Back to Journals » Journal of Inflammation Research » Volume 19
MAIT Cells Maintain Intestinal Homeostasis Through IL-22 in an Experimental Colitis
Authors Peng Y, Zheng C, Wang Y, Huang F, Shao Y, Liu L, Zhao Q ![]()
Received 17 October 2025
Accepted for publication 22 April 2026
Published 29 May 2026 Volume 2026:19 570529
DOI https://doi.org/10.2147/JIR.S570529
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Shouya Feng
Yanan Peng,1,2,* Chunlan Zheng,1,2,* Youwei Wang,1,2,* Fengxing Huang,1,2 Yu Shao,1,2 Lan Liu,1,2,* Qiu Zhao1,2,*
1Department of Gastroenterology, Zhongnan Hospital of Wuhan University, Wuhan, 430071, People’s Republic of China; 2Hubei Clinical Center and Key Laboratory of Intestinal and Colorectal Diseases, Wuhan, 430071, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qiu Zhao, Email [email protected] Lan Liu, Email [email protected]
Objective: Mucosa-associated invariant T (MAIT) cells are a unique subset of innate-like lymphocytes abundant in the gastrointestinal tract, yet their role in maintaining intestinal homeostasis remains incompletely understood. We investigated how MAIT cells contribute to epithelial barrier integrity and the molecular mechanisms underlying this process during experimental colitis.
Methods: Acute colitis was induced by dextran sulfate sodium (DSS) in wild-type (WT) and MR1-deficient (MR1−/−) mice. Disease severity was quantified via weight loss, colon length, and histopathology. Barrier function was assessed by FITC-dextran permeability and expression of tight junction proteins (ZO-1 and Claudin-1). Single-cell RNA sequencing (scRNA-seq) was employed to identify MAIT-cell- associated mediators, followed by rescue experiments with recombinant IL-22 or CCL2.
Results: MAIT cells accumulated in the inflamed colon and displayed a tissue-specific activated phenotype during DSS-induced colitis. MR1−/− mice developed more severe colitis than WT mice, with aggravated mucosal injury, increased inflammatory cytokine production, disrupted tight junction expression, enhanced intestinal permeability, and increased bacterial translocation. MAIT cell deficiency was associated with markedly reduced colonic IL-22 expression and impaired IL-22 production across multiple immune populations, including Th22 cells, Th17 cells, ILC3s, and CD4⁺ T cells. Recombinant IL-22 partially alleviated colitis and improved barrier-associated protein expression, indicating that IL-22 is an important downstream mediator of MAIT cell-dependent protection. Mechanistically, single-cell RNA sequencing identified CCL2 as a MAIT-associated soluble factor induced during colitis. In vivo CCL2 supplementation restored AHR/CYP1A1 expression, enhanced IL-22 production in Th22 and other lymphocyte subsets, and ameliorated epithelial barrier damage and disease severity in MR1−/− mice.
Conclusion: Our study identifies a novel MAIT–CCL2–AHR–IL-22 regulatory axis that that contributes to epithelial barrier protection during acute experimental colitis. MAIT cells appear to act not only as effector cells but also as upstream coordinators of IL-22-dependent mucosal repair, providing a mechanistic basis for targeting this pathway in intestinal inflammatory disease.
Keywords: inflammatory bowel disease, acute experimental colitis, MAIT cell, IL-22, CCL2, intestinal barrier
Introduction
Inflammatory bowel diseases (IBDs), encompassing ulcerative colitis (UC) and Crohn’s disease (CD), are chronic relapsing inflammatory disorders of the gastrointestinal tract with increasing global incidence.1,2 Although their etiology is multifactorial, IBD is widely considered to arise from dysregulated immune responses to the intestinal microbiota in genetically susceptible individuals, leading to persistent inflammation and epithelial barrier dysfunction.3,4 Achieving a delicate balance between innate and adaptive immune responses in the gut is a key factor in treating mucosal inflammation and tissue damage in IBDs. Increasing evidence indicates that, beyond conventional adaptive immunity, unconventional T cell populations positioned at mucosal interfaces are critical regulators of tissue homeostasis and barrier repair.5
Mucosa-associated invariant T (MAIT) cells are a subset of innate-like lymphocytes that recognize riboflavin-derived metabolites presented by the major histocompatibility complex class I-related molecule (MR1).6–8 MAIT cells are highly enriched in mucosal tissues and are developmentally shaped by early-life microbial exposure,9 as demonstrated by the marked reduction of MAIT cells in germ-free mice.10,11 The use of MR1 tetramers loaded with riboflavin metabolites enables reliable identification of MAIT cells across tissues.12 In addition to their antimicrobial functions, MAIT cells exhibit considerable functional plasticity and can adopt distinct effector programs depending on the inflammatory context. These include pro-inflammatory responses characterized by IFN-γ and IL-17A production, as well as tissue-protective programs associated with barrier repair.13–16
Recent studies have further highlighted the importance of MAIT cell–microbiome interactions in human IBD. Circulating MAIT cells are often reduced in patients, whereas they accumulate and become activated in inflamed intestinal tissues, consistent with recruitment to sites of barrier disruption and microbial translocation.17–20 In parallel, alterations in microbial composition and metabolic activity during intestinal inflammation can influence the availability of MR1 ligands and shape MAIT cell responses. Beyond antigen recognition, emerging evidence suggests that microbial metabolic cues contribute to the regulation of MAIT cell function and downstream cytokine programs, providing an additional layer of control over mucosal immunity.16,17,21 Notably, DSS-induced colitis, although primarily driven by epithelial injury, recapitulates key features of microbiota-associated inflammation observed in human IBD, thereby offering a relevant model to investigate microbiome-dependent MAIT cell responses.
IL-22 is a member of the IL-10 cytokine family and plays a central role in maintaining epithelial barrier integrity.22,23 Unlike many immune mediators, IL-22 primarily acts on non-hematopoietic cells, including intestinal epithelial cells, where it promotes proliferation, enhances tight junction formation, and induces antimicrobial peptide production.22–24 Multiple immune cell populations, such as Th22 cells, Th17 cells, innate lymphoid cells, and γδ T cells, can produce IL-22 in a context-dependent manner.25–27 While IL-22 is generally considered protective in acute mucosal injury, its function is tightly regulated, and dysbalanced IL-22 responses have also been implicated in chronic inflammation.23,28
Given the close association of MAIT cells with microbial sensing and mucosal immunity, and the key role of IL-22 in epithelial repair, a potential functional link between these two components is biologically plausible but remains insufficiently defined. In particular, it is unclear whether MAIT cells regulate IL-22 responses indirectly through interactions with other immune cell populations during intestinal inflammation. In this study, we used DSS-induced colitis in WT and MR1−/− mice to investigate the role of MAIT cells in intestinal homeostasis. By combining transcriptomic analysis and in vivo functional experiments, we explored the mechanisms by which MAIT cells influence IL-22 production and epithelial barrier integrity during acute colitis.
Materials and Methods
Animal and DSS-Induced Acute Colitis Model
Specific pathogen-free (SPF) male C57BL/6J mice (6–8 weeks old, 15–20g) were purchased from Beijing Weitonglihua Animal Experimental Technology Co, LTD. MR1−/− mice on a C57BL/6 background were provided by Prof. Xiong Ma (Renji Hospital, Shanghai Jiao Tong University). Animals were housed in the SPF Animal Laboratory under the standard 12h light/dark cycle at 22°C and had free access to food and water. All procedures were approved by the Ethics Committee of Zhongnan Hospital of Wuhan University and followed ARRIVE 2.0 guidelines.
After 1 week of acclimatization, acute colitis was induced by administration of 2% DSS (36–50 kDa) in drinking water for 7 consecutive days. Mice were randomly assigned into four groups (n = 5–7 per group): WT control, WT+DSS, MR1−/− control, and MR1−/−+DSS. Randomization was performed using a computer-generated random number table Body weight, stool consistency, and rectal bleeding were monitored daily to calculate the disease activity index (DAI). Investigators responsible for scoring and histological evaluation were blinded to group allocation. On day 7, mice were euthanized with sodium pentobarbital (150 mg/kg, i.p), and serum and colon tissues were collected.
To minimize experimental bias, investigators responsible for animal handling and outcome assessment were blinded to group allocation during disease activity scoring, histopathological evaluation, and biochemical analyses. Group identities were revealed only after completion of data analysis.
In vivo Recombinant Protein Administration
For rescue experiments, MR1−/− mice were randomly assigned to receive intraperitoneal injections of either sterile phosphate-buffered saline (PBS) or recombinant murine IL-22 (GenScript, USA) at a dose of 30 μg per mouse diluted in 100 μL PBS. The IL-22 dose and administration schedule were selected based on previously published studies demonstrating effective epithelial protection and barrier restoration in DSS-induced colitis models.29,30 Injections were administered once daily starting on day 1 of DSS exposure and continued throughout the 7-day DSS treatment period.
To investigate the role of the MAIT–CCL2 axis, a separate cohort of MR1−/− DSS-treated mice received recombinant murine CCL2 (Novoprotein,China) at a dose of 100ng per mouse diluted in 100 μL PBS via intraperitoneal injection.31 CCL2 administration was initiated on day 1 of DSS exposure and continued once daily for seven consecutive days. Control mice received an equal volume of PBS following the same schedule. At the experimental endpoint (day 7), colonic tissues and lamina propria mononuclear cells (LPMCs) were harvested for subsequent analysis.
Inclusion and Exclusion Criteria for Mice in DSS-Induced Acute Colitis Models
For mice in DSS-induced acute colitis models, inclusion criteria include specific pathogen-free (SPF) inbred strains (preferably C57BL/6), 6–8 weeks of age, 15–20 g body weight (with less than 10% weight difference among group members), no pre-existing diseases/enteric pathogens/prior interventions (eg., antibiotics), and gender matching or stratification; exclusion criteria cover baseline abnormalities (out-of-range weight/temperature, microbiota dysbiosis, genetic mutations) and post-DSS issues (≥20% weight loss in 72 h/severe symptoms like persistent diarrhea, <70% DSS water intake, accidental death, extra-intestinal pathology, and protocol non-compliance).
Colon Histopathological Score
After direct colonic resection from mice, specimens were fixed overnight in 4% formaldehyde at room temperature. Paraffin-embedded tissues were sectioned for hematoxylin and eosin (HE) staining. Histopathological grading was independently assessed by three pathologists using blinded scoring based on the Neurath criteria.32
Liver CFU Measurement
Fresh livers were weighted and homogenized with 3mL sterile PBS to prepare a tissue homogenate, which were plated on Blood Agar Base (Huankai microbial, China). Eighteen hours after incubation, the CFUs were counted and calculated as count per gram weight of the liver.
Intestinal Permeability Assay
Mice were fasted overnight before intragastric administration of FITC-dextran (Sigma, USA). 3 hours later, the blood was collected and centrifuged at 12000g at 4°C, and the supernatant fluorescence intensity was detected by a multi-function enzyme labeling instrument (BIORAD, USA).
Immunofluorescence Staining
The colonic samples were fixed in formaldehyde for 2–3 days and embedded in paraffin. The sectioned tissue was blocked BSA solution at room temperature for 30 minutes and incubated overnight with the primary antibody ZO-1 (Absin, China), the secondary antibody FITC-labeled goat anti-rabbit (CST, USA) was bound. Then the nucleus was stained with a DAPI staining solution. Finally, the sections were sealed and detected under fluorescence microscope.
