Back to Journals » Drug Design, Development and Therapy » Volume 19
Magnesium Isoglycyrrhizinate Alleviates Alectinib-Induced Hepatotoxicity by Inhibiting Mitochondrial Damage-Mediated Pyroptosis
Authors Chen Y, Wang C, Wu Y, Zeng Y, Xie S, Weng J, Guo L, Fu J, Zhou T, Zhang X, Zhou Z ![]()
Received 8 March 2025
Accepted for publication 14 July 2025
Published 22 July 2025 Volume 2025:19 Pages 6219—6233
DOI https://doi.org/10.2147/DDDT.S523455
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Muzammal Hussain
Yizhang Chen,1,2,* Chenxiang Wang,1,* Yuna Wu,3,* Yuhan Zeng,1,4 Shangjing Xie,5 Jialu Weng,5 Lei Guo,1,2 Jing Fu,1 Tao Zhou,1 Xiuhua Zhang,4 Ziye Zhou4,5
1Department of Pharmacy, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China; 2School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China; 3The Eye Hospital, School of Ophthalmology & Optometry, Wenzhou Medical University, Wenzhou, People’s Republic of China; 4Clinical Research Center, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China; 5Zhejiang Key Laboratory of Intelligent Cancer Biomarker Discovery and Translation, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ziye Zhou, Clinical Research Center and Zhejiang Key Laboratory of Intelligent Cancer Biomarker Discovery and Translation, The First Affiliated Hospital of Wenzhou Medical University, Nanbaixiang Street, Ouhai District, Wenzhou, 325000, People’s Republic of China, Tel +86-577-55579593, Email [email protected] Xiuhua Zhang, Clinical Research Center, The First Affiliated Hospital of Wenzhou Medical University, Nanbaixiang Street, Ouhai District, Wenzhou, 325000, People’s Republic of China, Tel +86-577-55579590, Email [email protected]
Introduction: Alectinib is a widely used first-line ALK inhibitor for fusion-positive non-small cell lung cancer. However, its clinical use is limited by hepatotoxicity, and its mechanism remains unclear. This study aims to elucidate how alectinib induces liver injury and to explore a potential protective strategy.
Methods: In vitro, AML-12 hepatocytes were incubated with alectinib to determine cell viability and morphology by using CCK-8 assay and optical microscopy, respectively. Necrosis was assessed by flow cytometry after Annexin V-FITC/PI staining. Mitochondrial damage was analyzed by measuring membrane potential, ultrastructure, and respiratory chain complex activities using JC-1 staining under fluorescence microscopy, transmission electron microscopy, and assay kits, respectively. Intracellular reactive oxygen species (ROS) levels were detected using DCFH-DA staining and flow cytometry. Pyroptosis- and oxidative stress-related proteins (NLRP3, GSDMD-N, P20, cleaved IL-1β, Nrf2, HO-1) were quantified by Western blot. In vivo, C57BL/6J mice were divided into control, alectinib treatment, and alectinib plus magnesium isoglycyrrhizinate (MgIG) treatment groups. Serum ALT and AST were measured to assess liver function. Hepatic oxidative stress was evaluated by SOD and MDA levels. Inflammatory cytokines including IL-1β and TNF-α were measured by corresponding kits. Liver histopathology was examined by hematoxylin-eosin staining.
Results: We found alectinib could induce the death of AML-12 hepatocytes. Alectinib impaired both the function and structure of mitochondria and caused a significant increase in ROS levels. The excessive accumulation of ROS triggered oxidative stress and finally resulted in cell pyroptosis in AML-12 cells. MgIG was found to alleviate mitochondrial damage and reduce ROS levels, restore the Nrf2/HO-1 signaling pathway, thereby inhibiting oxidative stress and pyroptosis caused by alectinib.
Conclusion: Alectinib induces elevated ROS levels in hepatocytes by damaging mitochondria and causing oxidative stress in hepatocytes, which results in cell pyroptosis and ultimately hepatotoxicity, whereas MgIG can treat alectinib-induced hepatic injury by restoring mitochondrial function and structure.
Keywords: alectinib, mitochondria damage, oxidative stress, pyroptosis, magnesium isoglycyrrhizinate
Introduction
Lung cancer is the second most frequently occurred cancer globally and has the highest cancer-associated death rate among all cancers.1 Approximately 85% of lung cancer patients were diagnosed as non-small cell lung cancer (NSCLC).2 Nearly 5% of patients with NSCLC exhibit anaplastic lymphoma kinase (ALK) rearrangement.3 Alectinib is the second-generation ALK-tyrosine kinase inhibitor (ALK-TKI). In 2015, the US Food and Drug Administration (FDA) approved alectinib for treating ALK fusion-positive NSCLC patients with advanced brain metastases either resistant to crizotinib or becoming more severe after receiving crizotinib therapy. In 2017, the FDA ratified alectinib as a first-line medication for advanced ALK fusion-positive NSCLC. In the approved clinical trials of ALK-TKIs, liver injury has been frequently reported as a serious safety issue related to the drug itself.4 This is despite the fact that alectinib has been used globally for many years with good clinical efficacy, but some studies have found that alectinib causes liver injury in patients, including elevated levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase and bilirubin were present in 51%, 34%, 47% and 39% of patients, with grade 3 and higher serious adverse events in 3.6%, 4.8%, 1.2% and 2.4% of patients, respectively.5 Another real-world study showed that approximately 4.35% of the 12,000 patients treated with alectinib experienced significant liver damage,6 which greatly reduced clinical benefits for patients with usage of alectinib.
Drug induced liver injury (DILI) is one kind of serious adverse drug reactions. This creates the onset of adverse consequences, which can lead to poor medication adherence and worsened prognosis, exacerbate existing injuries, and result in the need for additional treatment. The occurrence of DILI may be associated with a variety of mechanisms, including mitochondria dysfunction, increased reactive oxygen species (ROS) levels, presence of elevated apoptosis and necrosis, and is estimated to be 25% of safety failures in the clinic.7,8 The specific mechanism of DILI caused by alectinib has not yet been clarified, which caused a lack of corresponding preventive and therapeutic strategies in the clinic. Therefore, the aim of this study is to investigate the specific mechanism of alectinib-mediated hepatotoxicity.
Pyroptosis is a lytic and inflammatory programmed cell death, typically triggered by inflammasomes and executed by gasdermin proteins.9 The pathogenesis of many diseases is related to pyroptosis, such as inflammatory diseases,10 metabolic diseases,11 nervous system diseases,12 cardiovascular diseases,13 and liver diseases.14 Oxidative stress refers to a tendency towards oxidation in vivo, leading to inflammatory infiltration of neutrophils, increased secretion of proteases, and the production of large amounts of oxidative intermediates, which is considered as an important factor causing aging and disease, such as osteoporosis,15 heart failure16 and inflammatory diseases.17,18 Mitochondria are important organelle that maintain cellular material and energy and are sensitive to damage.19 Mitochondrial damage is now understood to play a role in adverse drug reactions such as acetaminophen20 and doxorubicin.21 Our study aims to explore the relationship between the three factors and find the effective therapeutic target.
