Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Major depressive disorder: mechanism-based prescribing for personalized medicine
Authors Saltiel P, Silvershein D, Lapolla T
Received 26 August 2014
Accepted for publication 12 December 2014
Published 31 March 2015 Volume 2015:11 Pages 875—888
DOI https://doi.org/10.2147/NDT.S73261
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Roger Pinder
Philip F Saltiel,1 Daniel I Silvershein2
1Department of Psychiatry, New York University School of Medicine/Langone Medical Center New York University Behavioral Health Programs, New York University Pearl Barlow Center for Memory Evaluation and Treatment, New York, NY, USA; 2Department of Medicine, New York University School of Medicine/Langone Medical Center, New York, NY, USA
Abstract: Individual patients with depression present with unique symptom clusters – before, during, and even after treatment. The prevalence of persistent, unresolved symptoms and their contribution to patient functioning and disease progression emphasize the importance of finding the right treatment choice at the onset and the utility of switching medications based on suboptimal responses. Our primary goal as clinicians is to improve patient function and quality of life. In fact, feelings of well-being and the return to premorbid levels of functioning are frequently rated by patients as being more important than symptom relief. However, functional improvements often lag behind resolution of mood, attributed in large part to persistent and functionally impairing symptoms – namely, fatigue, sleep/wake disturbance, and cognitive dysfunction. Thus, patient outcomes can be optimized by deconstructing each patient’s depressive profile to its component symptoms and specifically targeting those domains that differentially limit patient function. This article will provide an evidence-based framework within which clinicians may tailor pharmacotherapy to patient symptomatology for improved treatment outcomes.
Keywords: MDD, tailored pharmacotherapy, patient-specific profile, individualized pharmacotherapy
Defining depression
Depression is the most common psychiatric disorder and the leading cause of disability worldwide.1,2 In the US, about 7%–9% of the adult population experiences a major depressive episode (MDE) each year and an estimated 8 million (3.4%) meet criteria for major depressive disorder (MDD).3–5 The cognitive, emotional, and physical symptoms of depression translate to considerable impairments in psychosocial functioning across physical, social, and educational/occupational domains.6,7 Indirect workplace costs alone, characterized by low productivity (presenteeism) and days missed (absenteeism), account for over 60% of the total economic burden of depression and twice as much as that attributed to direct medical costs.8
The likelihood of long-term treatment success is improved with early and accurate diagnosis, continual multidimensional assessment, and rational pharmacotherapy tailored to the patient’s symptomatology, coexisting disorders, and treatment needs.9 Yet achieving these outcomes is confounded by the personal and multidimensional nature of the disease itself. There are no validated biological tests that can be used to diagnose depression. Further, without objective outcomes measures, clinicians must gauge treatment response and make clinical decisions over time based on subjective impressions of patient-reported symptoms.9–11
Validated assessment tools (eg, Montgomery–Åsberg Depression Rating Scale, Hamilton Rating Scale for Depression [HAM-D24]) based on core criteria (Table 1)12 can facilitate the categorical diagnosis of depression and help track the presence and severity of symptoms at each visit.11,13,14 However, clinicians may find it more practical to ask about symptoms directly through the course of the patient interview. Direct questioning combined with a clinical impression formed by the patient’s speech, affect, and appearance can help define how individual symptoms adversely affect patient-specific functioning and quality of life (QoL). If the symptom profile of an MDE is not properly assessed before and during a well-orchestrated antidepressant trial, ongoing symptoms may not be easily distinguished from treatment-related side effects or from those due to comorbid psychiatric or medical conditions.15–18 About half of patients who report normal functioning consider themselves to be in remission from depression despite persistent depressive symptoms.19 Therefore, understanding the relationship between the symptoms of depression and how they adversely affect patient functioning is essential for optimized clinical decision making.
 | Table 1 DSM-5 criteria for MDE |
Defining recovery
The gold standard for treatment is the full resolution of symptoms and associated improvements in function and QoL.9,11,14 While response to treatment implies a clinically meaningful degree of symptom reduction (typically defined as ≥50% reduction in pretreatment symptom severity), remission and recovery require that the symptoms of depression be absent or close to it (Figure 1).20–22 Many clinical studies define remission as low scores on rating scales, which is not equivalent to an asymptomatic state.23,24 Further, as depressed mood and loss of interest generally overshadow other symptoms of depression, when mood improves, patients may misguidedly be considered in remission. In fact, most patients considered ‘in remission’ do not actually achieve complete resolution from all symptoms, even after multiple treatment steps, and often show greater depressive illness burden as symptoms persist.24–27 The Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study of nearly 4,000 ‘real-world’ outpatients reported a cumulative remission rate of 67% after four treatment steps; however, about 70% relapsed within 1 year.25 Figure 1 depicts a schematic representation of the progressive nature of depression, and illustrates the need for achieving and sustaining full symptom recovery early in disease pathogenesis.22