Western Blotting
Mice colon was milled with the radioimmunoprecipitation assay (RIPA) buffer (Beyotime, China), phenylmethylsulfonylfluoride (Thermo, USA), and protease inhibitor ((Beyotime, China). The extracted protein was denaturation with loading buffer and separated by sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE), then transferred to a PVDF membrane (Beyotime, China). Blocked with 5% skim milk for 2 hours. The primary antibody Zo-1 (Absin, China; 1:1000), Claudin1 (Absin, China; 1:1000), IL-22 (Affinity, China; 1:1000), AHR (ABclonal, UK; 1:1000), CYP1A1 (ABclonal, UK; 1:1000) and GAPDH (CST, USA; 1:1000) incubated overnight, and appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (CST, USA, 1:2000) incubated for 2 hours at room temperature.
Quantitative Real-Time PCR
Total RNA was extracted from colon with TRIzol reagent according to the manufacturer’s instructions. Then, the RNA samples were reverse transcribed into cDNA using a reverse transcription kit (Roche, USA). The target gene was amplified and detected by quantitative fluorescence PCR (ABI, USA).
Lamina Propria Mononuclear Cells Isolation
The colon was shredded (0.5mm). The tissues were treated with RPMI 1640 containing 0.5mmol/L EDTA (Biosharp, China) 30 minutes. Then digested with RPMI 1640 containing 0.5mg/mL collagenase VIII (Sigma, USA), 100 μg/mL DNase I (Sigma, USA), and 10% fetal bovine serum (Gibco, USA) 20 minutes at 37 °C. The supernatant was centrifuged at 500g for 10 minutes at 4 °C. The precipitates were stratified by 80% and 40% Percoll (GE life, USA) with no brake centrifugation, and the cells between the boundary of two Percoll gradients were the lamina propria mononuclear cells.
Flow Cytometry and Gating Strategy
The collected lamina propria mononuclear cells were centrifuged at 500g for 10 minutes at 4 °C. The precipitates were re-suspended with 100μL staining buffer (BD Biosciences, USA). For MAIT cell identification, cells were stained with flow antibody APC-Cy7-conjugated CD3 (BD Pharmingen, USA), FITC-conjugated TCR β (BD Pharmingen, USA), PE-conjugated MR1-tetramer (NIH Tetramer Core Facility, USA) for 30 min at 4 °C. Th17 cells were defined as viable CD45⁺CD3⁺CD4⁺ lymphocytes expressing IL-17A.Th22 cells were defined as viable CD45⁺CD3⁺CD4⁺ lymphocytes expressing IL-22 but not IL-17A, thereby distinguishing them from Th17 cells capable of co-producing IL-22. ILC3 were defined as viable CD45⁺ lymphocytes lacking CD3 and CD4 expression while co-expressing CD127 and the transcription factor RORγt.
For intracellular cytokine/chemokine detection, cells were stimulated with PMA (50 ng/mL) and ionomycin (500 ng/mL) in the presence of a protein transport inhibitor (brefeldin A or monensin) for 4 h at 37 °C. Cells were then fixed and permeabilized using Cytofix/Cytoperm buffer (BD Biosciences) and stained with PerCP-Cy5.5–conjugated anti-IL-22, PE-Cy7-conjugated anti-IL-17A, and APC-conjugated anti-CCL2 antibodies for 30 min at 4 °C. The cells were analyzed by flow cytometry.
Magnetic Enrichment of MAIT Cells Using MR1 Tetramer
The collected mononuclear cells in lamina propria were centrifuged for 10 minutes at 4 °C, re-suspended with 100 μ l running buffer (MiltenyiBiotec, Germany), added with PE-conjugated MR1-tetramer antibody (NIH Tetramer Core Facility, USA), incubated at 4 °C for 30 minutes, Anti-PE beads (MiltenyiBiotec, Germany) were added, incubated for 15 minutes. 500g centrifuge for 10 minutes at 4 °C, cell suspension was added to the separation column fixed on the magnetic frame, and the adsorbed cells in the separation column are called MAIT cells.
Enzyme-Linked Immunosorbent Assay (ELISA)
The serum, tissue-milled supernatant, and cell culture supernatant were collected, 50μL supernatant was added to the enzyme plate, incubated at 37 °C for 30 minutes, the chromogenic agent was added and incubated at 37 °C for 15 minutes. After adding the terminating solution, the absorbance of each hole was detected by an enzyme labeling instrument, and the concentration of each group was calculated according to the standard curve.
Single-Cell RNA Sequencing (scRNA-Seq) Analysis
Publicly available scRNA-seq datasets of colonic immune cells (GSE264408) were analyzed to identify differentially expressed genes (DEGs) in MAIT cells during colitis. Data integration, scaling, and clustering were performed using the Seurat R package (v4.0). MAIT cells were identified based on the expression of canonical markers (Zbtb16, Il18r1, and Rora). Candidate soluble mediators were screened based on fold-change and secretome database filtering, identifying Ccl2 as a top candidate significantly downregulated in MR1−/−DSS colonic tissues.
Statistical Analysis
All data are presented as mean ± standard deviation (SD). All experiments were repeated independently at least three times unless otherwise specified. Statistical analyses were performed using GraphPad Prism 9 (GraphPad Software, USA). Normality was assessed using the Shapiro–Wilk test. For comparisons between two groups, an unpaired two-tailed Student’s t-test was used for normally distributed data, whereas the Mann–Whitney U-test was applied for non-normal data. For multiple group comparisons, one-way ANOVA with Tukey’s post hoc test or the Kruskal–Wallis test with Dunn’s correction was used as appropriate. A P value < 0.05 was considered statistically significant.
Results
MAIT Cells Redistribute to the Inflamed Colon and Exhibit a Tissue-Specific Activated Phenotype
WT mice were fed with 2.0% dextran sodium sulfate (DSS) in drinking water for 7 consecutive days to induce acute colitis. After sacrifice, mononuclear cells were isolated from colonic lamina propria, spleen, and peripheral blood. MAIT cells were identified by flow cytometry as CD3+TCRβ+MR1−tetramer+ and CD3+TCRβ+6-FP− (Figure 1A). In DSS-treated mice, the proportion of MAIT cells were significantly increased in the colon, while significantly decreased in the spleen and peripheral blood compared to WT controls (Figure 1B and C). This reciprocal change suggests a redistribution of MAIT cells from systemic compartments to the inflamed intestine during acute colitis. Moreover, the proportion of colonic CD4+ and CD8+MAIT cells in mice from the DSS treatment group was significantly elevated, while that of DN MAIT cells decreased significantly, suggesting a potential functional shift in the colonic MAIT cell pool under inflammatory conditions induced by DSS (Figure 1B and D). However, neither splenic nor circulating MAIT cells showed similar changes in the constituent ratio.
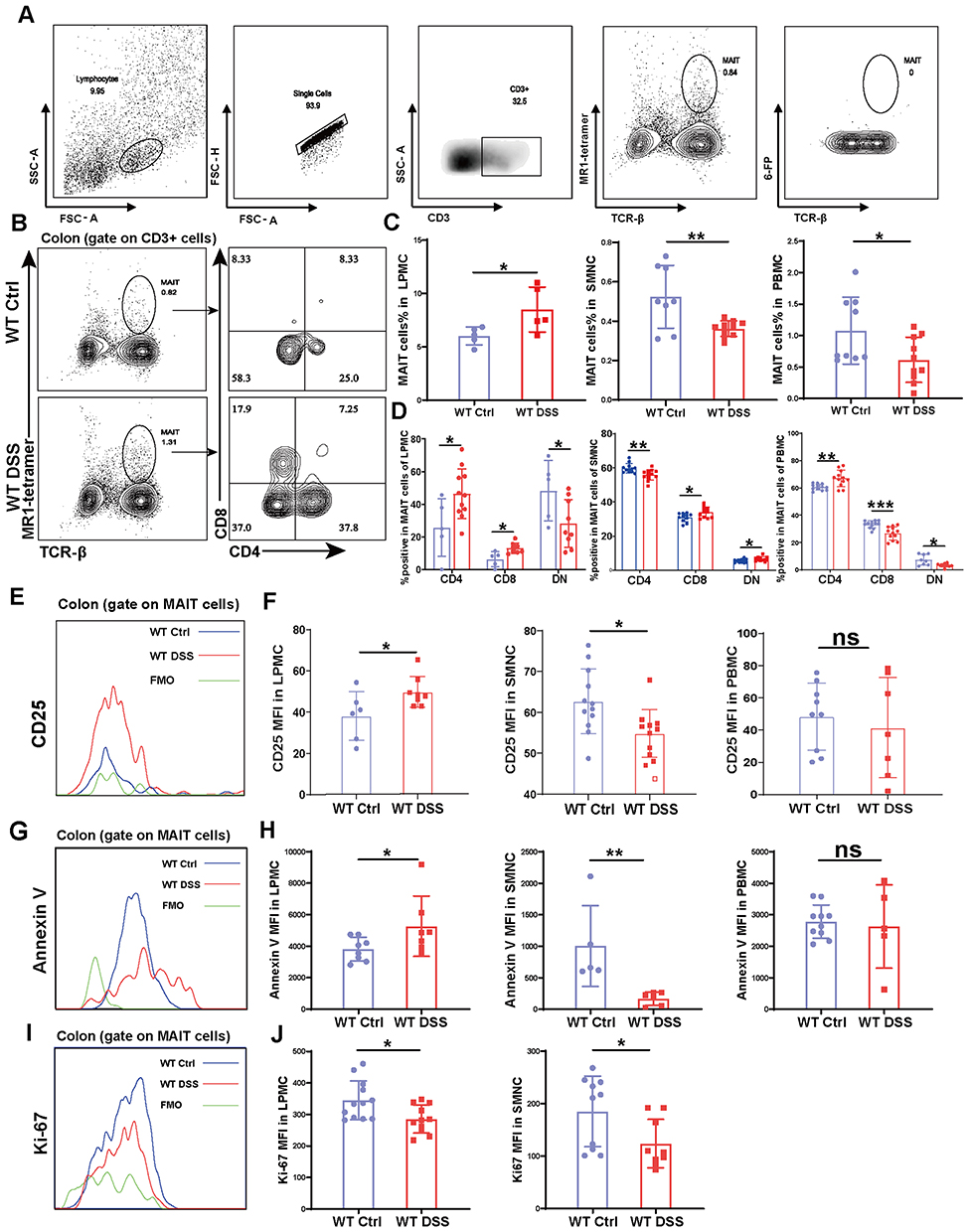 |
Figure 1 MAIT cells accumulate in the colon during DSS-induced colitis through redistribution from systemic pools. (A) Gating strategy for the identification of MAIT cells as CD3⁺TCRβ⁺MR1-tetramer⁺ and CD3⁺TCRβ⁺6-FP− populations by flow cytometry; (B and C) Proportion and absolute number of MAIT cells in the colon, spleen, and peripheral blood of WT and DSS-treated WT mice; (B and D) Subset composition of colonic MAIT cells showing increased frequencies of CD4⁺ and CD8⁺ subsets and a decrease in double-negative (DN) MAIT cells following DSS treatment; (E and F) Activation status assessed by CD25 expression on MAIT cells from colon, spleen, and peripheral blood; (G–J) Apoptosis (Annexin V⁺) and proliferation (Ki-67⁺) of MAIT cells across indicated tissues. Data are presented as mean ± SD; n = 5–10 mice per group. Statistical significance was determined using unpaired two-tailed Student’s t-test. ***p<0.001, **p<0.01, * p<0.05. Abbreviation: ns, not significant. |
We further assessed the activation status of MAIT cells across tissues. Colonic MAIT cells from DSS-treated mice showed markedly increased expression of CD25, indicating an activated phenotype (Figure 1E and F). In contrast, splenic MAIT cells exhibited reduced CD25 expression, and peripheral blood MAIT cells showed a similar—though statistically insignificant—trend (Figure 1F). These findings imply compartment-specific activation during colitis.