DILI in the clinical setting is preternatural and irregular, which made it could not be identical therapy. Currently, most of the clinical responses to DILI are immediate drug discontinuation and hepatoprotective drug therapy, and even liver transplantation is required for severe DILI.22 Natural products had been proven to be effective in treating various diseases, particularly inflammatory diseases and DILI.23–25 Magnesium isoglycyrrhizinate (MgIG) is synthesized by isomerization and salification from 18β‐glycyrrhizic acid, which is a triterpenoid extracted from Glycyrrhiza glabra. Some studies showed that MgIG could inhibit oxidation and inflammation and used for improvement of liver function.26–31 Based on its potential therapeutic mechanism, we investigated the effect of MgIG on the hepatic injury induced by alectinib.
Materials and Methods
Drugs and Reagents
Alectinib (purity ≥ 98%) was obtained from Aladdin Biochemical Technology Co., Ltd (Shanghai, China). Cell culture grade dimethyl sulfoxide (DMSO; purity ≥ 99.5%) was purchased from Solarbio Science & Technology Co., Ltd. (Beijing, China). N‐acetyl‐L‐cysteine (NAC; purity ≥ 98%) were purchased from MedChemExpress Co., Ltd. (New Jersey, United States). Cell culture grade phosphate‐buffered saline (PBS) was purchased from Solarbio Science & Technology Co., Ltd. (Beijing, China). MgIG (5 mg/mL) was purchased from Zhengda Tianqing Pharmaceutical Group Co., Ltd. (Jiangsu, China). 0.25% Trypsin-EDTA was purchased from Thermo Fisher Scientific (China) Co., Ltd. (Shanghai, China). 100x Penicillin/Streptomycin was purchased from Thermo Fisher Scientific (China) Co., Ltd. (Shanghai, China).
Cell Culture
AML-12 cells were obtained from the ATCC Center (Virginia, United States). The catalog number of cell line is CRL-2254. Cells were cultured in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12; Gibco, Shanghai, China) supplemented with 10% foetal bovine serum (FBS; Gibco, Shanghai, China) and 1% P/S in a humidified cell incubator (Thermo Fisher Scientific, Massachusetts, United States) with 5% CO2 at 37°C. Mycoplasma contamination was monitored weekly during cell culture. 10-cm plates were used for culturing the cells. Once the cells had grown to approximately 80–90% of the plate’s capacity, they were passaged to a new plate at a ratio of 1:3. Cells within passages 3–10 were utilized for all experiments to ensure consistency and viability.
Cell Viability Assay
AML-12 cells were treated with 0–30 μM alectinib, 10 mM NAC, 1 mg/mL MgIG, 10 μM alectinib plus 10 mM NAC pretreatment, 10 μM alectinib plus 1 mg/mL MgIG pretreatment, respectively. Cell viability was evaluated using CCK-8 assay (MedChemExpress, New Jersey, United States). 5×103 AML-12 cells were added into each well of a 96-well plate. After 24 h of alectinib treatment, the supernatant was removed and then added 100 μL of CCK-8 solution (10 μL CCK-8 and 90 μL culture medium) to each well of the 96-well plate. After 0.5 h incubation in a humidified cell incubator, the optical density (OD) value was measured at a wavelength of 450 nm using a micro-plate reader (Thermo Fisher Scientific, Massachusetts, United States). Based on the OD value, cell viability was calculated.
Cell Death Analysis
AML-12 cells were treated with 5 μM alectinib, 10 μM alectinib, respectively. An Annexin V-FITC apoptosis detection kit (Beyotime, Shanghai, China) was used to gauge necrosis and apoptosis. After treatment with alectinib for 24 h, hepatocytes were digested by trypsin solution without EDTA and washed with pre‐chilled PBS for three times. Then, the hepatocytes were resuspended in a 210 μL binding buffer with 5 μL Annexin V-FITC and 10 μL propidium iodide in it, and incubated for 15 min. Blank samples were used to adjust the voltage of FSC axis and SSC axis, and the single-stained samples were used to adjust the compensation. Then, hepatocytes were analyzed immediately.
Western Blotting
AML-12 cells were treated with 5 μM alectinib, 10 μM alectinib, 10 μM alectinib plus 10 mM NAC pretreatment, 10 μM alectinib plus 1 mg/mL MgIG pretreatment, respectively. Protein was extracted from AML-12 cells by adding protease inhibitor (Beyotime, Shanghai, China) and phosphatase inhibitor (Beyotime, Shanghai, China) with RIPA lysate (Beyotime, Shanghai, China). Samples containing 25 µg of protein were separated on 10% gel and transferred to a 0.45 µm PVDF membrane (Millipore, Massachusetts, United States). Afterwards, the PVDF membrane was closed with 50 g/L skim milk powder, and incubated with antibodies against GSDMD, P20 and c-IL-1β (1:1000, Affinity Biosciences, Jiangsu, China), NF-κB (P65), p-P65 (1:1000, Cell Signaling Technology, Massachusetts, United States), NLRP3 (1:1000, Abcam, Massachusetts, United States), Nrf2, HO-1 and β‐actin (1:1000, Proteintech, Hubei, China) overnight at 4°C. After washed by tris buffered saline with Tween-20 (TBST) for three times, the PVDF membrane was incubated with the secondary antibody (1:10000, Proteintech, Hubei, China) for 1 h at room temperature. Luminescence was then detected using the enhanced chemiluminescence system.
Mitochondrial Membrane Potential (MMP) Assay
An MMP assay kit with JC-1 (Beyotime, Shanghai, China) was used to determined the variation of MMP. AML-12 cells were treated with 5 μM alectinib, 10 μM alectinib, 10 μM alectinib plus 10 mM NAC pretreatment, 10 μM alectinib plus 1 mg/mL MgIG pretreatment, respectively. One well was incubated with 10 μM carbonyl cyanide m-chlorophenyl hydrazone (CCCP) for 30 min as a positive control. Then, the supernatant of each well was removed, and 1 mL culture medium and 1 mL 1 × JC-1 working solution were added in each well. After 30 min incubation in a humidified cell incubator, the supernatant was removed and washed with pre‐chilled 1 × JC-1 binding buffer for at least twice, and then added 2 mL culture medium to maintain the cell viability. Then hepatocytes were observed using a fluorescence microscope.
Transmission Electron Microscopy
The cells with different treatments (vehicle group, 10 μM alectinib treatment group, 10 μM alectinib treatment plus 1 mg/mL MgIG pretreatment group, respectively) from the 6-well cell culture plate were collected and fixed with transmission electron microscopy fixation fluid at 4°C overnight. The samples were gradient dehydrated with ethanol, embedded in epoxy resin and sectioned at a thickness of 80 nm. Finally, ultrathin sections were double-stained by uranium acetate and lead citrate, and examined under a H‐7500 electron microscope (Hitachi, Tokyo, Japan).
ROS Quantification Assay
AML-12 cells were treated with 5 μM alectinib, 10 μM alectinib, 10 μM alectinib plus 10 mM NAC pretreatment, 10 μM alectinib plus 1 mg/mL MgIG pretreatment, respectively. After removing the supernatant, AML-12 cells were rinsed twice with PBS and incubated with 2′,7′-Dichlorodihydrofluorescein diacetate (DCFH‐DA; MedChemExpress, New Jersey, United States) working solution in a humidified cell incubator for at least 30 min. After washed with DMEM without FBS for three times, cells were collected and the ROS levels were quantified using FITC values by flow cytometer.
