Back to Archived Journals » Hypoxia » Volume 2
Macrophage-mediated response to hypoxia in disease
Authors Tazzyman S, Murdoch C ![]() , Yeomans J, Harrison J, Muthana M
, Yeomans J, Harrison J, Muthana M
Received 19 August 2014
Accepted for publication 10 September 2014
Published 15 November 2014 Volume 2014:2 Pages 185—196
DOI https://doi.org/10.2147/HP.S49717
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Simon Tazzyman,1 Craig Murdoch,2 James Yeomans,1 Jack Harrison,1 Munitta Muthana3
1Department of Oncology, 2School of Clinical Dentistry, 3Department of Infection and Immunity, University of Sheffield, Sheffield, UK
Abstract: Hypoxia plays a critical role in the pathobiology of various inflamed, diseased tissues, including malignant tumors, atherosclerotic plaques, myocardial infarcts, the synovia of rheumatoid arthritic joints, healing wounds, and sites of bacterial infection. These areas of hypoxia form when the blood supply is occluded and/or the oxygen supply is unable to keep pace with cell growth and/or infiltration of inflammatory cells. Macrophages are ubiquitous in all tissues of the body and exhibit great plasticity, allowing them to perform divergent functions, including, among others, patrolling tissue, combating invading pathogens and tumor cells, orchestrating wound healing, and restoring homeostasis after an inflammatory response. The number of tissue macrophages increases markedly with the onset and progression of many pathological states, with many macrophages accumulating in avascular and necrotic areas, where they are exposed to hypoxia. Recent studies show that these highly versatile cells then respond rapidly to the hypoxia present by altering their expression of a wide array of genes. Here we review the evidence for hypoxia-driven macrophage inflammatory responses in various disease states, and how this influences disease progression and treatment.
Keywords: macrophage, hypoxia, inflammation, cytokine
Introduction
Macrophages are extremely versatile cells that adopt a distinct phenotype in response to a changing microenvironment. These cells are derived from circulating monocytes that exit the vasculature and invade into the surrounding tissues where they differentiate under the influence of local signals into resident tissue macrophages. Resident macrophages have a variety of roles; they patrol tissues for damaged or apoptotic cells, which they clear by phagocytosis, they identify and eliminate such invading pathogens as bacteria, fungi, and virally infected cells, they scavenge lipoproteins, and they are also responsible for regulating tissue oxygenation by influencing the formation of new blood vessels and modulating vascular permeability.1 Although tissue macrophages are anatomically distinct from one another, and have different transcriptional profiles and functional capabilities, they are all required for the maintenance of homeostasis. However, these reparative and homeostatic functions can be subverted by chronic insults, resulting in a causal association of macrophages with disease states. One such insult is hypoxia, which usually occurs due to an inefficient blood supply or where there is an imbalance between the number of actively respiring cells and the supply of oxygen at a particular localized tissue site. Of course, in such diseases as tumors, both these things occur simultaneously. When macrophages experience hypoxia, they respond to it rapidly with altered gene expression, mediated, in part, via upregulation of the transcription factors, hypoxia-inducible factors 1 and 2 (HIF-1 and HIF-2).2,3 These consist of a distinct hypoxia-inducible α subunit and a common, constitutively expressed β subunit. HIF-1α and HIF-2α protein stability is negatively regulated by prolyl hydroxylase domain protein-(PHD) dependent