To explore mechanisms underlying the accumulation of MAIT cells in the colon, we assessed apoptosis and proliferation. Colonic MAIT cells from DSS mice showed increased apoptosis and reduced proliferation (Figure 1G–J). In the spleen, both apoptosis and proliferation were decreased, while peripheral blood MAIT cells showed reduced apoptosis without significance (Figure 1H and J). These results suggest that colonic MAIT cells in WT DSS mice exhibit high apoptosis and low proliferation. The observed increase in colonic MAIT cells despite heightened apoptosis and reduced local proliferation suggests that their accumulation is likely driven by migration from systemic pools rather than in situ expansion. This redistribution highlights the role of tissue-specific homing in maintaining intestinal homeostasis during inflammation.33
MAIT Cells Exhibit Compartment-Specific Activation and Effector Profiles in Colitis
To further characterize the functional state of MAIT cells during colitis, we assessed their cytokine and effector molecule profiles by flow cytometry. We observed a striking tissue-compartmentalized functional divergence between colon and spleen. Compared to WT controls, MAIT cells isolated from the colon of DSS-treated mice exhibited a pro-inflammatory effector profile, characterized by significantly enhanced secretion of IL-17a and IFNγ, alongside elevated expression of the degranulation marker CD107A and cytolytic molecule Granzyme B (GraB). Notably, this was accompanied by a significant reduction in Perforin expression (Figure 2A and B). In contrast, splenic MAIT cells from the same DSS-treated mice displayed distinct and somewhat suppressed phenotype: production of IL-17a, IFNγ, and GraB was significantly decreased, while Perforin and CD107A were increased (Figure 2C).
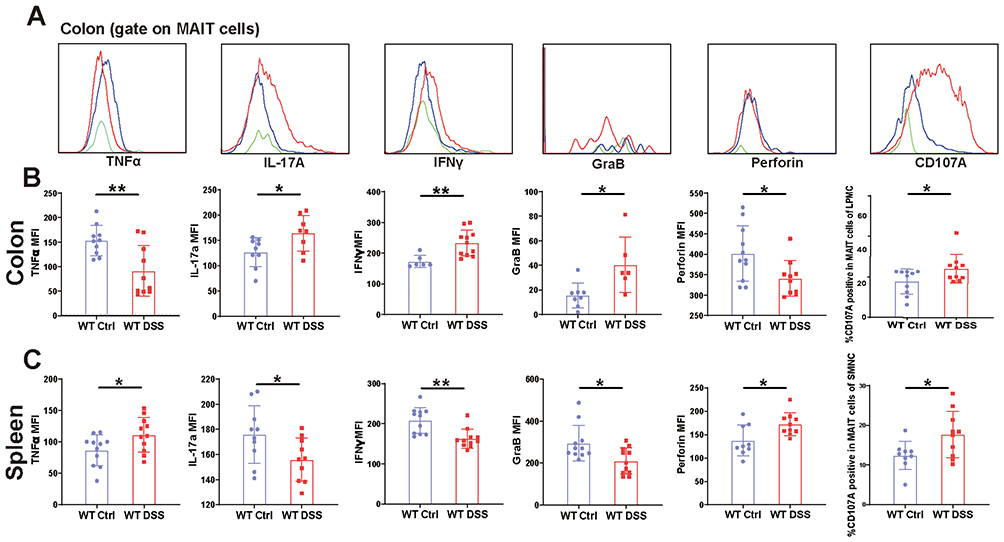 |
Figure 2 MAIT cells exhibit a tissue-compartmentalized functional dichotomy in DSS-induced colitis. (A) Flow cytometric analysis of cytokine production and effector molecule expression in colonic MAIT cells from DSS-treated and control (WT) mice. Expression levels of TNFα, IL-17a, IFNγ, perforin, Granzyme B, and CD107A in colon (B) and splenic (C) MAIT cells. Data are presented as mean ± SD; n = 6–11 mice per group. Statistical significance was determined using unpaired two-tailed Student’s t-test.**p <0.01, * p<0.05. |
This functional dichotomy suggests that the systemic immune response in the spleen may be actively regulated to prevent excessive inflammation, whereas colonic MAIT cells are fully engaged in the local inflammatory battle. The co-expression of IL-17a/IFNγ and GraB/CD107A in the colon indicates that MAIT cells are capable of mounting a multifaceted attack, potentially against invading microbes or damaged epithelium. The concurrent downregulation of Perforin, a pore-forming protein that can cause significant tissue damage, may represent a critical self-regulatory mechanism to limit collateral damage to the already compromised intestinal barrier—a state often associated with activation-induced cell exhaustion.
MAIT Cell Deficiency Aggravates Acute Colitis
To definitively validate the role of MAIT cells in acute colitis, we compared the severity of DSS-induced colitis between WT and MR1−/− mice, which lack MAIT cells. As the colitis is progressing, mice in the DSS group exhibited weight loss, reduced appetite, lethargy, decreased activity, and bloody stools. Strikingly, MR1−/− mice developed significantly more severe disease than their WT counterparts, as evidenced by exacerbated weight loss and higher clinical disease activity scores (Figure 3A and B). This heightened severity was further confirmed by a greater reduction in colon length and (Figure 3C and D), and and histopathological analysis revealed near-total loss of crypt architecture, extensive inflammatory infiltration, and severe edema in MR1−/− DSS mice (Figure 3E).
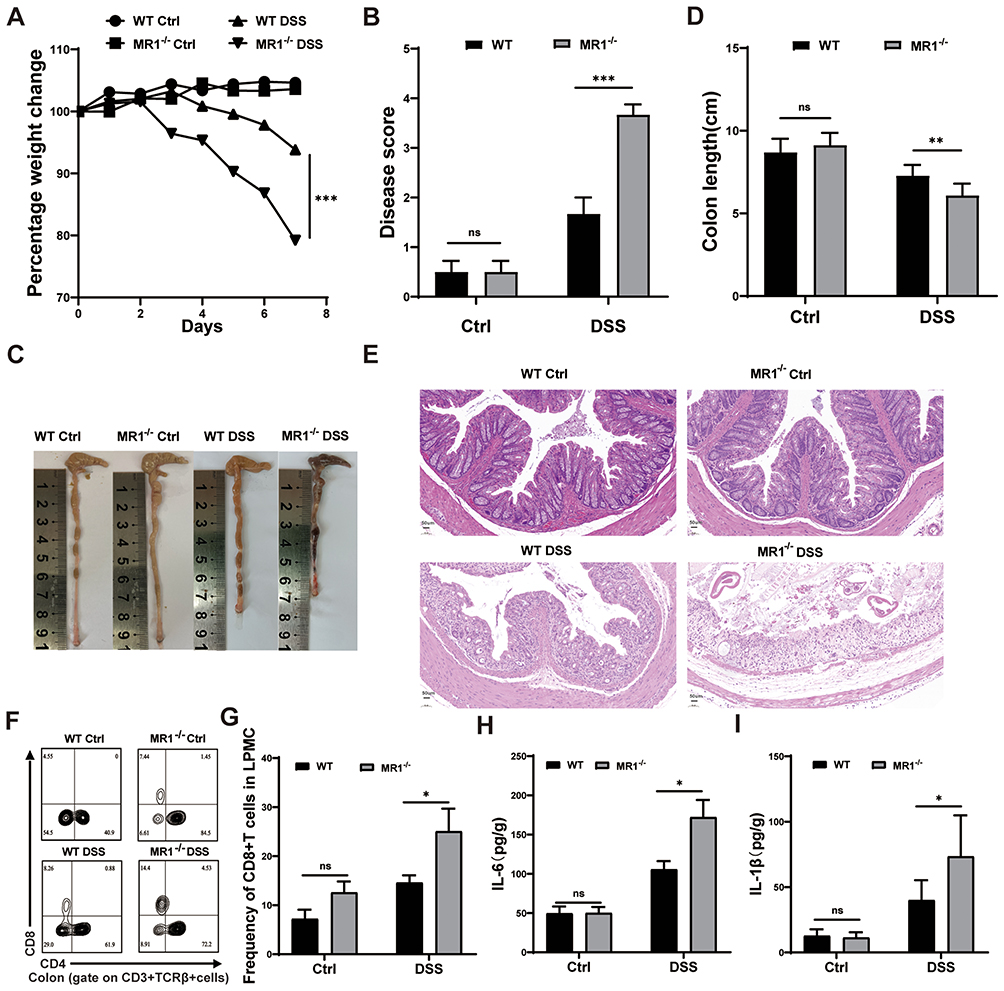 |
Figure 3 MAIT cell deficiency aggravates colonic inflammatory response in acute colitis. (A) Changes in mouse body weight; (B) Performance of mouse disease activity score; (C) Comparison of colonic lengths in each group of mice; (D) Statistical graph of colon lengths in each group of mice; (E) HE staining of the colon of the four groups of mice; (F and G) Flow cytometry analysis of CD8+ T cell content in the colon of the four groups of mice; (H and I) ELISA detection of IL-6 and IL-1β levels in colon tissues of each group of mice. Data are presented as mean ± SD; n = 5–7 mice per group. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test.***p <0.001, **p<0.01, * p<0.05. |
To explore the potential immune mechanisms underlying this exacerbated phenotype, we profiled the colonic immune landscape. Flow cytometric analysis revealed a significant expansion of intestinal CD8+ T cells in MR1−/− DSS mice compared to WT DSS mice (Figure 3F and G). Concomitantly, ELISA assays confirmed that the absence of MAIT cells led to a markedly enhanced inflammatory response, with significantly higher levels of the pro-inflammatory cytokines IL-1β and IL-6 in colonic tissues (Figure 3H and I). These results demonstrate that MAIT cell deficiency not only exacerbates intestinal inflammation but also is associated with a dysregulated adaptive immune response, characterized by the expansion of CD8+ T cells. This suggests that MAIT cells may play a crucial immunomodulatory role in restraining overt CD8+ T cell activation and limiting the production of damaging inflammatory cytokines during colitis.
MAIT Cell Deficiency Worsens Intestinal Barrier Damage
To investigate the potential protective role of MAIT cells in preserving the intestinal barrier, we assessed multiple parameters of barrier integrity in WT and MR1−/− mice with DSS-induced colitis. Immunofluorescence analysis revealed a marked reduction in the expression of the tight junction protein ZO-1 in the colon of MR1−/− DSS mice compared to WT DSS controls (Figure 4A). This observation was confirmed by Western blot, which demonstrated significantly lower protein levels of both ZO-1 and Claudin-1 in MR1−/− DSS mice (Figure 4B and C), indicating compromised tight junction integrity.