Evaluation of NADH-CoQ Reductase Activity
AML-12 cells were treated with 5 μM alectinib, 10 μM alectinib, 10 μM alectinib plus 10 mM NAC pretreatment, 10 μM alectinib plus 1 mg/mL MgIG pretreatment, respectively. NADH-CoQ reductase activity was measured using the NADH-CoQ reductase activity assay kit (Solarbio, Beijing, China). After hepatocytes were digested by trypsin, 5 × 106 of cells were added with 1 mL mitochondria isolation reagent, then the hepatocytes were homogenized (30 strokes) under ice bath and performed gradient centrifugation. Finally, the mitochondria were lysed by ultrasonic and NADH-CoQ reductase activity was assessed spectrophotometrically at 340 nm.
Evaluation of CoQ-Cytochrome C Reductase Activity
AML-12 cells were treated with 5 μM alectinib, 10 μM alectinib, 10 μM alectinib plus 10 mM NAC pretreatment, 10 μM alectinib plus 1 mg/mL MgIG pretreatment, respectively. CoQ-cytochrome C reductase activity was measured using the CoQ-cytochrome C reductase activity assay kit (Solarbio, Beijing, China). After hepatocytes were digested by trypsin, 5 × 106 of cells were added with 1 mL mitochondria isolation reagent, then the hepatocytes were homogenized (30 strokes) under ice bath and performed gradient centrifugation. Finally, the mitochondria were lysed by ultrasonic and CoQ-cytochrome C reductase activity was assessed spectrophotometrically at 550nm.
In vivo Experiment
Healthy male C57BL/6 mice (SPF-grade, 7-week-old, 20 ± 2 g) were obtained from the Laboratory Animal Center of Wenzhou Medical University. All operations in this study followed the Guideline for Ethical Review of Laboratory Animal Welfare (China, 2018), and the protocol was approved by the Review of Laboratory Animal Ethics Committee of the First Affiliated Hospital of Wenzhou Medical University (approval number: WYYY-AEC-2024-220). The mice were maintained in a 12-hour light-dark cycle (light on at 7:00) to mimic the natural environment. Mice were kept under laboratory conditions with 25°C for 7 days before the experiment to minimize suffering. Then, mice were randomly assigned into three groups by analysts using online random number generators: control group (n=6), alectinib treatment group (n=6), and alectinib plus MgIG treatment group (n=6) to ensure the reliability of the study. In alectinib plus MgIG treatment group, mice were treated by intraperitoneal injection of MgIG (25 mg/kg) daily followed 2 h later by gavage alectinib (150 mg/kg); in the alectinib treatment group, the 5 mL/kg of saline was administered intraperitoneally daily and 2 h later the 150 mg/kg of alectinib was given by gavage; in the control group, the 5 mL/kg of saline was administered intraperitoneally daily and 2 h later the 10 mL/kg of 0.1% carboxymethylcellulose sodium (used for dissolving alectinib) was given by gavage. To minimize the potential confounders, mice were kept in the same environment, and the same person carried out the operation. To minimize the influence of subjective biases, neither operators nor analysts are aware of each other’s experiments. After 2 weeks, the mice were euthanized for collecting blood and liver tissues to evaluate hepatic injury by histopathology staining and determining transaminases, inflammatory factors and oxidative stress markers.
Histopathology Staining
Liver tissues from mice of each group were soaked in 4% paraformaldehyde for 24 h and rinsed under running water overnight, then made a gradient dehydration by ethanol. The liver tissues were soaked in xylene twice for 1 h each, and embedded in paraffin. The blocks were cut into 4 µm slices, and the slices were placed in warm water and then transferred to slides for drying. The slices were baked in an oven at 66°C for 4 h, then made a gradient rehydration, rinsed twice with water. Then, staining with hematoxylin for 5 min, washed with dilute hydrochloric acid and rinsed twice with water, then stained with eosin. The slices were made a gradient dehydration by ethanol and soaked in xylene twice and then sealed with neutral resin. The liver damage was assessed by comprehensive scoring of liver histopathology with Camargo32 criteria (Table 1).
 |
Table 1 The Scoring Criteria for Liver Histopathology |
Transaminase Assessment
The ALT and AST levels were assessed by ALT assay kit and AST assay kit (Nanjing Jiancheng, Jiangsu, China). 20 μL stroma was added in control well, with 20 μL stroma and 5 μL plasma were added in determination well. 20 μL 2,4-Dinitrophenylhydrazine and 5 μL plasma were added in control well, with 20 μL 2,4-Dinitrophenylhydrazine was added in determination well after 30 min at 37°C. 200 μL 0.4% NaOH was added in control and determination wells after 20 min. Microplate reader was used to gauge the absorbance at 505 nm (ALT) and 510 nm (AST) after 15 min at room temperature.
Determination of Inflammatory Factors and Oxidative Stress Markers
The liver tissue of mice was added with nine times amount of 0.9% NaCl to prepare 10% tissue homogenate in the ice bath. Then homogenate was centrifuged at 8000 g, 4°C for 10 min to obtain supernatant. IL-1β, TNF-α, SOD and MDA were determined by corresponding assay kits (Solarbio, Beijing, China), respectively. Plasma was used to determine IL-1β and TNF-α levels, supernatant of homogenate was used to determine SOD activities and MDA levels according to the kit instructions, respectively.
Statistical Analysis
Statistical analysis was conducted with GraphPad Prism v8.0.2 (IBM, California, United States). All results were obtained from at least independent 3 samples (five samples in CCK-8 experiment; six samples in mice). When the group data followed a normal distribution, the data were calculated as means ± standard deviation (SD) and the analysis of multiple groups comparisons conducted by Tukey’s test; when variance between groups was nonnormal distribution, the data were calculated as median and the analysis of multiple groups comparisons conducted by Kruskal–Wallis rank sum test. P < 0.05 expressed statistically significant.
Results
Alectinib Induced Hepatocytes Pyroptosis
Firstly, we evaluated the cytotoxicity of alectinib and found that alectinib caused significant toxicity to AML-12 cells in vitro, with an IC50 value of 10.19 μM for 24 h treatment (Figure 1A). Flow cytometry result found that alectinib primarily caused hepatocytes necrosis (Figure 1B). Meanwhile, it could be observed that spherical vacuolization appeared in the alectinib-treated cells by the light microscope, which reminded us of the morphological features of pyroptosis (Figure 1C). In order to further validate the mechanism of alectinib’s damage on hepatocytes, Western blotting assay was conducted (Figure 1D), and the results showed that alectinib could significantly up-regulate the expressions of the inflammasome NLRP3, the pyroptosis executing protein GSDMD-N, the pyroptosis signaling molecule P20, and the inflammatory factor activated IL-1β in hepatocytes, respectively. The classical cellular pyroptosis pathway starts at the inflammasome NLRP3, its activation allows caspase1 to form P20. The active P20 could cut GSDMD to form GSDMD-N, which would induce the perforation of cell membrane and rupture the cells, leading to inflammation. Furthermore, the active P20 could form active IL-1β, which could converge inflammatory cells and aggravate inflammatory response.33,34 These findings suggested that alectinib could cause the pyroptosis of hepatocytes via the classical cellular pyroptosis pathway and in turn produces liver injury.
