Back to Journals » International Journal of Nanomedicine » Volume 15
Magnetically Directed Enzyme/Prodrug Prostate Cancer Therapy Based on β-Glucosidase/Amygdalin
Authors Zhou J, Hou J, Rao J, Zhou C ![]() , Liu Y, Gao W
, Liu Y, Gao W
Received 14 December 2019
Accepted for publication 9 June 2020
Published 29 June 2020 Volume 2020:15 Pages 4639—4657
DOI https://doi.org/10.2147/IJN.S242359
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Jie Zhou,1,2 Jing Hou,1,2 Jun Rao,2,3 Conghui Zhou,2,4 Yunlong Liu,5 Wenxi Gao1,2
1Department of Urology, Hubei Provincial Hospital of Traditional Chinese Medicine, Wuhan, Hubei, People’s Republic of China; 2Hubei Province Academy of Traditional Chinese Medicine, Wuhan, Hubei, People’s Republic of China; 3Clinical Laboratory, Hubei Provincial Hospital of Traditional Chinese Medicine, Wuhan, Hubei, People’s Republic of China; 4Department of Pharmaceutical Sciences, Hubei Provincial Hospital of Traditional Chinese Medicine, Wuhan, Hubei, People’s Republic of China; 5Department of Urology, the First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, People’s Republic of China
Correspondence: Jie Zhou Email [email protected]
Background: β-Glucosidase (β-Glu) can activate amygdalin to kill prostate cancer cells, but the poor specificity of this killing effect may cause severe general toxicity in vivo, limiting the practical clinical application of this approach.
Materials and Methods: In this study, starch-coated magnetic nanoparticles (MNPs) were successively conjugated with β-Glu and polyethylene glycol (PEG) by chemical coupling methods. Cell experiments were used to confirm the effects of immobilized β-Glu on amygdalin-mediated prostate cancer cell death in vitro. Subcutaneous xenograft models were used to carry out the targeting experiment and magnetically directed enzyme/prodrug therapy (MDEPT) experiment in vivo.
Results: Immobilized β-Glu activated amygdalin-mediated prostate cancer cell death. Tumor-targeting studies showed that PEG modification increased the accumulation of β-Glu-loaded nanoparticles in targeted tumor tissue subjected to an external magnetic field and decreased the accumulation of the nanoparticles in the liver and spleen. Based on an enzyme activity of up to 134.89 ± 14.18mU/g tissue in the targeted tumor tissue, PEG-β-Glu-MNP/amygdalin combination therapy achieved targeted activation of amygdalin and tumor growth inhibition in C57BL/6 mice bearing RM1 xenografts. Safety evaluations showed that this strategy had some impact on liver and heart function but did not cause obvious organ damage.
Conclusion: All findings indicate that this magnetically directed enzyme/prodrug therapy strategy has the potential to become a promising new approach for targeted therapy of prostate cancer.
Keywords: magnetic nanoparticles, β-glucosidase, amygdalin, prostate cancer, magnetically directed enzyme/prodrug therapy
Introduction
Prostate cancer remains the most common cancer among men in the United States.1 Its incidence is gradually increasing in Asians.2 The most common treatment options, with or without hormone administration, include active surveillance (i.e., watchful waiting), prostatectomy, radiation and chemotherapy. Surgery and radiation are usually followed by complications such as urinary incontinence, irritable bowel syndrome and impotence. Chemotherapy has had very little clinical success thus far due to palliative responses and induced systemic toxicity caused by the lack of target selectivity and poor potency of existing small molecule drugs.3,4 Hence, a noninvasive and highly efficacious but less toxic pharmacological treatment method is urgently needed.
Amygdalin can be extracted from bitter almond, which has been used as a traditional natural medicine to treat cancer for many years. Although there is still controversy regarding the anticancer effect of amygdalin, cancer patients in many countries continue to use it.5,6 Amygdalin can inhibit tumor growth through multiple mechanisms, such as blocking the cell cycle,7,8 inducing cell apoptosis and influencing cancer cell adhesion and invasion.9–11 Amygdalin can also inhibit the growth and invasion of both castration-sensitive and castration-resistant prostate cancer cells.12,13 However, the inhibitory effect of a small amount of amygdalin is weak, and large doses may cause systemic toxicity, which limits its clinical application.6,14
β-Glu can accelerate the hydrolysis of amygdalin into hydrogen cyanide, which can effectively kill tumor cells by inhibiting cytochrome C oxidase in mitochondria, resulting in a significant increase in the cell mortality rate.5 For example, the combination of amygdalin with β-Glu increases the liver cancer cell killing efficiency of amygdalin by 143 times.15 However, the killing effect of amygdalin combined with β-Glu lacks tumor cell specificity, and the produced hydrogen cyanide is toxic in vivo, especially for the nervous system and the cardiac system, which limits its practical clinical application. The specific activation of amygdalin by β-Glu in tumor tissue may be an effective method for decreasing the general toxicity and increasing the killing effect.
Targeted enzyme/prodrug strategies have been investigated as a means to improve the tumor selectivity of therapeutics with decreased side effects.16 The enzyme or its encoding gene is first delivered to the tumor site using a targeting carrier. After clearance of the enzyme from circulation, the prodrug is administered and then converted to an active anticancer drug, thus achieving enhanced anticancer efficacy and decreased systemic toxicity.17,18 Since the concept was first envisioned in 1974,19,20 a number of enzyme and prodrug coadministration strategies have been investigated and improved. Currently, enzyme carriers include antibodies, receptors, viruses, polymers and liposomes. Unfortunately, targeted enzyme prodrug strategies have not been translated to practical clinical applications due to various technical bottlenecks, including the low stability of bioactive carriers (such as antibodies) in vivo, the scarcity and heterogeneity of tumor-specific antigens, poor delivery efficiency, the immunogenicity of the carrier, monitoring difficulties, uncontrollable phagocytosis and pharmacokinetic characteristics.21,22 Amygdalin/β-Glu has been investigated as an enzyme/prodrug strategy. Syrigos et al. achieved the specific binding of β-Glu to bladder cancer cells by conjugating it with a tumor-associated monoclonal antibody (HMFG1). A specific HMFG1-β-glucosidase conjugate enhanced the cytotoxicity of amygdalin (36×).23 However, the combination effect has not been verified in vivo to date, perhaps due to the poor accumulation of β-Glu at the tumor site and the unsatisfactory therapeutic effect in vivo.24 Linamarase is a β-Glu that can hydrolyze linamarin (an amygdalin analog) to inhibit tumor growth.25 Some researchers have used transgenic tumor cells to express linamarase in the presence of linamarin to inhibit subcutaneous tumor growth.26,27 However, when the system was used in rats bearing glioma tumors in the brain, the mortality of the rats was significantly increased because of severe toxic effects.28 Therefore, targeted enzyme/prodrug strategies need to be further improved before clinical translation.
Magnetic nanoparticles are novel carriers for magnetic targeting. Their magnetic orientation, advantageous modifiability, satisfactory biocompatibility and relatively large surface-to-volume ratio make them promising in biomedical applications.29,30 Depending on the applied magnetic field and the enhanced permeation and retention (EPR) effect, a variety of carried substances, such as proteins and genes, can be delivered to tumor sites.31–33 Moreover, the imaging characteristics and modifiability of magnetic nanoparticles allow monitoring of their dynamic distribution in the body by magnetic resonance imaging (MRI) and other imaging methods.34–36
Therefore, we designed a magnetically directed enzyme/prodrug strategy based on amygdalin/β-Glu combination therapy. β-Glu was delivered to the targeted tumor site by magnetic nanoparticles, and amygdalin was activated at the tumor site, resulting in low systemic toxicity and increased tumor suppression efficiency. β-Glu has been successfully immobilized on magnetic nanoparticles and showed favourable activity.37,38 In a previous study, we also prepared β-Glu-loaded magnetic nanoparticles and confirmed their targeted delivery using a mouse model of subcutaneous 9L glioma.39 Moreover, polyethylene glycol (PEG) modification of β-Glu increased the in vivo stability of MNP-β-Glu and enhanced their accumulation in the targeted tumor tissue.40 However, the enzyme activity was not robust enough to activate amygdalin to inhibit tumor growth. The following issues may have contributed to this result: 1. The amount of β-Glu-loaded magnetic nanoparticles delivered to the tumor tissue was too small to activate amygdalin, and 2. 9L glioma cells were not sensitive to β-Glu/amygdalin therapy. In this study, to address these issues, we further improved the strategy. First, to prepare smaller MNP-β-Glu, the previously used glutaraldehyde crosslinking method was replaced by the sodium periodate oxidation method. Decreasing particle size might increase the stability of the particles and, more importantly, facilitate the accumulation of particles at the tumor site via the EPR effect. Second, the cytotoxicity of β-Glu/amygdalin was compared in different prostate cancer cells. The most sensitive cells were chosen as the model cells. In addition, considering the toxicity of the magnetic nanoparticles and the produced hydrogen cyanide, a local injection of amygdalin was used after the targeted accumulation of MNP-β-Glu-PEG to reduce the systemic toxicity of the amygdalin/MNP-β-Glu-PEG combination. As a result of the above improvements, the quantity of β-Glu-loaded particles that accumulated in the tumor tissue significantly increased. The enzyme activity of β-Glu in the targeted tumor tissue reached a level that activated amygdalin and inhibited RM1 tumor growth in C57BL/6 mice. These studies provide a basis for the clinical translation of magnetically targeted enzyme/prodrug strategies for prostate cancer.
Materials and Methods
Materials
All materials were obtained from commercial suppliers and used without further purification, unless otherwise noted. Starch-coated fluidMAG-D magnetite (Fe3O4) nanoparticles (D-MNP) were purchased from Chemicell® GmbH (25 g/L, Berlin, Germany). β-Glucosidase (β-Glu), sodium periodate, sodium borohydride, 4-nitrophenyl-β-D-glucopyranoside (GlcβNp), and sodium phosphate (mono- and dibasic) were obtained from Sigma-Aldrich (Milwaukee, MI, USA). NHS-PEG (20 kDa) was purchased from Laysan Bio, Inc (Alabama, USA). All deionized water (DI H2O) used in this study was produced by a Milli-Q A10 Biocel water purification system (Millipore, Massachusetts, USA).
