")
Back to Journals » Journal of Inflammation Research » Volume 16
Macrophage Polarization and the Regulation of Bone Immunity in Bone Homeostasis
Authors Hu K, Shang Z, Yang X, Zhang Y, Cao L
Received 31 May 2023
Accepted for publication 15 August 2023
Published 22 August 2023 Volume 2023:16 Pages 3563—3580
DOI https://doi.org/10.2147/JIR.S423819
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Kangyi Hu, Zhengya Shang, Xiaorui Yang, Yongjie Zhang, Linzhong Cao
Clinical College of Traditional Chinese Medicine, Gansu University of Chinese Medicine, Lanzhou, People’s Republic of China
Correspondence: Linzhong Cao, Clinical College of Traditional Chinese Medicine, Gansu University of Chinese Medicine, Lanzhou, 73000, People’s Republic of China, Email [email protected]
Abstract: Bone homeostasis is a dynamic equilibrium state of bone formation and absorption, ensuring skeletal development and repair. Bone immunity encompasses all aspects of the intersection between the skeletal and immune systems, including various signaling pathways, cytokines, and the crosstalk between immune cells and bone cells under both homeostatic and pathological conditions. Therefore, as key cell types in bone immunity, macrophages can polarize into classical pro-inflammatory M1 macrophages and alternative anti-inflammatory M2 macrophages under the influence of the body environment, participating in the regulation of bone metabolism and playing various roles in bone homeostasis. M1 macrophages can not only act as precursors of osteoclasts (OCs), differentiate into mature OCs, but also secrete pro-inflammatory cytokines to promote bone resorption; while M2 macrophages secrete osteogenic factors, stimulating the differentiation and mineralization of osteoblast precursors and mesenchymal stem cells (MSCs), and subsequently increase bone formation. Once the polarization of macrophages is imbalanced, the resulting immune dysregulation will cause inflammatory stimulation, and release a large amount of inflammatory factors affecting bone metabolism, leading to pathological conditions such as osteoporosis (OP), rheumatoid arthritis (RA), and steroid-induced femoral head necrosis (SANFH). In this review, we introduce the signaling pathways and related factors of macrophage polarization, as well as their relationships with immune factors, OB, OC, and MSC. We also discuss the roles of macrophage polarization and bone immunity in various diseases of bone homeostasis imbalance, as well as the factors regulating them, which may help to develop new methods for treating bone metabolic disorders.
Keywords: M1 macrophage, M2 macrophage, bone immunity, bone homeostasis
Introduction
The skeletal system is one of the important systems in the human body, providing structural support, protecting the body’s internal organs, and maintaining normal life activities of the human body.1 At the same time, mesenchymal stem cells (MSCs) in the bone marrow, as multi-lineage cells, can differentiate into osteoblasts (OBs), adipocytes, and chondrocytes, and together with OBs and OCs (OC), participate in bone metabolic balance, However, in various pathological microenvironments, cell-to-cell communication among different cells in the bone microenvironment is influenced by a variety of cellular factors. Insufficient mineralization of OB, overactivation of OC, and bone formation being less than bone resorption, lead to decreased bone density, increased bone fragility, destruction of the bone microstructure, and damage to the healthy skeleton.2 Therefore, the dynamic balance process between bone formation and bone resorption is important for maintaining bone mass and mineral stability. Macrophages, as highly plastic and dynamic innate immune cells, play a crucial role in bone immunity. They can engulf pathogens, cell fragments, and dead cells, among other exogenous and endogenous substances, and release various immune regulatory factors such as interleukins (IL) and tumor necrosis factor-alpha (TNF-α), making them significant regulators of chronic aseptic inflammation.3
In recent years, an increasing number of studies have shown that the bone immune response involves interactions between macrophages and bone cells. Macrophages can polarize into pro-inflammatory M1 macrophages and anti-inflammatory M2 macrophages, and the different phenotypes of macrophages and their cellular factors interact with MSCs, OCs, and OBs, participating extensively in the pathophysiology of bone tissues. M1 macrophages release various cytokines (such as TNF-α, IL-6, IL-1) and chemokines (such as CXCL2, CXCL8, and CXCL10) to promote the differentiation and maturation of OCs; while cytokines secreted by M2 macrophages (IL-10 and IL-4) inhibit the formation of OCs.4 At the same time, M1 macrophages can promote angiogenesis, and enhance the osteogenesis of early and mid-stage MSCs, while M2 macrophages secrete osteogenic factors, stimulating the mineralization of osteoblast precursors and MSCs.5 However, if macrophage polarization is imbalanced and M1 macrophages are activated for extended periods, a large number of inflammatory factors are released, creating a chronic inflammatory environment that can disrupt bone homeostasis. Diseases characterized by the deterioration of bone tissue microstructure and abnormal bone metabolism, such as osteoporosis (OP), steroid-induced avascular necrosis of the femoral head (SANFH), and rheumatoid arthritis (RA), which pose a serious threat to human health, cannot avoid the stimulus of chronic inflammation.6–8 As immune effector cells, macrophages are closely related to the processes of bone resorption and bone formation and participate in the development of bone homeostasis disorders in various ways.
In this review, we primarily focus on the impact of macrophage polarization and its signaling pathways on bone metabolism-related cells such as OBs, OCs, and MSCs. We find that a variety of cytokines produced during the process of macrophage polarization, such as ILs and TNF, are related to bone metabolism. Furthermore, we are interested in the impact of macrophage polarization on bone homeostasis disorders under a chronic inflammatory environment. This review aims to summarize the regulatory role of macrophage polarization and bone immunity in bone metabolic balance, which may help maintain bone homeostasis in an inflammatory environment. Targeting the polarization of M1/M2 as a treatment for bone homeostasis disorders may be an effective method.
Polarization of Macrophages
One group of macrophages consists of inflammatory macrophages that are biochemically differentiated from monocytic eukaryocytes and are transported to the site of inflammation via the bloodstream; the other group consists of tissue-resident macrophages. These tissue-specific macrophages engulf dead cells, debris, and endogenous and exogenous antigenic material, orchestrate the inflammatory process, and recruit other macrophages as needed9 Macrophages are highly plastic. As a highly plastic and dynamic cell population, macrophages are capable of changing their phenotype and altering their function to adapt to specific biological functions and pathophysiological processes in reaction to different triggers from the environment, a process known as macrophage polarization.10
There are two types of macrophage polarization: classical macrophage polarization (M1) and alternative macrophage polarization (M2). The Th1 cytokine interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), the bacterial product lipopolysaccharide (LPS), viral infection, and other stimuli that cause macrophage polarization are the main stimulators of M1 macrophages.11 IL-1, IL-6, IL-12, TNF-α, nitric oxide (NO), reactive oxygen species (ROS), and other pro-inflammatory and pro-immune cytokines are released in considerable quantities.12 Important for pathogen elimination and host protection against infections, M1 macrophages are therefore potent pro-inflammatory and antimicrobial agents. However, when M1 cells are chronically activated, the body is exposed to a chronic inflammatory environment, resulting in chronic inflammation.13 Therefore, to prevent the chronic inflammatory response caused by prolonged M1 cell activation, macrophages are polarized into M2 macrophages, which exert a significant anti-inflammatory effect.14 M2 macrophages are mainly activated by substances such as Th2 cells, immune complex.11 M2 macrophages can be classified into different subpopulations based on their activation pathways. The M2a subpopulation is activated by IL-4 and IL-13, whereas the M2b subpopulation is induced by immune complexes or Toll-like receptor agonists. The M2c subpopulation is induced by IL-10 and macrophage colony-stimulating factor (M-CSF), and the M2d subpopulation is induced by M-CSF, Toll-like receptor antagonists, IL-10, and other factors.15 The activation of M2 macrophages leads to the secretion of anti-inflammatory factors such as IL-10, TGF-β, VEGF, and small amounts of IL-12. These factors also increase the expression of chemokines and proteins such as Arg1, BMP-2, and BMP-4.16 As a result, M2 macrophages have a dual role in removing damaged cells, debris, dead cells, and apoptotic neutrophils while also promoting tissue regeneration and vascular repair17 (Figure 1).
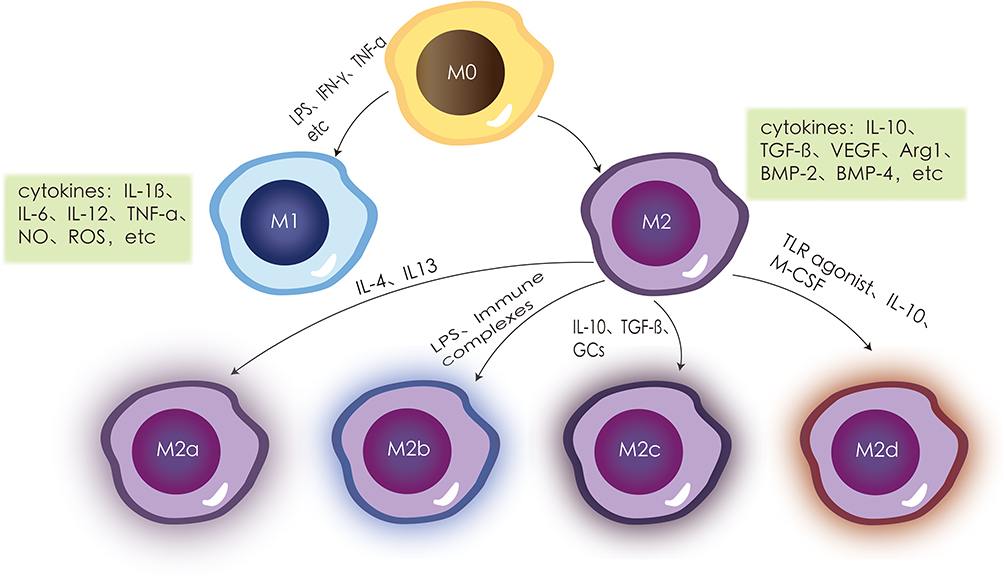 |
Figure 1 Macrophages are polarized into the M0 state and can be induced into pro-inflammatory M1 macrophages by stimulatory factors such as LPS, IFN-γ, TNF-α, secreting a large number of inflammatory cytokines such as IL-1β, IL-6, IL-12, TNF-α, nitric oxide (NO), and reactive oxygen species (ROS). In contrast, anti-inflammatory M2 macrophages are activated by substances such as Th2 cells and immune complexes, releasing a large number of anti-inflammatory factors like IL-10, transforming growth factor β (TGF-β), vascular endothelial growth factor (VEGF), and very low levels of pro-inflammatory cytokines such as IL-12. M2 is further divided into four subtypes: the M2a subgroup activated by IL-4 and IL-13, the M2b subgroup induced by immune complexes or Toll-like receptor agonists, the M2c subgroup activated by IL-10, and the M2d cell subgroup polarized by factors like macrophage colony-stimulating factor (M-CSF), Toll-like receptor antagonists, and IL-10. Note: Adapted from Deng L, Jian Z, Xu T, et al. Macrophage Polarization: An Important Candidate Regulator for Lung Diseases. Molecules. 2023;28(5):2379.18 Creative Commons Attribution (CC BY) license (https:// creativecommons.org/licenses/by/ 4.0/). |
M1 cells are primarily activated during the acute infection phase and require large amounts of energy for antimicrobial activity, so glycolysis is the best metabolic pathway for inhibiting fatty acid uptake and oxidation.19 The sustained anti-inflammatory effect of M2 cells requires long-term energy, so oxidative glucose metabolism and fatty acid oxidation are the best metabolic pathways for M2 cells.20 M2 cells require sustained long-term energy to exert their long-term anti-inflammatory effects. When the M1-M2 metabolism of macrophages is unbalanced, it is easy for M1 to be abnormally activated for a long time, M1 cannot be converted to M2, and the body turns into a chronic inflammatory state, which leads to immune dysregulation, promotes the emergence of metabolic bone disease and interferes with the balance between osteogenesis and bone metabolism.