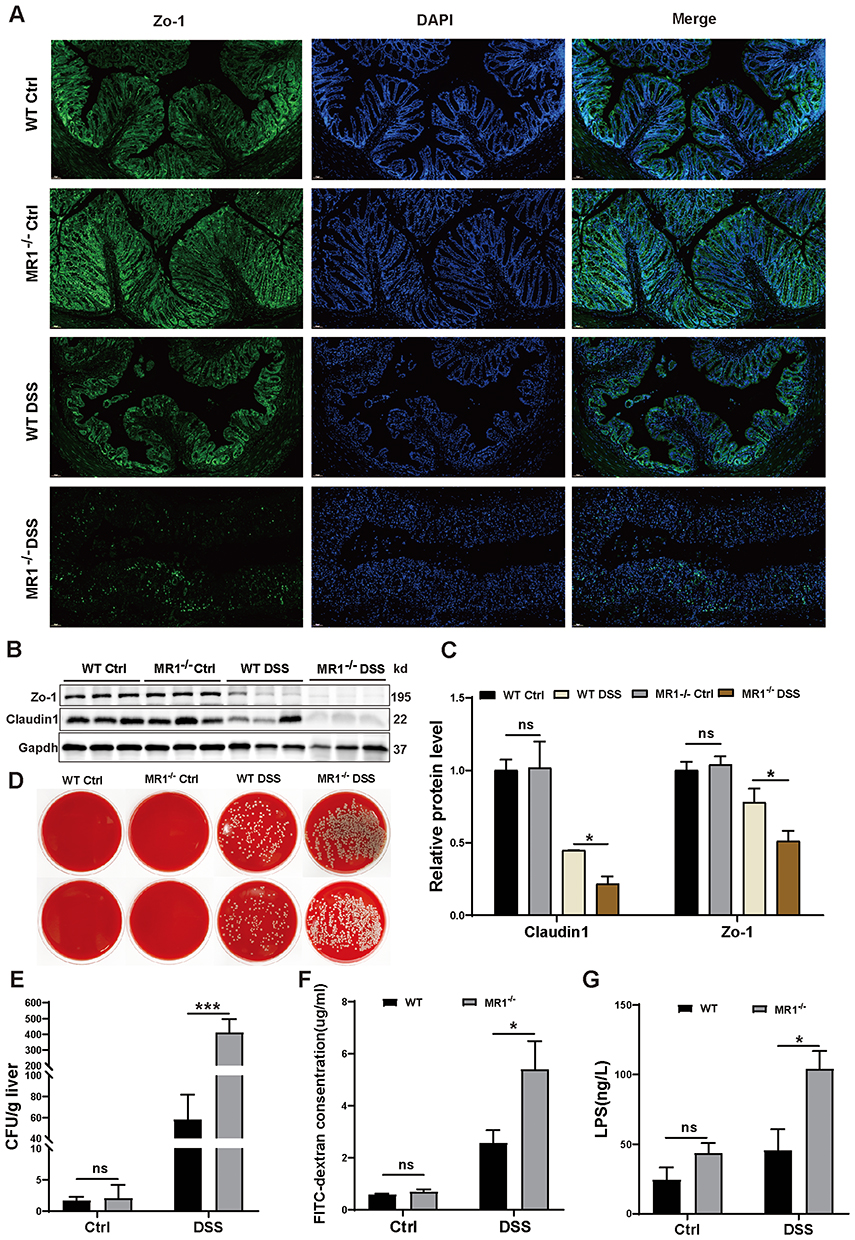 |
Figure 4 Loss of MAIT cells leads to increased intestinal permeability in mice. (A) Changes in ZO-1 expression in the colon of the four groups of mice. ZO-1 (green), DAPI nuclear staining (blue), magnification 20X; (B) Representative Western Blotting images of Zo-1 and Claudin1 in the colon of each group of mice; (C) Statistical graph of Western Blotting; (D) Representative images of colony formation on blood agar plates from liver homogenates of each group of mice; (E) Statistical graph of colony formation counts in each group of mice; (F) Concentration of FITC-dextran in the peripheral blood of each group of mice; (G) ELISA detection of lipopolysaccharide (LPS) levels in the serum of each group of mice. Data are presented as mean ± SD; n = 5–7 mice per group. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test.***p<0.001, **p<0.01, * p<0.05. Abbreviation: ns, not significant. |
We next evaluated functional consequences of this impaired barrier. Assessment of bacterial translocation to the liver showed significantly more bacterial colonies in MR1−/− DSS mice compared to WT DSS mice (Figure 4D and E), while no colonies were detected in mice without DSS treatment. Consistent with this finding, MR1−/− DSS mice exhibited significantly higher serum levels of LPS (Figure 4G), indicating increased systemic translocation of microbial products. Furthermore, the FITC-dextran assay confirmed enhanced intestinal permeability in MR1−/− DSS mice compared to WT DSS controls (Figure 4F). Collectively, these results demonstrate that MAIT cell deficiency leads to more severe impairment of intestinal barrier function in DSS-induced colitis, characterized by loss of tight junction proteins, increased intestinal permeability, and enhanced bacterial translocation. This provides compelling evidence that MAIT cells play a crucial role in maintaining intestinal barrier integrity during inflammatory stress.
MAIT Cell Deficiency Leads to Decreased Expression of IL-22
To elucidate the mechanism by which MAIT cells confer protection in colitis, we focused on IL-22, a cytokine critical for epithelial repair and barrier integrity, previously implicated in MAIT cell function17 and shown to inhibit DSS-induced inflammation.34 We first assessed IL-22 production by MAIT cells directly ex vivo. Flow cytometric analysis revealed a modest but detectable increase in IL-22 mean fluorescence intensity (MFI) within colonic MAIT cells from WT DSS mice compared to controls (Figure 5A and B). To confirm active secretion, MAIT cells were purified from the lamina propria of WT Ctrl and WT DSS mice and cultured for 48 hours. ELISA analysis of the supernatant confirmed that MAIT cells from inflamed colons secreted significantly higher levels of IL-22 (Figure 5C), validating the functional relevance of the ex vivo MFI data. The most pronounced differences, however, were observed at the tissue level. In MR1−/− DSS mice, both IL-22 mRNA and protein levels in colonic tissues were significantly lower than those in WT DSS mice (Figure 5D, E and H).
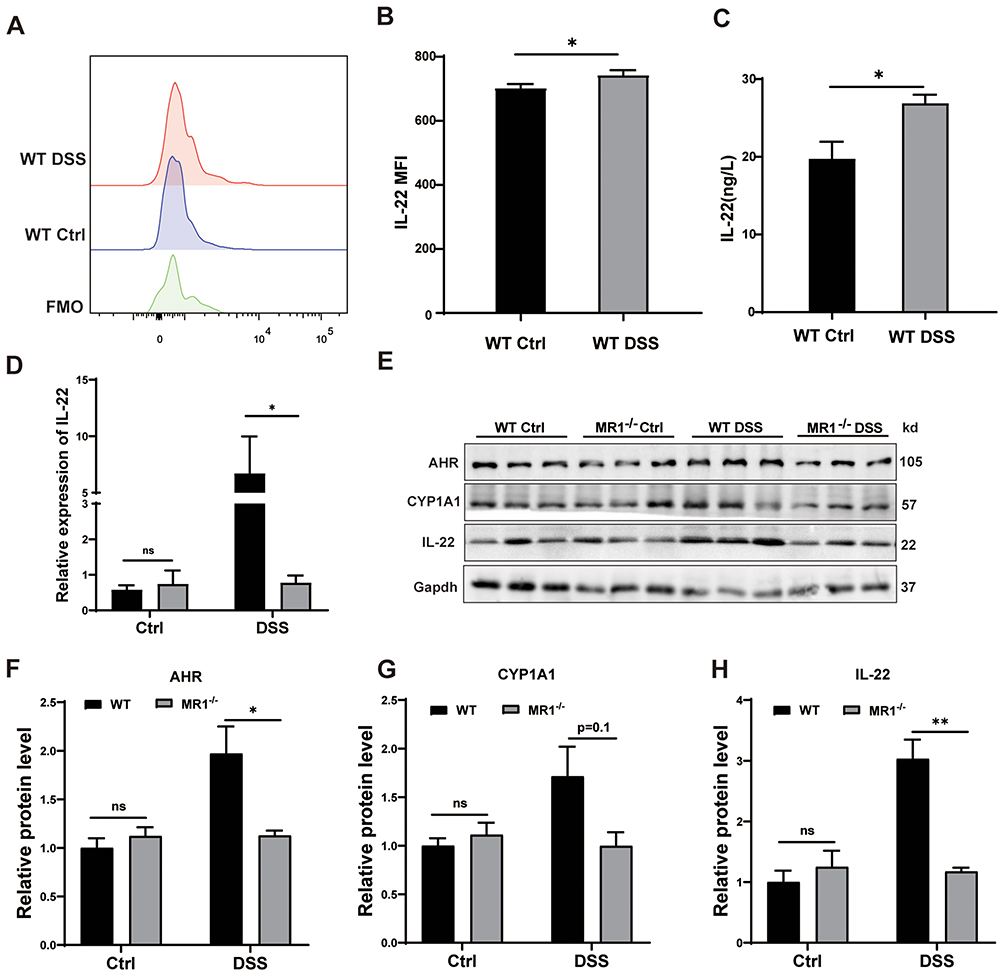 |
Figure 5 MAIT cell deficiency leads to decreased expression of IL-22 in mice. (A) Representative histograms of IL-22 expression on colonic MAIT cells from WT mice treated with DSS (red), WT control (blue), and Fluorescence Minus One (FMO, green); (B) Detection of IL-22 expression of MAIT by flow cytometry; (C) Detection of IL-22 secreted by MAIT cells by ELISA kit; (D) mRNA levels of IL-22 in colon tissues of the four groups of mice; (E) Protein levels of AHR, CYP1A1, and IL-22 in colon tissues of the four groups of mice; (F-H) Statistical graphs of Western Blotting results for AHR, CYP1A1, and IL-22. Data are presented as mean ± SD; n = 6 mice per group. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test.***p<0.001, **p<0.01, *p<0.05. Abbreviation: ns, not significant. |
Since the transcription of IL-22 is potently regulated by the Aryl hydrocarbon Receptor (AHR), we analyzed this pathway. Protein levels of AHR and its downstream target, CYP1A1 (a marker of AHR activation), were both significantly reduced in the colons of MR1−/− DSS mice (Figure 5E–G). Considering the established protective role of IL-22 in intestinal homeostasis, we conclude that the absence of MAIT cells leads to a blunted AHR-IL-22 axis. This deficiency in a key reparative pathway provides a mechanistic explanation for the exacerbated inflammation and barrier damage observed in MAIT cell-deficient mice.
