. 2016;37(6):7383–7394. | ||
Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9(7):517–531. | ||
Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39(2):91–100. | ||
Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–112. | ||
Wang XS, Shankar S, Dhanasekaran SM, et al. Characterization of KRAS rearrangements in metastatic prostate cancer. Cancer Discov. 2011;1(1):35–43. | ||
Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–352. | ||
Chen X, Shi J, Zhong J, et al. miR-1, regulated by LMP1, suppresses tumour growth and metastasis by targeting K-ras in nasopharyngeal carcinoma. Int J Exp Pathol. 2015;96(6):427–432. | ||
Liu R, Li J, Lai Y, Liao Y, Liu R, Qiu W. Hsa-miR-1 suppresses breast cancer development by down-regulating K-ras and long non-coding RNA MALAT1. Int J Biol Macromol. 2015;81:491–497. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.