Signaling Pathways and Factors Related to Macrophage Polarization
Macrophage polarization is a complex process that involves the interplay of various signaling pathways, cytokines, and microenvironmental factors. Recent research has uncovered several key signaling pathways involved in this process, including the Janus tyrosine kinase (JAK)-signal transducer and activator of transcription (STAT) JAK/STAT signaling pathway, Toll-like receptors (TLRs), nuclear factor kappa B (NF-κB) TLRs/NF-κB signaling pathway, phosphatidylinositol 3-kinase (PI3K)-protein kinase B PI3K/AKT signaling pathway, Notch signaling pathway, Hypoxia-inducible factor 1 (HIF-1), c-Jun N-terminal kinase (JNK) pathway, and cyclic adenosine (cAMP) pathway. These pathways are all involved in regulating macrophage polarization.
The precise roles of these signaling pathways and factors in macrophage polarization vary depending on the microenvironment. In some cases, they may act alone, while in other cases, they may act in concert with other factors to regulate macrophage polarization. The broad range of signaling pathways and factors associated with macrophage polarization underscores the crucial role it plays in maintaining bone homeostasis (Figure 2).
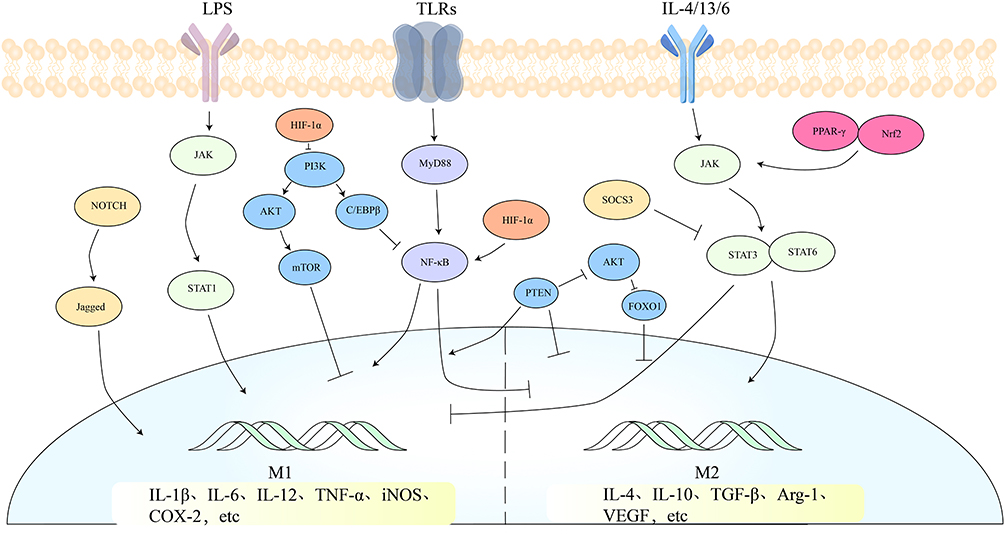 |
Figure 2 Related to macrophage polarization, there are mainly five signaling pathways: the JAK/STAT signaling pathway, the TLRs/NF-κB signaling pathway, the PI3K/AKT signaling pathway, the TLRs/NF-κB signaling pathway, the Notch signaling pathway, and the HIF-1 signaling pathway. These pathways either individually or in conjunction with other pathways regulate macrophage polarization. |
JAK/STAT Signaling Pathway
The JAK/STAT pathway is a highly conserved signaling pathway that constitutes an important cascade of signaling and cytokine transduction factors in the body. It is linked to the emergence of numerous diseases and plays a crucial role in the immune system and cell growth.21 The JAK/STAT signaling pathway is composed of cellular receptors, JAK proteins, and STAT proteins. JAK1, JAK2, JAK3, and TYK2 are the four big molecular proteins that make up the JAK family.22 JAKs have seven homologous domains and JAK kinases have two nearly identical structural domains that transfer phosphate groups, JH1 is a carboxy-terminal catalytic or kinase structural domain and JH2 is a pseudokinase (PK) or kinase-like structural domain that regulates the activity of the kinase structural domain. The Src-homology 2 (SH2) domain, which is made up of half of JH3 and half of JH4, and the FERM domain, which is made up of half of JH4, JH5, JH6, and JH7, control the binding of JAK to the box1/2 proximal area of the cytokine receptor membrane.23
The STAT family of proteins is composed of seven members: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6.24 These proteins contain a DNA-binding region, an SH3 structural domain, an SH2 structural domain, and a transcriptional activation region (TAD) located at the protein’s C-terminus. After being phosphorylated at tyrosine sites, these proteins can dimerize through their individual SH2 phosphotyrosine interactions and translocate to the nucleus, where they regulate the transcription of a wide range of genes.25
Regarding the role of the JAK/STAT signaling pathway in macrophage polarization, Tomoya found that STAT1 phosphorylation was enhanced in LPS and IFN-γ macrophages and that gene expression of IL-6, NO synthase, and cyclooxygenase (COX-2) was enhanced in the cells, demonstrating that LPS + IFN-γ stimulation enhances macrophage activation through JAK/STAT1 signaling.26 In M1, Wu discovered the high expression of the chemokine CXCL9, which was significantly enriched in the JAK/STAT signaling pathway. However, over time, CXCL9 expression decreased, resulting in the inhibition of the JAK/STAT signaling pathway. Consequently, M1 macrophages underwent a gradual differentiation into M2 macrophages.27 By stimulating the STAT3 signaling pathway, IL-4 encourages M2 phenotype, Li found that in IL-13- and IL-4-induced polarization to M2 macrophages, etanercept decreased TNF-α and IL-6/12/23 levels, increased IL-4/10 levels, decreased the Th17/Treg ratio, and promoted differentiation of M1 macrophages into M2 macrophages.28 An IL-4-mediated signaling pathway is primarily regulated by STAT6, which in turn plays a crucial role in the modulation of M2. Wang’s study demonstrated that adipose-derived stem cell-secreted exosomes (ADSC-Exo) significantly increased the phosphorylation levels of JAK and STAT6 in macrophages, thereby activating the JAK/STAT6 pathway and inducing M2 polarization.29 Inhibition or knockdown of peroxisome proliferator-activated receptor γ (PPAR-γ) and nuclear factor erythroid-related factor 2 (Nrf2) decreases the accumulation of M2 macrophage markers IL-19 and Arg1, and STAT6 can increase JAK2 phosphorylation and promote M2 macrophage polarization by triggering the Nrf2-associated antioxidant transcription and the PPAR-associated anti-inflammatory pathway.30 Xu demonstrated that scorpion venom polypeptide (SVP) could prevent M2 polarization of macrophages and reduce levels of M2 polarization-associated cytokines and fibrosis factors by inhibiting protein phosphorylation in the JAK/STAT6 pathway.31 The SOCS family of cytokine signal transduction inhibitors are suppressors of the STAT family that play a crucial part in limiting the inflammatory response. Song’s research showed that upregulating the expression of SOCS3 inhibited the phosphorylation of JAK2 and STAT3, thereby blocking the JAK2/STAT3 pathway. This blockage resulted in the suppression of M1 macrophage polarization and inflammatory responses.32
TLRs/NF-κB Signaling Pathway
A well-known regulation route of the inflammatory response, the TLRs/NF-B is implicated in several inflammatory activities. TLRs belong to the superfamily of innate immunity receptors, and pattern recognition receptors (PRRs), and are important inflammatory recognition receptors. TLRs activate NF-κB signaling by recognizing specific PAMPs or other cytokines, enhancing the expression of genes associated with inflammation as a result, starting the inflammatory response.33 The signaling pathway of Toll-like receptors (TLRs) and NF-κB is implicated in regulating macrophage polarization, TLR4/NF-κB pathway is one of the earliest and most well-established pathways identified as promoting M1 macrophage polarization. High expression levels of TLR4 have been shown to induce increased M1 and decreased M2 both in vitro and in vivo.34 MyD88, a binding protein common to all TLRs except TLR3, activates NF-κB. TLRs stimulate NF-κB entry into the nucleus via MyD88, which increases M1 polarizing factor and decreases M2 polarizing factor in macrophages, increasing the M1/M2 ratio and accelerating the body’s inflammatory response.35 Using a mouse model of colitis, Kang observed significant increases in the levels of signaling molecules TLR2 and TLR4, as well as downstream proteins MyD88 and NF-κB. Correspondingly, levels of proinflammatory factors CLL-2, IL-1β, and IL-6, and the percentage of M1 also increased.36 NF-κB, a downstream factor of TLRs, was activated and entered the nucleus to regulate gene expression and induce inflammatory responses. NF-κB induced polarization of M1 macrophages, which was attenuated by inhibition of NF-κB expression and thus of M1 trademark genes IL-6, iNOS, and TNF-α.37 In LPS-induced M1 polarization, levels of IL-1β, IL-6, TNF-α, COX-2, and NO were increased, whereas LPS strongly promoted fluorescent expression of NF-κBp65 in the nucleus and increased nuclear translocation of NF-κB, but LEE found that after recombinant human KAI1 second extracellular domain protein (rhKAI1) inhibited NF-κB factors, these factors remained stable until they were removed. The mechanism of action is that Glu214 from rhKAI1 residues strongly interacts with Lys 360 from TLR4 residues, inhibiting transcription of NF-κB, a downstream factor of TLR4, resulting in reduced levels of M1 polarization and anti-inflammatory effects in LPS-polarized macrophages.38 Tian’s research indicates that EGCG has the ability to indirectly regulate macrophages’ transformation from M1 to M2 phenotype. By reducing NF-κB activation in macrophages, EGCG helps to restore their immunomodulatory capacity.39
PI3K/AKT Signaling Pathway
The PI3K/AKT pathway is a crucial intracellular signaling pathway that responds to extracellular signals, promoting cellular functions, including metabolism, proliferation, cell survival, growth, and motility. A heterodimer of two subunits called PI3K, which binds a variety of cytokines and phosphorylates phosphatidylinositol biphosphate (PIP2) to create phosphatidylinositol triphosphate (PIP3), is made up of the catalytic p110 subunit and the regulatory p85 subunit. PIP3 acts as an important lipid second messenger, activating PDK1 and AKT, whereas phosphatase and tensin (PTEN) homologs, negative regulators of AKT, dephosphorylate PIP3 to PIP2, thereby reducing AKT activation.40 There are three different isoforms of AKT, which are AKT1, AKT2, and AKT3. These isoforms are all serine/threonine protein kinases. AKT acts as a downstream target protein of PI3K and becomes activated in response to PI3K stimulation. It plays a crucial role in macrophage polarization.41 Upon activation of the PI3K/AKT signaling pathway, the expression of surface antigens of M1 macrophages, including CD11c, CD16, and CD86, is significantly reduced. This leads to a decrease in the secretion of inflammatory factors and an inhibition of M0 polarization towards M1.42 In M1 macrophages treated with PI3K inhibitors, protein levels of p-AKT and the AKT downstream factor p-mTOR were significantly reduced, the PI3K/AKT pathway was inhibited, CD206 and iNOS levels in M1 were enhanced, and pro-inflammatory factors IL-12, TNF-α, and IL-6 were increased, promoting M1 polarization and accelerating inflammatory factor expression.12,43 Phosphorylation of AKT activates degradation of the NF-κB inhibitor kinase IKB, NF-κB is then allowed to leave the cytoplasm and move into the nucleus, where it begins to express genes that it targets to support cell survival, thereby promoting M1 by inhibiting PI3K signaling and activating NF-κB signaling.44 Wang discovered that macrophages express chemokine-like receptor 1 (CMKLR1), which stimulates the activation of C/EBPβ through the PI3K/AKT signaling pathway. This process effectively restricts the activation of NF-κB, promoting the polarization of anti-inflammatory M2 cells and inhibiting the polarization of pro-inflammatory M1 macrophages.45 Activation of the PTEN/FOXO1 pathway, a negative regulator of AKT, inhibits the transcription factor function of FOXO1 and results in the phosphorylation of AKT, which causes FOXO1 to move from the nucleus to the cytoplasm. This process leads to the suppression of TLR4 expression, thereby inhibiting the polarization of M2 macrophage.46,47 The WU study showed that in mice with periodontitis, PTEN were more prominent than in other species. In mice with periodontitis, PTEN expression was significantly higher than in controls, and high PTEN expression promoted classical macrophage polarization (M1 polarization). By inhibiting PTEN overexpression, Akt1 activation was promoted, whereas Akt2 expression was inhibited. Inhibition of PTEN in macrophages thus induced an alternative polarization of macrophages (M2 polarization). Furthermore, overexpression of Akt2 rescues the effect of PTEN on NF-κB, and inhibition of PTEN causes macrophages toward the M2 phenotype via differentially regulating Akt1 and Akt2.48