Animals and Cell Lines
The mouse prostate cancer cell line RM1 and human prostate cancer cell lines PC3 (castration-resistant) and LNCaP (castration-sensitive) were obtained from Myhalic Biotechnology Co., Ltd. (Wuhan, Hubei, China). RM1 and PC3 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium with 100 IU/mL penicillin, 10% fetal bovine serum (FBS) and 100 μg/mL streptomycin at 37°C with 5% CO2. LNCaP cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 100 IU/mL penicillin, 10% FBS and 100 μg/mL streptomycin at 37°C with 5% CO2.
C57 mice (20±2.0 g) were provided by the Hubei Provincial Centers for Disease Control and Prevention (Wuhan, Hubei, China). The animals were given food and water ad libitum and were allowed to acclimatize for 1 week before the experiments. Experimental conditions and procedures involving animals were approved by the Institutional Animal Ethics Committee (IAEC) of Hubei University of Chinese Medicine and carried out in accordance with the laboratory animal use guidelines of the IAEC. Animal handling followed the National Animal Welfare Law of China.
Preparation of MNP-β-Glu-PEG
As shown in Figure 1A, hydroxyl groups in the sugar rings of D-MNP with a cross-linked starch coating were partially oxidized by periodate to generate activated aldehyde functional groups,41 which further reacted with the amino groups of β-Glu via Schiff’s base formation to achieve β-Glu-loaded MNPs. The formed hydrazone bonds were further reduced and stabilized with the mild reducing agent NaBH4 to avoid the dissociation of conjugated β-Glu in acidic environments.
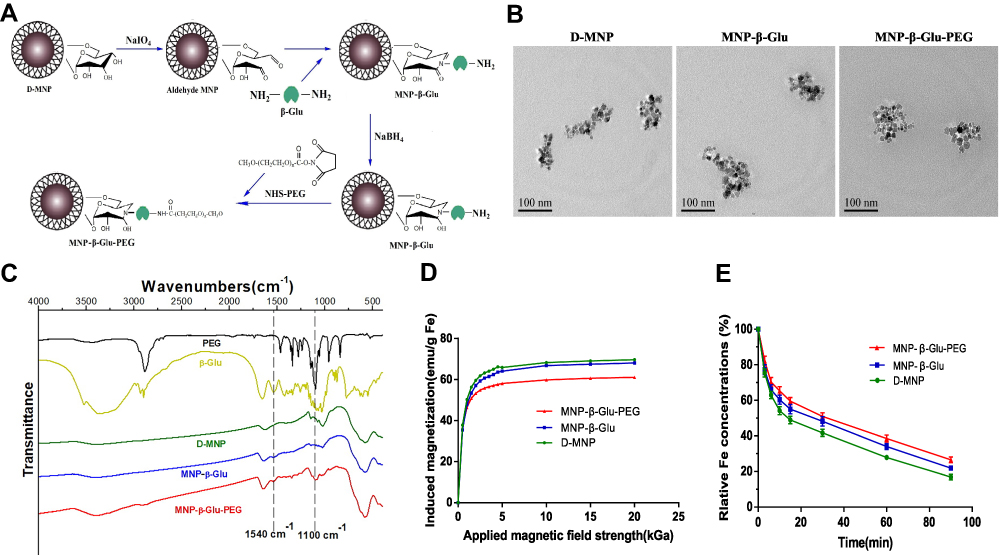 |
Figure 1 Preparation process and characterization analyses of MNP-β-Glu-PEG. (A) Schematic diagram of the preparation process. (B) Representative TEM images of D-MNP, MNP-β-Glu and MNP-β-Glu-PEG. Various particles showed no significant morphological differences. (C) Infrared spectrum analysis. A characteristic β-Glu peak located at 1540 cm−1 is visible in the MNP-β-Glu spectrum. Characteristic β-Glu and PEG peaks located at 1540 cm−1 and 1100 cm−1 are visible in the MNP-β-Glu-PEG spectrum. (D) The magnetization properties of D-MNP, MNP-β-Glu and MNP-β-Glu-PEG. Neither hysteresis nor remnant magnetization was observed for all particles. (E) Magnetophoretic mobility curves for D-MNP, MNP-β-Glu and MNP-β-Glu-PEG (n=3). Relative Fe concentrations for the nanoparticle suspensions indicate the ratio of noncaptured nanoparticles in the magnetic field. |
In brief, 2.4 mg of D-MNP were mixed with 240 μL of 0.2 M sodium acetate and then incubated with 50 μL of 0.1 M periodate at 4°C for 30 min in the dark to allow partial oxidation of the starch. The aldehyde nanoparticles were concentrated to 0.8 mL by magnetic separation for 10 min to remove the free periodate. The resulting solution was dropped into 5 mL of β-Glu water solution (0.8 mg/mL), and the pH was adjusted to 10.0. The mixture was then incubated with shaking at 4°C for 24 hours in the dark on a rolling shaker. The hydrazone linkages were reduced to more stable secondary amine bonds by the addition of 40 μL of 5 mg/mL sodium borohydride per milliliter of conjugation mixture and incubation for 2 hours at 4°C. The chemical reagents and unconjugated β-Glu were removed using a Dynal magnetic separator (Invitrogen, California, USA) at 4°C to obtain the resulting β-Glu-loaded iron oxide nanoparticles denoted as MNP-β-Glu.
MNP-β-Glu were further modified with PEG by amidation. In detail, 320 μL of MNP-β-Glu (40 mg Fe/mL) were dropped into 640 μL of NHS-PEG (~38 mg) aqueous solution. After 3 hours of shaking at room temperature, the mixture was then incubated with shaking at 4°C for 12 hours in the dark on a rolling shaker. The product was washed 4 times with fresh DI H2O using a Dynal magnetic separator to obtain the resulting PEG-modified β-Glu-loaded iron oxide nanoparticles denoted as MNP-β-Glu-PEG.
Characteristics of MNP-β-Glu-PEG
Representative TEM images of samples were acquired using scanning transmission electron microscopy-bright field (STEM-BF) imaging with an accelerated voltage of 200 kV using a Tecnai G220S-TWIN electron microscope (FEI, Czech). The infrared spectra of all samples were collected according to a previously published method.40 The magnetization properties and magnetophoretic mobility of all MNPs were measured according to a previously published method.40
Measurement of Enzyme Activity and Fe Concentration
GlcβNp was used as the substrate to measure free and conjugated β-Glu activity throughout this study.39 One β-Glu activity unit (1 U) was defined as the amount of enzyme required to hydrolyze 1 μmol of the substrate per minute under the testing conditions. Inductively coupled plasma optical emission spectroscopy (ICP-OES) was carried out throughout the preparation to measure the iron content of all MNP samples using an Optima DV 2000 spectrometer (Perkin Elmer, Massachusetts, USA).40
Cytotoxicity of Amygdalin in Prostate Cancer Cells
The cytotoxicity of amygdalin in RM1, PC3 and LNCaP cells was assessed with a Cell Counting Kit-8 (Yeasen Corporation, Shanghai, China) assay according to the manufacturer’s instructions. Briefly, prostate cancer cells were seeded in a 96-well plate and allowed to adhere overnight. The cells were then incubated in quadruplicate for 24 hours with the following agents at preset amygdalin concentrations: (1) amygdalin alone; (2) amygdalin plus β-Glu (0.3 U/mL); (3) amygdalin plus MNP suspension (0.725 mg Fe/mL); and (4) amygdalin plus MNP-β-Glu-PEG suspension (0.3 U/mL). The cells were then incubated with fresh medium for 24 hours. CCK-8 reagent was added to each well, and the cells were incubated for an additional 4 hours. The absorbance values at 450 nm were measured using a microplate reader (SpectraMax M5, Molecular Devices LLC, Sunnyvale, CA, USA). Cells treated with PBS were used as the negative control. Complete medium without cells was used as the blank control. The cytotoxicity of amygdalin was expressed as the percentage of cell survival, which was calculated using the following formula:
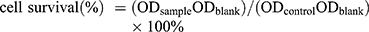
Flow Cytometry Analysis
RM1, PC3 and LNCaP cells (5×105cells/well) were seeded in 6-well plates for 24 hours. Then, the following agents were added: (1) amygdalin (10 mg) alone or (2) amygdalin (10 mg) plus MNP-β-Glu-PEG (0.3 U). After 24 hours, cell apoptosis and necrosis were determined by annexin V-FITC/PI double staining and flow cytometry analysis. In brief, the cells were collected, washed twice in ice-cold PBS, and then resuspended in 100 μL of binding buffer containing 5 μL of annexin V-FITC and 5 μL of PI provided with the annexin V-FITC/PI Apoptosis Detection Kit (Yeasen Corporation, Shanghai, China). The cells were incubated at 37°C for 15 min in the dark and analyzed with a FACS Calibur (BD Biosciences).
DNA Fragmentation Assay
To test whether the decrease in cell viability observed after treatment with amygdalin/MNP-β-Glu-PEG was due to necrosis or apoptosis, DNA fragmentation assays were performed. Cells were cultured in medium containing the following agents: PBS, 5 mg/mL amygdalin, 5 mg/mL amygdalin + 0.15 U/mL β-Glu, and 5 mg/mL amygdalin + 0.15 U/mL MNP-β-Glu-PEG. After 24 hours, the cells were collected, and DNA was extracted using a DNA Extraction Kit (TIANGEN Corporation, Beijing, China) according to the manufacturer’s instructions. The collected samples were detected using 1% agarose gel electrophoresis at 100 V for 30 min. Cells treated with PBS were used as controls.
AO/EB Double-Staining Assay
RM1 cells were seeded in 6-well plates for 24 hours. Then, the following reagents were added: PBS, 1 mg/mL amygdalin, 5 mg/mL amygdalin, 1 mg/mL amygdalin + 0.15 U/mL β-Glu, 5 mg/mL amygdalin + 0.15 U/mL β-Glu, 1 mg/mL amygdalin + 0.15 U/mL MNP-β-Glu-PEG, 5 mg/mL amygdalin + 0.15 U/mL MNP-β-Glu-PEG, and cisplatin (0.75 mg/mL) in PBS. After 24 hours, the cells were collected, and 2 µL of 100 mg/L AO and 100 mg/L EB were added. Fluorescent staining was observed and photographed under a fluorescence microscope.