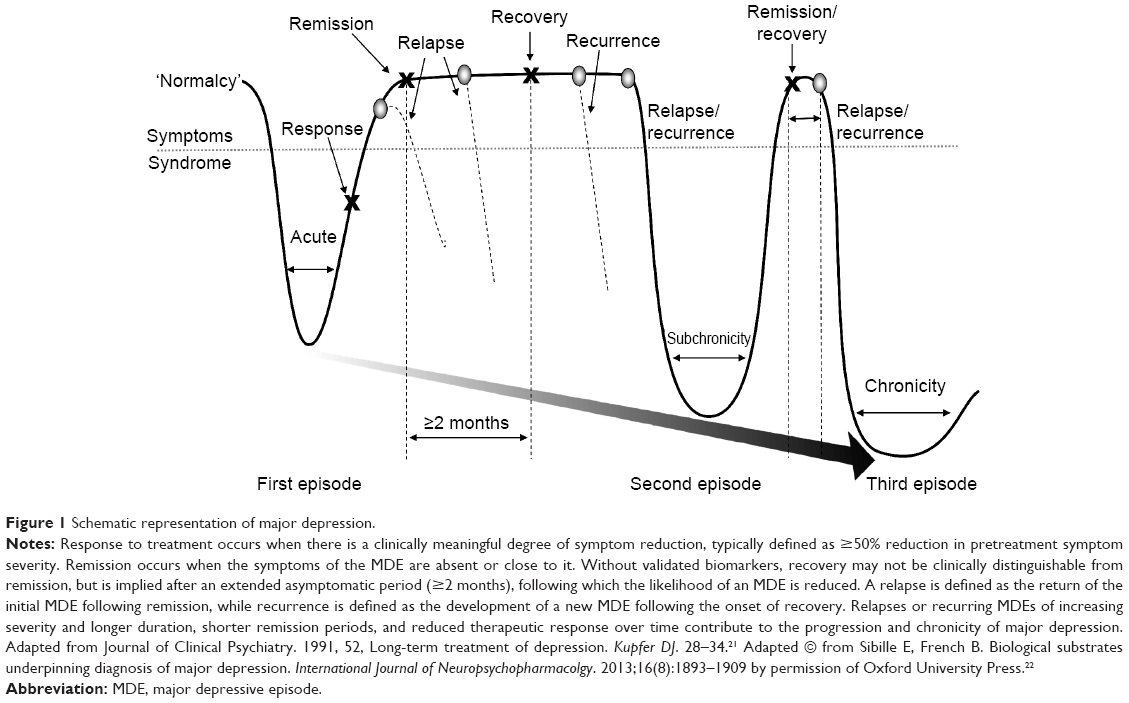 | Figure 1 Schematic representation of major depression. |
Symptoms that persist during remission are associated with relapse, recurrence, and ultimately, treatment resistance.15,27,28 During a 3-year prospective study of 267 depressed patients, Conradi et al assessed the weekly presence of individual symptoms during each phase of depression (Figure 2).29 Study participants who relapsed within 8 weeks reported a greater overall symptom severity during the remission period than those who remained in remission for greater than 8 weeks (ie, in recovery). Gradual accumulation of subthreshold symptoms over time appeared to trigger the relapse, suggesting that the initial MDE had not resolved and remained subsyndromal only temporarily. In fact, patients with unresolved depressive symptoms are three times as likely to relapse as patients with asymptomatic recovery.28
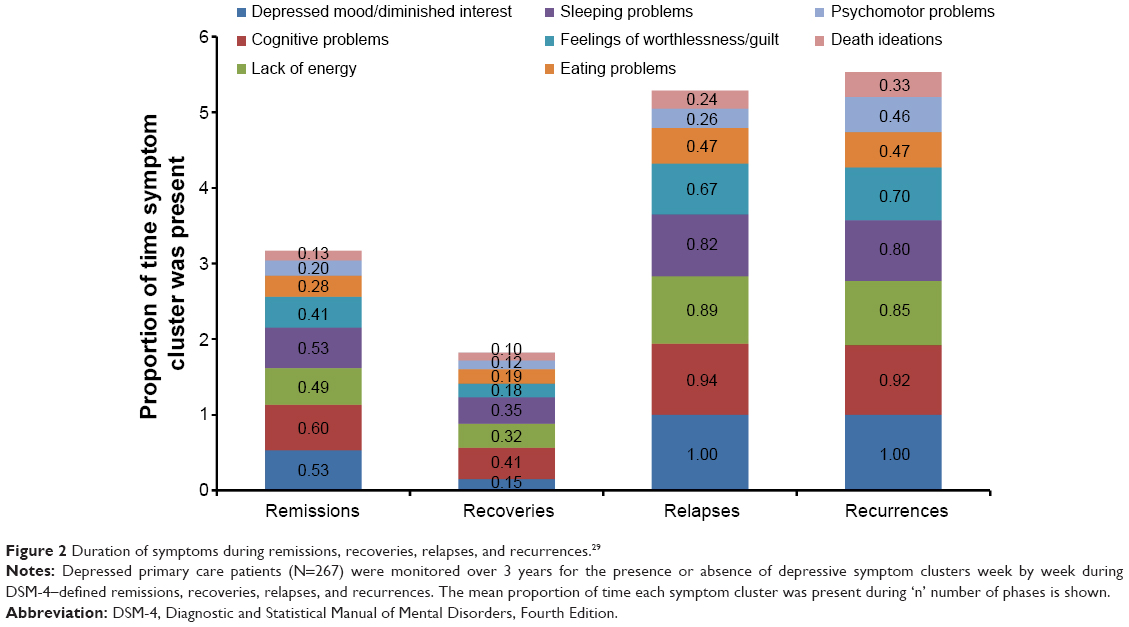 | Figure 2 Duration of symptoms during remissions, recoveries, relapses, and recurrences.29 |
The prevalence of unresolved symptoms and their contribution to disease progression emphasize the importance of finding the right treatment choice at the onset and switching medications if necessary based on suboptimal responses. Moreover, feelings of well-being and the return to premorbid levels of functioning are frequently rated by patients as more important than symptom relief, yet functional improvements often lag behind resolution of mood.6,30 Reasons for delayed functional recovery are only partly characterized; however, the symptoms that most impair function are the same symptoms that commonly persist despite treatment, namely fatigue, sleep/wake disturbance, and cognitive dysfunction.6,18,29,31–33 For example, patient self-reports frequently identify fatigue and low energy, insomnia, and concentration and memory problems as especially disruptive to occupational and global functioning.6,32 In the seminal study by Conradi et al (Figure 2), patients exhibited problems with cognition 60% of the time during periods of remission compared with 41% during periods of recovery.29 Similarly, a lack of energy was reported nearly half of the time during remission but only one-third of the time during recovery. These data suggest that antidepressant selection based, in part, on the most disabling patient-reported symptoms may help optimize treatment outcomes and add to a growing body of evidence on the importance of initial and ongoing individualized assessment.9 Table 2 highlights the MDD-associated symptoms that may co-occur during depressive episodes or persist during remission.34–47 While additional studies are needed to gauge the most effective treatments for these symptoms, Table 2 includes select approaches with demonstrated efficacy.34–47
 | Table 2 Possible pharmacotherapeutic approaches for the treatment of select symptoms in MDD |
The neurobiology of depression
The biological basis of mood disorders, in general, includes genetic, epigenetic, biochemical, and psychosocial factors.48–50 Precisely how these factors interact over time is not firmly established, but recent insights into the structure and function of discrete brain regions offer an opportunity to characterize the neural basis of depression and its idiosyncratic course, clinical presentation, and treatment responsiveness.51,52 Underlying each depressive symptom may in fact be a unique mechanism comprising multiple malfunctioning neural circuits.53,54 Structural and/or functional alterations have been identified in brain regions involved in emotional processing as well as cognitive control, learning, and/or memory formation.51,55
Notably, reduced hippocampal volume is common among depressed patients and directly correlates with frequency and length of depressive episodes.52,56 The hippocampus plays a salient role in explicit memory formation, homeostatic adaptation to stressful stimuli, and emotion processing.55 Hippocampal volume reductions were found to occur after disease onset, emphasizing the importance of early treatment to minimize disease progression.57 Further, discrete brain regions of depressed patients – including the prefrontal and limbic areas – demonstrate differential metabolic rates, reflecting aberrant neural activity thought to contribute to depressive symptomatology.58 In addition to structural changes, alterations in neural function in the brain are also implicated in the neurobiology of MDD. A recent neuroimaging study that investigated reduced gray matter volume in the parietal–temporal regions and functional alterations in the temporal regions and cerebellum in patients with MDD suggested that structural and functional deficits contributed independently to the neurobiology of the disease.59 While the relationship between altered brain structure and function and MDD symptoms remains an active area of research, the data increasingly suggest that core criterion symptoms of depression can be mapped to these and other regions of the brain (Figure 3).60–62
 | Figure 3 Neurotransmitters and their hypothetically malfunctioning brain circuits in regions associated with the diagnostic symptoms for depression. |