MAIT Cell Deficiency Decreased Secretion of IL-22 Among Various Immune Cells
Given that IL-22 is predominantly produced by immune cells, we next sought to determine whether MAIT cells influence IL-22 production by other key lymphocyte populations in the colonic lamina propria. Strikingly, a comparative analysis revealed that the deficiency of MAIT cells resulted in a significantly impaired capacity for IL-22 production across multiple innate and adaptive immune cell types. Specifically, Th22 cells, Th17 cells, ILC3s, and conventional CD4⁺ T cells all exhibited markedly reduced levels of IL-22 in MR1−/− DSS mice compared to WT DSS controls (Figure 6A–E). Notably, this global deficit in IL-22 secretion occurred independently of major shifts in the abundance of these populations. Compared with WT DSS controls, MR1−/− DSS mice exhibited a significantly decreased proportion of Th22 cells. Although the absolute number of Th22 cells showed a decreasing trend, this difference did not reach statistical significance (Figure 6F and J). The proportion and absolute number of Th17 cells were only marginally affected by the absence of MAIT cells. (Figure 6G and K). Intriguingly, both the proportion and number of ILC3s and CD4⁺ T cells were even increased in MR1−/− DSS mice (Figure 6H, I, L and M), suggesting that the reduction in IL-22 is not due to a loss of these cells but rather a specific defect in their function. These results indicate that MAIT cells are essential for the maintenance of robust IL-22 production within the colonic immune landscape during inflammation. The presence of MAIT cells appears to license or enable multiple other lymphocyte populations to secrete this critical reparative cytokine, potentially through direct cell-cell interactions or the provision of soluble factors. The specific regulatory mechanisms underlying this crosstalk, however, remain a key question for future investigation and represent a limitation of the present study.
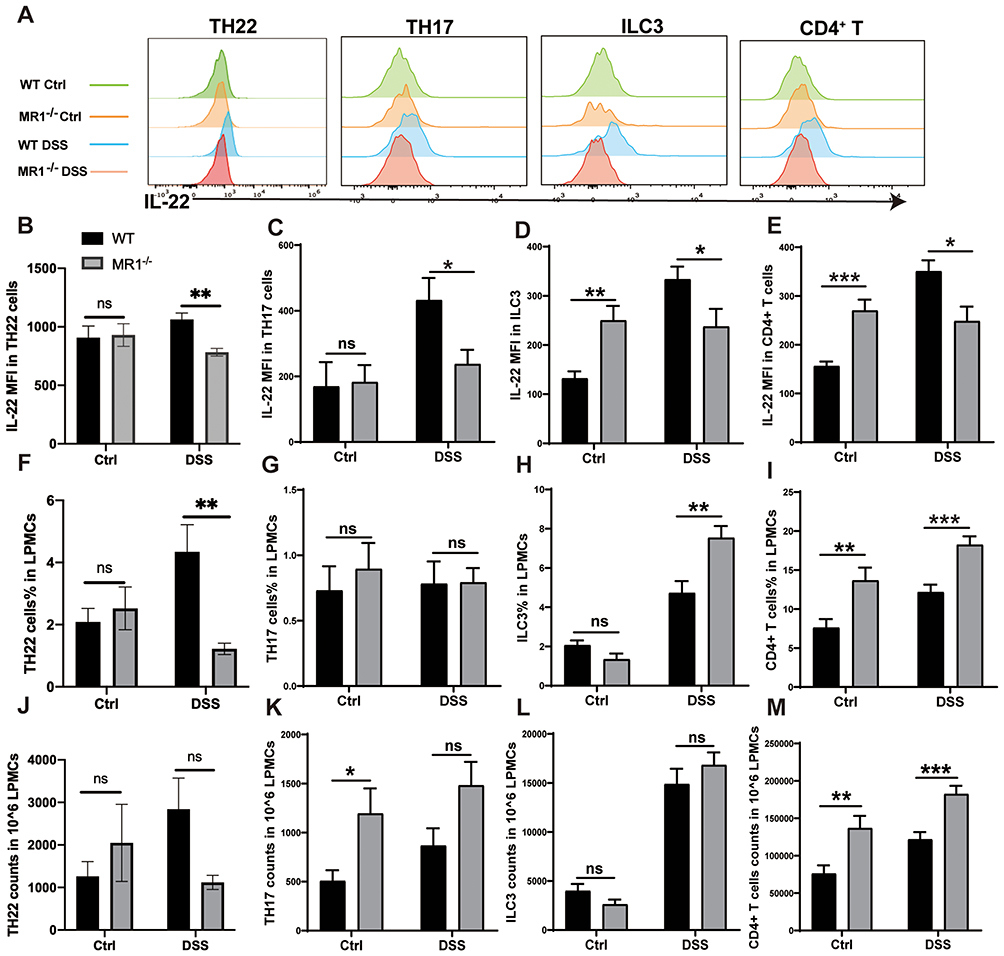 |
Figure 6 MAIT cell deficiency leads to decreased secretion of IL-22 by various immune cells. (A) Flow cytometry histograms showing IL-22 secretion by different immune cells; (B–E) Statistical analysis graphs of IL-22 secretion by different immune cells; (F–I) Proportions of different immune cells in the colonic lamina propria; (J–M) Absolute numbers of different immune cells in the colonic lamina propria. Data are presented as mean ± SD; n = 6 mice per group. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test.***p<0.001, **p<0.05, * p<0.01. |
Supplementing IL-22 Alleviates Acute Colitis in MAIT Cell Deficiency Mice
To directly investigate whether the protective role of MAIT cells is mediated primarily through IL-22, we administered recombinant IL-22 to MR1−/− mice with DSS-induced colitis. Exogenous IL-22 supplementation led to a significant but partial amelioration of the disease phenotype. Treated mice (MR1−/− DSS+IL-22) exhibited reduced weight loss, less colon shortening, and improved histological scores compared to untreated MR1−/− DSS controls (Figure 7A–F). Furthermore, IL-22 administration enhanced the expression of the tight junction proteins ZO-1 and claudin-1 (Figure 7G and H), and increased protein levels of AHR and its target CYP1A1 (Figure 7I), indicating a partial restoration of barrier integrity and associated signaling pathways. These results demonstrate that IL-22 is a key, but not sole, mediator of MAIT cell-dependent protection. The fact that exogenous IL-22 did not fully reverse the severe colitis phenotype of MR1−/− mice to WT levels indicates that MAIT cells contribute to intestinal homeostasis through additional mechanisms beyond IL-22. This partial rescue suggests that other MAIT-derived effector molecules are likely involved. These could include direct cytolytic activity via granzyme B and perforin, which can eliminate infected or stressed epithelial cells, or the secretion of other immunomodulatory cytokines such as IFN-γ, IL-17, or TNF-α. Additionally, MAIT cells may engage in critical cell-cell interactions that are not replicated by cytokine administration alone, such as modulating the function of antigen-presenting cells or other T cell subsets through surface receptors. The production of tissue repair factors like amphiregulin (AREG) represents another potential pathway. Therefore, while IL-22 is a crucial component, the complete protective function of MAIT cells appears to be multifaceted, involving a synergy of IL-22-dependent and IL-22-independent mechanisms.
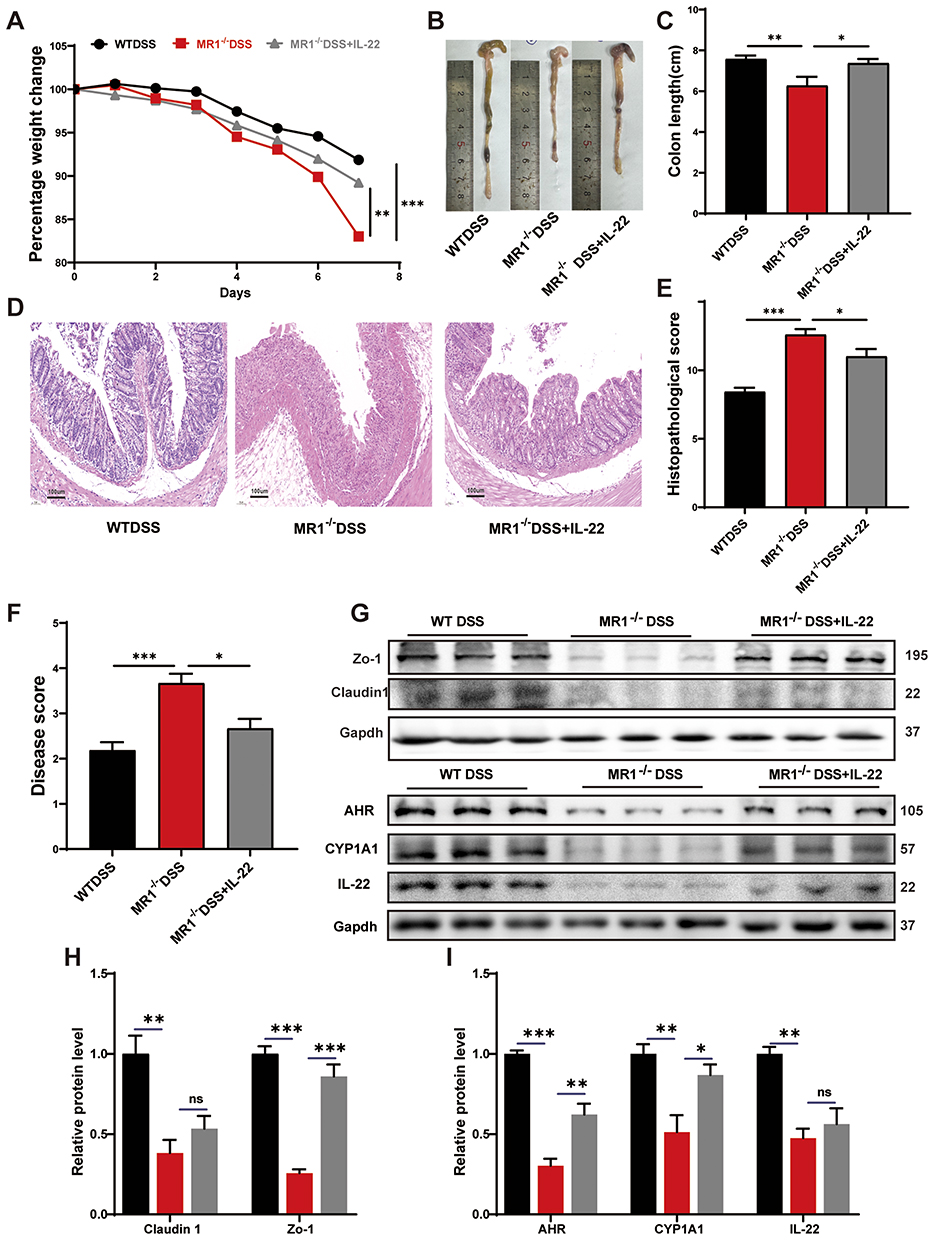 |
Figure 7 Supplementing IL-22 alleviates colitis caused by MAIT cell deficiency. (A) Changes in mouse body weight; (B) Comparison of colonic lengths in each group of mice; (C) Statistical graph of colon lengths in each group of mice; (D) Staining situation of colon tissues in each group of mice; (E) Statistical graph of pathological scores of colon tissues in each group of mice; (F) Performance of mouse disease activity score; (G–I) Representative images and statistical graphs of Western Blotting in colon tissues of each group of mice. Data are presented as mean ± SD; n = 6 mice per group. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test.***p<0.001, **p<0.01, * p<0.05. |
MAIT Cell–Derived CCL2 Promotes IL-22 Production During DSS-Induced Colitis
To further investigate the mechanism underlying the reduced IL-22 production observed in MAIT-deficient mice, we analyzed a publicly available single-cell RNA-seq dataset of acute colitis (GSE264408). MAIT cells were identified and their transcriptional profiles were compared between WT and DSS-treated mice (Figure 8A–C). Differential expression analysis revealed several upregulated genes encoding secreted proteins in MAIT cells during DSS-induced colitis, including Il1b, Ccl2, Saa3, Serpina3n, and Igfbp4 (Figure 8D). Among these candidates, Ccl2 showed the most prominent induction pattern and was therefore selected for further investigation, given its known role in immune cell recruitment and intercellular communication. To further validate this finding, we next assessed CCL2 expression in MAIT cells by flow cytometry and found that the proportion of CCL2-expressing MAIT cells was significantly increased in WT DSS mice compared with WT controls (Figure 8E). These results suggested that CCL2 may represent a MAIT-associated soluble mediator induced during intestinal inflammation.