Notch Signaling Pathway
The Notch is a highly conserved pathway that plays a vital role in regulating the differentiation and function of various cells and participating in a wide range of fundamental tissue processes. It is also a crucial pathway involved in mediating the inflammatory response.49 Notch ligands can be divided into two types: Delta-type receptors and Jagged-type receptors. The γ-secretase complex initiates a series of proteolytic cleavages that activate the Notch intracellular structural domain (NICD). NICD is translocated into the nucleus, where it binds to the JK site nuclear recombination recognition sequence binding protein (RBP-J) and the Mastermind-like nuclear effector, activating downstream target genes Hey, Hes, and other basic-helix-loop-helix families of transcriptional helix-loop-helix (bHLH) repressors.50 These repressors contribute to the polarization of macrophages. Dll4 interacts with RBP-J to facilitate the polarization of M1 macrophages by binding to the Notch1 receptor, activating downstream-secretase and ADAM proteases, and allowing NICD to enter the nucleus.51 In the context of LPS and IL-4-induced differentiation of bone marrow macrophages into M1 and M2, respectively, upregulation of NICD, Hes5, and Hes1 was associated with LPS-mediated differentiation of macrophage cells into M1 cells. mRNA levels of MCP-1, iNOS, IL-12, and IL-6 released by M1 were elevated, but mRNA levels of inflammatory cytokines released by M2 were not. Inflammatory cytokine levels were elevated, but they were reversed after the use of the Notch signaling inhibitor DAPT.52 Inhibition of Notch1 overexpression accelerated the polarization of M2 macrophages.53 In a model of acute liver injury, Notch1 and NICD expression were significantly upregulated in liver macrophages. Levels of proinflammatory factors TNF-α, IL-6, and IL-1β were higher in the Notch1-overexpressing group than in the Notch1-deficient group. Notch1 deficiency in the bone marrow inhibited YAP signaling and promoted M2 macrophage polarization in the damaged liver. However, YAP overexpression increased the expression of the Notch ligand gene jagged1 and promoted activation of the Notch1 pathway, accompanied by increased expression of TNF-α, Il-1β, Mcp-1, and Cxcl-1, which accelerated macrophage polarization to the M1 phenotype.54
Hypoxia-Inducible Factor HIF-1
HIF-1, HIF-2, and HIF-3 are the three members of the HIF family. When cells are subjected to hypoxia, HIF-1 is stabilized and moved to the nucleus where it forms a heterodimer with HIF-1. Physiological and pathological processes, including angiogenesis, cellular metabolism, and inflammation, are regulated by heterodimerization, which binds to the hypoxia-responsive element (HRE).55 NF-κB is the most important signaling factor that activates HIF-1α in addition to hypoxia, and NF-κB inhibitors are phosphorylated in hypoxic environments to release NF-κB, which in turn upregulates HIF-1α transcription. In addition to NF-κB, it participates in the regulation of the PI3K/AKT signaling pathway, which directly or indirectly controls the release of proinflammatory factors and inflammatory responses, including IL-1β, IL-6, and TNF-α.56 M1 macrophages rely on glycolysis to produce ATP, and hypoxia is the main driving factor of glycolysis. HIF-1 activates the expression of glycolytic enzymes such as hexokinase II (HKII), phosphofructokinase (PFKFB3) and blocks the flow of glucose through the TCA cycle and OXPHOS, which reduces the level of M2 polarization.57 In a mouse model of LPS-induced acute respiratory distress syndrome (ARDS), Chen discovered that patients with ARDS had increased levels of inflammatory cytokines and an imbalance in Th17/Treg and M1/M2 ratios. This was determined by HIF-1α immunoprecipitation and ubiquitination analysis. Chen also found that SPP1 leads to reduced ubiquitination and degradation of HIF-1α in ARDS. But inhibited M2 polarization by upregulating HIF-1α. Therefore, the ubiquitination-dependent degradation of HIF-1α decreased M2 polarization and increased the M1/M2 ratio.58 Li’s research showed that in RA, proinflammatory M1 macrophages could activate the signaling cascade between NF-κB and HIF-1α to produce reactive ROS, further enhancing M1 and exacerbating the development of RA. By inhibiting the HIF-1α/NF-κB cascade, the transformation of M1 to M2 can be effectively achieved, which helps with macrophage repolarization and reduces inflammation in the body.59
The Role of Macrophage Polarization in the Regulation of Bone Homeostasis
Osteoimmunology, as a cross-discipline, focuses on how the immune system and skeletal system interact. Immune cells and OBs share cytokines and signaling molecules in the intraosseous environment. Under physiological and pathological conditions, osteocytes and immune cells collaborate to maintain the intraosseous environment’s equilibrium. Abnormal immune function affects the consistency of the atmosphere within the skeletal system; Inversely, alterations in the homeostasis of the bone marrow’s internal environment can impact immune cells and immunological chemicals, resulting in anomalies in the immune system. Macrophages, a significant class of immune cells, are crucial for the control of bone homeostasis (Figure 3).
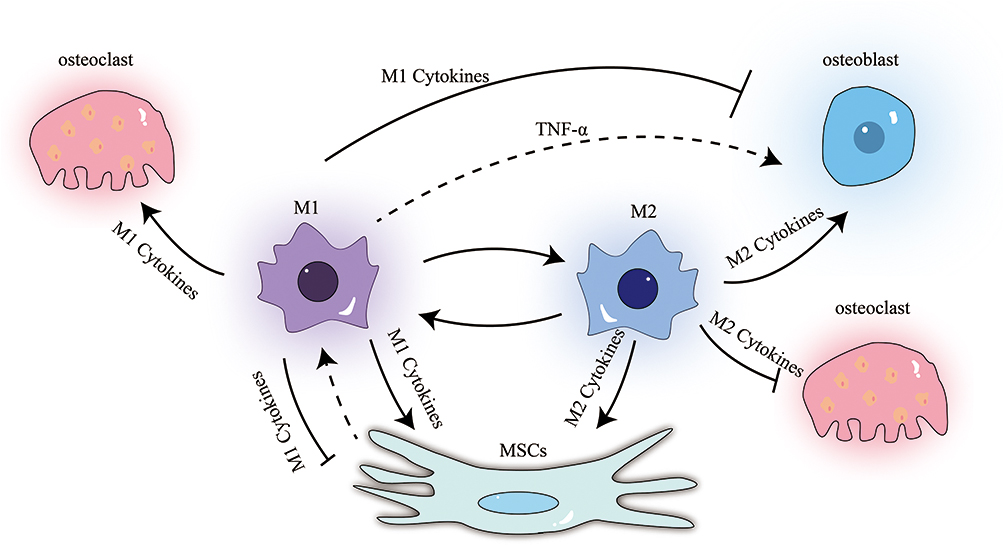 |
Figure 3 Relationship between M1, M2, OBs, OCs and BMSCs: M1 cells promote the formation of OCs, and depending on the cytokines they secrete, they may have a promoting or inhibiting effect on OBs and BMSCs. At the same time, BMSCs seem to have a regulatory effect on M1 as well. M2 cells inhibit the formation of OCs and have a promoting effect on OBs and BMSCs. |
Effect of Macrophages on Osteoblast
OB is a key cell in bone formation, derived from MSCs, and is crucial for bone processes’ synthesis, secretion, and mineralization.60 Macrophages can affect bone function directly or indirectly since they are present on the surface of the bone, near the mature bone cells, in addition to the bone marrow.61 Studies have confirmed that depletion of CD169+ macrophages affects bone repair and that CD169+ are necessary for the maintenance of bone tissue, providing important anabolic support during bone homeostasis and repair.62 Macrophages are essential to the maintenance of OB. Upon timely transition of macrophages from M1 to M2 phenotype, OB exhibits enhanced proliferation, adhesion, and extracellular mineralization capacity, as well as osteogenic gene phenotypes including Runx2, ALP, COL1A1, OPN, and OCN.63 Simultaneously inducing macrophage M2 polarization and activator release reduces bone damage.64
Current studies have shown that macrophage regulation of OB relies primarily on specific cytokines secreted by macrophages. M1 macrophages, as pro-inflammatory cells, secrete a large number of TNF-α and IL-6 during maturation, all of which stimulate OB activity. TNF-α, as a mononuclear factor for M1 phenotypic polarization released by macrophages, has TNFR1 and TNFR2 receptors, both expressed in immune cells, with TNFR1 activation leading to apoptosis and TNFR2 activation to cell proliferation. In the skeletal system, TNF-α inhibits bone formation by inhibiting OB function and reducing OB alkaline phosphatase activity. Early on, TNF-α prevents OB formation by increasing RUNX2 expression through the regulation of Smurf1 and Smurf2, which degrade Runx2 protein. In addition, TNF-α inhibits OB differentiation by suppressing IGF-1.65 Zhao observed a slight increase in TNF-α and IL-6 expression in MG-63 OB, along with a decrease in ALP, collagen-I, Runt-related transcription factor 2 (RUNX2), and osteoprotegerin (OPG) expression.66 It has also been noted that TNF-α may, in a dose-dependent manner, boost osteogenic differentiation. Additionally, it has been shown that low levels of TNF-α can increase RUNX2 expression, thereby promoting osteogenesis.67 As a member of the interleukin family, IL-6 binds to the IL-6 receptor, resulting in dimerization of the IL-6 signaling transmembrane receptor subunit of the cell surface protein (gp130) and subsequent activation of JAK/STAT to generate downstream pathways involved in the regulation of osteogenesis. IL-6 and its soluble receptors activate the SHP2/MEK2/ERK and SHP2/PI3K/Akt2 pathways, negatively regulating OB differentiation by significantly reducing ALP activity, OB gene expression and mineralization in a dose-dependent manner.68 On the other hand, IL-6 and its receptors can also promote OB differentiation through alternative mechanisms. In fact, in primary rat OB cultures, exogenous IL-6 and IL-6sR have been demonstrated to synergistically enhance the action of the osteogenic protein BMP-7, leading to an increase in ALP activity and the development of mineralized bone nodules.69
It has been discovered that M2 macrophages are essential for OB differentiation and osteogenic activity. Upon triggering M2 polarization of macrophages, BMP-2 and Osx mRNA expression is upregulated, which accelerates OB proliferation and promotes bone regeneration.70 Moreover, M2 cells secrete not only anti-inflammatory factors but also BMP-2 and BMP-4. BMP-2, for example, as demonstrated to induce the differentiation and proliferation of undifferentiated MSCs into OBs, promote OB differentiation and maturation, participate in the growth and development of bone and cartilage, and accelerate the reconstruction process, thus accelerating the repair of bone defects and demonstrating strong osteogenic induction. By binding to the BMP receptor, BMP-2 activates phosphorylation of SMAD1, and induces translocation of Runx2 to the nucleus, this leads to an increase in the expression of ALP and osteocalcin in cells, promoting osteogenesis.71 As a result, macrophages and the cytokines they emit can control how functionally active OBs are; which is crucial for the control of bone production.