The structure and function of these brain regions are modulated by monoaminergic neurotransmission. The serotonin (5-hydroxytryptamine, 5HT), norepinephrine (NE), and dopamine (DA) systems originate from the brainstem and each project to select brain regions (Figure 3).60,61,63,64 The interdependent biological actions of 5HT, NE, and DA are mediated by their cognate transporters and receptors.64–66 For example, the serotonin 5HT2A and 5HT2C receptor subtypes inhibit postsynaptic connections on the NE and DA systems, respectively.64 D2-like receptor subtypes and alpha-1 adrenoceptors can positively influence 5HT neurotransmission while presynaptic alpha-2 adrenoceptors are thought to dampen 5HT signaling (Figure 4).64,67–69 Further, in addition to directly influencing extracellular monoamine levels, postsynaptic 5HT receptors (eg, 5HT1A, 5HT3, 5HT7) modulate inhibitory gamma-aminobutyric acid (GABA) interneurons, which in turn affect the release of 5HT itself, acetylcholine (ACh), NE, DA, and glutamate (Glu) (Figure 4).67,69,70
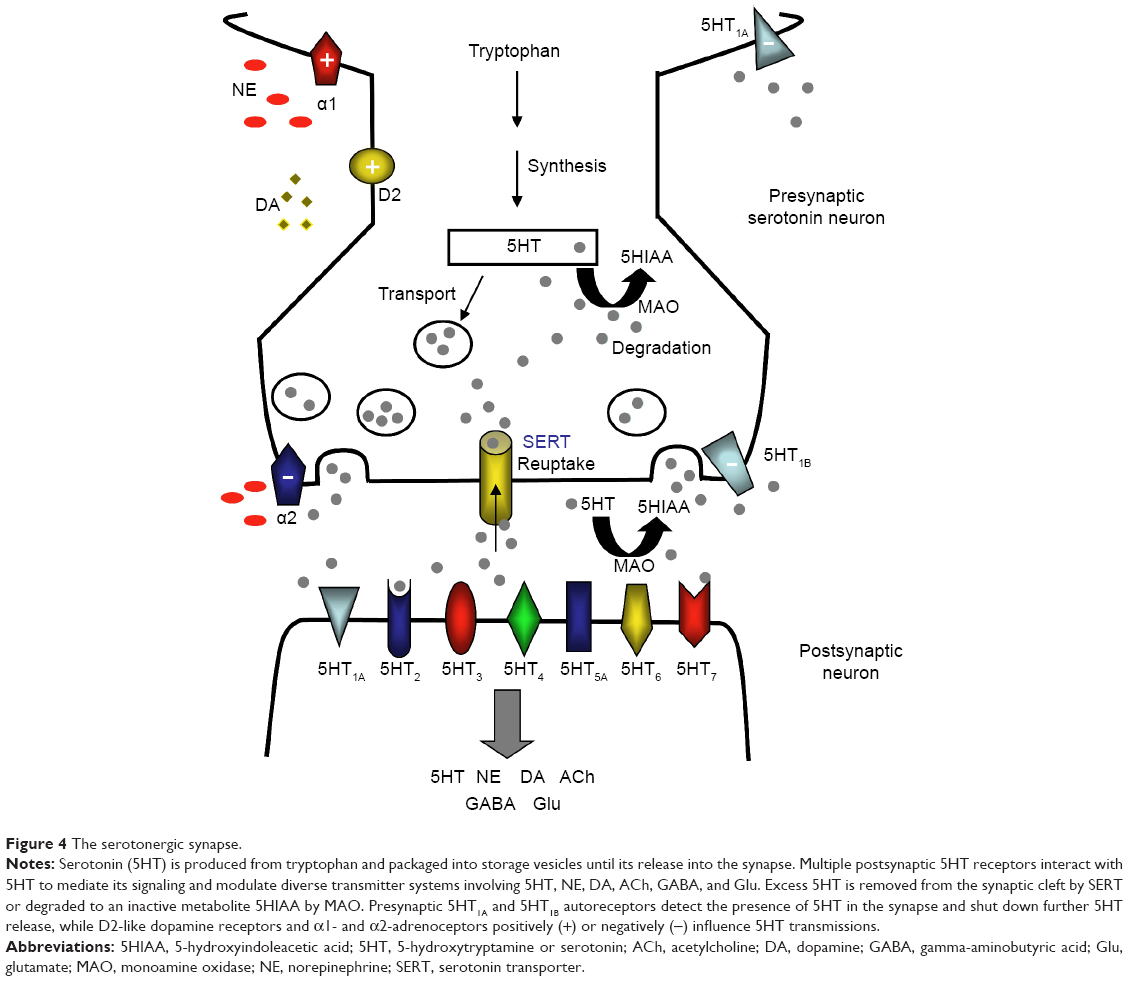 | Figure 4 The serotonergic synapse. |
Interconnected monoaminergic neurotransmission provides a neural basis for mood, reward, pleasure, motivation, and related cognitive and executive functions.64 Critically, disturbances in the functional connectivity in these and related neural networks appear to be integrally involved in the onset and progression of major depression.64 Aberrant DA and NE signaling is thought to adversely influence motivation, an essential driver of goal-directed behaviors and related executive functions.65,66 Impaired 5HT neurotransmission forms, at least in part, the pathophysiologic basis for anhedonia, guilt, and similar ‘negative affects’ of depression.61 Further, 5HT and NE have descending spinal pathways that mediate physical fatigue and related somatic symptoms.71 Of note, numerous studies have demonstrated an increased risk for depression linked to genetic polymorphisms of 5HT, NE, or DA receptor subtypes and transporters.48
While the particulars are extraordinarily complex and still only partly understood, monoamine signaling clearly exerts profound ‘downstream’ effects – both inhibitory and stimulatory – on interconnected neural networks, disturbances of which can manifest as depressive symptoms and contribute to disease progression.52 Importantly, these and related biochemical lesions provide a mechanistic rationale for antidepressant therapy. Emerging data now support the very real prospect of individualized pharmacotherapy. For example, neuroimaging studies have shown that antidepressants can reverse functional changes observed in depressed patients.58 These and related advances in translational research are beginning to characterize the neurobiological correlates of antidepressant treatment responses.64 While we are not yet at a stage to characterize lesions on a patient-by-patient basis, we can nevertheless build a conceptual framework within which clinicians can compare individual antidepressant medications and, based on empirical observations of treatment response, adjust therapeutic strategies accordingly.
Monoaminergic pathways as drug targets
Current first-line treatment options include selective serotonin reuptake inhibitors (SSRIs), serotonin–norepinephrine reuptake inhibitors (SNRIs), and norepinephrine–dopamine reuptake inhibitors (NDRIs).9,14 While the precise mechanisms by which these agents achieve their antidepressant effects are only partly characterized, subtle changes in monoaminergic neural systems clearly play an important role. More precisely, these drugs selectively inhibit the serotonin transporter (SERT), the NE transporters (NET), and/or the DA transporters (DAT), and thereby increase synaptic concentrations of their respective monoamines, which in turn affect the activity of interconnected neurotransmitter systems. However, there may be some mechanistic overlap between classes of antidepressants, and individual agents demonstrate subtle yet clinically relevant pharmacologic differences (Table 3 and Figure 5).63,72–74 And while all SSRIs chiefly inhibit SERT, select agents also influence NE and DA neuronal activity – for example, paroxetine weakly inhibits NET and sertraline weakly inhibits DAT.72,75 Further, although paroxetine and fluvoxamine have similar half-lives, paroxetine, which also binds to cholinergic receptors and NET, is ten times as likely to elicit withdrawal reactions (ie, SSRI discontinuation syndrome).77
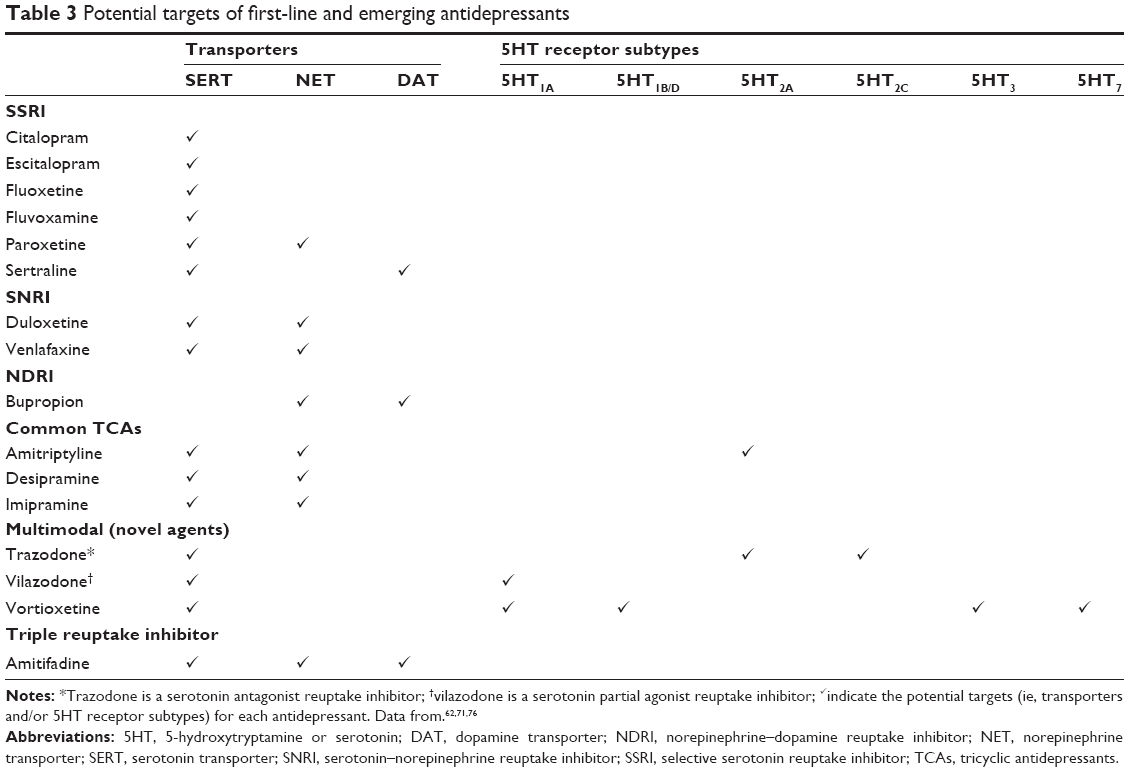 | Table 3 Potential targets of first-line and emerging antidepressants |
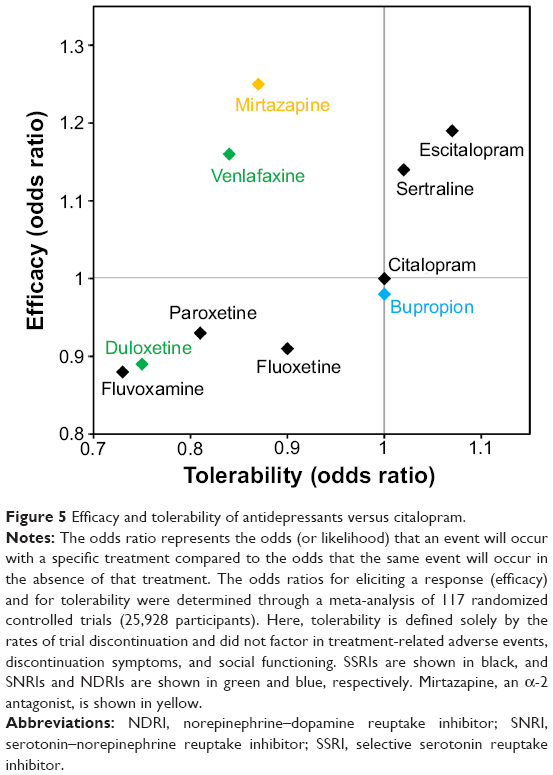 | Figure 5 Efficacy and tolerability of antidepressants versus citalopram. |
The clinical manifestation of these pharmacologic differences was recently illustrated in a meta-analysis of 117 randomized controlled trials (25,928 participants) comparing clinical response and discontinuation rates of antidepressants.73 Using citalopram as the reference compound, the odds ratio (OR) for an SSRI initiating a treatment response ranged from a lower likelihood with escitalopram (OR: 0.68) to a higher likelihood with reboxetine (OR: 1.72); while tolerability ranged from a lower likelihood of tolerance with mirtazapine (OR: 0.42) to a higher likelihood of tolerance with escitalopram (OR: 1.17).73 Differences were also observed between the two SNRIs – duloxetine and venlafaxine – included in the study (Figure 5).73 These data highlight the importance of balancing the known efficacy of a drug with its side effect profile, and the potential benefits gained from switching to another antidepressant, even within the same class.