Western Blot Analysis
Caspase-3, Bax and Bcl-2 protein expression was measured to identify the effect of β-Glu/amygdalin on prostate cancer cells. RM1 cells were seeded in a 6-well plate at a density of 5×103 cells/well for 24 hours. The following agents were added: PBS, 5 mg/mL amygdalin, 5 mg/mL amygdalin + 0.15 U/mL β-Glu, and 5 mg/mL amygdalin + 0.15 U/mL MNP-β-Glu-PEG. After 24 hours, the cells were collected and lysed in RIPA buffer (Bioswamp, Myhalic Biotechnology Co., Ltd., Wuhan, China). The extracted total protein (~20 µg) was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to a polyvinylidene difluoride membrane (Amersham, Little Chalfont, UK), and immunoblotted with specific antibodies against Bcl-2, Bax and cleaved caspase-3 (Bioswamp, Myhalic Biotechnology Co., Ltd., Wuhan, China). After incubation with an HRP-conjugated secondary antibody (Bioswamp, Myhalic Biotechnology Co., Ltd., Wuhan, China) at room temperature for 1 hour, immunoreactive bands were visualized by luminescence detection using SuperSignal™ West Pico PLUS chemiluminescent substrate (Thermo Fisher Scientific, MA, USA), and images were obtained using an Image Quant LAS 4000 imaging system (GE Healthcare Life Sciences, MA, USA). Band intensities were quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA) and normalized to that of GAPDH.
Tumor-Targeted Delivery and Distribution of MNP-β-Glu-PEG
Because the combined administration showed a more pronounced inhibitory effect on prostate cancer RM1 cells, we selected RM1 cells as implant cells to establish subcutaneous xenograft models for subsequent magnetic targeting experiments in vivo. Briefly, C57BL/6 mice (weight, 20–25 g) were implanted with RM1 cells (5×106) in the right flank. Magnetic targeting experiments were conducted when the tumor volume reached approximately 50 mm3.
According to the previous literature, magnetic targeting of tumors was achieved by a small cylindrical magnet attached to the pole of 3 tandem DYOYO-52 permanent magnets.39 The magnetic field density at the pole face of the small magnet was adjusted to approximately 800 mT. Twenty-four mice were randomized into 4 groups to ensure a similar average tumor size. Three groups were then injected with MNP-β-Glu-PEG, MNP-β-Glu or MNP suspension at a dose of 12 mg Fe/kg through the lateral tail vein and were retained in a magnetic field for 2 hours. Untreated mice were used as the control group. MRI experiments were carried out using a 7T Direct Drive small animal imaging system (Varian, Walnut Creek, CA, USA). T2-weighted images of mice were acquired prior to injection and 2 hours post injection. After the MRI scans, the mice were euthanized immediately by CO2 inhalation overdose. Tissue samples of targeted tumors, livers and spleens were obtained for further analysis.
The samples obtained for histology were fixed and encased in paraffin blocks. Tissue sections (5 μm) were cut from paraffin blocks and subsequently affixed to microscopy slides. The sections were rehydrated and incubated at 37 °C for 4 hours with 10% (g/mL) K4Fe(CN)6 in 5% (V/V) HCl (freshly made). Prussian blue-stained slides were rinsed in DI H2O and subsequently stained with Fast Red for 30 min at room temperature. Stained slides were visualized with a microscope and photographed (Nikon, Melville, NY, USA).
The Fe concentrations in the tumor, liver and spleen tissues were measured by electron spin resonance spectroscopy (ESR). Briefly, the sample (~50 mg) was homogenized on ice in 500 μL of lysis buffer (Tropix Inc., Bedford, MA, USA) using a pestle tissue grinder. The tissue homogenate was transferred to a glass capillary tube and then analyzed using electron spin resonance spectroscopy at −128°C to calculate the Fe concentration of tissues, as described previously.42
To minimize background endogenous β-Glu activity, the β-Glu activity in the excised tissue samples was assayed using a modified spectrophotometric method. In brief, tissue samples (~50 mg) were homogenized at 0°C in 500 µL of lysis buffer (Tropix Inc., Bedford, MA, USA) using a pestle tissue grinder. The samples were dispersed by sonication at 30% amplitude for 5 seconds. Citrate-phosphate buffer (20 µL, 0.1 mol/L, pH 6.0) containing GlcβNp (0.1 M) was added, and the samples were incubated at 37°C for 1 hour. Immediately following incubation, the reaction mixture was mixed with 0.5 mL of carbonate buffer (0.2 mol/L, pH 10.2) and then centrifuged at 15,000 r.p.m. for 20 min. The supernatant was collected, and its optical density was analyzed on a microplate reader at 405 nm to calculate the β-Glu activity of the tissue samples. The β-Glu activity in the tissue samples from the control group was also measured and subtracted from those of the experimental groups to determine the net activity induced by the targeted β-Glu.
Tumor Growth Inhibition Experiment
Thirty-six C57BL/6 mice were used to establish the tumor model according to the method described above. When the tumor volume reached approximately 20 mm3, the animals were randomized into 6 groups according to the injected agent, which was administered through the lateral tail vein with/without magnetic targeting. Group A (the model group) were injected with normal saline, Group B was injected with normal saline, combined with magnetic targeting for 2 hours. Group C was injected with β-Glu solution at an enzyme activity of 5 U/kg, combined with magnetic targeting for 2 hours. Group D was injected with MNP-β-Glu-PEG suspension at an enzyme activity of 5 U/kg without magnetic targeting. Group E was injected with MNP-β-Glu suspension at an enzyme activity of 5 U/kg, combined with magnetic targeting for 2 hours. Group F was injected with MNP-β-Glu-PEG suspension at an enzyme activity of 5 U/kg, combined with magnetic targeting for 2 hours. Then, 0.1 mL of amygdalin saline solution (320 mg/kg) was immediately injected directly into the core of the tumor in B-F group mice. The dosage regimen was repeated every 2 days, and serial changes in tumor volume were estimated by measuring the diameter of each tumor using a digital caliper. Tumor volume was calculated as length × width2 × 0.5. The mice were weighed every day, and general health was also observed. To evaluate heart, liver and kidney function, prior to administration and euthanasia, 300 µL blood samples were collected via the vena angularis to measure the creatinine (Cr), urea nitrogen (BUN), alanine aminotransferase (ALT), aspartate amino transferase (AST), lactate dehydrogenase (LDH) and creatine kinase (CK) levels using an ADVIA® 2400 automatic biochemical analysis system (Siemens Corporate, German). Three weeks after treatment, the animals were sacrificed by cervical dislocation, and the tumor, heart, lungs, liver, spleen and kidneys were removed for histological analysis.
The extracted tumors and organs were fixed in 4% paraformaldehyde solution and subsequently embedded in paraffin. The paraffin-embedded tumor tissue was sectioned (4 µm thick) at the largest tumor area. To validate the tumor growth inhibition effect of amygdalin plus MNP-β-Glu-PEG, apoptotic cells in tumor tissues were detected by terminal deoxynucleotidyl transferase (dUTP) - mediated nick end labeling (TUNEL) using an in situ Apoptosis Detection Kit (Abcam, Cambridge, UK) according to the manufacturer’s instructions. The number of cells positive for TUNEL staining was determined by counting at least 1000 neoplastic nuclei subdivided into 10 randomly selected fields at 400× magnification. The apoptosis index was calculated according to the following formula: number of positive cells/total cell count×100%. The paraffin-embedded organ tissues were sectioned and stained with hematoxylin and eosin (H&E).
Statistical Analysis
Data are presented as the mean±standard deviation unless indicated otherwise. SPSS 17.0 (SPSS Inc., Illinois, USA) was used for data analysis. Statistical evaluation of numerical variables was performed using Student’s t-test for 2 groups and ANOVA for multiple groups. A p-value < 0.05 was considered statistically significant.
Results
Synthesis and Characterization of MNP-β-Glu-PEG
The main tumor targeting principle of many drug delivery systems is based on the EPR effect of nanoparticles. In general, particles < 300nm are suitable for the EPR effect.22 In addition, particle size is also an important parameter related to particle clearance from the blood circulation.43,44 Previously, we used glutaraldehyde as a crosslinking agent to prepare β-Glu-loaded MNPs. The prepared MNP-β-Glu was larger than 250 nm and agglomerated easily, possibly because the bilateral functional aldehyde terminal ends of glutaraldehyde facilitated particle crosslinking.39,40 Figure 1A shows the synthesis process of MNP-β-Glu-PEG in this experiment. β-Glu was directly conjugated on the surface of starch-coated nanoparticles by amino aldehyde group reactions. Unilateral functional aldehyde ends relieved the crosslinking phenomenon.
The immobilized β-Glu showed a higher Km (3.21±0.17 mmol l−1) than (2.22±0.31 mmol l−1) did free β-Glu (Table 1), showing that the immobilized enzyme possesses better affinity for the substrate GlcβNp. The maximum reaction rate of MNP-β-Glu was found to be 27.8±3.14 μmol min−1 mg−1, which is approximately 86% of that of free enzyme (32.5±2.44 μmol min−1 mg−1). However, as shown in Figure 1B and Table 1, PEGylation influenced morphology and enzyme activity very little. Based on the activity (6 U/mg) and molecular weight (79 kB) of β-Glu, the enzyme activity (0.421 U/mg Fe) of MNP-β-Glu, and the concentration (1.8*1015/g) of particles, it is estimated that more than 300 β-Glu molecules were coupled to a single MNP. Thus, the MNPs showed satisfactory loading efficiency. Figure 1C shows an absorption peak at 1540 cm−1 for MNP-β-Glu, which is the characteristic absorption peak for β-Glu. In addition, the characteristic absorption peak (1100 cm−1) for PEG appeared in the MNP-β-Glu-PEG absorption spectrum. These results further confirmed the successful coupling of the 3 components.