 |
Figure 1 Alectinib induced hepatocytes pyroptosis. (A) AML-12 cells were treated with different concentrations of alectinib for 24 h and cell survival was determined by CCK-8 reagent (n = 5). (B) Apoptosis and necrosis of AML-12 cells were detected by flow cytometry after 24 h treatment with different concentrations of alectinib (n = 3). (C) Images for observation of AML-12 cells under light microscope (scale bar: 100 μm; the arrows point to pyroptosis hepatocytes). (D) The Western blotting results of GSDMD-N, full-length GSDMD (GSDMD-FL), NLRP3, P20, c-IL-1β and β-actin, respectively (n = 3). The data are expressed as mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 vs vehicle group by Tukey’s test. |
Alectinib Induced Oxidative Stress in Hepatocytes via Mitochondrial Damage Causing Excessive Accumulation of ROS
In order to investigate the mechanism of pyroptosis caused by alectinib, we examined the morphology of organelles by transmission electron microscopy, which revealed that alectinib caused mitochondrial swelling and loss of structural integrity in AML-12 cells (Figure 2A). The MMP assay showed that alectinib could decrease the MMP level of hepatocytes (Figure 2B). Further validation of NADH-CoQ reductase and CoQ-cytochrome C reductase levels in mitochondria revealed that alectinib could inhibit the activities of NADH-CoQ reductase (Figure 2C) and CoQ-cytochrome C reductase (Figure 2D). The disruption of mitochondrial function and structure could cause the dysregulation of ROS levels.8,35 And the result of flow cytometry consistently showed an elevated level of ROS (Figure 2E), suggesting that the mitochondrial damage caused by alectinib led to an excessive accumulation of intracellular ROS. Based on the elevation of p-P65, we further determined the levels of its upstream regulators. Western blotting results revealed that alectinib increased the expression levels of Nrf2 and HO-1 in hepatocytes, respectively (Figure 2F). These findings suggested that alectinib caused oxidative stress in hepatocytes via mitochondrial damage.
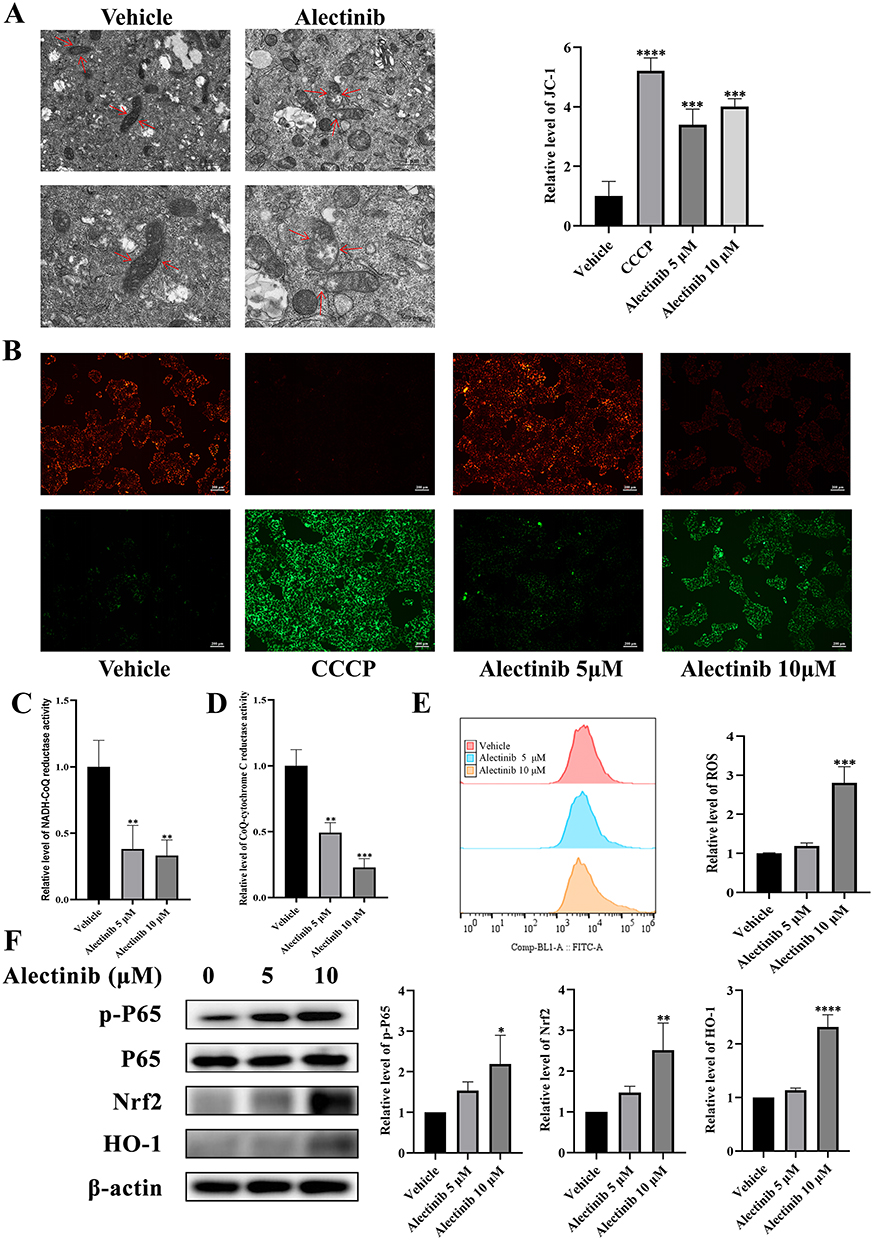 |
Figure 2 Alectinib induced oxidative stress in hepatocytes via mitochondrial damage causing excessive accumulation of ROS. (A) Transmission electron microscopy images of mitochondrial morphology (scale bar: 1 μm and 500 nm; the arrows point to mitochondrial) in hepatocytes treated with or without alectinib (10 μM). (B) The results obtained from JC-1 fluorescence images (scale bar: 200 μm) of AML-12 cells treated with different concentrations of alectinib for 24 h (CCCP was used as the positive control, n = 3). (C) The relative levels of NADH-CoQ reductase activities of AML-12 cells treated with different concentrations of alectinib for 24 h (n = 3). (D) The relative levels of CoQ-cytochrome C reductase activities of AML-12 cells treated with different concentrations of alectinib for 24 h (n = 3). (E) The flow cytometry results of AML-12 cells after 24 h of different concentration alectinib treatment (n = 3). (F) The Western blotting results of protein p-P65, P65, Nrf2, HO-1 and β-actin, respectively (n = 3). The data are expressed as mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 vs vehicle group by Tukey’s test. |
Alectinib-Induced Excessive Accumulation of ROS Caused Hepatocyte Pyroptosis
To further explore the role of ROS accumulation in alectinib-induced hepatotoxicity, we used NAC as ROS scavenger to verify the recovery of liver injury. AML-12 cells were pretreated with 10 mM of NAC for 2 h before treatment with alectinib. Cell viability assay demonstrated that NAC was nontoxic to AML-12 cells and alleviated alectinib-induced cell death (Figure 3A). Flow cytometry proved NAC could significantly reduce ROS levels in alectinib-treated hepatocytes (Figure 3B). The MMP assay revealed a significant reduction of MMP in hepatocytes with alectinib treatment; this reduction was effectively reversed by NAC pretreatment (Figure 3C). Furthermore, the activities of NADH-CoQ reductase (Figure 3D) and CoQ-cytochrome C reductase (Figure 3E) were elevated significantly in hepatocytes with NAC treatment. Western blotting results indicated that alectinib elevated the levels of Nrf2, HO-1 and the phosphorylation of p65, facilitating oxidative stress in hepatocytes (Figure 3F). The elevation of NLRP3, GSDMD-N, P20, and c-IL-1β protein expression levels demonstrated that alectinib further promoted hepatocyte pyroptosis (Figure 3G). And NAC could resist oxidative stress and further alleviated hepatocyte pyroptosis (Figure 3F and G). These findings suggested that excessive accumulation of ROS expressed a direct role in the hepatic injury caused by alectinib, and targeting ROS might be a reasonable treatment option.