In fact, recent studies have demonstrated that the initial choice of antidepressant medication and self-reported symptomatic improvement over 2 weeks are predictive of treatment outcomes.78,79 Yet the criteria by which to select the initial antidepressant have not been well-defined. This does not translate, however, into a strictly arbitrary choice. Careful alignment of patient symptomatology with the biochemical pharmacology of antidepressants can help clinicians tailor therapy. While symptom resolution is the primary driver, the efficacy of a chosen antidepressant should be weighed against other patient-specific factors such as functional goals and expectations, comorbid conditions, and concerns about side effects (eg, weight gain, sedation) and treatment costs.
Mechanism-based prescribing
Interrelationships between clinical symptoms, affected neural systems, and antidepressant mechanisms of action may help clinicians customize pharmacotherapy to a patient’s unique clinical profile.54 As noted, fatigue, sleep/wake disturbances, and cognitive dysfunction are among the most troublesome symptoms, as they commonly persist after antidepressant therapy and have a disparate impact on patient functioning.18,29,31,80 Clinicians may wish to identify and specifically target these and other baseline symptoms to facilitate asymptomatic remission and thereby minimize the associated risk of relapse and recurrence.
Results from STAR*D and other studies underscore the importance of therapeutic adjustments tailored to initial and ongoing monitoring.27 Most patients do not respond to the initial antidepressant regimen or are nonadherent because of side effects.27,81 Nonadherence with antidepressant medications is as high as 52% in patients with MDD,82 with inefficacy, sexual dysfunction, and weight gain the most cited reasons for discontinuation.81 Physicians may wish to adjust the dose of the initial antidepressant, switch to an alternative medication, or consider adjunctive treatment.83 Nearly one-third of patients who fail an initial SSRI treatment may respond well to another antidepressant.27 Adjunctive therapy predominantly includes psychotherapy and combination drug treatment. A second antidepressant with a different mechanism of action or an atypical antipsychotic can be added to the treatment plan.9,14 Evidence-based adjunctive treatment strategies have been reviewed elsewhere.9,84,85 As treatment options continue to evolve, many primary care providers prefer to consult psychiatrists for guidance on switch and augmentation treatment strategies.86
The simplicity and safety of monotherapy are frequently cited by patients and clinicians who prefer to avoid combination therapy or adjunctive treatment with atypical antipsychotics. Novel monotherapeutic agents that engage multiple targets may therefore provide an attractive option with the potential to ameliorate functionally impairing symptoms and reduce side effects. Interestingly, genetic variations explain an estimated 50% of an antidepressant’s efficacy.48 These and related data support the development of multimodal agents that target multiple gene products, each a validated target for therapeutic intervention that may maximize the likelihood of eliciting a response.54
Triple reuptake inhibitors block SERT, NET, and DAT (Table 3), and may address potential disturbances across three neurotransmitter circuits and their associated depressive symptoms simultaneously (Figure 3).87 However, the degree of activity at each transporter needs to be considered – too much DAT inhibition may render the drug potentially abusable, whereas too little SERT inhibition may provide insufficient antidepressant action. Amitifadine inhibits SERT, NET, and DAT with a potency ratio of 1:2:8, respectively, and is currently in clinical trials.88
Multimodal drugs interact with both transporters and membrane-bound receptors, employing two distinct modes of neural signaling that may improve symptomatic resolution and reduce treatment-related side effects. The recently approved drugs vilazodone and vortioxetine exhibit SERT inhibition and 5HT1A agonism (Table 3).89,90 5HT1A receptors play a critical autoregulatory role in the synthesis and release of serotonin. Continuous treatment with SSRIs desensitizes 5HT1A autoreceptors, leading to enhanced serotonergic neurotransmission. However, 5HT1A desensitization generally requires 2 weeks, which may account for the delayed onset of traditional SSRIs.91 Multimodal agents with 5HT1A agonist activity should therefore accelerate treatment response. Preclinical studies support this model: vortioxetine treatment desensitizes 5HT neuronal firing after only 1 day (vs 14 days with fluoxetine).92 In one clinical trial, reduction in depressive symptoms was observed after 1 week of treatment with vilazodone, although the findings were not replicated in a second study.93 Vortioxetine is also a 5HT1D, 5HT3, and 5HT7 receptor antagonist and 5HT1B receptor partial agonist. Interestingly, current and emerging research suggests that 5HT7 receptors play an important if partly characterized role in cognition and sleep, and are thought to reflect their effects on Glu and serotonin signaling in cortical and subcortical circuits, among other areas of the brain.69,94–97 Through these multimodal mechanisms, vilazodone and vortioxetine can modulate downstream effects of 5HT on interconnected neurotransmitter systems. Further, clinical studies with vilazodone and vortioxetine have demonstrated favorable side effect profiles with minimal reports of sexual dysfunction and weight gain.96–100 Additional long-term studies are required to further evaluate the clinical relevance of these pharmacologic properties.
Cognition
The diminished ability to think or concentrate, or indecisiveness is one of the core diagnostic features of depression (Table 1)12 and among the most frequently persisting and functionally debilitating symptoms following treatment.29,80,101 All domains of cognition are worse in patients with depression than in healthy controls, and many – including immediate memory, attention, ideation fluency, and visuospatial function and learning – continue to adversely affect life functioning despite improvement of depressive symptoms.80,102,103 In a recent systematic review, nine of eleven studies reported that patients with remitted unipolar depression had decreased performance on neuropsychological tests compared with never depressed controls.104 Sustained cognitive impairments following an MDE frequently lead to frustration, low self-esteem, and impaired interpersonal relationships in the family, workplace, and social network.6,105 Further, cognitive dysfunction is associated with the cumulative duration of depressive episodes, emphasizing the need for early detection and targeted treatment.106
While additional studies are required to elucidate the neurochemical basis of persistent cognitive deficits, recent imaging studies suggest that hypoactive frontal and prefrontal cortices contribute to the diminished ability to think, concentrate, or make decisions.107,108 Numerous neurotransmitters – including 5HT, DA, NE, ACh, and Glu – shape the activity of procognitive circuits and provide insights into mechanism-based treatments that may be more likely to reduce cognitive impairment in depression.53,64,70 For example, the decreased dopaminergic activity associated with depressive symptoms suggests that increasing DA activity may reduce cognitive impairment in depression.54 Indeed, drug combinations with DA agonists like pramipexole as add-on treatments to antidepressants in patients with affective disorders have been shown to improve cognition in some studies.109 Glu, especially, is essential for cognitive processing, and therapeutic agents that modulate Glu transmission, such as the N-methyl-D-aspartate antagonists memantine and ketamine, have demonstrated antidepressant-like properties; follow-up studies in depressed patients have shown conflicting neurocognitive effects, however.110,111 5HT action across discrete 5HT receptor subtypes is thought to modulate GABAergic interneurons that influence Glu circuits involved in cognitive functions.70 Vortioxetine, with known effects on 5HT1A, 5HT1B, 5HT1D, 5HT3, and 5HT7, has demonstrated procognitive effects in various placebo-controlled clinical trials.90 In a recent study, vortioxetine-treated patients demonstrated significant improvements versus placebo in the digit symbol substitution test, an objective measure of cognitive performance, as well as in verbal learning and memory (as measured by improved scores in the Rey Auditory Verbal Learning Test [RAVLT]); the active reference duloxetine only showed significant improvement in RAVLT scores versus placebo.112 Both duloxetine (ΔHAM-D24 −5.5, P<0.0001) and vortioxetine (ΔHAM-D24 −3.3, P=0.0011) demonstrated significant antidepressant effects versus placebo.112
Cognitive function and its impact on patient function should be assessed in all patients with depression and monitored throughout treatment. While clinicians generally gauge the presence or absence of symptoms based on patient report, assessment tools, such as the six-item British Columbia Cognitive Complaints Inventory (BC-CCI),113 may help if the patient is unresponsive. The independent and lasting effects that cognitive impairment has on functional improvement highlight the importance of treating cognitive impairment at baseline and for defining recovery. Further, these insights reinforce the need for well-designed studies with active comparator arms to establish definitive conclusions about differential procognitive benefits between medications.