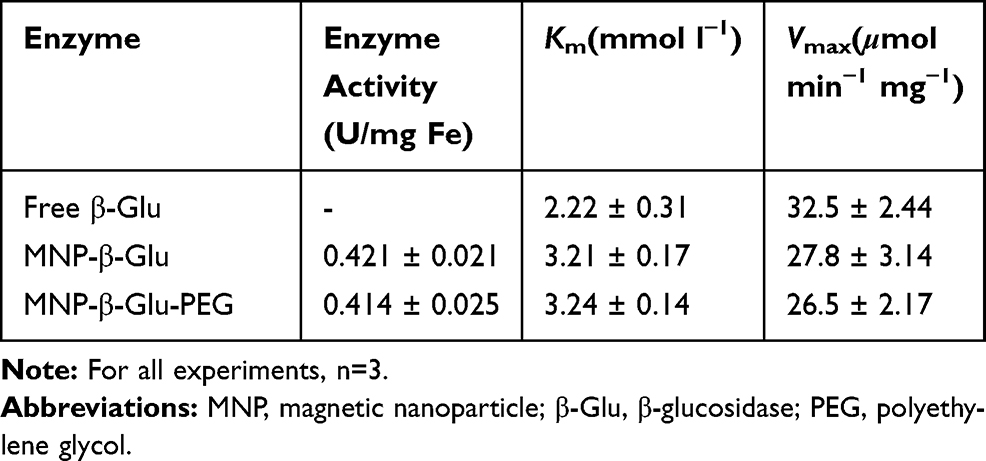 |
Table 1 Activity of β-Glu and β-Glu-Loaded Magnetic Nanoparticles |
The induced magnetization curves (Figure 1D) displayed neither hysteresis nor remnant magnetization for D-MNP, MNP-β-Glu and MNP-β-Glu-PEG, and the saturation magnetization values of these MNPs were 69.7, 68.1 and 61.1 emu/g Fe, respectively. The migration half-time (Mt1/2) was defined as the time required to reduce the Fe concentration by 50%, which represents the responsiveness of the nanoparticles to magnetic fields. Magnetophoretic mobility curves (Figure 1E) showed Mt1/2 values of approximately 31, 28 and 14 min for MNP-β-Glu-PEG, MNP-β-Glu and D-MNP, respectively. These results showed that MNP-β-Glu and MNP-β-Glu-PEG possessed tropism to an external magnetic field, although the response intensity was decreased by β-Glu loading and PEG modification.
All these results indicated that both β-Glu and the MNPs remained relatively stable during the preparation process. The physical and chemical properties of the prepared MNP-β-Glu-PEG meet the needs for further experimental applications.
Cell Experiments
As shown in Figure 2A–C, amygdalin displayed no significant inhibitory effect on prostate cancer cells until the concentration exceeded 10 mg/mL. β-Glu alone showed no significant effect on cell growth (Figure S1). However, combined administration of amygdalin with only 0.3 U/mL β-Glu significantly increased the inhibitory effect of amygdalin (p < 0.01). Compared with the amygdalin alone group, the IC50 of the amygdalin + β-Glu group for RM1, PC3 and LNCaP cells decreased 272.83-fold (0.3±0.11 mg/mL vs. 81.85±4.33 mg/mL), 11.18-fold (8.37±0.73 mg/mL vs. 93.55±4.72 mg/mL) and 42.5-fold (2.08±0.33 mg/mL vs. 88.39±3.79 mg/mL), respectively. In addition, the IC50 of the amygdalin + MNP-β-Glu-PEG group decreased 264.03-fold (0.31±0.1 mg/mL vs. 81.85±4.33 mg/mL), 10-fold (9.35±0.69 mg/mL vs. 93.55±4.72 mg/mL) and 35.36-fold (2.5±0.24 mg/mL vs. 88.39±3.79 mg/mL), respectively, with values close to those of the amygdalin + β-Glu group, suggesting that MNP-β-Glu-PEG can hydrolyze amygdalin in the same way as β-Glu and enhance the inhibitory effect on prostate cancer cells.
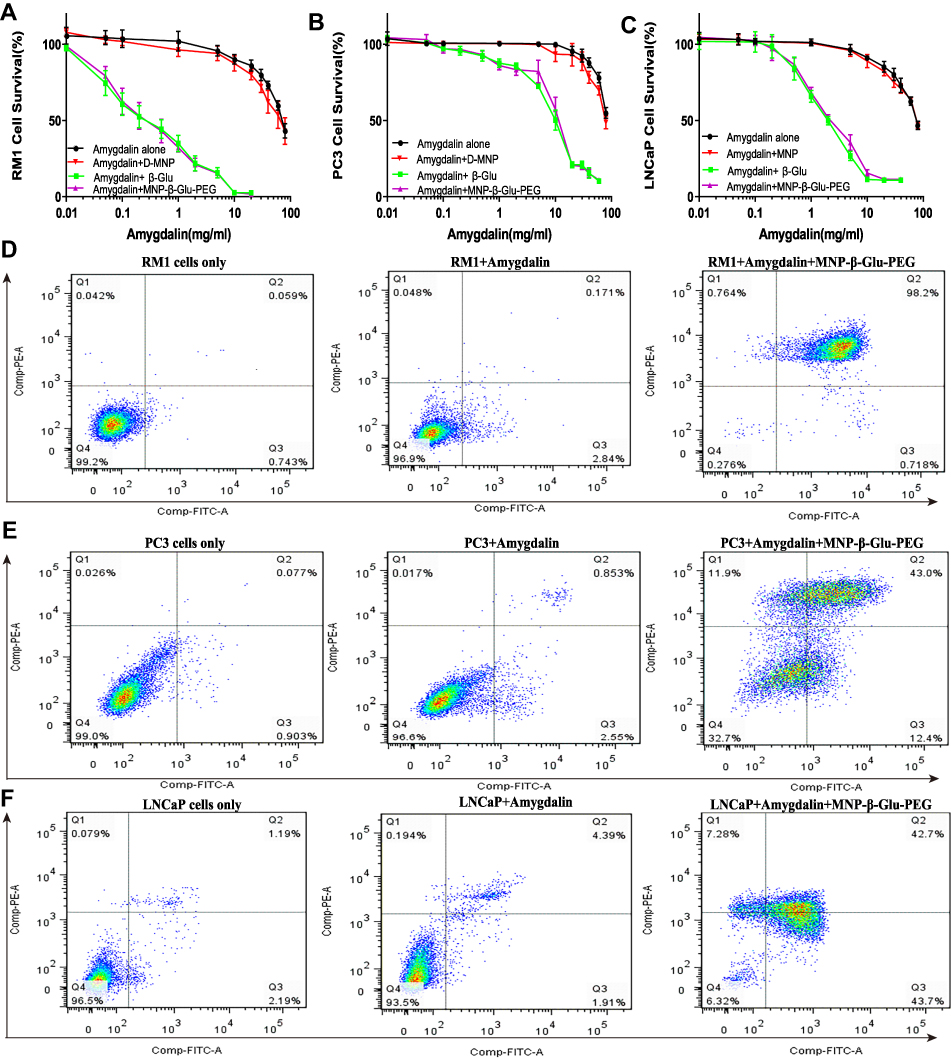 |
Figure 2 Proliferation inhibition and apoptosis analyses of prostate cancer cells. (A–C) The growth inhibition effects of amygdalin, amygdalin/MNP, amygdalin/β-Glu and amygdalin/MNP-β-Glu-PEG on RM1 cells, PC3 cells and LNCaP cells. Data show the mean±standard deviation of measurements conducted in quadruplicate. (D–F) Representative annexin V-FITC/PI flow cytometry analysis of RM1, PC3 and LNCaP cells after amygdalin or amygdalin/MNP-β-Glu-PEG treatment. Cells were defined as viable (PI−, annexin V−, lower left quadrant), early apoptotic (PI−, annexin V+, lower right quadrant), late-stage apoptotic (PI+, annexin V+, upper right quadrant) or necrotic (PI+, annexin V−, upper left quadrant). |
It was reported that β-Glu (3.7 U/mL) can increase the killing ability of amygdalin in HepG2 cells by 143.16-fold.15 Figure S2 shows that a low concentration (18.75 mU/mL) of β-Glu increased the inhibitory effect of amygdalin on RM1 cells and that the IC50 of amygdalin decreased 44.73-fold (1.83±0.50 mg/mL vs. 81.85±4.33 mg/mL), which suggested that even accumulation of a small amount of β-Glu in tumor tissues likely improves the tumor inhibition efficiency of amygdalin.
Flow cytometry analysis (Figure 2D–F) showed that amygdalin increased the proportions of necrotic and apoptotic cells among total RM1, PC3 and LNCaP cells (0.72±0.44% vs. 3.04±1.18%, 0.96±0.66% vs. 3.30±1.13%, and 2.67±1.00% vs. 6.59±2.38%, p < 0.05). However, combined with MNP-β-Glu-PEG, the proportions of necrotic and apoptotic cells were significantly increased to 92.05±4.78%, 53.04±5.28% and 86.35±5.25%, respectively (p < 0.001). Therefore, similar to β-Glu, MNP-β-Glu-PEG can promote the ability of amygdalin to induce prostate cancer cell apoptosis or necrosis. According to a previous report,26 cells treated with β-Glu/amygdalin should concentrate in the necrotic cell quadrant (PI-positive, annexin V-negative). However, in this study, the ratio of late-stage apoptotic cells (PI-positive, annexin V-positive cells) was highest in the MNP-β-Glu-PEG/amygdalin group. The possible reason might be that phosphatidylserine in necrotic prostate cancer cells conjugated with annexin V to increase the detection of PI-positive, annexin V-positive cells.
AO/EB staining showed, morphologically, the effect of combined administration on prostate cancer cells. Most of the cells treated with cisplatin showed obvious characteristics of apoptosis, such as pyknotic and fragmented nuclei. However, in the cells treated with β-Glu/amygdalin or MNP-β-Glu-PEG/amygdalin, some characteristics of necrotic cells, such as cell distension and cytomembrane breaks, could be observed, and only a small number of cells showed characteristics of apoptosis (Figure 3A). These results indicated that both the apoptosis pathway and the necrotic pathway may be involved in the mechanism of prostate cancer cell death resulting from combined administration. One of the classic features of apoptosis is the cleavage of genomic DNA into 180–200 bp oligonucleosomal fragments. In the DNA fragmentation assay, long DNA fragments aggregated in cells treated with PBS or amygdalin. A distinct DNA ladder was not observed for cells treated with β-Glu/amygdalin or MNP-β-Glu-PEG/amygdalin. Instead, only a smear was observed, which usually indicates the presence of necrotic cells (Figure 3B).
 |
Figure 3 Mechanism of prostate cancer cell death resulting from amygdalin combined with β-Glu or MNP-β-Glu-PEG treatment. (A) AO/EB staining analyzing the effects of amygdalin combined with β-Glu or MNP-β-Glu-PEG on prostate cancer. Green indicates live cells; cells with orange pyknotic nuclei in the cisplatin group are apoptotic cells; yellow swollen cells in the combined drug administration groups (β-Glu/amygdalin or MNP-β-Glu-PEG/amygdalin) are necrotic cells; and red indicates cells in the late phases of apoptosis/necrosis. 400×. (B) Band 1 is the control group (PBS solution), band 2 is the amygdalin group, band 3 is the amygdalin + β-Glu group, and band 4 is the amygdalin + MNP-β-Glu-PEG group. (C) 1 is the control group, 2 is the amygdalin group, 3 is the amygdalin + β-Glu group, and 4 is the amygdalin + MNP-β-Glu-PEG group. The relative amounts of protein expression were calculated according the grayscale values and were showed in the histogram. (compared with the control group, *p < 0.05, **p < 0.01, compared with the amygdalin group, #p < 0.01, n=4). |
Western blot analysis showed that the expression of cleaved caspase-3, Bcl-2 and Bax was significantly higher in the amygdalin group than in the control group (p < 0.05) and that the expression of cleaved caspase-3, Bcl-2 and Bax in the combined drug administration groups (β-Glu/amygdalin or MNP-β-Glu-PEG/amygdalin) showed a further significant increase compared to that in the amygdalin group (Figure 3C) (p < 0.01). In addition, Bcl-2 and Bax increased more than cleaved caspase-3, suggesting that amygdalin and the combination of amygdalin and β-Glu may activate the Bcl-2/Bax pathway during tumor cell death and that reduced ATP production due to respiratory chain blockage hindered further activation cascades after the activation of the caspase-3 apoptotic pathway. In summary, amygdalin administration and combined drug administration can cause changes in the expression of apoptosis-related proteins, but the activation of the apoptotic pathway may not be the main cause of death for prostate cancer cells. The main mechanism of increased cell death could involve hydrocyanic acid, which is a product of combined drug administration and can interact with cytochrome C to block the respiratory chain and inhibit ATP production, thus inducing cell death.