 |
Figure 3 Alectinib-induced excessive accumulation of ROS caused hepatocyte pyroptosis. (A) AML-12 cells were pretreated with or without 10 mM NAC before 10 μM alectinib treatment and cell survival was determined by CCK-8 reagent (n = 5). (B) The flow cytometry results of AML-12 cells after 24 h of 10 μM alectinib treatment which is pretreated with 10 mM of NAC or not (n = 3). (C) The results obtained from JC-1 fluorescence images (scale bar: 200 μm) of AML-12 cells pretreated with or without 10 mM NAC before 10 μM alectinib treatment for 24 h (CCCP was used as the positive control, n = 3). (D) The relative levels of NADH-CoQ reductase activities of AML-12 cells after 24 h of 10 μM alectinib treatment which is pretreated with 10 mM of NAC or not (n = 3). (E) The relative levels of CoQ-cytochrome C reductase activities of AML-12 cells after 24 h of 10 μM alectinib treatment which is pretreated with 10 mM of NAC or not (n = 3). (F) The results obtained from Western blotting of protein p-P65, P65, Nrf2, HO-1 and β-actin, respectively (n = 3). (G) The results obtained from Western blotting of protein GSDMD-N, GSDMD-FL, NLRP3, P20, c-IL-1β and β-actin, respectively (n = 3). The data are expressed as mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 vs vehicle group by Tukey’s test; #p < 0.05, ##p < 0.01, ###p < 0.001 and ####p < 0.0001 vs 10 μM alectinib treatment group by Tukey’s test. |
MgIG Alleviated Alectinib-Induced Pyroptosis in Hepatocyte by Treating Mitochondrial Damage
To verify whether MgIG could prevent and treat alectinib-induced liver injury, AML-12 cells were pretreated with 1 mg/mL of MgIG for 2 h before treated with alectinib. Cell viability assay showed that MgIG was nontoxic to hepatocytes and significantly improved the survival rate of hepatocytes with alectinib treatment (Figure 4A). Transmission electron microscopy observed that the mitochondrial damage caused by alectinib was effectively alleviated by MgIG (Figure 4B). The fluorescence microscopy result confirmed that MgIG could significantly elevate the MMP level in hepatocytes treated by alectinib (Figure 4C). And the activities of NADH-CoQ reductase (Figure 4D) and CoQ-cytochrome C reductase (Figure 4E) were also increased significantly by MgIG. Flow cytometry result exhibited MgIG could significantly attenuate intracellular ROS levels (Figure 4F). Western blotting results proved that MgIG could cause a significant reduction in the levels of Nrf2, HO-1, and phosphorylation of P65, suggesting that MgIG alleviated the oxidative stress induced by alectinib (Figure 4G). Furthermore, expression levels of NLRP3, GSDMD-N, P20, and c-IL-1β were significantly down-regulated, suggesting that MgIG effectively mitigated alectinib-induced hepatocytes pyroptosis (Figure 4H). These results suggested MgIG could prevent and treat mitochondrial damage to alleviate alectinib-induced pyroptosis.
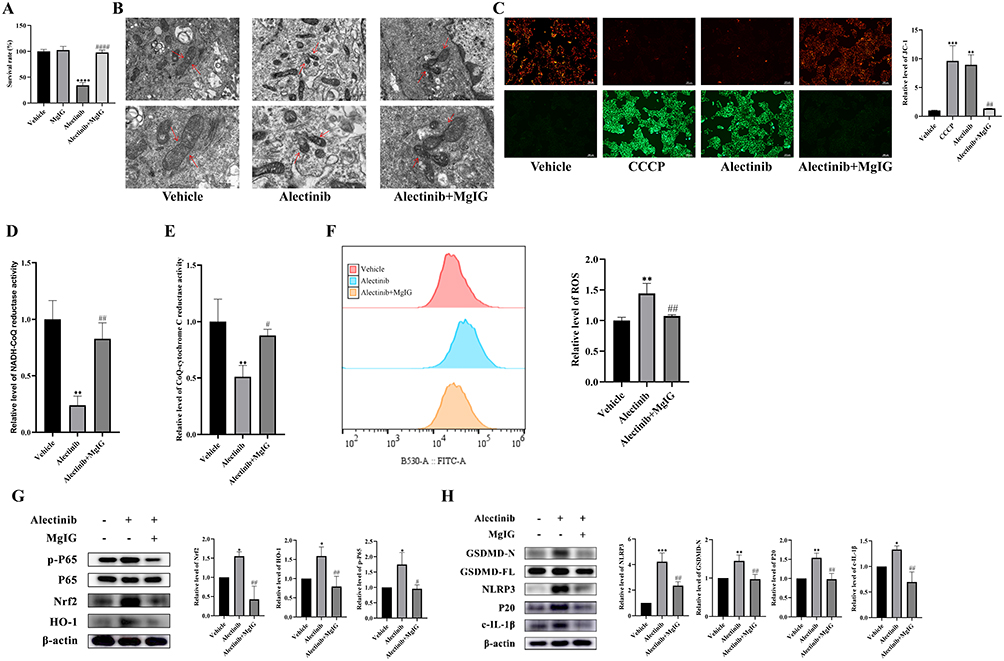 |
Figure 4 MgIG alleviated alectinib-induced pyroptosis in hepatocyte by treating mitochondrial damage. (A) AML-12 cells were pretreated with or without 1 mg/mL MgIG before 10 μM alectinib treatment for 24 h and cell survival was determined by CCK-8 reagent (n = 5). (B) Transmission electron microscopy images of mitochondrial morphology (scale bar: 1 μm and 500 nm; the arrows point to mitochondrial) in hepatocytes pretreated with or without 1 mg/mL MgIG before 10 μM alectinib treatment. (C) The results obtained from JC-1 fluorescence images (scale bar: 200 μm) of AML-12 cells pretreated with or without 1 mg/mL MgIG before 10 μM alectinib treatment for 24 h (CCCP was used as the positive control, n = 3). (D) The relative levels of NADH-CoQ reductase activities of AML-12 cells after 24 h of 10 μM alectinib treatment which is pretreated with 1 mg/mL of MgIG or not (n = 3). (E) The relative levels of CoQ-cytochrome C reductase activities of AML-12 cells after 24 h of 10 μM alectinib treatment which is pretreated with 1 mg/mL of MgIG or not (n = 3). (F) The results obtained from flow cytometry of AML-12 cells after 24 h of 10 μM alectinib treatment which is pretreated with 1 mg/mL of MgIG or not (n = 3). (G) The results obtained from Western blotting of protein p-P65, P65, Nrf2, HO-1 and β-actin, respectively (n = 3). (H) The results obtained from Western blotting experiments of protein GSDMD-N, GSDMD-FL, NLRP3, P20, c-IL-1β and β-actin, respectively (n = 3). The data are expressed as mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 vs vehicle group by Tukey’s test; #p < 0.05, ##p < 0.01 and ####p < 0.0001 vs 10 μM alectinib treatment group by Tukey’s test. |
MgIG Alleviated Alectinib-Induced Liver Injury in Mice
Compared with those in vehicle group, the levels of ALT and AST elevated significantly in alectinib treatment group, and the levels of ALT and AST reduced significantly by MgIG treatment (Figure 5A and B). The inflammatory factors IL-1β (Figure 5C) and TNF-α (Figure 5D) elevated significantly after alectinib treatment, while IL-1β and TNF-α reduced significantly with MgIG treatment subsequently. In addition, SOD level (Figure 5E) and MDA level (Figure 5F) in liver tissue of mice proved obvious increase after alectinib treatment and reduction with MgIG treatment subsequently. Liver histopathology indicated inflammatory cells infiltration, spotty necrosis, hepatocytes edema and acidophilic degeneration were found in alectinib treatment group, while significant mitigation of the liver injury was observed in alectinib plus MgIG treatment group (Figure 5G), and the scores for liver histopathology showed statistically significant difference (Figure 5H). These findings evidenced that MgIG could alleviate alectinib-induced liver injury in vivo, which in agreement with in vitro results.