Effect of Macrophages on Osteoclast
OC is a key cell mediating bone resorption and plays an important role in bone metabolism. Factors include nuclear factor kappa receptor activator and macrophage colony-stimulating factor (M-CSF) to promote the OC precursor. OCs and macrophages are derived from the same progenitor and compete to differentiate into their respective cell types. This competition can ultimately impact bone resorption by regulating the maturation and function of OCs. Additionally, macrophages themselves can regulate bone resorption by impacting OC maturation and function. In a mouse model of chronic inflammatory bone loss, researchers found that mesenchymal stromal cell therapy, which secretes IL-4, can polarize M1 into M2 anti-inflammatory cells. This treatment significantly reduces the number of OCs and effectively decreases local bone loss.72 In a periodontitis model using diabetic rats, higher levels of macrophage infiltration and M1 macrophage polarization were observed compared to control groups. Additionally, when M1/M2 macrophages were treated with high levels of glucose, the supernatants increased the formation of OCs compared to cells cultured at normal glucose levels. However, when N-acetylcysteine was added in combination with high glucose treatment, ROS levels were reduced and M1/M2 polarization was reversed. This effectively ameliorated symptoms of alveolar bone loss and reduced the number of bone lesions.73
M1 macrophages secrete proinflammatory factors like TNF-α and IL-1β to mature OCs precursors and form the OCs, However, M2 macrophages prevent the development of OCs by OCs precursor cells by secreting the IL-4 and IL-10. TNF-α is the most potent OCs-forming factor secreted by M1 and, in addition to directly promoting OCs precursor formation, it also stimulates RANKL secretion on the surface of other cells. It promotes the formation of OC. There is a strong correlation between TNF-α and both RANK and estrogen levels in patients with OP. Through the activation of the NF-κB and PI3K/Akt signaling pathways in in vitro tests, TNF-α was revealed to synergistically increase RANKL-induced OCs formation.74 In OBs that were cultured with TNF-α, there was a higher expression of RANKL mRNA compared to the control group. Furthermore, the number of TRAP-positive cells and the percentage of RANKL-positive cells increased, and MAPK phosphorylation was upregulated, which activated the NF-κB-like pathway in these cells. As a result, there was an increase in OC production and enhancement of bone resorption.75 Furthermore, TNF-α affected OC without the help of the RANKL/RANK/OPG system. TNF-α was only moderately produced by RANKL-induced macrophages. TLR4 inhibitors had no impact on the OC era generated by RANKL. While TNF-α was linked to the increase in OC differentiation in OCs treated with LPS, the RANKL/RANK/OPG axis was not.76 In contrast, the AMPK/SIRT1/NF-κB pathway inhibited the conversion of macrophages to a proinflammatory M1 phenotype and suppressed bone tissue differentiation and bone resorption by reducing TNF-α secretion.77 IL-1 is the most important and notable proinflammatory factor of the interleukin family and promotes OC production, which in turn stimulates bone resorption.78 IL-1 also induces the release of TNF-α and IL-6, which stimulate bone formation.79 IL-1β has been found to increase the expression of iNOS, IGF2, as well as the chemokines stromal cell-derived factor 1 (SDF-1) and C-X3-C motif ligand 1 (CX3CL1) in non-OCs of mouse bone marrow cells. This, in turn, causes a rise in the production of OCs.80 In a mouse model of arthritis, IL-1β blockers were found to be more effective in reducing the disease during the early stages of arthritis than in established arthritis. This was particularly evident in the prevention of bone erosion, where blocking IL-1β resulted in a reduction of OC production and bone resorption.81 IL-6, one of the OC factors, and STAT3 can be activated by the JAK pathway via IL-6, thereby inducing OC formation.82 Studies have shown that via modifying the NF-κB, ERK, and JNK signaling pathways, IL-6/sIL-6R can control bone cell differentiation and activity induced by RANKL. Therefore, IL-6 may have a dual role in the development of OP, operating as both a pro-resorption and an osteoprotective factor, depending on the level of RANKL in the local microenvironment.83 In a diabetic rat model, the JAK2/STAT3 signaling pathway was used to regulate IL-6 levels, this caused peri-implant bone resorption in peri-implantitis and a decline in the number of OCs.84 Anti-inflammatory M2 macrophages secrete IL-4 and IL-10, which inhibit the differentiation of OC precursors into OC and reduce the amount of OCs by suppressing pro-inflammatory factors like TNF-α, IL-1, and IL-6. IL-4 and TNF-α also decrease the levels of anti-tartaric acid phosphatase 5b (TRACP5b), a bone resorption marker, in a mouse model.85 IL-4 can inhibit the differentiation of OC precursors into OCs in vitro and in vivo. This inhibition is achieved through downregulating RANKL expression in OB and upregulating OPG by inhibiting NF-κB, the MAPK signaling pathway.86 IL-4 also directly inhibits OC formation by binding to the peroxisome proliferator-activated receptor γ1 on OC precursors.87 Moreover, when TNF-α was used to induce OC production independently of RANKL, IL-4 inhibited OC differentiation through a STAT6-dependent mechanism.88
The Effect of Macrophages on Bone Marrow Mesenchymal Stem Cells
Bone marrow mesenchymal stem cells (BMSCs) are stromal stem cells of the bone marrow that have the capacity to differentiate into osteogenic, lipogenic, chondrogenic, and vascular cells. Macrophages influence BMSC migration, differentiation, proliferation, and apoptosis in addition to helping MSCs differentiate into osteogenic MSCs, which in turn helps with bone neogenesis. Then again, MSCs can also influence macrophage polarization and function. Thus, macrophages and MSCs interact to regulate bone homeostasis.
The polarization of macrophages from M1 to M2 stimulated osteogenic differentiation in BMSCs to enhance osteogenesis by upregulating the expression of osteogenic genes (Runx-2, OCN, OPN), in addition, boosting the expression of angiogenic genes (VEGF and bFGF) to increase angiogenesis.89 Cai observed that when macrophages and BMSCs were co-cultured, the macrophages shifted towards the M2 phenotype and secreted TGF-β1, which stimulated BMSC migration, proliferation, and osteogenic differentiation.90 Conversely, when macrophages were co-cultured with human MSCs, the MSCs exhibited immunomodulatory activity, suppressing the M1 and enhancing the M2. The addition of macrophages enhanced the osteogenic differentiation of MSCs, irrespective of their polarization status. Interestingly, pro-inflammatory M1 macrophages were the most effective in promoting new bone formation.91 However, hypoxia-induced M1 macrophage-derived exosomes led to a decline in BMSCs’ viability and migratory capacity, and increased apoptosis.92 In another study by Wang, a diabetic model was used to show that AGEs induced M1 macrophage polarization, leading to increased levels of TNF-α, iNOS, and IL-6, activation of NF-κB, and altered ALP activity and matrix mineralization in BMSCs. AGEs also decreased the expression of ALP, OCN, OPN, and OSX, inhibiting osteogenesis in BMSCs.93 The study’s findings are presented below, When BMSCs derived from M0, typical M1, low inflammatory M1, and M2 macrophages were treated with M1 and low inflammatory M2 macrophages, significant increases in migration, osteogenic differentiation, and mineralization were observed. Among the macrophages tested, low inflammatory M1 macrophages induced the highest autophagy levels in BMSCs. Autophagy was found to be responsible for the observed effects on BMSC migration and osteogenic differentiation, and interference with autophagy eliminated these effects.94 The study also indicates that M1 macrophages have differential effects on BMSCs, and whether they promote or inhibit osteogenesis may depend on the type and amount of inflammatory factors they produce. In BMSCs with low or early levels of inflammation, M1 macrophages may be critical in promoting bone repair.
The M2 polarization of macrophages in bone regeneration, which is well known for its anti-inflammatory qualities and capacity to promote tissue repair and bone production. M2 macrophages can promote osteogenesis in vitro and protect alveolar bone from resorption in vivo by inhibiting OC production. M2-Exos has also been found to promote osteogenic differentiation by upregulating the IL-10 cytokine program in BMSCs and macrophages through direct delivery of exosomal IL-10 mRNA. This activates the IL-10/IL-10R cellular pathway, regulating cell differentiation and bone metabolism.95 In diabetic OP model mice, drug treatment increased the proportion of M2 F4/80+ and CD206+ phenotype cells and decreased the level of the proinflammatory factor IL-1β secreted by M1 macrophages. It also increased the level of the anti-inflammatory factor TGF-β1 secreted by M2 macrophages, which is considered a key factor in promoting BMSC migration and osteogenic differentiation. The drug may promote osteogenic differentiation of BMSCs by activating the PI3K/AKT pathway to increase the surface area of mineralized nodules in the cells.96 In addition, the anti-inflammatory factor IL-4 can increase M2 macrophage polarization and reduce apoptosis in vivo. It also increases the expression of TGF-β1, activating the TGF-β1/Smad pathway in BMSCs and promoting osteogenic differentiation.97 To summarize, the M2 plays a critical role in bone regeneration, and various factors IL-10, TGF-β1, and IL-4 can promote osteogenic differentiation in different ways.
Macrophages and MSCs are known to mutually influence each other’s polarization and metabolism. On one hand, MSCs boost the expression of anti-inflammatory genes while decreasing the expression of pro-inflammatory genes, can promote the M1 to the M2 type. On the other hand, macrophages can promote the osteogenic differentiation of MSCs.98 Under inflammatory conditions, MSCs produce prostaglandin E2 (PGE2), which accelerates M2 macrophage polarization and increases the expression of IL-10 while suppressing the inflammatory response. PGE2 also acts as a lipid signaling molecule that stimulates bone resorption and formation, leading to an increase in bone mass and strength. M2 macrophages enhance BMSC proliferation and osteogenic differentiation by increasing a protein’s expression in pathways related to osteogenic differentiation, such as Runx2, OCN, and ALP.99
Macrophage Therapy for Diseases Associated with an Imbalance of Bone Homeostasis
Immune cells and immune-derived cytokines are involved in bone homeostasis. The complex cross-talk between the skeletal and immune systems also exists in various clinically relevant diseases, including OP, SANFH, RA and other bone homeostasis imbalance diseases. Macrophages, as immune cells, participate in the pathophysiology of various metabolic bone diseases with their different phenotypes. Both in vivo and in vitro studies have confirmed that regulating the level of macrophage polarization can regulate the immune homeostasis of these diseases.