Targeted Tumor Delivery of β-Glu-Loaded MNPs by an External Magnetic Field
According to the MRI characteristics of tumors and iron particles, tumor tissues appear as high-contrast regions in T2-weighted images, while iron particle aggregation appears as low-contrast regions. As shown in Figure 4A, after 2 hours of magnetic targeting, the subcutaneous tumor regions of the MNP-β-Glu-PEG group exhibited significant hypodense shadows. However, no significant hypodense shadows were observed in the MNP and MNP-β-Glu groups after magnetic targeting, suggesting that MNP-β-Glu-PEG accumulate more easily at the tumor site with an external magnetic field.
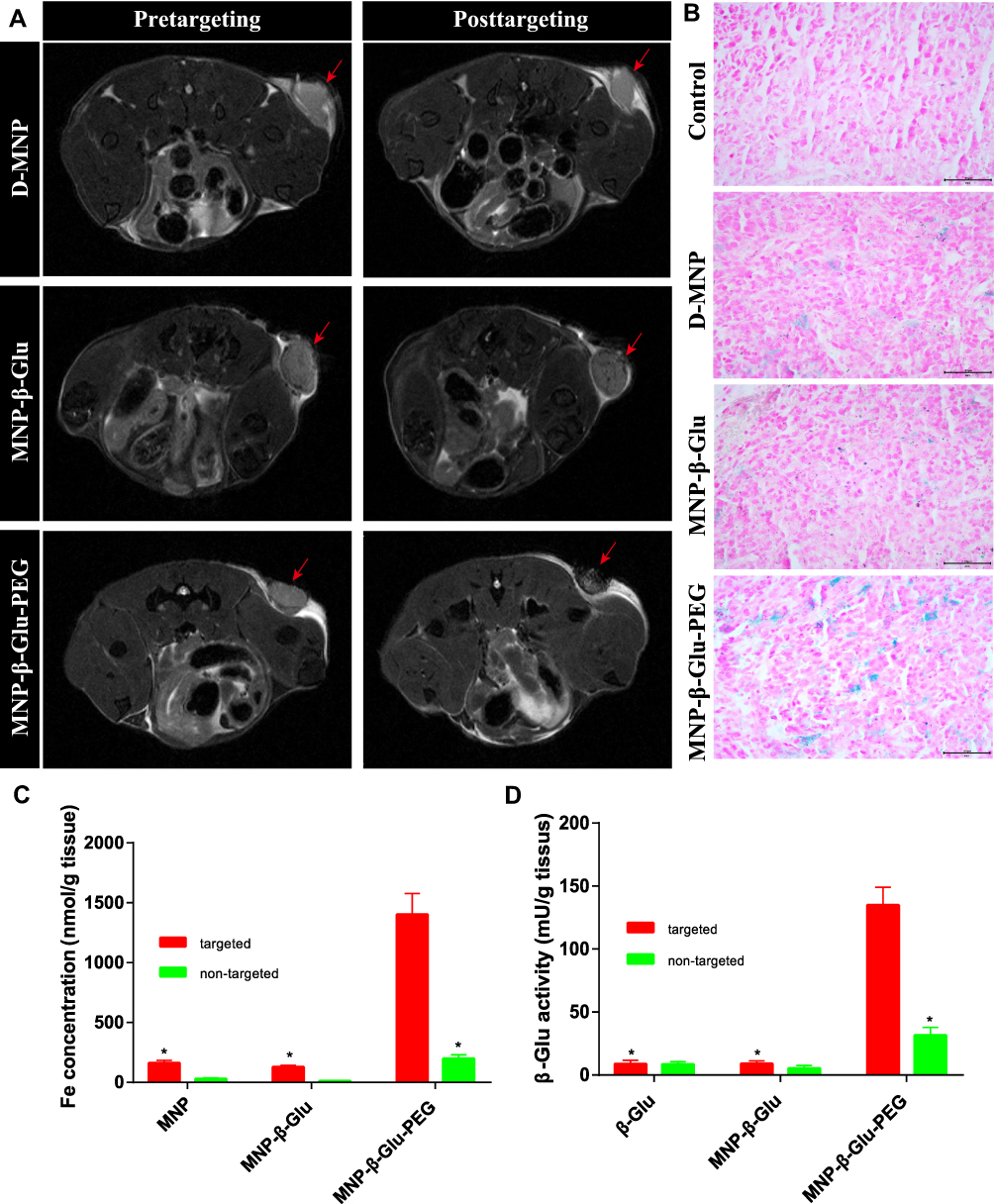 |
Figure 4 Targeted accumulation of β-Glu-loaded MNPs at tumor sites. (A) Representative MR images of particle aggregation at subcutaneous tumor sites with an external magnetic field after D-MNP, β-Glu-MNP or MNP-β-Glu-PEG administration. The red arrows indicate the locations of subcutaneous tumors. Significant hypodense shadows appear in the posttargeted tumor tissue of the MNP-β-Glu-PEG group. (B) Prussian blue staining of tumor tissues after D-MNP, MNP-β-Glu or MNP-β-Glu-PEG administration. Blue staining indicates iron particles. Scale bar: 50μm. (C) Fe concentrations and (D) enzymatic activities of targeted/nontargeted tumor tissues after D-MNP, β-Glu, MNP-β-Glu or MNP-β-Glu-PEG administration (compared with the targeted MNP-β-Glu-PEG group, *p < 0.01, n=6). |
The Prussian blue staining results for the tumor tissue sections were consistent with the MRI results. Figure 4B shows that a higher level of blue staining was found in the tumor tissues from the MNP-β-Glu-PEG group than in those from the MNP and MNP-β-Glu groups. Overall, these results indicated that PEGylation increased MNP accumulation in tumor tissues via magnetic targeting.
The accumulation of β-Glu-loaded nanoparticles at the tumor site is a prerequisite for the targeted activation of amygdalin. As shown in Figure 4C, magnetic targeting significantly increased the accumulation of MNP-β-Glu-PEG in tumor tissues (1401.86±176.30 nmol/g tissue vs. 198.48±32.48 nmol/g tissue) (p < 0.01). The Fe concentration of the MNP-β-Glu-PEG group after magnetic targeting was significantly different from that of the MNP-β-Glu group (1401.86±176.30 nmol/g tissue vs. 129.49±12.27 nmol/g tissue) (p < 0.01) after magnetic targeting. In summary, both magnetic targeting and PEGylation were positive factors for the targeted accumulation of particles at the tumor site.
The activity level of β-Glu in tumor tissue is the decisive factor for the local activation of amygdalin and tumor growth inhibition. Figure 4D shows that the β-Glu activity in tumor tissues of the MNP-β-Glu-PEG group after magnetic targeting reached approximately 134.89±14.18 mU/g tissue, which was approximately 4.3-fold greater than that (31.65±6.09 mU/g tissue) of the MNP-β-Glu-PEG group without magnetic targeting and was also approximately 14.8-fold and 15-fold greater than the activity levels (9.12±2.13 mU/g tissue and 8.99±2.70 mU/g tissue) of the MNP-β-Glu and β-Glu groups after magnetic targeting, respectively. As mentioned above, the magnitude of enzyme activity in tumor tissues of the MNP-β-Glu-PEG group after magnetic targeting was significantly higher than those reported in the literature.39,40 Considering that 18.75 mU/mL β-Glu can significantly increase the cytotoxicity of amygdalin to prostate cancer cells, the β-Glu activity (134.89±14.18 mU/g tissue) of tumor tissues in the MNP-β-Glu-PEG group after magnetic targeting should definitely be adequate for the planned in vivo feasibility study of MDEPT.