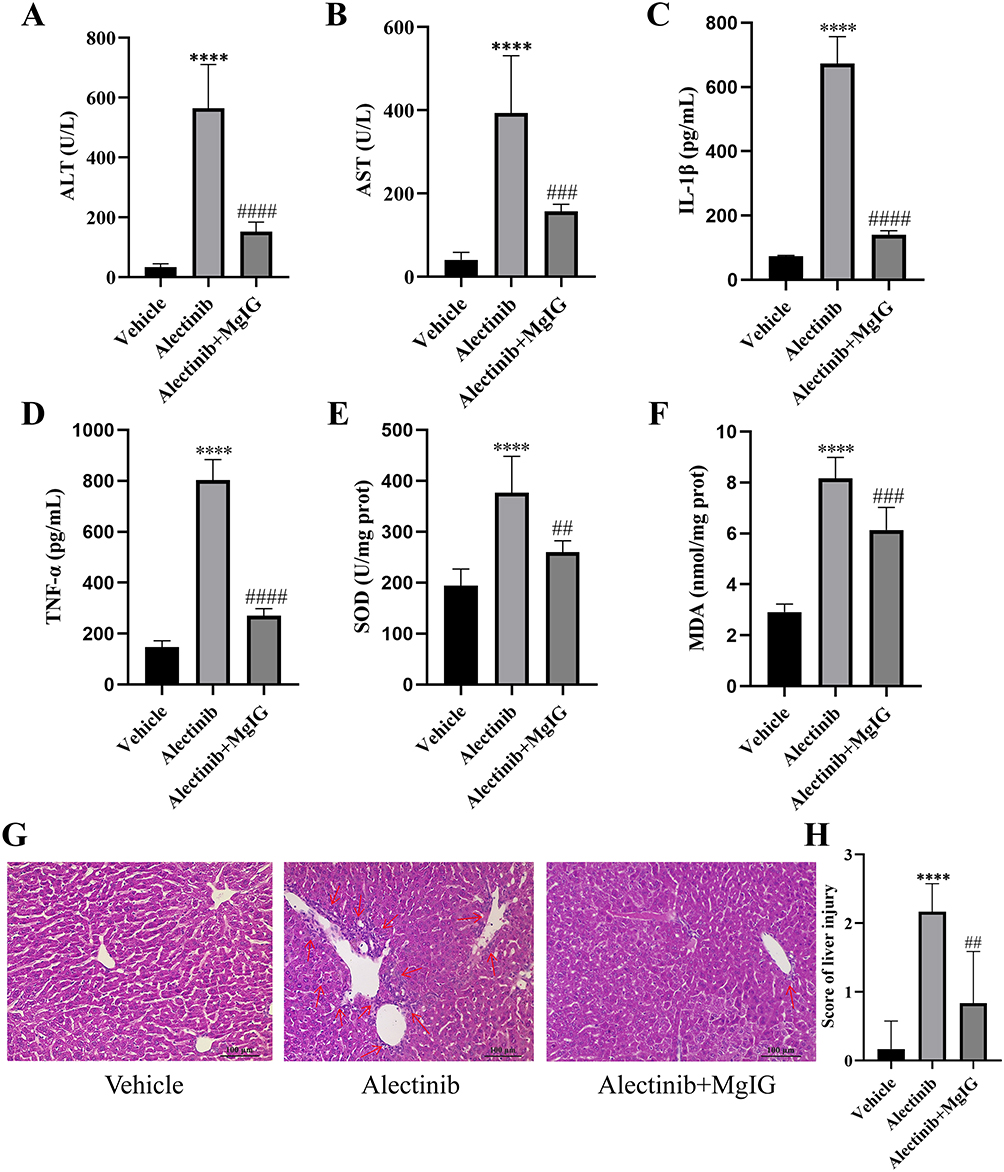 |
Figure 5 MgIG alleviates alectinib-induced liver injury in mice. (A) The plasma ALT level of mice pretreated with or without 25 mg/kg MgIG before 150 mg/kg alectinib treatment for 14 days (n = 6). (B) The plasma AST levels of mice pretreated with or without 25 mg/kg MgIG before 150 mg/kg alectinib treatment for 14 days (n = 6). (C) The plasma IL-1β levels of mice pretreated with or without 25 mg/kg MgIG before 150 mg/kg alectinib treatment for 14 days (n = 6). (D) The plasma TNF-α levels of mice pretreated with or without 25 mg/kg MgIG before 150 mg/kg alectinib treatment for 14 days (n = 6). (E) The liver SOD activities of mice pretreated with or without 25 mg/kg MgIG before 150 mg/kg alectinib treatment for 14 days (n = 6). (F) The liver MDA levels of mice pretreated with or without 25 mg/kg MgIG before 150 mg/kg alectinib treatment for 14 days (n = 6). (G) The pathological images (scale bar: 100 μm) of the liver pretreated with or without 25 mg/kg MgIG before 150 mg/kg alectinib treatment for 14 days. (H) The results obtained from the hematoxylin-eosin staining of the liver pretreated with or without 25 mg/kg MgIG before 150 mg/kg alectinib treatment for 14 days (n = 6). The data are expressed as mean ± SD; ****p < 0.0001 vs vehicle group by Tukey’s test; ##p < 0.01, ###p < 0.001 and ####p < 0.0001 vs alectinib treatment group by Tukey’s test. |
Discussion
Alectinib is an effective ALK fusion-positive NSCLC targeted drug and widely used in clinical. The mechanism of alectinib-induced hepatotoxicity is currently unclear in the field. Our study suggests that alectinib causes excessive accumulation of ROS by damaging mitochondria, which puts hepatocytes into an oxidative stress state and further causes hepatocyte pyroptosis. Our study elucidated the mechanism of alectinib-induced hepatotoxicity and fills the gap in the field. We found that MgIG regulated mitochondrial dysfunction caused by alectinib, so as to decrease ROS accumulation. MgIG further alleviated the oxidative stress and finally reversed the pyroptosis. The consistent in vivo experiment results support MgIG as a medicine for alectinib-induced hepatotoxicity.
Oxidative stress is an imbalance state between oxidation and anti-oxidation. The mitochondrial respiratory chain is the foremost source of intracellular ROS.36 Thus, oxidative stress may be associated with mitochondrial damage.37–39 Nrf2 is the major regulator of the anti-oxidation, and Nrf2/HO-1 pathway can be activated to resist the oxidative stress in DILI.40,41 Our study found that alectinib treatment decreased the activities of NADH-CoQ reductase, CoQ-cytochrome C reductase, and the levels of MMP, resulting in increase of the ROS levels. Excessive accumulation of ROS further promotes expressions of Nrf2, HO-1, and the phosphorylation of P65, which is also a characteristic factor of pyroptosis to generate NLRP3 inflammasome.42 Therefore, alectinib damages mitochondria to cause the excessive accumulation of ROS, which puts hepatocytes into an oxidative stress state, resulting in hepatocyte pyroptosis.