Macrophages and Osteoporosis
OP, is a systemic metabolic disease characterized by the destruction of bone microarchitecture, imbalance of bone homeostasis, and reduced bone density. It is classified as primary (postmenopausal OPand age-related OP) or secondary, with more than 200 million people worldwide suffering from OP, making it a global public health problem.100 The osteogenic capacity of patients with OP is lower than their bone resorption capacity, resulting in bone loss, increased bone fragility, and cumulative risk of fracture, with chronic inflammation being the primary cause of the disease.101 To activate adaptive immune responses and get rid of external irritants, M1 macrophages release pro-inflammatory mediators in the early stages. In the middle to late stages, M2 macrophages release anti-inflammatory mediators to reduce excessive inflammation and encourage bone and tissue regeneration. In patients with OP, there is chronic activation of M1 macrophages in the body, which leads to a persistent state of inflammation. Although M1 macrophages are important for bone resorption, they also promote the production of OC and hinder cellular osteogenesis by secreting pro-inflammatory and chemotactic factors. Research studies have revealed that the proportion of M1/M2 macrophage phenotypes increases in osteoporotic models, and lowering this ratio is a promising strategy for treating OP. Therefore, regulating the balance between M1 and M2 macrophages could be a potential therapeutic approach for managing OP.102 Decreased estrogen, an important factor in the postmenopausal OP, results in abnormal macrophage function in immune regulation, and estrogen deficiency results in increased IL-6 and TNF-α, which stimulate stable, long-term polarization of macrophages to the M1 phenotype, thereby stimulating OC formation leading to bone loss.103 Exosomes derived from M1 macrophages that were treated for OPhave been found to induce JNK phosphorylation and activate the JNK signaling pathway, upregulate miR-98 expression in cells, and at the same time inhibit osteogenic differentiation of OB, which further worsens bone loss and makes symptoms of OP more severe.104 As immune cells, macrophages are phagocytic in nature. In a high-resolution in vitro confocal imaging model of OPthat used mixed OCs-macrophage cultures on bone matrix, BATOON observed that macrophages positioned adjacent to the basolateral functional secretory domain of the cementing line effectively cleared degraded bone byproducts. The presence of bone particles in the co-cultured macrophages was not due to direct macrophage resorption but rather reflected indirect resorption of the bone matrix. Indirect resorption of resorption byproducts released from the OC, suggests that bone macrophages support and promote OC-mediated resorption in the OP mouse model.105 The use of TET in the OP rabbit model by FANG decreased the levels of MMP-9, PPAR-γ, RANKL, β-CTX, and TRACP-5b and increased the expression of OPG, ALP, and OC while inhibiting the production of IL-1, TNF-α, and IL-6 and promoting the M1 to M2 polarization of macrophages, suggesting that TET may promote M2 macrophage polarization via.106 ZHANG found that the use of berberine could inhibit OC production in vitro by downregulating the gene and protein expression levels of RANKL-induced CTSK, MMP-9, NFATc1, CD44, and DC-STAMP and revealed the formation of OC in bone could be considerably observed using berberine in an in vivo animal model of OP. The use of berberine in an in vivo model of OP mice resulted in a significant reduction in OP symptoms, leading to the conclusion that bone macrophage-induced OC production can be effectively treated by inhibiting OC production.107 Avicularin was found to reduce the expression of inflammatory cytokines and decrease the levels of CD86 and iNOS around implants in osteoporotic (OP) mice. This effect was achieved by blocking NF-κB phosphorylation, suggesting that avicularin has anti-inflammatory properties. Specifically, it inhibits the polarization of M1 cells through NF-κB inhibition, which helps to attenuate the inflammatory bone loss caused by macrophages of M1 phenotype.108 Chronic inflammation, a key driver of bone loss, is closely linked to macrophage polarization, and an imbalance in macrophage polarization leads to an accumulation of inflammatory factors, affecting bone metabolic homeostasis and exacerbating the development of bone loss. Therefore, targeting macrophage polarization to induce a dynamic M1/M2 balance could be a novel therapeutic strategy for OP.
Macrophages and Steroid-Induced Femoral Head Necrosis
The SANFH is caused by the unregulated use of glucocorticoids, which cause programmed osteogenic cell death, resulting in a lower rate of bone formation than bone resorption. This leads to an imbalance of bone homeostasis in the femoral head, as well as ischemic necrosis of the femoral head due to glucocorticoid damage to the bone microvasculature and localized complications of aseptic inflammation.8 Under the stimulation of chronic inflammation, necrotic bone forms, preventing normal bone reconstruction and repair and allowing the further development of SANFH. Macrophages, as major players in the immune system, have an important regulatory function in chronic inflammation and also regulate physiological cell functions, such as lipogenic osteogenic differentiation of BMSCs, osteogenic properties of OBs, and vascular endothelial cell turnover and repair, as well as the removal of apoptotic or necrotic cellular debris. Based on clinical data, it has been found that there is a significant infiltration of macrophages in the necrotic zone and adjacent areas of patients with ONFH. With significantly more macrophages present in SANFH patients compared to those with other risk factors.109 Additionally, the greatest number of inflammatory cells found in the cancellous bone tissue of the femoral head affected by osteonecrosis were macrophages.110 The expression of IL-1β, TNF-α, and IL-6 was likewise markedly enhanced in the ONFH piglet model, and the necrotic bone stimulated the inflammatory response by activating TLR4 to stimulate M1 polarization of macrophages, a large number of M1 macrophages infiltrated the necrotic area of SANFH.111 Glucocorticoids have an extremely potent inhibitory effect on immune system cells, regulating macrophage survival, migration, and proliferation by binding to glucocorticoid receptors in different ways. Glucocorticoids not only counteract the expression of proinflammatory genes in cells, but they also induce M2 polarization and enhance the production of IL-10 and TGF-β in THP1 monocytes treated with dexamethasone (Dex), while suppressing the production of IL-1β, TNF-α, and IL-6.112 It also increases the expression of NLRP3 inflammatory vesicles in macrophages113 and acts synergistically with cytokines to increase the production of pro-inflammatory mediators.114 Immune factors can be transmitted from the local lesion to the blood in the early stages of SANFH due to enhanced TNF- activity and significant infiltration of M1 macrophages in the osteonecrosis zone.115 Curcumin interferes with the JAK1/2-STAT1 pathway to inhibit M1 macrophage polarization and reduce M1 infiltration and systemic inflammation in the femoral head of a mouse model of SANFH. Additionally, curcumin also reduces osteocyte apoptosis and SANFH symptoms.116 Astragaloside, on the other hand, promotes the repolarization of M1 to M2 and reduces the proportion of M1 macrophages in SANFH. This reduces TNF-α and IL-1β levels, decreases osteocyte apoptosis, and alleviates arthritic symptoms, inflammatory cytokines, and other SANFH symptoms.117 In conclusion, macrophage polarization and autoimmunity play a very important role in the development of SANFH. In long-term inflammation, pro-inflammatory factors secreted by M1 macrophages promote the rate of bone resorption, which leads to bone loss and accelerates the process of SANFH. By inhibiting M1 macrophage polarization and promoting repolarization to M2 macrophages, this can help reduce inflammation, alleviate arthritic symptoms, and decrease osteocyte apoptosis in the femoral head, so that intervention in M1 macrophage polarization could be one of the treatment methods for SANFH.
Macrophages and Rheumatoid Arthritis
An aggressive form of arthritis is a defining feature of the chronic systemic inflammatory diseaseRA.118 It is characterized by erosive arthritis, patients with active RA have abnormal bone metabolism, with large amounts of inflammatory factors leading to an imbalance in the OB/OC ratio, resulting in bone loss. M1/M2 macrophage polarization imbalances play a role in the emergence of RA. Recent research has demonstrated that M1 macrophage infiltration in RA corresponds with osteogenesis of joint damage and that substantial numbers of M1 macrophages are present in arthritic models.119 Elimination of macrophage infiltration in inflamed tissues may contribute to the treatment of RA. Under the inflammatory conditions of RA, many bone loss-mediating IL-1β, IL-6, TNF-α, and HIF-1α, are produced, all of which are associated with M1 macrophages, which promote RANKL production and mediate bone cell hyperactivation. In RA, synovial tissue expresses the chemokine CCL21 in high numbers, and CCL21 stimulates an increase in the number of M1 macrophages, leading to elevated IL-6 and IL-23 transcription. IL-6 and IL-23 are involved in the maintenance of Th17 polarization, which plays an important role in OC production, and therefore, CCL21-induced promotion of Th17 by M1 cytokine polarization stimulates of OC production and exacerbation of OC production, exacerbation of erosive arthritis in RA.120 JAK/STAT is a key pathway of bone destruction in a rat model of RA, characterized by a high expression state of OCs and M1.121
In a rat model of RA, there is a high expression state of OCs and M1. Targeting increased apoptosis of M1 macrophages and reducing their number leads to a decrease in the expression levels of TNF-α and IL-1β. This approach also increases the number and thickness of bone trabeculae, thereby reducing the symptoms of bone loss to some extent, halting the progression of bone damage, and restoring normal bone homeostasis.122 Pun inhibits the activation of receptors in the NF-κB pathway and transforms M1 to M2 after PUN treatment. In the RA model, the expression of iNOS and other proinflammatory cytokines released by M1 macrophages is reduced in the PUN-treated group. However, the concomitant increase in the expression of anti-inflammatory markers of M2 macrophages, such as Arg-1 and IL-10, contributes to the treatment of RA.123 Macrophages are crucial immune cells in the development of RA, and understanding the dialectic between immune cells and skeletal cells and elucidating the mechanisms of macrophage polarization in RA may be effective in combating RA.
Discussion and Conclusion
Macrophages are one of the main functional cells of the immune system, playing an irreplaceable role in pathogen recognition, clearance, and innate and acquired immune responses. Depending on the microenvironment, they can polarize into two phenotypes, M1 and M2, and play different roles. Normal bone homeostasis is very important for bone development and maintenance. As a complex and dynamic process, various mechanisms coordinate to regulate bone homeostasis. In the past, the focus of bone homeostasis research was on the association between OBs and OCs. However, with the development of interdisciplinary integration, it has been found that the skeletal system and the immune system interact. As immune cells, macrophages can interact with OBs, OCs, and bone marrow stromal cells. An increasing number of studies have shown that macrophage polarization is an important factor in regulating bone metabolism and a target for treating bone homeostasis disorders. The mechanisms by which macrophages regulate bone homeostasis are complex, and different phenotypes affect bone homeostasis in different ways. As a pro-inflammatory cell, M1 macrophages may promote bone formation in the acute phase, but in chronic inflammation, M1 exacerbates bone resorption, decreases osteogenic ability, and leads to bone homeostasis imbalance. On the other hand, M2 macrophages release anti-inflammatory factors in the later stages of inflammation, reducing the body’s inflammatory response. At the same time, they induce OBs differentiation and increase bone mineralization, promoting bone formation, effectively reducing bone loss, and maintaining bone homeostasis. Therefore, by regulating macrophage polarization and promoting bone regeneration through immune cell factors, adjusting bone homeostasis may be an effective therapeutic target for promoting bone repair.