The Distribution of β-Glu-Loaded Particles in the Liver and Spleen
The organ distribution of enzyme-loaded particles is important for evaluating delivery efficiency and predicting systemic toxicity. As previously reported,45 most of the MNPs accumulated in liver and spleen tissues, and only a small number were located in other organs. Therefore, we performed Prussian blue staining and ESR analysis of liver and spleen tissues. As shown in Figure 5A, a substantial amount of blue staining was observed in liver and spleen tissue sections from both the MNP-β-Glu and MNP-β-Glu-PEG groups. ESR results also showed that the Fe concentration in liver and spleen tissues was significantly higher than that in tumor tissues after intravenous injection. It has been reported that PEG can inhibit uptake by the RES phagocytosis system, thereby reducing the accumulation of particles in the liver and spleen.46 In Figure 5A, blue staining was more significant in liver and spleen tissue sections from the MNP-β-Glu group than in liver and spleen tissue sections from the MNP-β-Glu-PEG group. Further ESR analyses (Figure 5B) confirmed that PEG modification significantly decreased the distribution of β-Glu-loaded particles in liver and spleen tissues (p < 0.01). Moreover, compared with that of the MNP-β-Glu group, the enzyme activity per mg of liver/spleen tissues was significantly lower (p < 0.01) in the MNP-β-Glu-PEG group (Figure 5C). Nevertheless, intravenous administration of the prodrug amygdalin still likely caused severe liver and spleen toxicity. Therefore, following MNP-β-Glu-PEG administration and magnetic targeting, amygdalin was administered through local injection to reduce systemic toxicity.
 |
Figure 5 Organ distribution of MNP-β-Glu and MNP-β-Glu-PEG. (A) Prussian blue staining of liver and spleen tissue sections after MNP-β-Glu or MNP-β-Glu-PEG administration. Scale bar: 50μm. (B) ESR analyses of liver and spleen tissues from the MNP-β-Glu and MNP-β-Glu-PEG groups (compared with MNP-β-Glu-PEG group/liver tissue,*p < 0.01, n=6; compared with MNP-β-Glu-PEG group/spleen tissue, #p < 0.01, n=6). (C) Enzyme activity analyses of liver and spleen tissues from the MNP-β-Glu and MNP-β-Glu-PEG groups (compared with MNP-β-Glu-PEG group/liver tissue, *p < 0.01, n=6; compared with MNP-β-Glu-PEG group/spleen tissue, #p < 0.01, n=6). |
Growth Inhibition of Subcutaneous RM1Xenografts
The results of the in vivo experiments are shown in Figure 6A and B. After amygdalin injection, tumor growth was reduced in the MNP-β-Glu-PEG group after magnetic targeting. However, in the MNP-β-Glu-PEG group without magnetic targeting, the MNP-β-Glu group after magnetic targeting, the β-Glu group and the control group, the tumors gradually increased in size, displaying very similar growth curves. At the end of the experiment, the tumor volume in the MNP-β-Glu-PEG group after magnetic targeting was 630.64±84.52 mm3, which was significantly (p < 0.01) less than those in the MNP-β-Glu-PEG group without magnetic targeting (1772.98±183.66 mm3), the MNP-β-Glu group after magnetic targeting (1891.70±101.44 mm3), the β-Glu group (2028.77±206.37 mm3) and the control group (2284.09±268.50 mm3) (Figure 6B).
 |
Figure 6 Magnetically directed enzyme/prodrug therapy with MNP-β-Glu-PEG/amygdalin in vivo. (A) A representative image showing mice with RM1 xenografts treated with AMY, β-Glu/AMY, MNP-β-Glu-PEG/AMY, MNP-β-Glu/MT/AMY or MNP-β-Glu-PEG/MT/AMY (17 days after treatment). (B) Volume (mm3) of the RM1 xenograft in mice treated according to the scheme described above. Tumor sizes were measured using a caliper on the indicated days (*p < 0.001; based on a two-tailed t-test, assuming unequal variance, n=6). (C) Representative apoptosis images and cell apoptosis index analyses of tumor tissues for all groups (compared with the control group, *p < 0.01; compared with the MNP-β-Glu-PEG/MT/AMY group, **p < 0.01). The apoptotic cells are brown. Scale bar: 50μm. β-Glu, MNP-β-Glu and MNP-β-Glu-PEG were administered via the tail vein, and the magnetic targeting time was 2 hours. Abbreviations: AMY, amygdalin; β-Glu, β-glucosidase; MT, magnetic targeting. |
To elucidate the possible mechanisms of tumor growth inhibition, tumor tissue sections were evaluated by the TUNEL assay. Representative micrographs are shown in Figure 6C. The apoptosis index (4.26±0.88%) of tumor tissue in the control group was minimal. The apoptotic index (45.15±7.82%) of the tumor tissue in the MNP-β-Glu-PEG group after magnetic targeting was significantly higher than those of the other groups (p < 0.01) (Figure 6C). No significant differences in the apoptosis index were observed in the MNP-β-Glu-PEG group without magnetic targeting, the MNP-β-Glu group after magnetic targeting and the β-Glu group (p > 0.05).
In summary, the tumor growth inhibition test showed that the application of an external magnetic field increased the delivery of MNP-β-Glu-PEG to tumor sites and allowed it to accumulate to a quantity sufficient to activate the prodrug amygdalin, thereby inducing RM1 cell necrosis and inhibiting subcutaneous tumor growth.
Toxicity Analysis
Figure 7A–D illustrates changes in body weight and cardiac, hepatic and renal function indexes over time in each group of mice. The mice in each group were in good general condition during drug administration, and no sudden death or other death occurred. Figure 7A shows that the weights of the tumor-bearing mice injected with normal saline gradually increased, while the weights of the control group and the amygdalin + β-Glu group gradually decreased after the fifth day of administration. Some mice had diarrhea in the two groups, suggesting that amygdalin does affect the growth of mice, possibly due to gradual accumulation in the body, producing certain toxic side effects. The weights of mice in the amygdalin + MNP-β-Glu-PEG group and those of mice in the amygdalin + MNP-β-Glu group after magnetic targeting also showed a gradually increasing trend initially and then a gradual decrease, which may also be related to the toxic effects of amygdalin. However, the decreasing weight trend was weaker than that in the control group, suggesting that the partial hydrolysis of amygdalin may reduce its toxic side effects. In contrast, after magnetic targeting, the amygdalin + MNP-β-Glu-PEG group exhibited slow tumor growth and a gradual increase in body weight, which may be because the complete hydrolysis of amygdalin led to significantly reduced toxic side effects in mice.
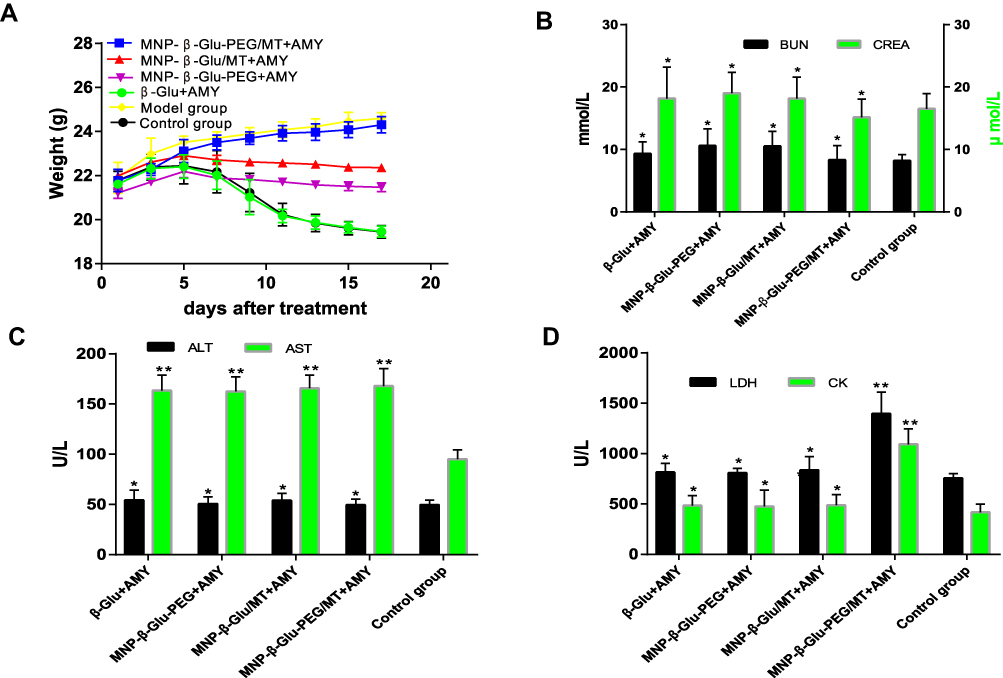 |
Figure 7 Toxic effects of combined administration to mice. (A) Body weight changes after administration (17 days after treatment, n=6). (B) Effects of administration on the kidney function (BUN, Cr) of mice (compared with the control group, *p > 0.05, n=6). (C) Effects of administration on the liver function (ALT, AST) of mice (compared with the control group, *p > 0.05, **p < 0.01, n=6). (D) Effects of administration on the heart function (LDH, CK) of mice (compared with the control group, *p > 0.05, ** p < 0.01, n=6). |
The biochemical indexes (Figure 7C) showed that AST was significantly higher in all drug administration groups than in the control group (p < 0.01), suggesting that drug intervention had a certain effect on liver function. Compared with those in the control group, CK and LDH were significantly increased in the amygdalin + MNP-β-Glu-PEG group after magnetic targeting (Figure 7D) (p < 0.01), suggesting that combined drug administration had greater effects on cardiac function, probably because hydrocyanic acid can produce significant toxicity to the heart. Pathological analysis of HE-stained heart, liver, spleen, lung and kidney sections showed no obvious pathological changes, such as tissue necrosis (Figure S3), suggesting that the treatment strategy did not cause pathological damage to various organs of the body.
Discussion
Since the anticancer effect of amygdalin was discovered, it has been widely studied as an alternative tumor drug. Although amygdalin alone may inhibit tumor growth through various mechanisms, its inhibition efficiency is low, and one study found that the inhibition efficiency at a concentration of 10 mg/mL was approximately 2 times that of the control group.12 In this study, low concentrations of amygdalin had no obvious killing effect on the 3 prostate cancer cell types within 24 hours, and only high concentrations of amygdalin (> 10 mg/mL) inhibited tumor cell growth. It is impossible to achieve such a high concentration of amygdalin for in vivo application.6 Therefore, it is necessary to use β-Glu in combination with amygdalin to improve the killing efficiency. The hydrocyanic acid produced after the combined administration directly caused cell necrosis, which significantly increased the inhibition efficiency of amygdalin in the 3 prostate cancer cell types, reducing the IC50 by several dozen-fold. Flow cytometry analysis also showed that β-Glu loaded on the MNPs activated amygdalin to inhibit tumor cell growth and that the effect was similar to that of free β-Glu. It has been reported that the combination of amygdalin and β-Glu kills liver cancer cells by inducing apoptosis,15 while other experimental studies have suggested that the main mechanism by which combined drug administration kills tumor cells is not by inducing apoptosis but by directly causing cell necrosis.26 In this study, DNA electrophoresis and AO/EB fluorescence staining confirmed that after combined drug administration, tumor cells died mainly through the necrotic pathway. Western blot experiments confirmed that both amygdalin alone and combined drug administration could induce changes in the expression of apoptosis-related proteins, suggesting that the BAX/Bcl-2 mitochondrial apoptosis pathway may be involved in the process of cell death. Therefore, apoptosis and necrosis may be present simultaneously during combined drug administration-mediated tumor cell killing. We analyzed the reasons. HCN produced by amygdalin activation inhibits cytochrome oxidase in the mitochondrial respiratory chain, blocks oxidative phosphorylation, and induces ATP depletion.47 Usually, maintaining a certain level of ATP is required for the execution of apoptotic programs because it is a highly regulated process involving a number of ATP-dependent steps. An adequate ATP level is necessary for the activation of the apoptosis pathway.48 In this treatment strategy, although the apoptosis pathway was activated, a precipitous drop in ATP levels converted the cells to the necrosis pathway. Therefore, compared with other chemotherapy drugs, amygdalin/β-Glu combination therapy strategies are superior. First, macromolecular chemotherapeutic drugs require receptor-mediated internalization to exert their effects. However, the produced HCN possesses favorable diffusivity and can easily enter into tumor cells, thus avoiding difficulties related to drug internalization. In addition, common chemotherapy drugs inhibit cancer cells through the apoptosis pathway and may cause apoptosis resistance.49 Combination therapy strategies induce cancer cell death independently of the apoptosis pathway and thus may have potential for cancer therapy.