Fatigue
Fatigue or loss of energy is a defining criterion for depression (Table 1).12 The severity of fatigue reported by patients with MDD independently correlates with the severity of depression (P=0.028),111 which often persists following resolution of depressed mood, and markedly impairs psychosocial functioning and QoL.29,114–117
The prevalence of fatigue and loss of energy despite antidepressant treatment highlights the need for additional treatment options targeting the diverse neurobiology underlying psychomotor retardation, painful somatic symptoms, and executive dysfunction. Treatment-related side effects can complicate treatment as well. Tricyclic antidepressants (TCAs) and some SSRIs and SNRIs demonstrate sedative effects and can induce or exacerbate symptoms of fatigue and related functional impairments.117
Descending 5HT and NE fibers in the spinal cord may regulate the perception of physical tiredness, while mental fatigue is regulated at the cortical level by several key neurotransmitters (eg, NE, DA, ACh, histamine).53 In addition, both 5HT and DA projections to the striatum and 5HT and NE projections to the cerebellum regulate psychomotor functioning (Figure 3).115,118 Inhibition of NET and/or DAT by venlafaxine, bupropion, and sertraline (Table 3) can modulate NE and/or DA neurotransmission, offering advantages over SERT inhibition alone. When patients present with fatigue at baseline, clinicians should select antidepressants less likely to have sedating effects (eg, venlafaxine, bupropion, sertraline). A pooled analysis showed that bupropion is associated with lower levels of residual fatigue compared with the SSRIs.119 An alternative treatment strategy is to use adjunctive therapy that targets fatigue directly with medications by increasing NE and/or DA neurotransmission; agents such as psychostimulants, the NE reuptake inhibitor atomoxetine, or lower doses of atypical antipsychotics may alleviate fatigue by promoting improved nighttime sleep quality and quantity.54,115,117 Clinical studies evaluating the effects of multimodal agents on fatigue and related outcomes have been encouraging, though additional longitudinal studies of agents with comparable antidepressant efficacy are needed with larger patient populations.
Sleep/wake
Disruptions in circadian rhythms and the sleep/wake cycle are experienced in nearly all patients with depression.120 Even after treatment, sleep/wake problems – particularly insomnia or hypersomnia – commonly persist.18,29 Depression is associated with both long and short sleep durations, as well as over- or underestimation of sleep time.121 Compared with patients demonstrating good sleep/wake quality, those with sleep/wake problems and comorbid depression have significantly worse QoL, more functional impairments, and when accompanied by nightmares, increased risk for suicidal ideation and suicide attempts.122 Moreover, sleep/wake disturbances – including insomnia and sedation – have also been reported as side effects of common antidepressants (eg, paroxetine, venlafaxine).123,124 These data highlight the essential need to assess sleep/wake quality and quantity before, during, and even after treatment with antidepressants.
Sleep/wake disturbance can be mapped to dysfunction in the hypothalamus (Figure 3). States of arousal are regulated by the hypothalamic sleep/wake switch and brainstem monoamine projections to the cortex.125 Serotonin, histamine, and GABA each play a role in regulating normal wakefulness and sleep and can be modulated with appropriate treatment. Specifically, serotonin 5HT2A and 5HT7 receptors have been linked to sleep, circadian rhythm, and mood.94,121 The 5HT2A antagonist properties of trazodone (Table 3) likely contribute to its known sleep-promoting effects.126,127 Doxepin, a TCA approved for the treatment of depression, anxiety, and insomnia, antagonizes histamine H1 receptors and is beneficial as an add-on treatment with antidepressant therapy in depressed patients with comorbid insomnia or anxiety.128,129 Hypnotic medications (eg, zolpidem, zaleplon) are GABAA receptor allosteric modulators that potentiate GABA neurotransmission to promote sleep.128 When sleep problems emerge with treatment, switching to another antidepressant or adding a hypnotic medication may be beneficial.9 Further, sedating antidepressants should be avoided in patients with hypersomnia. Additional studies are needed – particularly well-designed head-to-head studies – to evaluate the differential effects of antidepressants on sleep/wake disturbances. Interestingly, the therapeutic effects of melatonin receptor agonists like agomelatine (not currently approved in the US), which have sleep-promoting, antidepressant, and anxiolytic properties, are currently an active area of research.129–134 However, the implications of melatonergic agents for clinical practice have yet to be fully realized.
Conclusion
A complete baseline assessment of depressive symptoms before initiating treatment provides a patient-specific profile from which the therapeutic plan may emerge. Clarifying the prior medication history is essential to differentiate between unresolved residual symptoms, comorbid conditions, and treatment-emergent side effects. By understanding the nature and magnitude of functional impairment, physicians can formulate treatment regimens based on realistic treatment goals. Further, the symptoms and symptom clusters can, with some degree of precision, be aligned with the pharmacologic actions of current and emerging antidepressants. An operational understanding of the neural systems and targets engaged by each agent at clinically relevant doses can help physicians customize pharmacotherapy to a patient’s unique constellation of symptoms. Such mechanism-based pharmacotherapy is the linchpin of personalized treatment, and provides a rational basis on which to make initial therapeutic choices for newly diagnosed patients, and those struggling with unremitting symptoms.
Acknowledgment
Medical writing assistance, supported financially by Takeda Pharmaceuticals, was provided by Bomina Yu, PhD, of inVentiv Medical Communications during the preparation of this manuscript.
Disclosure
Philip F Saltiel, MD, has received honoraria from AstraZeneca and Eli Lilly and Company for lectures and presentations. Daniel I Silvershein, MD, has received honoraria from King Pharmaceuticals, Inc. and Pfizer Inc. for lectures and presentations.