Biological materials with immunomodulatory properties have now been developed for use in bone tissue engineering. Olwyn developed an immunomodulatory scaffold containing bone-like nanohydroxyapatite particles (BMnP), which can induce the formation of M2 macrophages and increase the production of the anti-inflammatory cytokine IL-10. In vitro experiments also confirmed that macrophages treated with nanoparticles enhance the osteogenic effect of MSCs in vitro, promoting an increase in bone mass.124 Li developed a gastrodin-biodegradable polyurethane/nanohydroxyapatite composite biomaterial. This biomaterial can trigger the polarization of M2 macrophages and accelerate vascular generation.125 Huang found that chitooligosaccharides (COS) can regulate macrophage polarization, enhance the osteogenic differentiation of BMSCs, and promote bone formation.126 Meanwhile, researchers have also developed some bioactive drugs to correct bone homeostasis. Liu found that small extracellular vesicles derived from bone marrow mesenchymal stem cells (BMSC-sEV) influence macrophage polarization, thereby treating bone loss in periodontitis.127 Zhu used crocin to suppress inflammation induced by titanium particles and enhance the osteogenic differentiation of BMSCs by inducing the polarization of M2 macrophages.128 This provides new insights into the regulation of bone homeostasis by macrophages.
In general, the polarization of macrophages and bone immunity play a very important role in the occurrence and development of bone homeostasis disorders. Pro-inflammatory macrophages and inflammatory cell factors promote the generation of OCs, while anti-inflammatory macrophages can reduce bone loss. Therefore, regulating the direction of macrophage polarization can improve the pathogenesis of bone homeostasis imbalance and effectively treat such diseases.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The work was supported by National Natural Science Foundation of China (No: 81860859, 82160915).
Disclosure
The authors have no relevant financial or non-financial interests to disclose.
References
1. Chen X, Zhi X, Yin Z, et al. 18β-glycyrrhetinic acid inhibits osteoclastogenesis in vivo and in vitro by blocking RANKL-mediated RANK–TRAF6 interactions and NF-κB and MAPK signaling pathways. Front Pharmacol. 2018;9:647. doi:10.3389/fphar.2018.00647
2. Li J, Yin Z, Huang B, Xu K, Su J. Stat3 signaling pathway: a future therapeutic target for bone-related diseases. Front Pharmacol. 2022;13:897539. doi:10.3389/fphar.2022.897539
3. Muñoz J, Akhavan NS, Mullins AP, Arjmandi BH. Macrophage polarization and osteoporosis: a review. Nutrients. 2020;12(10). doi:10.3390/nu12102999
4. Sun Y, Li J, Xie X, et al. Macrophage-osteoclast associations: origin, polarization, and subgroups. Front Immunol. 2021;12:778078. doi:10.3389/fimmu.2021.778078
5. Chen K, Jiao Y, Liu L, et al. Communications between bone marrow macrophages and bone cells in bone remodeling. Front Cell Dev Biol. 2020;8:598263. doi:10.3389/fcell.2020.598263
6. Muñoz M, Robinson K, Shibli-Rahhal A. Bone health and osteoporosis prevention and treatment. Clin Obstet Gynecol. 2020;63(4):770–787. doi:10.1097/grf.0000000000000572
7. Molendijk M, Hazes JM, Lubberts E. From patients with arthralgia, pre-RA and recently diagnosed RA: what is the current status of understanding RA pathogenesis? RMD Open. 2018;4(1):e000256. doi:10.1136/rmdopen-2016-000256
8. Wang A, Ren M, Wang J. The pathogenesis of steroid-induced osteonecrosis of the femoral head: a systematic review of the literature. Gene. 2018;671:103–109. doi:10.1016/j.gene.2018.05.091
9. Yadav S, Dwivedi A, Tripathi A. Biology of macrophage fate decision: implication in inflammatory disorders. Cell Biol Int. 2022;46(10):1539–1556.
10. Razi S, Yaghmoorian Khojini J, Kargarijam F, et al. Macrophage efferocytosis in health and disease. Cell biochemistry and Function. 2023;41(2):152–165. doi:10.1002/cbf.3780
11. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol. 2019;10:1084.
12. Tian T, Wang Z, Chen L, Xu W, Wu B. Photobiomodulation activates undifferentiated macrophages and promotes M1/M2 macrophage polarization via PI3K/AKT/mTOR signaling pathway. Laser med sci. 2023;38(1):86. doi:10.1007/s10753-022-01777-z
13. He X, Tan S, Shao Z, Wang X. Latitudinal and longitudinal regulation of tissue macrophages in inflammatory diseases. Genes Dis. 2022;9(5):1194–1207.
14. Gharavi AT, Hanjani NA, Movahed E, Doroudian M. The role of macrophage subtypes and exosomes in immunomodulation. Cell Mol Biol Lett. 2022;27(1):83.
15. Li P, Ma C, Li J, et al. Proteomic characterization of four subtypes of M2 macrophages derived from human THP-1 cells. J Zhejiang Univ Sci B. 2022;23(5):407–422.
16. García-García A, Pigeot S, Martin I. Engineering of immunoinstructive extracellular matrices for enhanced osteoinductivity. Bioact Mater. 2023;24:174–184.
17. Zou Z, Lin H, Li M, Lin B. Tumor-associated macrophage polarization in the inflammatory tumor microenvironment. Front Oncol. 2023;13:1103149.
18. Deng L, Jian Z, Xu T, et al. Macrophage Polarization: An Important Candidate Regulator for Lung Diseases. Molecules. 2023;28(5):2379.
19. Shanley LC, Mahon OR, O’Rourke SA, et al. Macrophage metabolic profile is altered by hydroxyapatite particle size. Acta Biomater. 2023;160:311–321. doi:10.1016/j.actbio.2023.01.058
20. Sun X, Gao Y, Li Z, He J, Wu Y. Magnetic responsive hydroxyapatite scaffold modulated macrophage polarization through PPAR/JAK-STAT signaling and enhanced fatty acid metabolism. Biomaterials. 2023;295:122051. doi:10.1016/j.biomaterials.2023.122051
21. Liu J, Wang F, Luo F. The role of JAK/STAT pathway in fibrotic diseases: molecular and cellular mechanisms. Biomolecules. 2023;13(1). doi:10.3390/biom13010119
22. Owen KL, Brockwell NK, Parker BS. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers. 2019;11(12). doi:10.3390/cancers11122002
23. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. 2021;6(1):402. doi:10.1038/s41392-021-00791-1
24. Verhoeven Y, Tilborghs S, Jacobs J, et al. The potential and controversy of targeting STAT family members in cancer. Semin Cancer Biol. 2020;60:41–56. doi:10.1016/j.semcancer.2019.10.002
25. Ernst S, Müller-Newen G. Nucleocytoplasmic shuttling of STATs. A target for intervention? Cancers. 2019;11(11). doi:10.3390/cancers11111815
26. Kawakami T, Koike A, Maehara T, Hayashi T, Fujimori K. Bicarbonate enhances the inflammatory response by activating JAK/STAT signalling in LPS + IFN-γ-stimulated macrophages. J Biochem. 2020;167(6):623–631. doi:10.1093/jb/mvaa010
27. Wu L, Sun S, Qu F, et al. CXCL9 influences the tumor immune microenvironment by stimulating JAK/STAT pathway in triple-negative breast cancer. Cancer Immunol Immunother. 2022. doi:10.1007/s00262-022-03343-w
28. Li X, Jiang M, Chen X, Sun W. Etanercept alleviates psoriasis by reducing the Th17/Treg ratio and promoting M2 polarization of macrophages. Immun Inflamm Dis. 2022;10(12):e734. doi:10.1002/iid3.734
29. Wang X, Chen S, Lu R, et al. Adipose-derived stem cell-secreted exosomes enhance angiogenesis by promoting macrophage M2 polarization in type 2 diabetic mice with limb ischemia via the JAK/STAT6 pathway. Heliyon. 2022;8(11):e11495. doi:10.1016/j.heliyon.2022.e11495
30. Tu Y, Liu J, Kong D, et al. Irisin drives macrophage anti-inflammatory differentiation via JAK2-STAT6-dependent activation of PPARγ and Nrf2 signaling. Free Radic Biol Med. 2023. doi:10.1016/j.freeradbiomed.2023.03.014
31. Xu L, Zhang Y, Dai Q, et al. Scorpion venom polypeptide governs alveolar macrophage M1/M2 polarization to alleviate pulmonary fibrosis. Tissue Cell. 2022;79:101939. doi:10.1016/j.tice.2022.101939
32. Song M, Cui X, Zhang J, et al. Shenlian extract attenuates myocardial ischaemia-reperfusion injury via inhibiting M1 macrophage polarization by silencing miR-155. Pharm Biol. 2022;60(1):2011–2024. doi:10.1080/13880209.2022.2117828
33. Tajalli-Nezhad S, Mohammadi S, Atlasi MA, et al. Calcitriol modulate post-ischemic TLR signaling pathway in ischemic stroke patients. J Neuroimmunol. 2023;375:578013. doi:10.1016/j.jneuroim.2022.578013
34. Zhu W, Xu R, Du J, et al. Zoledronic acid promotes TLR-4-mediated M1 macrophage polarization in bisphosphonate-related osteonecrosis of the jaw. FASEB J. 2019;33(4):5208–5219. doi:10.1096/fj.201801791RR
35. Wang Y, Wu Y, Sailike J, et al. Fourteen composite probiotics alleviate type 2 diabetes through modulating gut microbiota and modifying M1/M2 phenotype macrophage in db/db mice. Pharmacol Res. 2020;161:105150. doi:10.1016/j.phrs.2020.105150
36. Kang Z-P, Wang M-X, T-t W, et al. Curcumin alleviated dextran sulfate sodium-induced colitis by regulating M1/M2 macrophage polarization and TLRs signaling pathway. Evid Based Complement Alternat Med. 2021;2021:3334994. doi:10.1155/2021/3334994
37. Zhou L, Zhao H, Zhao H, et al. GBP5 exacerbates rosacea-like skin inflammation by skewing macrophage polarization towards M1 phenotype through the NF-κB signalling pathway. J Eur Acad Dermatol Venereol. 2023;37(4):796–809. doi:10.1111/jdv.18725
38. Lee H, Han J-H, Ahn K, et al. Recombinant human KAI1/CD82 attenuates M1 macrophage polarization on LPS-stimulated RAW264.7 cells via blocking TLR4/JNK/NF-κB signal pathway. BMB Rep. 2023;2023:1.