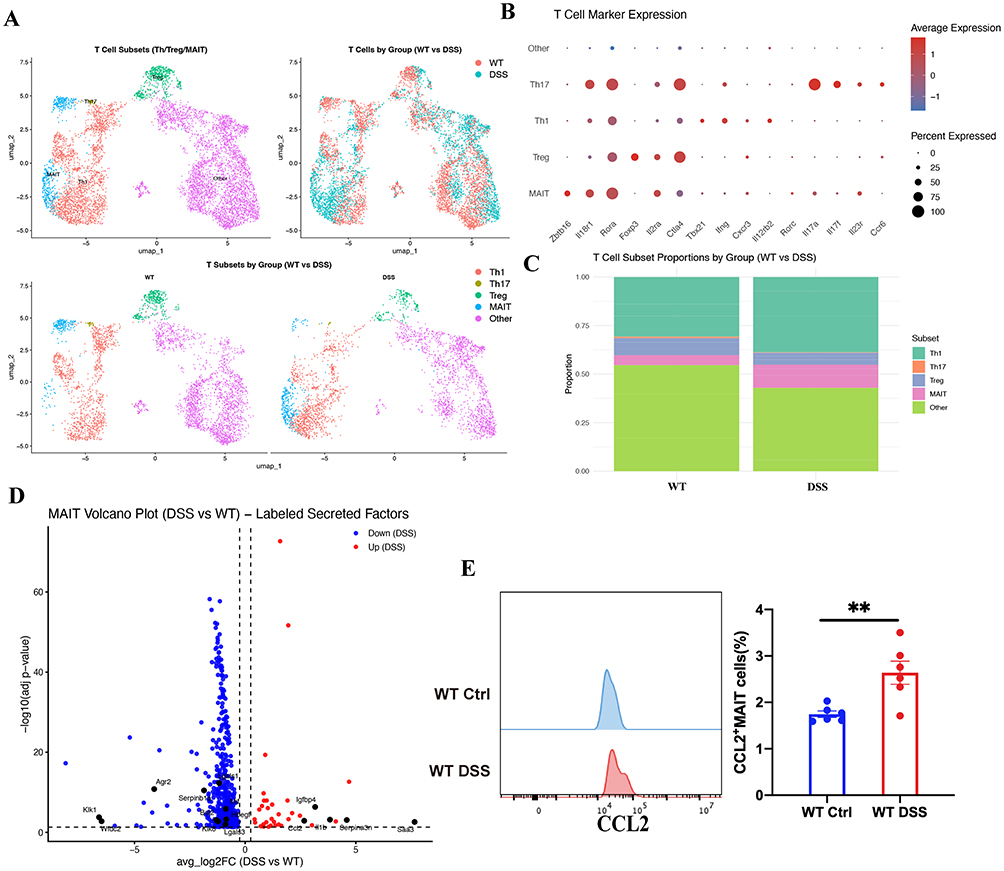 |
Figure 8 Identification of CCL2 as a MAIT-associated secreted factor during DSS-induced colitis. (A–C) Identification and annotation of MAIT cells from single-cell RNA-seq dataset (GSE264408), and comparison of transcriptional profiles between WT and DSS-treated mice; (D) Differentially expressed genes encoding secreted proteins in MAIT cells during DSS-induced colitis; (E) Flow cytometric analysis of CCL2 expression in colonic MAIT cells from WT control and WT DSS mice, with quantification of the proportion of CCL2⁺ MAIT cells. Data are presented as mean ± SD; n = 6 mice per group. Statistical significance was determined using unpaired two-tailed Student’s t-test. **p<0.01. |
We then asked whether MAIT-derived CCL2 contributes to the regulation of IL-22 production in vivo. Recombinant CCL2 was administered to MR1−/− mice during DSS-induced colitis. Flow cytometric analysis showed that CCL2 supplementation increased IL-22 production in multiple immune cell populations, including Th22 cells, Th17 cells, ILC3, and CD4+ T cells (Figure 9). Having observed that CCL2 restored IL-22 production across these immune cell populations, we next examined whether this effect was accompanied by improvement of colitis severity. Compared with untreated MR1−/− DSS mice, CCL2-treated mice showed less body weight loss during DSS exposure (Figure 10A). Consistently, colon shortening was attenuated following CCL2 treatment, as shown by both representative images and quantitative analysis of colon length (Figure 10B). Histological examination further supported a protective effect of CCL2. Colonic tissues from MR1−/− DSS mice displayed marked inflammatory cell infiltration, epithelial disruption, and mucosal injury, whereas these pathological changes were alleviated in the MR1−/− DSS+CCL2 group (Figure 10C). Accordingly, histopathological scores were significantly reduced after CCL2 supplementation (Figure 10D) and disease activity index (DAI) scores were also improved (Figure 10E). At the molecular level, Western blot analysis showed that CCL2 treatment increased the expression of the tight junction proteins ZO-1 and Claudin-1,along with AHR, CYP1A1, and IL-22 in colonic tissues (Figure 10F). These changes are consistent with restoration of epithelial barrier–associated signaling. Taken together, these findings indicate that CCL2, induced in MAIT cells during DSS colitis, promotes IL-22 production across multiple immune cell populations and is associated with improvement of epithelial barrier integrity and colitis severity in MR1−/− mice.
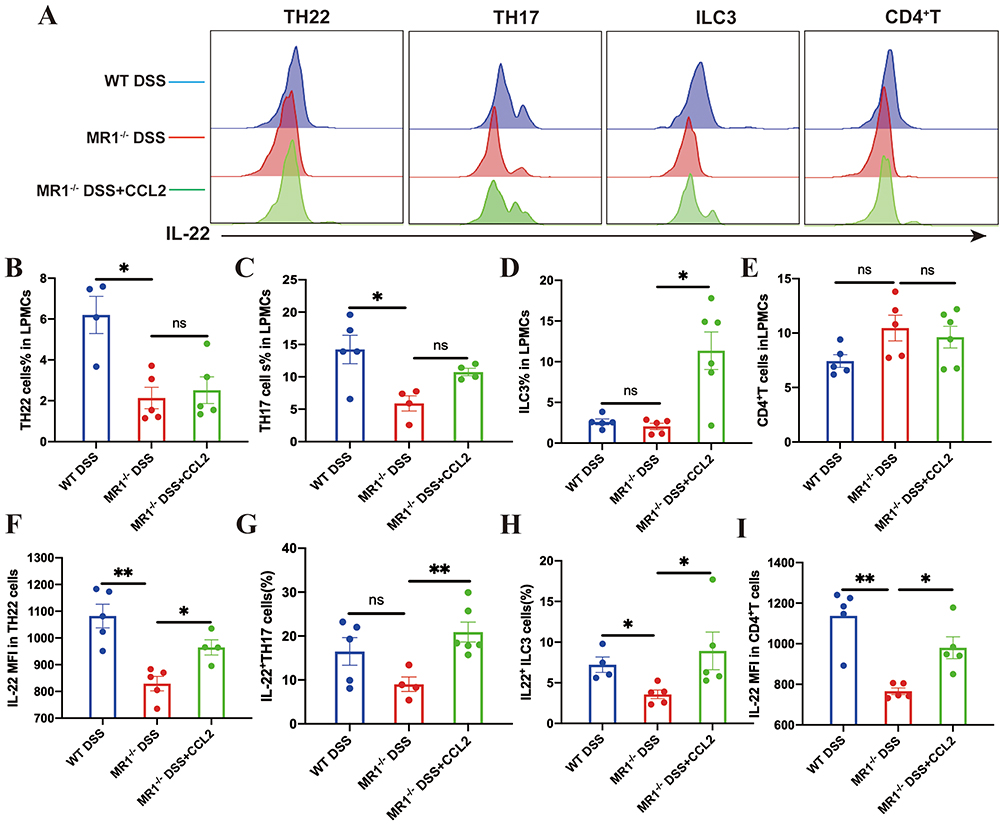 |
Figure 9 CCL2 supplementation restores IL-22 responses in multiple immune cell populations in MR1−/− mice. (A) Representative flow cytometry histograms of intracellular IL-22 expression in Th22 cells, Th17 cells, ILC3, and CD4⁺ T cells isolated from colonic lamina propria mononuclear cells; (B–E) Frequencies of Th22 cells, Th17 cells, ILC3, and CD4⁺ T cells among colonic lamina propria mononuclear cells in the indicated groups; (F–I) Quantification of IL-22 expression in Th22 cells, Th17 cells, ILC3, and CD4⁺ T cells across experimental groups. Data are presented as mean ± SD; n = 4–6 mice per group. Statistical significance was determined using one-way ANOVA followed by Tukey’s post hoc test. **p<0.01, *p<0.05. Abbreviation: ns, not significant. |
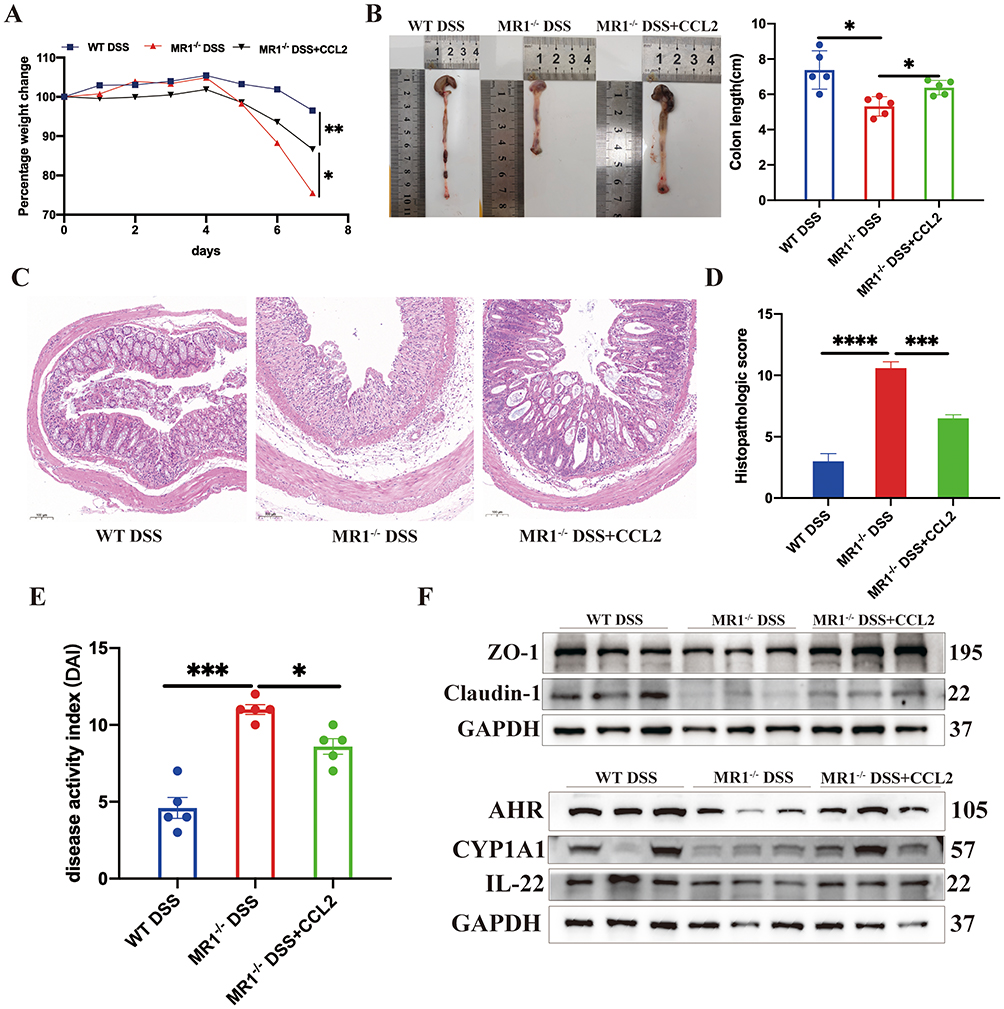 |
Figure 10 CCL2 supplementation ameliorates colitis and restores epithelial barrier–associated signaling in MR1−/− mice. (A) Changes in body weight during DSS-induced colitis; (B) Representative images and quantitative analysis of colon length; (C) Representative hematoxylin and eosin (H&E) staining of colonic tissues; (D) Histopathological scores of colonic inflammation; (E) Disease activity index (DAI) scores; (F) Representative Western blot images showing expression of ZO-1, Claudin-1, AHR, CYP1A1, and IL-22 in colonic tissues. Data are presented as mean ± SD; n = 5 mice per group. Statistical significance was determined using one-way ANOVA followed by Tukey’s post hoc test. ****p<0.0001, ***p<0.001, **p<0.01, *p<0.05. |
Discussions
MAIT cells are increasingly recognized as microbiota-responsive sentinels at mucosal surfaces,35 yet their precise contribution to intestinal barrier maintenance during acute inflammatory injury remains incompletely defined. Previous studies have shown that MAIT cells participate in antimicrobial defense, cytokine production, and tissue repair, and early adoptive transfer experiments suggested a protective role in TNBS-induced colitis.9,13,17,36 In line with these observations, our findings show that the absence of MAIT cells exacerbates disease severity in the DSS model, as evidenced by aggravated weight loss, rectal bleeding, colon shortening, and elevated levels of pro-inflammatory cytokines such as IL-6 and IL-1β in colonic tissues. These results support a protective role for MAIT cells in acute colitis and further extend current understanding of their function in maintaining mucosal homeostasis.