References
Penninx BW, Milaneschi Y, Lamers F, Vogelzangs N. Understanding the somatic consequences of depression: biological mechanisms and the role of depression symptom profile. BMC Med. 2013;11:129. | ||
Marcus M, Yasamy MT, van Ommeren M, Chisholm D, Saxena S; WHO Department of Mental Health and Substance Abuse. Depression: A Global Public Health Concern. Geneva, Switzerland: World Health Organization; 2012. Available from: http://www.who.int/mental_health/management/depression/who_paper_depression_wfmh_2012.pdf. Accessed August 12, 2013. | ||
Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617–627. | ||
Centers for Disease Control and Prevention (CDC). Current depression among adults – United States, 2006 and 2008. MMWR Morb Mortal Wkly Rep. 2010;59(38):1229–1235. | ||
Howden LM, Meyer JA. Age and Sex Composition: 2010. 2010 Census Briefs. Washington, DC: US Census Bureau; 2011. Available from: http://www.census.gov/prod/cen2010/briefs/c2010br-03.pdf. Accessed August 12, 2013. | ||
McKnight PE, Kashdan TB. The importance of functional impairment to mental health outcomes: a case for reassessing our goals in depression treatment research. Clin Psychol Rev. 2009;29(3):243–259. | ||
Kessler RC. The costs of depression. Psychiatr Clin North Am. 2012;35(1):1–14. | ||
Greenberg PE, Kessler RC, Birnbaum HG, et al. The economic burden of depression in the United States: how did it change between 1990 and 2000? J Clin Psychiatry. 2003;64(12):1465–1475. | ||
American Psychiatric Association. Practice Guideline for the Treatment of Patients With Major Depressive Disorder. 3rd ed. Arlington, VA: American Psychiatric Association; 2010. Available from: http://www.guideline.gov/content.aspx?id=24158. Accessed November 21, 2014. | ||
Mitchell J, Trangle M, Degnan B, et al. Adult Depression in Primary Care. Bloomington, MN: Institute for Clinical Systems Improvement; 2013 [updated September 2013]. Available from: https://www.icsi.org/_asset/fnhdm3/Depr-Interactive0512b.pdf. Accessed April 5, 2014. | ||
Patten SB, Kennedy SH, Lam RW, et al; Canadian Network for Mood and Anxiety Treatments (CANMAT). Canadian Network for Mood and Anxiety Treatments (CANMAT) clinical guidelines for the management of major depressive disorder in adults. I. Classification, burden and principles of management. J Affect Disord. 2009;117 Suppl 1:S5–S14. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington, DC: American Psychiatric Association; 2013. | ||
Ani C, Bazargan M, Hindman D, et al. Depression symptomatology and diagnosis: discordance between patients and physicians in primary care settings. BMC Fam Pract. 2008;9:1. | ||
The Department of Veterans Affairs, Department of Defense. Clinical Practice Guideline for Management of Major Depressive Disorder (MDD). Washington, DC: Department of Veterans Affairs, Department of Defense; 2009. Available from: http://www.healthquality.va.gov/mdd/mdd_full09_c.pdf. Accessed November 15, 2014. | ||
Paykel ES. Partial remission, residual symptoms, and relapse in depression. Dialogues Clin Neurosci. 2008;10(4):431–437. | ||
Egede LE. Major depression in individuals with chronic medical disorders: prevalence, correlates and association with health resource utilization, lost productivity and functional disability. Gen Hosp Psychiatry. 2007;29(5):409–416. | ||
Rubio JM, Markowitz JC, Alegria A, et al. Epidemiology of chronic and nonchronic major depressive disorder: results from the national epidemiologic survey on alcohol and related conditions. Depress Anxiety. 2011;28(8):622–631. | ||
McClintock SM, Husain MM, Wisniewski SR, et al. Residual symptoms in depressed outpatients who respond by 50% but do not remit to antidepressant medication. J Clin Psychopharmacol. 2011;31(2):180–186. | ||
Zimmerman M, McGlinchey JB, Posternak MA, Friedman M, Boerescu D, Attiullah N. Discordance between self-reported symptom severity and psychosocial functioning ratings in depressed outpatients: implications for how remission from depression should be defined. Psychiatry Res. 2006;141(2):185–191. | ||
Rush AJ, Kraemer HC, Sackeim HA, et al; ACNP Task Force. Report by the ACNP Task Force on response and remission in major depressive disorder. Neuropsychopharmacology. 2006;31(9):1841–1853. | ||
Kupfer DJ. Long-term treatment of depression. J Clin Psychiatry. 1991;52 Suppl:28–34. | ||
Sibille E, French B. Biological substrates underpinning diagnosis of major depression. Int J Neuropsychopharmacol. 2013;16(8):1893–1909. | ||
Zimmerman M, Posternak MA, Chelminski I. Defining remission on the Montgomery-Asberg depression rating scale. J Clin Psychiatry. 2004;65(2):163–168. | ||
Zimmerman M, Martinez J, Attiullah N, Friedman M, Toba C, Boerescu DA. Symptom differences between depressed outpatients who are in remission according to the Hamilton Depression Rating Scale who do and do not consider themselves to be in remission. J Affect Disord. 2012;142(1–3):77–81. | ||
Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–1917. | ||
Warden D, Rush AJ, Trivedi MH, Fava M, Wisniewski SR. The STAR*D Project results: a comprehensive review of findings. Curr Psychiatry Rep. 2007;9(6):449–459. | ||
Zisook S, Ganadjian K, Moutier C, Prather R, Rao S. Sequenced Treatment Alternatives to Relieve Depression (STAR*D): lessons learned. J Clin Psychiatry. 2008;69(7):1184–1185. | ||
Judd LL, Akiskal HS, Maser JD, et al. Major depressive disorder: a prospective study of residual subthreshold depressive symptoms as predictor of rapid relapse. J Affect Disord. 1998;50(2–3):97–108. | ||
Conradi HJ, Ormel J, de Jonge P. Symptom profiles of DSM-IV-defined remission, recovery, relapse, and recurrence of depression: the role of the core symptoms. Depress Anxiety. 2012;29(7):638–645. | ||
Zimmerman M, McGlinchey JB, Posternak MA, Friedman M, Attiullah N, Boerescu D. How should remission from depression be defined? The depressed patient’s perspective. Am J Psychiatry. 2006;163(1):148–150. | ||
Fava M, Graves LM, Benazzi F, et al. A cross-sectional study of the prevalence of cognitive and physical symptoms during long-term antidepressant treatment. J Clin Psychiatry. 2006;67(11):1754–1759. | ||
Lam RW, Michalak EE, Bond DJ, Tam EM, Axler A, Yatham LN. Which depressive symptoms and medication side effects are perceived by patients as interfering most with occupational functioning? Depress Res Treat. 2012;2012:630206. | ||
Banerjee S, Chatterji P, Lahiri K. Identifying the mechanisms for workplace burden of psychiatric illness. Med Care. 2014;52(2):112–120. | ||
Pae CU, Lim HK, Han C, et al. Fatigue as a core symptom of major depressive disorder: overview and the role of bupropion. Expert Rev Neurother. 2007;7(10):1251–1263. | ||
Demyttenaere K, De Fruyt J, Stahl SM. The many faces of fatigue in major depressive disorder. Int J Neuropsychopharmacol. 2005;8(1):93–105. | ||
Tritschler L, Felice D, Colle R, et al. Vortioxetine for the treatment of major depressive disorder. Expert Rev Clin Pharmacol. 2014;7(6):731–745. | ||
Schultz E, Malone DA Jr. A practical approach to prescribing antidepressants. Cleve Clin J Med. 2013;80(10):625–631. | ||
Thase ME, Chen D, Edwards J, Ruth A. Efficacy of vilazodone on anxiety symptoms in patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(6):351–356. | ||
Kurian BT, Greer TL, Trivedi MH. Strategies to enhance the therapeutic efficacy of antidepressants: targeting residual symptoms. Expert Rev Neurother. 2009;9(7):975–984. | ||
Jeon HJ, Fava M, Mischoulon D, et al. Psychomotor symptoms and treatment outcomes of ziprasidone monotherapy in patients with major depressive disorder: a 12-week, randomized, double-blind, placebo-controlled, sequential parallel comparison trial. Int Clin Psychopharmacol. 2014;29(6):332–338. | ||
Greer TL, Sunderajan P, Grannemann BD, Kurian BT, Trivedi MH. Does duloxetine improve cognitive function independently of its antidepressant effect in patients with major depressive disorder and subjective reports of cognitive dysfunction? Depress Res Treat. 2014;2014:627863. | ||
McIntyre RS, Lophaven S, Olsen CK. A randomized, double-blind, placebo-controlled study of vortioxetine on cognitive function in depressed adults. Int J Neuropsychopharmacol. 2014;17(10):1557–1567. | ||