39. Tian P, Zhao L, Kim J, et al. Dual stimulus responsive borosilicate glass (BSG) scaffolds promote diabetic alveolar bone defectsrepair by modulating macrophage phenotype. Bioact Mater. 2023;26:231–248. doi:10.1016/j.bioactmat.2023.02.023
40. Xie Y, Shi X, Sheng K, et al. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia. Mol Med Rep. 2019;19(2):783–791. doi:10.3892/mmr.2018.9713
41. Wang J, Hu K, Cai X, et al. Targeting PI3K/AKT signaling for treatment of idiopathic pulmonary fibrosis. Acta Pharm Sin B. 2022;12(1):18–32. doi:10.1016/j.apsb.2021.07.023
42. He X, Xiao J, Li Z, et al. Inhibition of PD-1 alters the SHP1/2-PI3K/Akt axis to decrease M1 polarization of alveolar macrophages in lung ischemia-reperfusion injury. Inflammation. 2023;46(2):639–654. doi:10.1007/s10753-022-01762-6
43. Tian T, Chen L, Wang Z, Zhu M, Xu W, Wu B. Sema3A drives alternative macrophage activation in the resolution of periodontitis via PI3K/AKT/mTOR signaling. Inflammation. 2023. doi:10.1007/s10753-022-01777-z
44. Wang Y, Zhang X, Wang J, et al. Inflammatory periodontal ligament stem cells drive M1 macrophage polarization via exosomal miR-143-3p-mediated regulation of PI3K/AKT/NF-κB signaling. Stem Cells. 2023;41(2):184–199. doi:10.1093/stmcls/sxac087
45. Wang C, Zhang M, Yan J, et al. Chemokine-like receptor 1 deficiency impedes macrophage phenotypic transformation and cardiac repair after myocardial infarction. Int J Cardiol. 2023;372. doi:10.1016/j.ijcard.2022.12.015
46. Yang B, Wang L, Tian Z. Silencing of RhoC induces macrophage M1 polarization to inhibit migration and invasion in colon cancer via regulating the PTEN/FOXO1 pathway. Int J Exp Pathol. 2023;104(1):33–42. doi:10.1111/iep.12460
47. Xu J, Chen P, Yu C, et al. Hypoxic bone marrow mesenchymal stromal cells-derived exosomal miR-182-5p promotes liver regeneration via FOXO1-mediated macrophage polarization. FASEB J. 2022;36(10):e22553. doi:10.1096/fj.202101868RRR
48. Wu X, Wang Y, Chen H, Wang Y, Gu Y. Phosphatase and tensin homologue determine inflammatory status by differentially regulating the expression of Akt1 and Akt2 in macrophage alternative polarization of periodontitis. J Clin Periodontol. 2023;50(2):220–231. doi:10.1111/jcpe.13730
49. Matsuno K. Notch signaling. Dev Growth Differ. 2020;62(1):3. doi:10.1111/dgd.12642
50. Meurette O, Mehlen P. Notch signaling in the tumor microenvironment. Cancer Cell. 2018;34(4):536–548. doi:10.1016/j.ccell.2018.07.009
51. Xu H, Zhu J, Smith S, et al. Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat Immunol. 2012;13(7):642–650. doi:10.1038/ni.2304
52. Wu Y, Liang M, Huang F, et al. Notch blockade specifically in bone marrow-derived FSP-1-positive cells ameliorates renal fibrosis. Cells. 2023;12(2). doi:10.3390/cells12020214
53. Chen X, Su C, Wei Q, Sun H, Xie J, Nong G. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate diffuse alveolar hemorrhage associated with systemic lupus erythematosus in mice by promoting M2 macrophage polarization via the microRNA-146a-5p/NOTCH1 axis. Immunol Invest. 2022;51(7):1975–1993. doi:10.1080/08820139.2022.2090261
54. Yang Y, Ni M, Zong R, et al. Targeting notch1-YAP circuit reprograms macrophage polarization and alleviates acute liver injury in mice. Cell Mol Gastroenterol Hepatol. 2023;15(5):1085–1104. doi:10.1016/j.jcmgh.2023.01.002
55. Ikeda H, Kakeya H. Targeting hypoxia-inducible factor 1 (HIF-1) signaling with natural products toward cancer chemotherapy. J Antibiot (Tokyo). 2021;74(10):687–695. doi:10.1038/s41429-021-00451-0
56. Suresh MV, Balijepalli S, Solanki S, et al. Hypoxia-inducible factor 1α and its role in lung injury: adaptive or maladaptive. Inflammation. 2023;46(2):491–508. doi:10.1007/s10753-022-01769-z
57. Díaz-Bulnes P, Saiz ML, López-Larrea C, Rodríguez RM. Crosstalk between hypoxia and ER stress response: a key regulator of macrophage polarization. Front Immunol. 2019;10:2951. doi:10.3389/fimmu.2019.02951
58. Chen L, Yang J, Zhang M, Fu D, Luo H, Yang X. SPP1 exacerbates ARDS via elevating Th17/Treg and M1/M2 ratios through suppression of ubiquitination-dependent HIF-1α degradation. Cytokine. 2023;164:156107. doi:10.1016/j.cyto.2022.156107
59. Li Y, Liang Q, Zhou L, et al. An ROS-responsive artesunate prodrug nanosystem co-delivers dexamethasone for rheumatoid arthritis treatment through the HIF-1α/NF-κB cascade regulation of ROS scavenging and macrophage repolarization. Acta Biomater. 2022;152:406–424. doi:10.1016/j.actbio.2022.08.054
60. Ponzetti M, Rucci N. Osteoblast differentiation and signaling: established concepts and emerging topics. Int J Mol Sci. 2021;22(13). doi:10.3390/ijms22136651
61. Schlundt C, Fischer H, Bucher CH, Rendenbach C, Duda GN, Schmidt-Bleek K. The multifaceted roles of macrophages in bone regeneration: a story of polarization, activation and time. Acta Biomater. 2021;133:46–57. doi:10.1016/j.actbio.2021.04.052
62. Batoon L, Millard SM, Wullschleger ME, et al. CD169+ macrophages are critical for osteoblast maintenance and promote intramembranous and endochondral ossification during bone repair. Biomaterials. 2019;196:51–66. doi:10.1016/j.biomaterials.2017.10.033
63. You Y, Wang W, Li Y, et al. Aspirin/PLGA coated 3D-printed Ti-6Al-4V alloy modulate macrophage polarization to enhance osteoblast differentiation and osseointegration. J Mater Sci Mater Med. 2022;33(10):73. doi:10.1007/s10856-022-06697-w
64. Shi C, Yuan F, Li Z, et al. MSN@IL-4 sustainingly mediates macrophagocyte M2 polarization and relieves osteoblast damage via NF-κB pathway-associated apoptosis. Biomed Res Int. 2022;2022:2898729. doi:10.1155/2022/2898729
65. Osta B, Benedetti G, Miossec P. Classical and paradoxical effects of TNF-α on bone homeostasis. Front Immunol. 2014;5:48. doi:10.3389/fimmu.2014.00048
66. Zhao M, Dai W, Wang H, et al. Periodontal ligament fibroblasts regulate osteoblasts by exosome secretion induced by inflammatory stimuli. Arch Oral Biol. 2019;105:27–34. doi:10.1016/j.archoralbio.2019.06.002
67. Glass GE, Chan JK, Freidin A, Feldmann M, Horwood NJ, Nanchahal J. TNF-alpha promotes fracture repair by augmenting the recruitment and differentiation of muscle-derived stromal cells. Proc Natl Acad Sci USA. 2011;108(4):1585–1590. doi:10.1073/pnas.1018501108
68. Kaneshiro S, Ebina K, Shi K, et al. IL-6 negatively regulates osteoblast differentiation through the SHP2/MEK2 and SHP2/Akt2 pathways in vitro. J Bone Miner Metab. 2014;32(4):378–392.
69. Yeh L-C-C, Zavala MC, Lee JC. Osteogenic protein-1 and interleukin-6 with its soluble receptor synergistically stimulate rat osteoblastic cell differentiation. J Cell Physiol. 2002;190(3):322–331.
70. Chen S, Ni S, Liu C, et al. Neglected immunoregulation: M2 polarization of macrophages triggered by low-dose irradiation plays an important role in bone regeneration. J Cell Mol Med. 2023. doi:10.1111/jcmm.17721
71. Wu M, Chen G, Li Y-P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016;4:16009. doi:10.1038/boneres.2016.9
72. Shen H, Kushioka J, Toya M, et al. Sex differences in the therapeutic effect of unaltered versus NFκB sensing IL-4 over-expressing mesenchymal stromal cells in a murine model of chronic inflammatory bone loss. Front Bioeng Biotechnol. 2022;10:962114. doi:10.3389/fbioe.2022.962114
73. Zhang B, Yang Y, Yi J, Zhao Z, Ye R. Hyperglycemia modulates M1/M2 macrophage polarization via reactive oxygen species overproduction in ligature-induced periodontitis. J Periodontal Res. 2021;56(5):1.
74. Zha L, He L, Liang Y, et al. TNF-α contributes to postmenopausal osteoporosis by synergistically promoting RANKL-induced osteoclast formation. Biomed Pharmacother. 2018;102:369–374. doi:10.1016/j.biopha.2018.03.080
75. Marahleh A, Kitaura H, Ohori F, et al. TNF-α directly enhances osteocyte RANKL expression and promotes osteoclast formation. Front Immunol. 2019;10:2925. doi:10.3389/fimmu.2019.02925
76. AlQranei MS, Senbanjo LT, Aljohani H, Hamza T, Chellaiah MA. Lipopolysaccharide- TLR-4 axis regulates osteoclastogenesis independent of RANKL/RANK signaling. BMC Immunol. 2021;22(1):23. doi:10.1186/s12865-021-00409-9
77. Ye Q, Xu H, Liu S, et al. Apoptotic extracellular vesicles alleviate Pg-LPS induced inflammatory responses of macrophages via AMPK/SIRT1/NF-κB pathway and inhibit osteoclast formation. J Periodontol. 2022;93(11):1738–1751. doi:10.1002/JPER.21-0657
78. Xing L, Carlson L, Story B, et al. Expression of either NF-kappaB p50 or p52 in osteoclast precursors is required for IL-1-induced bone resorption. J Bone Miner Res. 2003;18(2):260–269.
79. Kurihara N, Bertolini D, Suda T, Akiyama Y, Roodman GD. IL-6 stimulates osteoclast-like multinucleated cell formation in long term human marrow cultures by inducing IL-1 release. J Immunol. 1990;144(11):4226–4230.
80. Otsuka Y, Kondo T, Aoki H, et al. IL-1β promotes osteoclastogenesis by increasing the expression of IGF2 and chemokines in non-osteoclastic cells. J Pharmacol Sci. 2023;151(1):1–8. doi:10.1016/j.jphs.2022.10.007
81. Levescot A, Chang MH, Schnell J, et al. IL-1β-driven osteoclastogenic Tregs accelerate bone erosion in arthritis. J Clin Invest. 2021;131(18):1.
82. Deng Z, Zhang R, Li M, et al. STAT3/IL-6 dependent induction of inflammatory response in osteoblast and osteoclast formation in nanoscale wear particle-induced aseptic prosthesis loosening. Biomater Sci. 2021;9(4):1291–1300. doi:10.1039/d0bm01256d
83. Feng W, Liu H, Luo T, et al. Combination of IL-6 and sIL-6R differentially regulate varying levels of RANKL-induced osteoclastogenesis through NF-κB, ERK and JNK signaling pathways. Sci Rep. 2017;7:41411. doi:10.1038/srep41411
84. Liu -Q-Q, W-w W, Yang J, et al. A GP130-targeting small molecule, LMT-28, reduces LPS-induced bone resorption around implants in diabetic models by inhibiting IL-6/GP130/JAK2/STAT3 signaling. Mediators Inflamm. 2023;2023:9330439. doi:10.1155/2023/9330439
85. Fujii T, Kitaura H, Kimura K, Hakami ZW, Takano-Yamamoto T. IL-4 inhibits TNF-α-mediated osteoclast formation by inhibition of RANKL expression in TNF-α-activated stromal cells and direct inhibition of TNF-α-activated osteoclast precursors via a T-cell-independent mechanism in vivo. Bone. 2012;51(4):771–780. doi:10.1016/j.bone.2012.06.024
86. Palmqvist P, Lundberg P, Persson E, et al. Inhibition of hormone and cytokine-stimulated osteoclastogenesis and bone resorption by interleukin-4 and interleukin-13 is associated with increased osteoprotegerin and decreased RANKL and RANK in a STAT6-dependent pathway. J Biol Chem. 2006;281(5):2414–2429.