A central observation of this work is that MAIT cell deficiency leads to marked impairment of intestinal barrier function. MR1−/− mice exhibited reduced expression of tight junction proteins ZO-1 and Claudin-1, increased intestinal permeability, and enhanced bacterial translocation to the liver. These defects underscore the importance of MAIT cells in preserving epithelial integrity under inflammatory stress. Given that DSS-induced colitis is primarily driven by epithelial injury and barrier breakdown, our data position MAIT cells as key immune components linking microbial sensing to epithelial repair mechanisms.
Although MAIT cells displayed a mixed effector phenotype during colitis—producing IL-17A and IFN-γ alongside cytotoxic mediators15—our data indicate that their protective effect in this context is closely associated with regulation of IL-22. This point merits cautious interpretation, because both MAIT cells and IL-22 are increasingly recognized as context-dependent mediators whose functions may vary with disease stage and inflammatory milieu.28 IL-22 is a well-established epithelial-protective cytokine that promotes tight junction assembly, epithelial proliferation, and antimicrobial peptide production.24,37,38 Consistent with this role, we observed a profound reduction of IL-22 expression in colonic tissues of MR1−/− DSS mice. Importantly, this defect was not restricted to MAIT cells themselves but extended across multiple immune compartments, including Th17 cells, Th22 cells, ILC3s, and conventional CD4⁺ T cells. Notably, these changes occurred despite preserved or even increased frequencies of several IL-22–producing populations, indicating a functional impairment rather than numerical loss.
This global defect in IL-22 production highlights a previously unappreciated immunoregulatory role of MAIT cells: rather than serving solely as direct cytokine producers, MAIT cells function as coordinators that license IL-22 competence across the intestinal immune network. Supporting this concept, exogenous IL-22 administration partially ameliorated disease severity and barrier disruption in MR1−/− mice, confirming IL-22 as a key downstream effector of MAIT cell–mediated protection. The rescue, however, was incomplete, suggesting that MAIT cells contribute to intestinal homeostasis through both IL-22-dependent and IL-22-independent mechanisms, potentially including cytotoxic clearance of damaged cells and modulation of other immune subsets.
The integrity of the intestinal mucosal barrier is critical for preventing systemic dissemination of commensal bacteria and their products. Existing studies suggest that MAIT cells actively monitor intestinal dysbiosis to provide host protection.9,13,17 In the context of acute DSS-induced injury, our findings identify the MAIT–CCL2–IL-22 axis as a key mechanism through which these cells respond to microbial cues to reinforce epithelial integrity. At the same time, the DSS model is inherently biased toward epithelial damage and barrier repair. Therefore, while our data support a protective role for MAIT cells in acute injury, the function of this axis should be further evaluated in T-cell-driven or chronic models to define more fully the context-dependent role of MAIT cells in intestinal inflammation. Consistent with a barrier-protective function, MAIT cell deficiency resulted in increased bacterial translocation to the liver and elevated systemic levels of LPS and FITC-dextran, indicating heightened intestinal permeability.
IL-22 plays a critical role in regulating epithelial cell homeostasis and various aspects of the intestinal barrier.39 Studies indicate that IL-22 directly induces the expression of mucin in mucosal epithelial cells through the STAT3-dependent signaling pathway,38 and contributes to the clearance of intestinal pathogens,40 promotes tight junction expression, and enhances epithelial renewal, as evidenced by increased Ki-67-positive cells in IL-22 transgenic mouse intestinal crypts.41 Experimental approaches such as IL-22 microinjection and DNA vaccines containing the IL-22 locus have shown promise in reducing inflammatory infiltration in intestinal tissues in disease models.42 In addition, anti-TNF therapy has been reported to enhance IL-22 secretion in an AHR-dependent manner and to promote Th22 differentiation, leading to increased IL-22 production by CD4⁺ T cells.25 Innate lymphoid cells 3 (ILC3) also contribute to post-inflammatory epithelial barrier repair through IL-22 secretion.43,44 In our study, MR1−/− DSS mice exhibited impaired IL-22 production not only in MAIT cells but also in Th17 cells, Th22 cells, ILC3s, and CD4⁺ T cells, suggesting that MAIT cells exert a broader regulatory effect on IL-22-dependent mucosal immunity. Particularly, the reduction in Th22-derived IL-22 occurred without an obvious loss of Th22 cell abundance, indicating that MAIT cells primarily affect the functional capacity of these cells.
Although IL-22 is widely recognized for its epithelial-protective properties, excessive or prolonged IL-22 signaling may promote inflammation under certain conditions.45,46 In our acute DSS model, the partial rescue observed after recombinant IL-22 administration supports a protective role for IL-22 in acute epithelial injury, but does not imply that IL-22 is uniformly beneficial in all forms of colitis. Thus, both the action of IL-22 and the contribution of MAIT cells should be interpreted in a context-dependent manner.
Our findings support a model in which MAIT cells promote IL-22 production across multiple immune compartments through the secretion of CCL2 during acute colitis. Rather than representing a nonspecific consequence of intestinal inflammation, the reduction of IL-22 observed in MAIT-deficient mice can be mechanistically linked to impaired CCL2 signaling. This interpretation is supported by our rescue experiments, in which exogenous CCL2 partially restored IL-22 production in Th17, Th22, ILC3, and CD4⁺ T cells, accompanied by enhanced expression of epithelial barrier–associated proteins and attenuation of colonic tissue damage. Importantly, these results position CCL2 as a MAIT-associated soluble mediator that coordinates IL-22–dependent barrier-protective responses in DSS-induced colitis. Notably, this regulatory effect extends to Th22 cells, which are a principal source of IL-22 in the intestinal mucosa. Our data reveal that the absence of MAIT cells leads to a functional defect in Th22 cells rather than a loss of their population size, characterized by diminished IL-22 secretion. Mechanistically, MAIT cells appear to support the responsiveness of these cells to aryl hydrocarbon receptor (AHR) signaling. AHR is a ligand-activated transcription factor essential for IL-22 production, and our observation that CCL2 supplementation restores colonic AHR and CYP1A1 expression suggests that MAIT cells, via CCL2, help maintain an AHR-dependent effector program in Th22 and other lymphocytes. Although CCL2 is unlikely to be the sole mediator of MAIT cell function, the present data support a MAIT–CCL2–AHR–IL-22 regulatory axis contributing to mucosal homeostasis under acute inflammatory conditions. IL-22 function may also be modulated by endogenous antagonists such as IL-22 binding protein (IL-22BP), which was not directly addressed in the present study and warrants further investigation.
In addition to shaping IL-22 responses in immune cell subsets, the MAIT–CCL2–AHR axis may also contribute to epithelial protection by influencing pathways involved in barrier repair and oxidative stress control. Emerging evidence suggests functional crosstalk between IL-22 and AHR, linking immune-derived cytokine signals to microbial metabolite–driven programs that maintain mucosal homeostasis. In this context, the restoration of colonic AHR and CYP1A1 expression after CCL2 supplementation is consistent with activation of an epithelial protective program associated with IL-22/AHR signaling. AHR has also been implicated in the modulation of NF-κB–associated inflammatory responses and oxidative stress, including ROS production and detoxification pathways.47 Although the precise contribution of STAT3, NF-κB, and ROS signaling downstream of IL-22 was not directly examined in the present study, these pathways may provide an additional mechanistic link between MAIT cell–dependent immune regulation and epithelial repair during acute colitis.
In conclusion, this study identifies a MAIT–CCL2–IL-22 regulatory axis that contributes to epithelial barrier protection during acute colitis. Beyond its mechanistic significance, dysregulation of MAIT cells and IL-22 responses may hold diagnostic value in clinical IBD, as alterations in this axis could reflect disease activity, immune imbalance, and epithelial integrity in patients. From a therapeutic perspective, strategies aimed at restoring MAIT cell–associated IL-22 signaling may offer potential benefits for mucosal healing. In this context, emerging nanomedicine-based delivery systems provide a promising avenue to enhance the precision and safety of immunomodulatory interventions. Targeted or localized delivery of IL-22–related molecules, or agents that modulate MAIT cell function, may improve therapeutic efficacy while limiting off-target effects. Although further investigation is required, the integration of immune pathway modulation with advanced delivery technologies represents a forward-looking direction for the development of next-generation IBD therapies.