Singh AB, Bousman CA, Ng CH, Byron K, Berk M. Psychomotor depressive symptoms may differentially respond to venlafaxine. Int Clin Psychopharmacol. 2013;28(3):121–126. | ||
Cooper JA, Tucker VL, Papakostas GI. Resolution of sleepiness and fatigue: a comparison of bupropion and selective serotonin reuptake inhibitors in subjects with major depressive disorder achieving remission at doses approved in the European Union. J Psychopharmacol. 2014;28(2):118–124. | ||
Fava M, Thase ME, DeBattista C, Doghramji K, Arora S, Hughes RJ. Modafinil augmentation of selective serotonin reuptake inhibitor therapy in MDD partial responders with persistent fatigue and sleepiness. Ann Clin Psychiatry. 2007;19(3):153–159. | ||
Jacobsen PL, Mahableshwarkar AR, Chen Y, Chrones L, Clayton AH. Randomized, double-blind, head-to-head, flexible-dose study of vortioxetine vs escitalopram in sexual functioning in adults with well-controlled major depressive disorder experiencing treatment-emergent sexual dysfunction. Poster presented at: 29th CINP World Congress of Neuropsychopharmacology; June 22–26, 2014; Vancouver, Canada. | ||
Montgomery SA, Nielsen RZ, Poulsen LH, Häggström L. A randomised, double-blind study in adults with major depressive disorder with an inadequate response to a single course of selective serotonin reuptake inhibitor or serotonin-noradrenaline reuptake inhibitor treatment switched to vortioxetine or agomelatine. Hum Psychopharmacol. 2014;29(5):470–482. | ||
Weizman S, Gonda X, Dome P, Faludi G. Pharmacogenetics of antidepressive drugs: a way towards personalized treatment of major depressive disorder. Neuropsychopharmacol Hung. 2012;14(2):87–101. | ||
Aziz R, Steffens DC. What are the causes of late-life depression? Psychiatr Clin North Am. 2013;36(4):497–516. | ||
Nestler EJ. Epigenetic mechanisms of depression. JAMA Psychiatry. 2014;71(4):454–456. | ||
Price JL, Drevets WC. Neural circuits underlying the pathophysiology of mood disorders. Trends Cogn Sci. 2012;16(1):61–71. | ||
Moylan S, Maes M, Wray NR, Berk M. The neuroprogressive nature of major depressive disorder: pathways to disease evolution and resistance, and therapeutic implications. Mol Psychiatry. 2013;18(5):595–606. | ||
Stahl SM, Zhang L, Damatarca C, Grady M. Brain circuits determine destiny in depression: a novel approach to the psychopharmacology of wakefulness, fatigue, and executive dysfunction in major depressive disorder. J Clin Psychiatry. 2003;64 Suppl 14:6–17. | ||
Blier P. Neurotransmitter targeting in the treatment of depression. J Clin Psychiatry. 2013;74 Suppl 2:19–24. | ||
Kuhn M, Popovic A, Pezawas L. Neuroplasticity and memory formation in major depressive disorder: an imaging genetics perspective on serotonin and BDNF. Restor Neurol Neurosci. 2014;32(1):25–49. | ||
Koolschijn PC, van Haren NE, Lensvelt-Mulders GJ, Hulshoff Pol HE, Kahn RS. Brain volume abnormalities in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Hum Brain Mapp. 2009;30(11):3719–3735. | ||
McKinnon MC, Yucel K, Nazarov A, MacQueen GM. A meta-analysis examining clinical predictors of hippocampal volume in patients with major depressive disorder. J Psychiatry Neurosci. 2009;34(1):41–54. | ||
Kennedy SH, Evans KR, Kruger S, et al. Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. Am J Psychiatry. 2001;158(6):899–905. | ||
Guo W, Liu F, Yu M, et al. Functional and anatomical brain deficits in drug-naive major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2014;54:1–6. | ||
Lum CT, Stahl SM. Opportunities for reversible inhibitors of monoamine oxidase-A (RIMAs) in the treatment of depression. CNS Spectr. 2012;17(3):107–120. | ||
Stahl SM. Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications. 4th ed. New York, NY: Cambridge University Press; 2013. | ||
Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct. 2008;213(1–2):93–118. | ||
Blier P, El Mansari M. Serotonin and beyond: therapeutics for major depression. Philos Trans R Soc Lond B Biol Sci. 2013;368(1615):20120536. | ||
Hamon M, Blier P. Monoamine neurocircuitry in depression and strategies for new treatments. Prog Neuropsychopharmacol Biol Psychiatry. 2013;45:54–63. | ||
Moret C, Briley M. The importance of norepinephrine in depression. Neuropsychiatr Dis Treat. 2011;7 Suppl 1:9–13. | ||
Dunlop BW, Nemeroff CB. The role of dopamine in the pathophysiology of depression. Arch Gen Psychiatry. 2007;64(3):327–337. | ||
O’Leary OF, Cryan JF. Chapter 4.13 – The behavioral genetics of serotonin: relevance to anxiety and depression. In: Muller CP, Jacobs BL, eds. Handbook of Behavioral Neuroscience. Vol 21. London, UK: Elsevier; 2010:749–789. | ||
Aman TK, Shen RY, Haj-Dahmane S. D2-like dopamine receptors depolarize dorsal raphe serotonin neurons through the activation of nonselective cationic conductance. J Pharmacol Exp Ther. 2007;320(1):376–385. | ||
Adell A, Bortolozzi A, Diaz-Mataix L, Santana N, Celada P, Artigas F. Chapter 2.8 – Serotonin interactions with other transmitter systems. In: Muller CP, Jacobs BL, eds. Handbook of Behavioral Neuroscience. Vol 21. London, UK: Elsevier; 2010:259–276. | ||
Pehrson AL, Sanchez C. Serotonergic modulation of glutamate neurotransmission as a strategy for treating depression and cognitive dysfunction. CNS Spectr. 2014;19(2):121–133. | ||
Stahl S, Briley M. Understanding pain in depression. Hum Psychopharmacol. 2004;19 Suppl 1:S9–S13. | ||
Stahl SM, Lee-Zimmerman C, Cartwright S, Morrissette DA. Serotonergic drugs for depression and beyond. Curr Drug Targets. 2013;14(5):578–585. | ||
Cipriani A, Furukawa TA, Salanti G, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373(9665):746–758. | ||
Kennedy SH. A review of antidepressant therapy in primary care: current practices and future directions. Prim Care Companion CNS Disord. 2013;15(2). pii: PCC.12r01420. | ||
Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J Pharmacol Exp Ther. 1997;283(3):1305–1322. | ||
Ciraulo DA, Barnhill JG, Jaffe JH. Clinical pharmacokinetics of imipramine and desipramine in alcoholics and normal volunteers. Clin Pharmacol Ther. 1988;43(5):509–518. | ||
Renoir T. Selective serotonin reuptake inhibitor antidepressant treatment discontinuation syndrome: a review of the clinical evidence and the possible mechanisms involved. Front Pharmacol. 2013;4:45. | ||
Joel I, Begley AE, Mulsant BH, et al; IRL GREY Investigative Team. Dynamic prediction of treatment response in late-life depression. Am J Geriatr Psychiatry. 2014;22(2):167–176. | ||
Pan YJ, Liu SK, Yeh LL. Factors affecting early attrition and later treatment course of antidepressant treatment of depression in naturalistic settings: an 18-month nationwide population-based study. J Psychiatr Res. 2013;47(7):916–925. | ||
Greer TL, Kurian BT, Trivedi MH. Defining and measuring functional recovery from depression. CNS Drugs. 2010;24(4):267–284. | ||
Ashton AK, Jamerson BD, Weinstein WL, Wagoner C. Antidepressant-related adverse effects impacting treatment compliance: results of a patient survey. Curr Ther Res Clin Exp. 2005;66(2):96–106. | ||
Magura S, Rosenblum A, Fong C. Factors associated with medication adherence among psychiatric outpatients at substance abuse risk. Open Addict J. 2011;4:58–64. | ||
Preston TC, Shelton RC. Treatment resistant depression: strategies for primary care. Curr Psychiatry Rep. 2013;15(7):370. | ||
Patkar AA, Pae CU. Atypical antipsychotic augmentation strategies in the context of guideline-based care for the treatment of major depressive disorder. CNS Drugs. 2013;27 Suppl 1:S29–S37. | ||
Wright BM, Eiland EH 3rd, Lorenz R. Augmentation with atypical antipsychotics for depression: a review of evidence-based support from the medical literature. Pharmacotherapy. 2013;33(3):344–359. | ||
Chang TE, Jing Y, Yeung AS, et al. Effect of communicating depression severity on physician prescribing patterns: findings from the Clinical Outcomes in MEasurement-based Treatment (COMET) trial. Gen Hosp Psychiatry. 2012;34(2):105–112. | ||
Shao L, Li W, Xie Q, Yin H. Triple reuptake inhibitors: a patent review (2006–2012). Expert Opin Ther Pat. 2014;24(2):131–154. | ||
Tran P, Skolnick P, Czobor P, et al. Efficacy and tolerability of the novel triple reuptake inhibitor amitifadine in the treatment of patients with major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Psychiatr Res. 2012;46(1):64–71. | ||