One of the main purposes of targeted enzyme/prodrug strategies is to reduce the toxic effects of coadministration on normal tissues by targeted activation. The key is to administer the prodrug when the enzyme activity is at its highest in the tumor tissue and at its lowest in the circulation. This maximizes tumor suppression and minimizes systemic toxicity. Previous enzyme/prodrug strategies have been greatly limited by challenges in determining the amount of enzyme accumulation in tissues. Although the quantity of conjugates delivered to the tumor site can be indirectly displayed by fluorescent labeling,50 this method is affected by the amount and intensity of fluorescein, and the high background of in vivo tissue also leads to low accuracy. In this study, we demonstrated that in the application of MNP-loaded enzymes, the degree of particle aggregation at the tumor site can be monitored by MRI. Thus, due to the modifiability of the particles, particle aggregation at the tumor site should be monitored accurately by combining various imaging methods.34 These findings provide a better basis for assessing the timing of the administration of prodrugs. However, unlike the particles, the activity of the loaded enzyme will gradually decrease while being transported in the blood circulation, and the activity of the enzyme reaching the tumor site will gradually decrease with time. Therefore, the degree of particle aggregation at the tumor site cannot fully represent the enzyme activity at the tumor site. A method for dynamically determining enzyme activity in tumor tissue would further facilitate the application of this strategy.
The active targeting of targeting vectors such as antibodies requires binding of the antibody to a tumor-specific surface antigen. However, the antibody-coupled enzyme must first exit the blood vessels before entering the tumor tissue. This process is limited by many factors such as the diameter of capillaries and the hydrostatic pressure of the tumor tissue. Therefore, the quantity of antibody-enzyme conjugates entering the tumor tissue through tumor blood vessels is limited. Even if some antibody-coupled enzymes enter the tumor tissue, the expression of tumor-associated antigens is heterogeneous, and the amount of antigen on the surface of the tumor tissue that the conjugates can bind may be low. All such conditions will reduce the targeting efficiency of treatment strategies such as antibody-targeted enzyme prodrugs. In contrast, when using magnetic nanoparticles as a drug carrier, the targeted aggregation of the drug is unrelated to tumor cell antigens but is mainly related to the EPR effect caused by the large vascular fissure at the tumor site and the characteristics of the applied magnetic field. The intensity and duration of the applied magnetic field are highly controllable,51 and its effects are not dependent on EPR action,52 while the EPR effect depends largely on the vascular characteristics53 of tumor tissue and the stability of particles in blood.54 Therefore, improving the EPR effect by increasing the stability of the particles in the circulation becomes the primary means of increasing the accumulation of particles at the tumor site. In this study, the stability of the enzyme-loaded particles was significantly improved by PEG modification, and the amount of particles aggregation at the tumor site under the applied magnetic field was significantly higher than that of the non-PEG-modified enzymatic particles, while the β-Glu activity in the tumor tissue reached 134.89±14.18 mU/g tissue, which was also significantly higher than that of the latter. In particular, PEG modification significantly reduced the number of enzyme-loaded particles that accumulated in the liver and spleen, thereby reducing the organ toxicity caused by activation of amygdalin in the liver and spleen. Therefore, the use of PEG-modified enzyme-loaded particles in combination with magnetic targeting may be an effective method for increasing the amount of β-Glu accumulation at the tumor site.
MNP is often used to carry chemotherapeutic drugs because of their advantages, but accumulation in the liver and spleen may lead to serious toxic effects. The use of the MDEPT strategy may reduce the toxicity of chemotherapy drugs to the liver, and the amplification effect of enzyme activation and the bystander effect may increase the tumor cell killing efficiency. However, it is still possible that the administered prodrug is activated by enzyme-loaded particles in the liver. Moreover, the product hydrocyanic acid is a highly toxic small molecule that rapidly disperses to various important organs and is especially toxic to nerve cells and the heart. Studies have found that combined drug administration significantly inhibits the growth of tumor cells in vitro, but in vivo experiments on orthotopic glioma growth inhibition in rats demonstrated that combined drug administration significantly increased mortality in rats due to severe toxic effects.28 Therefore, the use of hydrocyanic acid-based enzyme/prodrug treatment strategies should minimize the toxic effects on the central nervous system and the cardiac system. In the experiment, the subcutaneous tumors are far from the important organs, which reduces the toxicity of hydrocyanic acid to other organs to some extent. Therefore, our experimental groups showed only the elevation of CK and LDH, representing cardiac function, but no death of mice occurred during the experimental procedure, and there was no obvious pathological change in the heart or other organs. Similar to subcutaneously transplanted tumors, the clinical lesion area of prostate cancer is far from important organs, and a magnetic field can easily be applied through the anus and is thus more appropriate for the clinical application of MDEPT.
Owing to their high safety, MNP have been approved by the FDA as an in vivo image contrast agent. This study confirmed the feasibility and advantages of using MNP as carriers for enzymes to achieve the targeted enzymatic activation of prodrugs. However, there are still many challenges for the clinical application of this therapy strategy: 1. Because MNPs accumulate in the liver and spleen, a very small fraction (approximately 1.4%) of the total i.v. administered dose of MNP-β-Glu-PEG reaches the tumor site, causing waste and increasing toxicity. 2. In this study, the level of hydrocyanic acid produced in the targeted tumor tissue may have been insufficient to achieve complete tumor growth inhibition. However, hydrocyanic acid disperses easily in tissues, and further increases in its production could enhance the possibility of toxicity in various organs. 3. Because the enzyme activity in the targeted tumor tissue cannot be maintained after a single drug administration, repeated targeted administration and repeated intratumoral injection of amygdalin are required, which increases the accumulation of enzyme-loaded particles in organs such as the liver and spleen and the probability of concurrent tumor tissue infection. The technology and procedures involved in this strategy require continued improvement to achieve clinical application for the treatment of prostate cancer.
Conclusions
In this study, we prepared β-Glu-loaded MNPs and then used in vitro experiments to confirm their ability to activate amygdalin-mediated prostate cancer cell death. In these experiments, tumor cell death induced by combination therapy was mainly mediated by cell necrosis. To further verify the feasibility of the MDEPT strategy, we used MNPs as carriers of β-Glu to achieve targeted delivery to subcutaneous tumor sites and found that PEG modification combined with magnetic targeting is an effective means of enhancing the accumulation of enzyme-loaded particles in tumor tissue. Accumulated enzyme-loaded particles in tumors activated the locally administered prodrug amygdalin, leading to significant inhibition of tumor growth. This treatment strategy showed only mild toxicity to the heart and liver. These results suggest that the MDEPT strategy based on amygdalin/β-Glu may have good prospects for clinical application in the treatment of prostate cancer.
Acknowledgments
The authors deeply thank Mr. Kangle Lv for performing SQUID measurements and MRI analyses (College of Resources and Environmental Science, South-Central University for Nationalities, Wuhan, Hubei, China). The authors also thank Ms. Juan Min for technical assistance with the flow cytometric analyses (Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan, Hubei, China). The authors are also grateful to American Journal Experts for English language editing of the manuscript.
Disclosure
The authors report no conflicts of interest in the work
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
2. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
3. Cornford P, Bellmunt J, Bolla M, et al. EAU-ESTRO-SIOG guidelines on prostate cancer. Part II: treatment of relapsing, metastatic, and castration-resistant prostate cancer. Eur Urol. 2017;71(4):630–642. doi:10.1016/j.eururo.2016.08.002
4. Aloysius H, Hu L. Targeted prodrug approaches for hormone refractory prostate cancer. Med Res Rev. 2015;35(3):554–585. doi:10.1002/med.21333
5. Blaheta RA, Nelson K, Haferkamp A, Juengel E. Amygdalin, quackery or cure? Phytomedicine. 2016;23(4):367–376. doi:10.1016/j.phymed.2016.02.004
6. Song Z, Xu X. Advanced research on anti-tumor effects of amygdalin. J Cancer Res Ther. 2014;10(Suppl 1):3–7. doi:10.4103/0973-1482.139743
7. Makarevic J, Rutz J, Juengel E, et al. Amygdalin blocks bladder cancer cell growth in vitro by diminishing cyclin A and cdk2. PLoS One. 2014;9(8):e105590. doi:10.1371/journal.pone.0105590
8. Juengel E, Thomas A, Rutz J, et al. Amygdalin inhibits the growth of renal cell carcinoma cells in vitro. Int J Mol Med. 2016;37(2):526–532. doi:10.3892/ijmm.2015.2439
9. Lee HM, Moon A. Amygdalin regulates apoptosis and adhesion in Hs578T triple-negative breast cancer cells. Biomol Ther(Seoul). 2016;24(1):62–66. doi:10.4062/biomolther.2015.172
10. Liczbiński P, Bukowska B. Molecular mechanism of amygdalin action in vitro: review of the latest research. Immunopharmacol. Immunotoxicol. 2018;40(3):212–218. doi:10.1080/08923973.2018.1441301
11. Saleem M, Asif J, Asif M, Saleem U. Amygdalin, from apricot kernels, induces apoptosis and causes cell cycle arrest in cancer cells: an updated review. Anticancer Agents Med Chem. 2018;18(12):1650–1655. doi:10.2174/1871520618666180105161136
12. Chang HK, Shin MS, Yang HY, et al. Amygdalin induces apoptosis through regulation of Bax and Bcl-2 expressions in human DU145 and LNCaP prostate cancer cells. Biol Pharm Bull. 2006;29(8):1597–1602. doi:10.1248/bpb.29.1597
13. Makarević J, Tsaur I, Juengel E, et al. Amygdalin delays cell cycle progression and blocks growth of prostate cancer cells in vitro. Life Sci. 2016;147:137–142. doi:10.1016/j.lfs.2016.01.039
14. Jaswal V, Palanivelu J, Ramalingam C. Effects of the Gut microbiota on amygdalin and its use as an anti-cancer therapy: substantial review on the key components involved in altering dose efficacy and toxicity. Biochem Biophys Rep. 2018;14:125–132. doi:10.1016/j.bbrep.2018.04.008
15. Zhou C, Qian L, Ma H, et al. Enhancement of amygdalin activated with β-D-glucosidase on HepG2 cells proliferation and apoptosis. Carohydr Polym. 2012;90(1):516–523. doi:10.1016/j.carbpol.2012.05.073
16. Rooseboom M, Commandeur JN, Vermeulen NP. Enzyme-catalyzed activation of anticancer prodrugs. Pharmacol Rev. 2004;56(1):53–102. doi:10.1124/pr.56.1.3
17. Sharma SK, Bagshawe KD. Antibody Directed Enzyme Prodrug Therapy (ADEPT): trials and tribulations. Adv Drug Deliv Rev. 2017;118:2–7. doi:10.1016/j.addr.2017.09.009
18. Zhang J, Kale V, Chen M. Gene-directed enzyme prodrug therapy. AAPS J. 2015;17(1):102–110. doi:10.1208/s12248-014-9675-7