Data Sharing Statement
The data generated during this study can be obtained from the corresponding author upon request.
Ethics Approval and Consent to Participate
All animal experiments were conducted under ethical conditions and approved by the Committee on the Ethics of Animal Experiments of Zhongnan Hospital of Wuhan University (authorization number 2021088).
Acknowledgments
We thank Jamie Rossjohn for the gift of the human MR1 tetramers produced by the NIH Tetramer Core Facility. We thank Professor Ma Xiong for donating MR1−/−mice.
Author Contributions
Yanan Peng, Chunlan Zheng, and Youwei Wang share the first authorship. Qiu Zhao and Lan Liu share the corresponding authorship. QZ; Conceptualization, Methodology, Writing – review & editing, Supervision, Validation, Project administration. LL; Conceptualization, Methodology, Writing – review & editing, Validation, Project administration. YP; Conceptualization, Methodology, Visualization, Writing – original draft, Validation, Project administration. CZ; Data curation, Investigation, Writing – original draft, Validation, Project administration. YW; Data curation, Methodology, Investigation, Validation, Project administration. YS; Data curation, Investigation,Validation, Project administration. FH; Data curation, Investigation, Validation, Project administration.
All authors took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (Grant/Award Number: 82303248); Key Project of Discipline and Platform Construction of Zhongnan Hospital of Wuhan University (Grant No. PTXM2023012 (Qiu Zhao)); National Key Clinical Specialty Construction Project (Grant No. GJZD2023002 (Qiu Zhao)); Natural Science Foundation of Hubei Province (Grant/Award Number: 2023AFB203).
Disclosure
All contributing authors declare no competing financial or personal interests.
References
1. Kaplan GG, Ng SC. Understanding and preventing the global increase of inflammatory bowel disease. Gastroenterology. 2017;152(2):313–321.e312. doi:10.1053/j.gastro.2016.10.020
2. Rosen MJ, Dhawan A, Saeed SA. Inflammatory bowel disease in children and adolescents. JAMA Pediatr. 2015;169(11):1053–21. doi:10.1001/jamapediatrics.2015.1982
3. Furey TS, Sethupathy P, Sheikh SZ. Redefining the IBDs using genome-scale molecular phenotyping. Nat Rev Gastroenterol Hepatol. 2019;16(5):296–311. doi:10.1038/s41575-019-0118-x
4. Adolph TE, Meyer M, Schwärzler J, Mayr L, Grabherr F, Tilg H. The metabolic nature of inflammatory bowel diseases. Nat Rev Gastroenterol Hepatol. 2022;19(12):753–767. doi:10.1038/s41575-022-00658-y
5. Linehan JL, Harrison OJ, Han SJ, et al. Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell. 2018;172(4):784–796.e718. doi:10.1016/j.cell.2017.12.033
6. Salou M, Legoux F, Lantz O. MAIT cell development in mice and humans. Mol Immunol. 2021;130:31–36. doi:10.1016/j.molimm.2020.12.003
7. Kjer-Nielsen L, Corbett AJ, Chen Z, et al. An overview on the identification of MAIT cell antigens. Immunol Cell Biol. 2018;96(6):573–587. doi:10.1111/imcb.12057
8. Constantinides MG, Belkaid Y. Early-life imprinting of unconventional T cells and tissue homeostasis. Science. 2021;374(6573):eabf0095. doi:10.1126/science.abf0095
9. Constantinides MG, Link VM, Tamoutounour S, et al. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science. 2019;366(6464). doi:10.1126/science.aax6624
10. Treiner E, Duban L, Bahram S, et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature. 2003;422(6928):164–169. doi:10.1038/nature01433
11. Koay HF, Gherardin NA, Enders A, et al. A three-stage intrathymic development pathway for the mucosal-associated invariant T cell lineage. Nat Immunol. 2016;17(11):1300–1311. doi:10.1038/ni.3565
12. Legoux F, Salou M, Lantz O. MAIT cell development and functions: the microbial connection. Immunity. 2020;53(4):710–723. doi:10.1016/j.immuni.2020.09.009
13. Leng T, Akther HD, Hackstein CP, et al. TCR and inflammatory signals tune human MAIT cells to exert specific tissue repair and effector functions. Cell Rep. 2019;28(12):3077–3091.e3075. doi:10.1016/j.celrep.2019.08.050
14. Hinks TSC, Zhang XW. MAIT cell activation and functions. Front Immunol. 2020;11:1014. doi:10.3389/fimmu.2020.01014
15. Loh L, Orlicky DJ, Spengler A, et al. MAIT cells exacerbate colonic inflammation in a genetically diverse murine model of spontaneous colitis. Mucosal Immunol. 2025;18(4):958–972. doi:10.1016/j.mucimm.2025.05.006
16. Jabeen MF, Hinks TSC. MAIT cells and the microbiome. Front Immunol. 2023;14:1127588. doi:10.3389/fimmu.2023.1127588
17. El Morr Y, Fürstenheim M, Mestdagh M, et al. MAIT cells monitor intestinal dysbiosis and contribute to host protection during colitis. Sci Immunol. 2024;9(96):eadi8954. doi:10.1126/sciimmunol.adi8954
18. Tominaga K, Yamagiwa S, Setsu T, et al. Possible involvement of mucosal-associated invariant T cells in the progression of inflammatory bowel diseases. Biomed Res. 2017;38(2):111–121. doi:10.2220/biomedres.38.111
19. Ju JK, Cho YN, Park KJ, et al. Activation, deficiency, and reduced IFN-γ production of mucosal-associated invariant T cells in patients with inflammatory bowel disease. J Innate Immun. 2020;12(5):422–434. doi:10.1159/000507931
20. Wu Z, Chen X, Han F, Leeansyah E. MAIT cell homing in intestinal homeostasis and inflammation. Sci Adv. 2025;11(6):eadu4172. doi:10.1126/sciadv.adu4172
21. Lei P, Yu H, Ma J, et al. Cell membrane nanomaterials composed of phospholipids and glycoproteins for drug delivery in inflammatory bowel disease: a review. Int J Biol Macromol. 2023;249:126000. doi:10.1016/j.ijbiomac.2023.126000
22. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21(2):241–254. doi:10.1016/j.immuni.2004.07.007
23. Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discov. 2014;13(1):21–38. doi:10.1038/nrd4176
24. Patnaude L, Mayo M, Mario R, et al. Mechanisms and regulation of IL-22-mediated intestinal epithelial homeostasis and repair. Life Sci. 2021;271:119195. doi:10.1016/j.lfs.2021.119195
25. Chen J, Yao J. Th22 cells and the intestinal mucosal barrier. Front Immunol. 2023;14:1221068. doi:10.3389/fimmu.2023.1221068
26. Basu R, O’Quinn DB, Silberger DJ, et al. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity. 2012;37(6):1061–1075. doi:10.1016/j.immuni.2012.08.024
27. Liang SC, Tan XY, Luxenberg DP, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203(10):2271–2279. doi:10.1084/jem.20061308
28. Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J Exp Med. 2010;207(6):1293–1305. doi:10.1084/jem.20092054
29. Zhao LP, Wu J, Quan W, et al. DSS-induced colitis activates the kynurenine pathway in serum and brain by affecting IDO-1 and gut microbiota. Front Immunol. 2022;13:1089200. doi:10.3389/fimmu.2022.1089200
30. Xu Y, Chen L, Hu X, et al. Brusatol ameliorates intestinal mucosal injury in ulcerative colitis via activating IL-22/STAT3 pathway. Int Immunopharmacol. 2025;153:114482. doi:10.1016/j.intimp.2025.114482
31. Maharshak N, Hart G, Ron E, et al. CCL2 (pM levels) as a therapeutic agent in Inflammatory Bowel Disease models in mice. Inflamm Bowel Dis. 2010;16(9):1496–1504. doi:10.1002/ibd.21254
32. Neurath MF, Fuss I, Kelsall BL, Stüber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182(5):1281–1290. doi:10.1084/jem.182.5.1281
33. Paiva RA, Salou M. MAIT and iNKT cells in tissue homeostasis and repair. Immunobiology. 2025;230(3):152917. doi:10.1016/j.imbio.2025.152917
34. Monteleone I, Rizzo A, Sarra M, et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. 2011;141(1):237–248,248.e231. doi:10.1053/j.gastro.2011.04.007
35. Godfrey DI, Koay HF, McCluskey J, Gherardin NA. The biology and functional importance of MAIT cells. Nat Immunol. 2019;20(9):1110–1128. doi:10.1038/s41590-019-0444-8
36. Chiba A, Murayama G, Miyake S. Characteristics of mucosal-associated invariant T cells and their roles in immune diseases. Int Immunol. 2021;33(12):775–780. doi:10.1093/intimm/dxab070
37. Keir M, Yi Y, Lu T, Ghilardi N. The role of IL-22 in intestinal health and disease. J Exp Med. 2020;217(3):e20192195. doi:10.1084/jem.20192195
38. Sugimoto K, Ogawa A, Mizoguchi E, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118(2):534–544. doi:10.1172/JCI33194
39. Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: pathophysiological basis for therapy. J Hepatol. 2020;72(3):558–577. doi:10.1016/j.jhep.2019.10.003
40. Stefanich EG, Rae J, Sukumaran S, et al. Pre-clinical and translational pharmacology of a human interleukin-22 IgG fusion protein for potential treatment of infectious or inflammatory diseases. Biochem Pharmacol. 2018;152:224–235. doi:10.1016/j.bcp.2018.03.031
41. Zwarycz B, Gracz AD, Rivera KR, et al. IL22 inhibits epithelial stem cell expansion in an ileal organoid model. Cell Mol Gastroenterol Hepatol. 2019;7(1):1–17. doi:10.1016/j.jcmgh.2018.06.008
42. Barmeyer C, Fromm M, Schulzke JD. Active and passive involvement of claudins in the pathophysiology of intestinal inflammatory diseases. Pflugers Arch. 2017;469(1):15–26. doi:10.1007/s00424-016-1914-6
43. Morel L, Domingues O, Zimmer J, Michel T. Revisiting the role of neurotrophic factors in inflammation. Cells. 2020;9(4):865. doi:10.3390/cells9040865
44. Ibiza S, García-Cassani B, Ribeiro H, et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature. 2016;535(7612):440–443. doi:10.1038/nature18644
45. Powell N, Pantazi E, Pavlidis P, et al. Interleukin-22 orchestrates a pathological endoplasmic reticulum stress response transcriptional programme in colonic epithelial cells. Gut. 2020;69(3):578–590. doi:10.1136/gutjnl-2019-318483
46. Mizoguchi A, Yano A, Himuro H, Ezaki Y, Sadanaga T, Mizoguchi E. Clinical importance of IL-22 cascade in IBD. J Gastroenterol. 2018;53(4):465–474. doi:10.1007/s00535-017-1401-7
47. Wan X, Zhang C, Lei P, et al. Precision therapeutics for inflammatory bowel disease: advancing ROS-responsive nanoparticles for targeted and multifunctional drug delivery. J Mater Chem B. 2025;13(10):3245–3269. doi:10.1039/D4TB02868F
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.