Singh M, Schwartz TL. Clinical utility of vilazodone for the treatment of adults with major depressive disorder and theoretical implications for future clinical use. Neuropsychiatr Dis Treat. 2012;8:123–130. | ||
Katona CL, Katona CP. New generation multi-modal antidepressants: focus on vortioxetine for major depressive disorder. Neuropsychiatr Dis Treat. 2014;10:349–354. | ||
Gardier AM, Malagié I, Trillat AC, Jacquot C, Artigas F. Role of 5-HT1A autoreceptors in the mechanism of action of serotoninergic antidepressant drugs: recent findings from in vivo microdialysis studies. Fundam Clin Pharmacol. 1996;10(1):16–27. | ||
Bétry C, Pehrson AL, Etiévant A, Ebert B, Sánchez C, Haddjeri N. The rapid recovery of 5-HT cell firing induced by the antidepressant vortioxetine involves 5-HT(3) receptor antagonism. Int J Neuropsychopharmacol. 2013;16(5):1115–1127. | ||
Richelson E. Multi-modality: a new approach for the treatment of major depressive disorder. Int J Neuropsychopharmacol. 2013;16(6):1433–1442. | ||
Bang-Andersen B, Ruhland T, Jorgensen M, et al. Discovery of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): a novel multimodal compound for the treatment of major depressive disorder. J Med Chem. 2011;54(9):3206–3221. | ||
Mørk A, Pehrson A, Brennum LT, et al. Pharmacological effects of Lu AA21004: a novel multimodal compound for the treatment of major depressive disorder. J Pharmacol Exp Ther. 2012;340(3):666–675. | ||
Hedlund PB, Huitron-Resendiz S, Henriksen SJ, Sutcliffe JG. 5-HT7 receptor inhibition and inactivation induce antidepressantlike behavior and sleep pattern. Biol Psychiatry. 2005;58(10):831–837. | ||
Weber S, Volynets V, Kanuri G, Bergheim I, Bischoff SC. Treatment with the 5-HT3 antagonist tropisetron modulates glucose-induced obesity in mice. Int J Obes (Lond). 2009;33(12):1339–1347. | ||
Alam MY, Jacobsen PL, Chen Y, Serenko M, Mahableshwarkar AR. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2014;29(1):36–44. | ||
Citrome L. Vortioxetine for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant – what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2014;68(1):60–82. | ||
Schwartz TL, Siddiqui UA, Stahl SM. Vilazodone: a brief pharmacological and clinical review of the novel serotonin partial agonist and reuptake inhibitor. Ther Adv Psychopharmacol. 2011;1(3):81–87. | ||
Hammar A, Sørensen L, Ardal G, et al. Enduring cognitive dysfunction in unipolar major depression: a test-retest study using the Stroop paradigm. Scand J Psychol. 2010;51(4):304–308. | ||
Baune BT, Miller R, McAfoose J, Johnson M, Quirk F, Mitchell D. The role of cognitive impairment in general functioning in major depression. Psychiatry Res. 2010;176(2–3):183–189. | ||
Jaeger J, Berns S, Uzelac S, Davis-Conway S. Neurocognitive deficits and disability in major depressive disorder. Psychiatry Res. 2006;145(1):39–48. | ||
Hasselbalch BJ, Knorr U, Kessing LV. Cognitive impairment in the remitted state of unipolar depressive disorder: a systematic review. J Affect Disord. 2011;134(1–3):20–31. | ||
Hammar A, Ardal G. Cognitive functioning in major depression – a summary. Front Hum Neurosci. 2009;3:26. | ||
Hasselbalch BJ, Knorr U, Hasselbalch SG, Gade A, Kessing LV. The cumulative load of depressive illness is associated with cognitive function in the remitted state of unipolar depressive disorder. Eur Psychiatry. 2013;28(6):349–355. | ||
Harvey PO, Fossati P, Pochon JB, et al. Cognitive control and brain resources in major depression: an fMRI study using the n-back task. Neuroimage. 2005;26(3):860–869. | ||
MacDonald AW 3rd, Cohen JD, Stenger VA, Carter CS. Dissociating the role of the dorsolateral prefrontal and anterior cingulate cortex in cognitive control. Science. 2000;288(5472):1835–1838. | ||
Dell’Osso B, Cremaschi L, Spagnolin G, Ketter TA, Altamura AC. Augmentative dopaminergic interventions for treatment-resistant bipolar depression: a focus on dopamine agonists and stimulants. J Psychopathol. 2013;19:327–340. | ||
Murrough JW, Wan LB, Iacoviello B, et al. Neurocognitive effects of ketamine in treatment-resistant major depression: association with antidepressant response. Psychopharmacology (Berl). 2013;231(3):481–488. | ||
Strzelecki D, Tabaszewska A, Barszcz Z, Józefowicz O, Kropiwnicki P, Rabe-Jabłońska J. A 10-week memantine treatment in bipolar depression: a case report. Focus on depressive symptomatology, cognitive parameters and quality of life. Psychiatry Investig. 2013;10(4):421–424. | ||
Katona C, Hansen T, Olsen CK. A randomized, double-blind, placebo-controlled, duloxetine-referenced, fixed-dose study comparing the efficacy and safety of Lu AA21004 in elderly patients with major depressive disorder. Int Clin Psychopharmacol. 2012;27(4):215–223. | ||
Iverson GL, Lam RW. Rapid screening for perceived cognitive impairment in major depressive disorder. Ann Clin Psychiatry. 2013;25(2):135–140. | ||
Ferentinos PP, Kontaxakis VP, Havaki-Kontaxaki BJ, Dikeos DG, Papadimitriou GN. Fatigue and somatic anxiety in patients with major depression. Psychiatriki. 2009;20(4):312–318. | ||
Fava M, Ball S, Nelson JC, et al. Clinical relevance of fatigue as a residual symptom in major depressive disorder. Depress Anxiety. 2014;31(3):250–257. | ||
Soyuer F, Şenol V. Functional outcome and depression in the elderly with or without fatigue. Arch Gerontol Geriatr. 2011;53(2):e164–e167. | ||
Targum SD, Fava M. Fatigue as a residual symptom of depression. Innov Clin Neurosci. 2011;8(10):40–43. | ||
MacHale SM, Lawŕie SM, Cavanagh JT, et al. Cerebral perfusion in chronic fatigue syndrome and depression. Br J Psychiatry. 2000;176:550–556. | ||
Papakostas GI, Nutt DJ, Hallett LA, Tucker VL, Krishen A, Fava M. Resolution of sleepiness and fatigue in major depressive disorder: a comparison of bupropion and the selective serotonin reuptake inhibitors. Biol Psychiatry. 2006;60(12):1350–1355. | ||
McClung CA. How might circadian rhythms control mood? Let me count the ways... Biol Psychiatry. 2013;74(4):242–249. | ||
Grandner MA, Drummond SP. Who are the long sleepers? Towards an understanding of the mortality relationship. Sleep Med Rev. 2007;11(5):341–360. | ||
Lai YC, Huang MC, Chen HC, et al. Familiality and clinical outcomes of sleep disturbances in major depressive and bipolar disorders. J Psychosom Res. 2014;76(1):61–67. | ||
Oswald I, Adam K. Effects of paroxetine on human sleep. Br J Clin Pharmacol. 1986;22(1):97–99. | ||
Salin-Pascual RJ, Galicia-Polo L, Drucker-Colin R. Sleep changes after 4 consecutive days of venlafaxine administration in normal volunteers. J Clin Psychiatry. 1997;58(8):348–350. | ||
Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24(12):726–731. | ||
Santos Moraes WA, Burke PR, Coutinho PL, et al. Sedative antidepressants and insomnia. Rev Bras Psiquiatr. 2011;33(1):91–95. | ||
Fagiolini A, Comandini A, Catena Dell’Osso M, Kasper S. Rediscovering trazodone for the treatment of major depressive disorder. CNS Drugs. 2012;26(12):1033–1049. | ||
Weber J, Siddiqui MA, Wagstaff AJ, McCormack PL. Low-dose doxepin: in the treatment of insomnia. CNS Drugs. 2010;24(8):713–720. | ||
Maclean L, Ahmedani BK. Sertraline and low-dose doxepin treatment in severe agitated-anxious depression with significant gastrointestinal complaints: two case reports. Prim Care Companion CNS Disord. 2011;13(4) pii: PCC.12r01420. | ||
Becker PM, Sattar M. Treatment of sleep dysfunction and psychiatric disorders. Curr Treat Options Neurol. 2009;11(5):349–357. | ||
De Berardis D, Marini S, Fornaro M, et al. The melatonergic system in mood and anxiety disorders and the role of agomelatine: implications for clinical practice. Int J Mol Sci. 2013;14(6):12458–12483. | ||
Kennedy SH, Cyriac A. A dimensional approach to measuring antidepressant response: implications for agomelatine. Psychology. 2012;3(10):864–869. | ||
Cardinali DP, Srinivasan V, Brzezinski A, Brown GM. Melatonin and its analogs in insomnia and depression. J Pineal Res. 2012;52(4):365–375. | ||
Laudon M, Frydman-Marom A. Therapeutic effects of melatonin receptor agonists on sleep and comorbid disorders. Int J Mol Sci. 2014;15(9):15924–15950. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.