Pyroptosis is inflammatory necrosis mediated by activation of inflammasome and is characterized by the release of cytokines associated with inflammation.9 In the classical cellular pyroptosis signaling pathway, activation of P65 phosphorylation promotes the expression of the inflammasome component NLRP3, which leads to the formation of caspase-1 P20 subunit and activation of the downstream execution protein GSDMD. Cleavage of GSDMD releases its N-terminal domain (GSDMD-N), which mediates the release of the inflammatory cytokine IL-1β and triggers inflammation.43–47 In our study, the levels of p-P65, NLRP3, P20, GSDMD-N, c-IL-1β were significant increase, respectively. These results showed that alectinib caused hepatocyte pyroptosis by P65/NLRP3/GSDMD pathway.
Previous studies found that natural product extracts expressed therapeutic effects on DILI,25,48 which has inspired us to develop a treatment strategy for alectinib-induced hepatotoxicity. As a hepatoprotective agent, MgIG is used in clinical applications for the treatment of chronic viral hepatitis, improvement of liver function abnormalities due to its anti-inflammatory, hepatocyte membrane protection and liver function improvement effects.26–31 MgIG was used in our study to prevent and treat alectinib-induced hepatotoxicity and expressed significant effect. We also elucidated the mechanism of MgIG to treat alectinib-induced hepatic injury, which provided a theoretical reference for the treatment of DILI with other drugs.
There were some limitations in our study. Firstly, primary human hepatocytes were not applied in the in vitro experiments because its inaccessibility, which may limit the relevance of our findings to human liver physiology. Secondly, the in vivo experimental content was relatively limited, which may not fully capture the complexity of the physiological responses in a whole-organism context. These limitations suggest that further studies incorporating human hepatocyte models and more comprehensive in vivo investigations are warranted to validate and extend our current findings.
Conclusion
Our research clarifies that the mechanism underlying alectinib-induced hepatotoxicity is mitochondrial damage-mediated pyroptosis. We also demonstrate that MgIG can prevent alectinib-induced hepatotoxicity by alleviating mitochondrial damage. These findings may furnish new clinical tactics for the prevention and therapy of hepatic injury caused by alectinib and may also supply potential research direction for DILI. There were some limitations in our study, we will perform further studies to validate and extend our current findings.
Data Sharing Statement
The data will be shared on reasonable request to the corresponding author.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Zhejiang Provincial Natural Science Foundation of China under Grant No. LYY19H310007 and Project of Wenzhou Science and Technology Bureau under Grant No. Y2020831.
Disclosure
The authors report no conflicts of interest and are responsible for the content of this article.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Liu G, Pei F, Yang F, et al. Role of autophagy and apoptosis in non-small-cell lung cancer. Int J Mol Sci. 2017;18(2):367. doi:10.3390/ijms18020367
3. Huang X. The potential role of HGF-MET signaling and autophagy in the war of Alectinib versus Crizotinib against ALK-positive NSCLC. J Exp Clin Cancer Res. 2018;37(1):33. doi:10.1186/s13046-018-0707-5
4. Shah RR, Morganroth J, Shah DR. Hepatotoxicity of tyrosine kinase inhibitors: clinical and regulatory perspectives. Drug Safety. 2013;36(7):491–503. doi:10.1007/s40264-013-0048-4
5. Genentech, Inc. Alecensa. 2015. Available from: www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf.
6. Yang Y, Tan S, Pu Y, Zhang J. Safety profile and hepatotoxicity of anaplastic lymphoma kinase tyrosine kinase inhibitors: a disproportionality analysis based on FDA adverse event reporting system database. Toxics. 2025;13(3):210. doi:10.3390/toxics13030210
7. Allison R, Guraka A, Shawa IT, Tripathi G, Moritz W, Kermanizadeh A. Drug induced liver injury - a 2023 update. J Toxicol Env Heal B. 2023;26(8):442–467. doi:10.1080/10937404.2023.2261848
8. Watkins PB. Drug safety sciences and the bottleneck in drug development. Clin Pharmacol Ther. 2011;89(6):788–790. doi:10.1038/clpt.2011.63
9. Rao Z, Zhu Y, Yang P, et al. Pyroptosis in inflammatory diseases and cancer. Theranostics. 2022;12(9):4310–4329. doi:10.7150/thno.71086
10. Vasudevan SO, Behl B, Rathinam VA. Pyroptosis-induced inflammation and tissue damage. Semin Immunopathol. 2023;69:101781. doi:10.1016/j.smim.2023.101781
11. Sharma BR, Kanneganti TD. NLRP3 inflammasome in cancer and metabolic diseases. Nat Immunol. 2021;22(5):550–559. doi:10.1038/s41590-021-00886-5
12. Hu Y, Wang B, Li S, Yang S. Pyroptosis, and its role in central nervous system disease. J Mol Biol. 2022;434(4):167379. doi:10.1016/j.jmb.2021.167379
13. Toldo S, Abbate A. The role of the NLRP3 inflammasome and pyroptosis in cardiovascular diseases. Nat Rev Cardiol. 2024;21(4):219–237. doi:10.1038/s41569-023-00946-3
14. Knorr J, Wree A, Feldstein AE. Pyroptosis in steatohepatitis and liver diseases. J Mol Biol. 2022;434(4):167271. doi:10.1016/j.jmb.2021.167271
15. Kimball JS, Johnson JP, Carlson DA. Oxidative stress and osteoporosis. J Bone Joint Surg Am Vol. 2021;103(15):1451–1461. doi:10.2106/jbjs.20.00989
16. van der Pol A, van Gilst WH, Voors AA, van der Meer P. Treating oxidative stress in heart failure: past, present and future. Eur J Heart Fail. 2019;21(4):425–435. doi:10.1002/ejhf.1320
17. Tang Y, Zhou X, Cao T, et al. Endoplasmic reticulum stress and oxidative stress in inflammatory diseases. DNA Cell Biol. 2022;41(11):924–934. doi:10.1089/dna.2022.0353
18. Dutta S, Sengupta P, Slama P, Roychoudhury S. Oxidative stress, testicular inflammatory pathways, and male reproduction. Int J Mol Sci. 2021;22(18):10043. doi:10.3390/ijms221810043
19. Lumpuy-Castillo J, Amador-Martínez I, Díaz-Rojas M, et al. Role of mitochondria in Reno-cardiac diseases: a study of bioenergetics, biogenesis, and GSH signaling in disease transition. Redox Biol. 2024;76:103340. doi:10.1016/j.redox.2024.103340
20. Jaeschke H, Duan L, Nguyen N, Ramachandran A. Mitochondrial damage and biogenesis in acetaminophen-induced liver injury. Liver Res. 2019;3(3–4):150–156. doi:10.1016/j.livres.2019.10.002