87. Bendixen AC, Shevde NK, Dienger KM, Willson TM, Funk CD, Pike JW. IL-4 inhibits osteoclast formation through a direct action on osteoclast precursors via peroxisome proliferator-activated receptor gamma 1. Proc Natl Acad Sci USA. 2001;98(5):2443–2448.
88. Moreno JL, Kaczmarek M, Keegan AD, Tondravi M. IL-4 suppresses osteoclast development and mature osteoclast function by a STAT6-dependent mechanism: irreversible inhibition of the differentiation program activated by RANKL. Blood. 2003;102(3):1078–1086.
89. Li S, Zhang L, Liu C, et al. Spontaneous immunomodulation and regulation of angiogenesis and osteogenesis by Sr/Cu-borosilicate glass (BSG) bone cement to repair critical bone defects. Bioact Mater. 2023;23:101–117. doi:10.1016/j.bioactmat.2022.10.021
90. Cai G, Lu Y, Zhong W, et al. Piezo1-mediated M2 macrophage mechanotransduction enhances bone formation through secretion and activation of transforming growth factor-β1. Cell Prolif. 2023:e13440. doi:10.1111/cpr.13440
91. Romero-López M, Li Z, Rhee C, et al. Macrophage effects on mesenchymal stem cell osteogenesis in a three-dimensional in vitro bone model. Tissue Eng Part A. 2020;26(19–20):1099–1111. doi:10.1089/ten.TEA.2020.0041
92. Qi Y, Zhu T, Zhang T, et al. M1 macrophage-derived exosomes transfer miR-222 to induce bone marrow mesenchymal stem cell apoptosis. Lab Invest. 2021;101(10):1318–1326. doi:10.1038/s41374-021-00622-5
93. Wang F, Kong L, Wang W, et al. Adrenomedullin 2 improves bone regeneration in type 1 diabetic rats by restoring imbalanced macrophage polarization and impaired osteogenesis. Stem Cell Res Ther. 2021;12(1):288. doi:10.1186/s13287-021-02368-9
94. Yang L, Xiao L, Gao W, et al. Macrophages at low-inflammatory status improved osteogenesis via autophagy regulation. Tissue Eng Part A. 2021. doi:10.1089/ten.TEA.2021.0015
95. Chen X, Wan Z, Yang L, et al. Exosomes derived from reparative M2-like macrophages prevent bone loss in murine periodontitis models via IL-10 mRNA. J Nanobiotechnology. 2022;20(1):110. doi:10.1186/s12951-022-01314-y
96. Lu Y, Liu S, Yang P, et al. Exendin-4 and eldecalcitol synergistically promote osteogenic differentiation of bone marrow mesenchymal stem cells through M2 macrophages polarization via PI3K/AKT pathway. Stem Cell Res Ther. 2022;13(1):113. doi:10.1186/s13287-022-02800-8
97. Zhang J, Shi H, Zhang N, Hu L, Jing W, Pan J. Interleukin-4-loaded hydrogel scaffold regulates macrophages polarization to promote bone mesenchymal stem cells osteogenic differentiation via TGF-β1/Smad pathway for repair of bone defect. Cell Prolif. 2020;53(10):e12907. doi:10.1111/cpr.12907
98. Yu K, Huangfu H, Qin Q, et al. Application of bone marrow-derived macrophages combined with bone mesenchymal stem cells in dual-channel three-dimensional bioprinting scaffolds for early immune regulation and osteogenic induction in rat calvarial defects. ACS Appl Mater Interfaces. 2022;14(41):47052–47065. doi:10.1021/acsami.2c13557
99. Jiang Q, Bai G, Liu X, et al. 3D GelMA ICC scaffolds combined with SW033291 for bone regeneration by modulating macrophage polarization. Pharmaceutics. 2021;13(11). doi:10.3390/pharmaceutics13111934
100. Yong EL, Logan S. Menopausal osteoporosis: screening, prevention and treatment. Singapore Med J. 2021;62(4):159–166. doi:10.11622/smedj.2021036
101. Zhang W, Gao R, Rong X, et al. Immunoporosis: role of immune system in the pathophysiology of different types of osteoporosis. Front Endocrinol. 2022;13:965258. doi:10.3389/fendo.2022.965258
102. Dou C, Ding N, Zhao C, et al. Estrogen deficiency-mediated M2 macrophage osteoclastogenesis contributes to M1/M2 ratio alteration in ovariectomized osteoporotic mice. J Bone Miner Res. 2018;33(5):899–908. doi:10.1002/jbmr.3364
103. Brunetti G, Storlino G, Oranger A, et al. LIGHT/TNFSF14 regulates estrogen deficiency-induced bone loss. J Pathol. 2020;250(4):440–451. doi:10.1002/path.5385
104. Yu L, Hu M, Cui X, et al. M1 macrophage-derived exosomes aggravate bone loss in postmenopausal osteoporosis via a microRNA-98/DUSP1/JNK axis. Cell Biol Int. 2021;45(12):2452–2463. doi:10.1002/cbin.11690
105. Batoon L, Millard SM, Raggatt LJ, et al. Osteal macrophages support osteoclast-mediated resorption and contribute to bone pathology in a postmenopausal osteoporosis mouse model. J Bone Miner Res. 2021;36(11):2214–2228. doi:10.1002/jbmr.4413
106. Shi F, Ni L, Gao Y-M. Tetrandrine attenuates cartilage degeneration, osteoclast proliferation, and macrophage transformation through inhibiting P65 phosphorylation in ovariectomy-induced osteoporosis. Immunol Invest. 2022;51(3):465–479. doi:10.1080/08820139.2020.1837864
107. Zhang C, Zhong Z, Sang W, et al. The dibenzyl isoquinoline alkaloid berbamine ameliorates osteoporosis by inhibiting bone resorption. Front Endocrinol. 2022;13:885507. doi:10.3389/fendo.2022.885507
108. Yang Y, Sheng D, Shi J, et al. Avicularin alleviates osteoporosis-induced implant loosening by attenuating macrophage M1 polarization via its inhibitory effect on the activation of NF-κB. Biomed Pharmacother. 2023;158:114113. doi:10.1016/j.biopha.2022.114113
109. Kamal D, Trăistaru R, Kamal CK, Alexandru DO, Ion DA, Grecu DC. Macrophage response in patients diagnosed with aseptic necrosis of the femoral head presenting different risk factors. Rom J Morphol Embryol. 2015;56(1):163–168.
110. Vicaş RM, Bodog FD, Fugaru FO, et al. Histopathological and immunohistochemical aspects of bone tissue in aseptic necrosis of the femoral head. Rom J Morphol Embryol. 2020;61(4):1249–1258. doi:10.47162/RJME.61.4.26
111. Adapala NS, Yamaguchi R, Phipps M, Aruwajoye O, Kim HKW. Necrotic bone stimulates proinflammatory responses in macrophages through the activation of toll-like receptor 4. Am J Pathol. 2016;186(11):2987–2999. doi:10.1016/j.ajpath.2016.06.024
112. Jeong H, Yoon H, Lee Y, et al. SOCS3 attenuates dexamethasone-induced M2 polarization by down-regulation of GILZ via ROS- and p38 MAPK-dependent pathways. Immune Netw. 2022;22(4):e33. doi:10.4110/in.2022.22.e33
113. Busillo JM, Azzam KM, Cidlowski JA. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J Biol Chem. 2011;286(44):38703–38713. doi:10.1074/jbc.M111.275370
114. Lannan EA, Galliher-Beckley AJ, Scoltock AB, Cidlowski JA. Proinflammatory actions of glucocorticoids: glucocorticoids and TNFα coregulate gene expression in vitro and in vivo. Endocrinology. 2012;153(8):3701–3712. doi:10.1210/en.2012-1020
115. Fang B, Wang D, Zheng J, et al. Involvement of tumor necrosis factor alpha in steroid-associated osteonecrosis of the femoral head: friend or foe? Stem Cell Res Ther. 2019;10(1):5. doi:10.1186/s13287-018-1112-x
116. Jin S, Meng C, He Y, et al. Curcumin prevents osteocyte apoptosis by inhibiting M1-type macrophage polarization in mice model of glucocorticoid-associated osteonecrosis of the femoral head. J Orthop Res. 2020;38(9):2020–2030. doi:10.1002/jor.24619
117. Jiang C, Zhou Z, Lin Y, et al. Astragaloside IV ameliorates steroid-induced osteonecrosis of the femoral head by repolarizing the phenotype of pro-inflammatory macrophages. Int Immunopharmacol. 2021;93:107345. doi:10.1016/j.intimp.2020.107345
118. Otón T, Carmona L. The epidemiology of established rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2019;33(5):101477. doi:10.1016/j.berh.2019.101477
119. Wang Q, Zhou X, Zhao Y, et al. Polyphyllin I ameliorates collagen-induced arthritis by suppressing the inflammation response in macrophages through the NF-κB pathway. Front Immunol. 2018;9:2091. doi:10.3389/fimmu.2018.02091
120. Van Raemdonck K, Umar S, Palasiewicz K, et al. CCL21/CCR7 signaling in macrophages promotes joint inflammation and Th17-mediated osteoclast formation in rheumatoid arthritis. Cell Mol Life Sci. 2020;77(7):1387–1399. doi:10.1007/s00018-019-03235-w
121. Hu L, Liu R, Zhang L. Advance in bone destruction participated by JAK/STAT in rheumatoid arthritis and therapeutic effect of JAK/STAT inhibitors. Int Immunopharmacol. 2022;111:109095. doi:10.1016/j.intimp.2022.109095
122. Deng C, Zhang Q, He P, et al. Targeted apoptosis of macrophages and osteoclasts in arthritic joints is effective against advanced inflammatory arthritis. Nat Commun. 2021;12(1):2174. doi:10.1038/s41467-021-22454-z
123. Ge G, Bai J, Wang Q, et al. Punicalagin ameliorates collagen-induced arthritis by downregulating M1 macrophage and pyroptosis via NF-κB signaling pathway. Sci China Life Sci. 2022;65(3):588–603. doi:10.1007/s11427-020-1939-1
124. Mahon OR, Browe DC, Gonzalez-Fernandez T, et al. Nano-particle mediated M2 macrophage polarization enhances bone formation and MSC osteogenesis in an IL-10 dependent manner. Biomaterials. 2020;239:119833. doi:10.1016/j.biomaterials.2020.119833
125. Li L, Li Q, Gui L, et al. Sequential gastrodin release PU/n-HA composite scaffolds reprogram macrophages for improved osteogenesis and angiogenesis. Bioact Mater. 2023;19:24–37. doi:10.1016/j.bioactmat.2022.03.037
126. Huang X, Chen M, Wu H, Jiao Y, Zhou C. Macrophage polarization mediated by chitooligosaccharide (COS) and associated osteogenic and angiogenic activities. ACS Biomater Sci Eng. 2020;6(3):1614–1629. doi:10.1021/acsbiomaterials.9b01550
127. Liu L, Guo S, Shi W, et al. Bone marrow mesenchymal stem cell-derived small extracellular vesicles promote periodontal regeneration. Tissue Eng Part A. 2021;27(13–14):962–976. doi:10.1089/ten.TEA.2020.0141
128. Zhu K, Yang C, Dai H, et al. Crocin inhibits titanium particle-induced inflammation and promotes osteogenesis by regulating macrophage polarization. Int Immunopharmacol. 2019;76:105865. doi:10.1016/j.intimp.2019.105865
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.