19. Xu G, McLeod HL. Strategies for enzyme/prodrug cancer therapy. Clin Cancer Res. 2001;7(11):3314–3324.
20. Schellmann N, Deckert PM, Bachran D, Fuchs H, Bachran C. Targeted enzyme prodrug therapies. Mini Rev Med Chem. 2010;10(10):887–904. doi:10.2174/138955710792007196
21. Sharma SK, Bagshawe KD. Translating antibody directed enzyme prodrug therapy(ADEPT) and prospects for combination. Expert Opin Biol Ther. 2017;17(1):1–13. doi:10.1080/14712598.2017.1247802
22. Bae YH, Park K. Targeted drug delivery totumors: myths, reality and possibility. J Control Release. 2011;153(3):198–205. doi:10.1016/j.jconrel.2011.06.001
23. Syrigos KN, Rowlinson-Busza G, Epenetos AA. In vitro cytotoxicity following specific activation of amygdalin by beta-glucosidase conjugated to a bladder cancer-associated monoclonal antibody. Int J Cancer. 1998;78(6):712–719. doi:10.1002/(SICI)1097-0215(19981209)78:6<712::AID-IJC8>3.0.CO;2-D
24. Syrigos KN, Epenetos AA. Antibody directed enzyme prodrug therapy (ADEPT): a review of the experimental and clinical considerations. Anticancer Res. 1999;19(1A):605–613.
25. Kousparou CA, Epenetos AA, Deonarain MP. Antibody-guided enzyme therapy of cancer producing cyanide results in necrosis of targeted cells. Int J Cancer. 2002;99(1):138–148. doi:10.1002/ijc.10266
26. Li J, Li H, Zhu L, et al. The adenovirus-mediated linamarase/linamarin suicide system: a potential strategy for the treatment of hepatocellular carcinoma. Cancer Lett. 2010;289(2):217–227. doi:10.1016/j.canlet.2009.08.016
27. García-Escudero V, Gargini R, Izquierdo M. Glioma regression in vitro and in vivo by a suicide combined treatment. Mol Cancer Res. 2008;6(3):407–417. doi:10.1158/1541-7786.MCR-07-0243
28. Girald W, Collin A, Izquierdo M. Toxicity and delivery methods for the linamarase/linamarin/glucose oxidase system, when used against human glioma tumors implanted in the brain of nude rats. Cancer Lett. 2011;313(1):99–107. doi:10.1016/j.canlet.2011.08.029
29. Cardoso VF, Francesko A, Ribeiro C, Bañobre-López M, Martins P, Lanceros-Mendez S. Advances in magnetic nanoparticles for biomedical applications. Adv Healthc Mater. 2018;7(5):1700845.
30. Wang D, Fei B, Halig LV, et al. Targeted iron-oxide nanoparticle for photodynamic therapy and imaging of head and neck cancer. ACS Nano. 2014;8(7):6620–6632. doi:10.1021/nn501652j
31. Chertok B, David AE, Yang VC. Magnetically-enabled and MR-monitored selective brain tumor protein delivery in rats viamagnetic nanocarriers. Biomaterials. 2011;32(26):6245–6253. doi:10.1016/j.biomaterials.2011.05.004
32. Wong J, Prout J, Seifalian A. Magnetic nanoparticles: new perspectives in drug delivery. Curr Pharm Des. 2017;23(20):2908–2917. doi:10.2174/1381612823666170215104659
33. Ulbrich K, Holá K, Šubr V, Bakandritsos A, Tuček J, Zbořil R. Targeted drug delivery with polymers and magnetic nanoparticles: covalent and noncovalent approaches, release control, and clinical studies. Chem Rev. 2016;116(9):5338–5431. doi:10.1021/acs.chemrev.5b00589
34. Xu C, Shi S, Feng L, et al. Long circulating reduced graphene oxide-iron oxide nanoparticles for efficient tumor targeting and multimodality imaging. Nanoscale. 2016;8(25):12683–12692. doi:10.1039/C5NR09193D
35. Bai J, Wang JT, Mei KC, Al-Jamal WT, Al-Jamal KT. Real-time monitoring of magnetic drug targeting using fibered confocal fluorescencemicroscopy. J Control Release. 2016;244(Pt B):240–246. doi:10.1016/j.jconrel.2016.07.026
36. Próspero AG, Quini CC, Bakuzis AF, et al. Real-time in vivo monitoring of magnetic nanoparticles in the bloodstream by AC biosusceptometry. J Nanobiotechnology. 2017;15(1):22. doi:10.1186/s12951-017-0257-6
37. Ricco R, Doherty CM, Falcaro P. Evaluation of coupling protocols to bind beta-glucosidase on magnetic nanoparticles. J Nanosci Nanotechnol. 2014;14(9):6565–6573. doi:10.1166/jnn.2014.9353
38. Verma ML, Chaudhary R, Tsuzuki T, Barrow CJ, Puri M. Immobilization of β-glucosidase on a magnetic nanoparticle improves thermostability: application in cellobiose hydrolysis. Bioresour Technol. 2013;135:2–6. doi:10.1016/j.biortech.2013.01.047
39. Zhou J, Zhang J, David AE, Yang VC. Magnetic tumor targeting of β- glucosidase immobilized iron oxide nanoparticles. Nanotechnology. 2013;24(37):375102. doi:10.1088/0957-4484/24/37/375102
40. Zhou J, Zhang J, Gao W. Enhanced and selective delivery of enzyme therapyto 9L-glioma tumor via magnetic targeting of PEG-modified, β-glucosidase-conjugated iron oxide nanoparticles. Int J Nanomedicine. 2014;9:2905–2917. doi:10.2147/IJN.S59556
41. Tseng SH, Chou MY, Chu IM. Cetuximab-conjugated ironoxide nanoparticles for cancerimaging and therapy. Int J Nanomedicine. 2015;10:3663–3685. doi:10.2147/IJN.S80134
42. Chertok B, Cole AJ, David AE, Yang VC. Comparison of electron spin resonance spectroscopy and inductively-coupled plasma optical emission spectroscopy for biodistribution analysis of iron-oxide nanoparticles. Mol Pharm. 2010;7(2):375–385. doi:10.1021/mp900161h
43. Albanese A, Tang PS, Chan WC. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng. 2012;14:1–16. doi:10.1146/annurev-bioeng-071811-150124
44. Chen X, Gao C. Influences of size and surface coating of gold nanoparticles on inflammatory activation of macrophages. Colloids Surf B Biointerfaces. 2017;160:372–380. doi:10.1016/j.colsurfb.2017.09.046
45. Arami H, Khandhar A, Liggitt D, Krishnan KM. In vivo delivery, pharmacokinetics, biodistribution and toxicity of iron oxide nanoparticles. Chem Soc Rev. 2015;44(23):8576–8607. doi:10.1039/C5CS00541H
46. Cole AJ, David AE, Wang J, Galbán CJ, Yang VC. Magnetic brain tumortargeting and biodistribution of long-circulating PEG-modified, cross-linkedstarch-coated iron oxide nanoparticles. Biomaterials. 2011;32(26):6291–6301. doi:10.1016/j.biomaterials.2011.05.024
47. Leavesley HB, Li L, Prabhakaran K, Borowitz JL, Isom GE. Interaction of cyanide and nitric oxide with cytochrome c oxidase: implications for acute cyanidetoxicity. Toxicol Sci. 2008;101(1):101–111. doi:10.1093/toxsci/kfm254
48. Tatsumi T, Shiraishi J, Keira N, et al. Intracellular ATP is required for mitochondrial apoptotic pathways in isolated hypoxic rat cardiacmyocytes. Cardiovasc Res. 2003;59(2):428–440. doi:10.1016/S0008-6363(03)00391-2
49. Fulda S. Tumor resistance to apoptosis. Int J Cancer. 2009;124(3):511–515. doi:10.1002/ijc.24064
50. Alam MK, E-Sayed A, Barreto K, Bernhard W, Fonge H, Geyer CR. Site-specific fluorescent labeling of antibodies and diabodies using SpyTag/SpyCatcher system for in vivo optical imaging. Mol Imaging Biol. 2019;21(1):54–66. doi:10.1007/s11307-018-1222-y
51. Liu YL, Chen D, Shang P, Yin DC. A review of magnet systems for targeted drug delivery. J Control Release. 2019;302:90–104. doi:10.1016/j.jconrel.2019.03.031
52. Park J, Kadasala NR, Abouelmagd SA, et al. Polymer-iron oxide composite nanoparticles for EPR-independent drug delivery. Biomaterials. 2016;101:285–295. doi:10.1016/j.biomaterials.2016.06.007
53. Mei KC, Bai J, Lorrio S, Wang JT, Al-Jamal KT. Investigating the effect of tumor vascularization on magnetic targeting in vivo using retrospective design of experiment. Biomaterials. 2016;106:276–285. doi:10.1016/j.biomaterials.2016.08.030
54. Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99(Pt A):28–51. doi:10.1016/j.addr.2015.09.012
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.