21. Tai P, Chen X, Jia G, et al. WGX50 mitigates doxorubicin-induced cardiotoxicity through inhibition of mitochondrial ROS and ferroptosis. J Transl Med. 2023;21(1):823. doi:10.1186/s12967-023-04715-1
22. Katarey D, Verma S. Drug-induced liver injury. Clin Med. 2016;16(Suppl 6):s104–s109. doi:10.7861/clinmedicine.16-6-s104
23. Feng Y, Pan M, Li R, et al. Recent developments and new directions in the use of natural products for the treatment of inflammatory bowel disease. Phytomedicine. 2024;132:155812. doi:10.1016/j.phymed.2024.155812
24. Zhang S, Yan F, Luan F, et al. The pathological mechanisms and potential therapeutic drugs for myocardial ischemia reperfusion injury. Phytomedicine. 2024;129:155649. doi:10.1016/j.phymed.2024.155649
25. Deng Y, Chu X, Li Q, et al. Xanthohumol ameliorates drug-induced hepatic ferroptosis via activating Nrf2/xCT/GPX4 signaling pathway. Phytomedicine. 2024;126:155458. doi:10.1016/j.phymed.2024.155458
26. Cao Y, Xia Y, Wang Y, Shi H, Wu Y, Lu Y. MgIG attenuates oxaliplatin-induced hepatotoxicity through suppression of connexin 43 in hepatic stellate cells. J Clin Transl Hepatol. 2023;11(3):584–594. doi:10.14218/jcth.2022.00048
27. Li M, Wang C, Yu Z, et al. MgIG exerts therapeutic effects on crizotinib-induced hepatotoxicity by limiting ROS-mediated autophagy and pyroptosis. J Cell Mol Med. 2022;26(16):4492–4505. doi:10.1111/jcmm.17474
28. Lu L, Hao K, Hong Y, et al. Magnesium isoglycyrrhizinate reduces hepatic lipotoxicity through regulating metabolic abnormalities. Int J Mol Sci. 2021;22(11):5884. doi:10.3390/ijms22115884
29. Fan Z, Li Y, Chen S, et al. Magnesium isoglycyrrhizinate ameliorates concanavalin A-induced liver injury by inhibiting autophagy. Front Pharmacol. 2021;12:794319. doi:10.3389/fphar.2021.794319
30. Chen C, Liu YH, Cheng SB, Wu SL, Zhai XJ. The hepatoprotective effects of XCHD and MgIG against methotrexate-induced liver injury and inflammation in rats through suppressing the activation of AIM2 inflammasomes. Pathol Res Pract. 2020;216(4):152875. doi:10.1016/j.prp.2020.152875
31. Jiang W, Xu S, Guo H, et al. Magnesium isoglycyrrhizinate prevents the nonalcoholic hepatic steatosis via regulating energy homeostasis. J Cell Mol Med. 2020;24(13):7201–7213. doi:10.1111/jcmm.15230
32. Camargo CA, Madden JF, Gao W, Selvan RS, Clavien PA. Interleukin-6 protects liver against warm ischemia/reperfusion injury and promotes hepatocyte proliferation in the rodent. Hepatology. 1997;26(6):1513–1520. doi:10.1002/hep.510260619
33. Luo T, Zhou X, Qin M, et al. Corilagin restrains NLRP3 inflammasome activation and pyroptosis through the ROS/TXNIP/NLRP3 pathway to prevent inflammation. Oxid Med Cell Longev. 2022;2022:1652244. doi:10.1155/2022/1652244
34. Wang Q, Wen W, Zhou L, et al. LL-37 improves sepsis-induced acute lung injury by suppressing pyroptosis in alveolar epithelial cells. Int Immunopharmacol. 2024;129:111580. doi:10.1016/j.intimp.2024.111580
35. Björnsson ES, Bergmann OM, Björnsson HK, Kvaran RB, Olafsson S. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology. 2013;144(7):1419–25, 1425.e1–3; quize 19–20. doi:10.1053/j.gastro.2013.02.006
36. Guénebaut V, Schlitt A, Weiss H, Leonard K, Friedrich T. Consistent structure between bacterial and mitochondrial NADH: ubiquinone oxidoreductase (complex I). J Mol Biol. 1998;276(1):105–112. doi:10.1006/jmbi.1997.1518
37. Ho HJ, Shirakawa H. Oxidative stress and mitochondrial dysfunction in chronic kidney disease. Cells. 2022;12(1):88. doi:10.3390/cells12010088
38. Garza-Lombó C, Pappa A, Panayiotidis MI, Franco R. Redox homeostasis, oxidative stress and mitophagy. Mitochondrion. 2020;51:105–117. doi:10.1016/j.mito.2020.01.002
39. Subramaniam SR, Chesselet MF. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog Neurobiol. 2013;106–107:17–32. doi:10.1016/j.pneurobio.2013.04.004
40. Dykens JA, Will Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov Today. 2007;12(17–18):777–785. doi:10.1016/j.drudis.2007.07.013
41. Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspect Med. 2011;32(4–6):234–246. doi:10.1016/j.mam.2011.10.006
42. Labbe G, Pessayre D, Fromenty B. Drug-induced liver injury through mitochondrial dysfunction: mechanisms and detection during preclinical safety studies. Fundam Clin Pharmacol. 2008;22(4):335–353. doi:10.1111/j.1472-8206.2008.00608.x
43. Li S, Sun Y, Song M, et al. NLRP3/caspase-1/GSDMD-mediated pyroptosis exerts a crucial role in astrocyte pathological injury in mouse model of depression. JCI Insight. 2021;6(23):e146852. doi:10.1172/jci.insight.146852
44. Sun L, Ma W, Gao W, et al. Propofol directly induces caspase-1-dependent macrophage pyroptosis through the NLRP3-ASC inflammasome. Cell Death Dis. 2019;10(8):542. doi:10.1038/s41419-019-1761-4
45. Peng L, Wen L, Shi QF, et al. Scutellarin ameliorates pulmonary fibrosis through inhibiting NF-κB/NLRP3-mediated epithelial-mesenchymal transition and inflammation. Cell Death Dis. 2020;11(11):978. doi:10.1038/s41419-020-03178-2
46. Qiu Z, He Y, Ming H, Lei S, Leng Y, Xia ZY. Lipopolysaccharide (LPS) aggravates high glucose- and hypoxia/reoxygenation-induced injury through activating ROS-dependent NLRP3 inflammasome-mediated pyroptosis in H9C2 cardiomyocytes. J Diabetes Res. 2019;2019:8151836. doi:10.1155/2019/8151836
47. Liu C, Yao K, Tian Q, et al. CXCR4-BTK axis mediate pyroptosis and lipid peroxidation in early brain injury after subarachnoid hemorrhage via NLRP3 inflammasome and NF-κB pathway. Redox Biol. 2023;68:102960. doi:10.1016/j.redox.2023.102960
48. Liu YY, Zhang Y, Shan GY, et al. Hinokiflavone exerts dual regulation on apoptosis and pyroptosis via the SIX4/Stat3/Akt pathway to alleviate APAP-induced liver injury. Life Sci. 2024;354:122968. doi:10.1016/j.lfs.2024.122968
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Mechanisms and Clinical Application Prospects of Curcumin in the Treatment of Sepsis
Zhang W, Liu S, Ma W, Huang D, Li Y
Journal of Inflammation Research 2025, 18:9627-9636
Published Date: 22 July 2025
Novel Perspectives in Hepatic Ischemia-Reperfusion Injury: The cGAS-STING Pathway
Chen R, Yang T, Jiang Z, Long Y, Qian B, Fu W
Journal of Inflammation Research 2025, 18:16427-16448
Published Date: 24 November 2025