hydroxylation, which leads to von Hippel–Lindau protein-(VHL) dependent ubiquitination and proteasome-dependent degradation under normoxic conditions.4,5 HIF-1α and HIF-2α transactivation function is negatively regulated by factor inhibiting HIF-1 (FIH-1) through FIH-1-dependent asparaginyl hydroxylation, which blocks binding of the coactivators, CREB-binding protein (CBP) and p300.6,7 Thus, the stability and activity of the HIFs are modulated by hydroxylation, such that changes in oxygen availability are transduced to the nucleus as changes in HIF activity (Figure 1).8
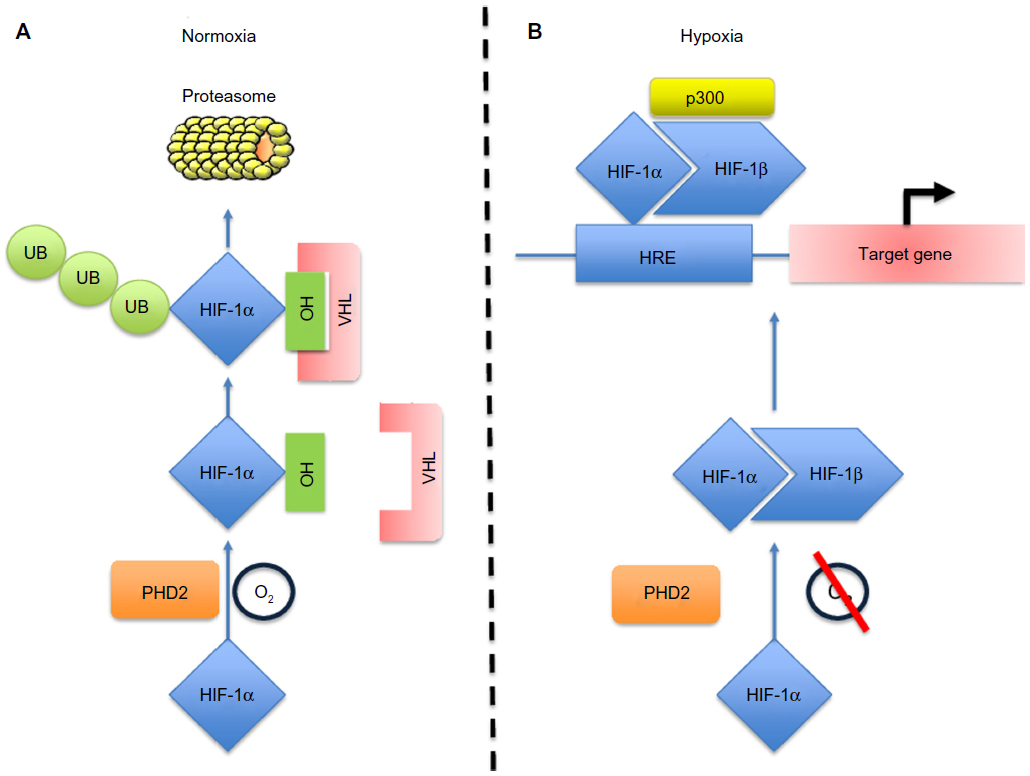 | Figure 1 Schematic diagram of the HIF-1α pathway. |
A number of studies have documented the distinct changes in gene expression that occur in macrophages when they experience hypoxia in vitro (see summary in Table 1). These include upregulation of molecules required for macrophage survival, tissue revascularization (and thus reoxygenation of the ischemic site), matrix remodeling, and recruitment and activation of more macrophages and/or other inflammatory cells.2,8,9 Importantly, a macrophage in a hypoxic tumor microenvironment is likely to express different genes than a macrophage in a hypoxic area in a wound, as other stimuli besides hypoxia will be present. In this review, we outline the responses of macrophages to hypoxia in a number of inflamed and/or diseased tissues. We then discuss the possible implications of these for their progression and treatment.
 | Table 1 Summary of select cytokines/proteases regulated by hypoxic macrophages |
Malignant tumors
One of the defining features of the tumor microenvironment is the presence of hypoxia. Within normal healthy tissue, oxygen levels are maintained within a typical range of 20–80 mmHg, due to a balance between supply and demand. In tumor tissues, this balance is disrupted by a combination of increased metabolic activity, due to rapidly dividing tumor cells, and a poor supply of oxygen from the poorly developed vasculature.10 This leads to some tumor regions experiencing oxygen levels below 10 mmHg or <1.3% oxygen. Use of hypoxic cell markers, like pimonidazole, has demonstrated the presence of both transient (avascular, nonnecrotic) and chronic (perinecrotic) areas of hypoxia in both human and experimental tumors.11,12
Tumor hypoxia is thought to play an important part in the education of tumor-associated macrophages (TAMs). Indeed, analysis of these hypoxic regions revealed a large infiltration of TAMs than has been observed in multiple tumor types, including breast, endometrial, ovarian, colorectal, prostate, and oral tumors.13–16 The accumulation of TAMs in these hypoxic areas is mediated by several mechanisms, including active recruitment to the tumor and inhibition of macrophage escape (Figure 2). In response to reduced oxygen tension, tumors release such hypoxia-induced chemoattractants as vascular endothelial growth factor (VEGF) A, endothelins, endothelial monocyte-activating polypeptide-II (EMAPII), angiopoietin 2, and CXCL12 (stromal cell-derived factor 1 [SDF-1]).17 For example, correlative studies have demonstrated that poorly vascularized areas of breast tumors express high levels of VEGF and have increased TAM numbers.18 More recently, the hypoxia-induced tumor-derived chemoattractant CXCL12 was shown to be the main driver for TAM accumulation in experimental murine gliomas, where it also increased their survival.19 CXCL12 binds to the macrophage cell surface receptor, CXCR4, which is upregulated through a HIF-1α-dependent mechanism in both monocytes and macrophages. Therefore, the hypoxia-dependent CXCL12/CXCR4 axis is likely to be important for the chemotaxis of TAMs to hypoxic tumor sites.20 Similarly, semaphorin 3A (Sema3A) also acts as an attractant for TAMs, by triggering VEGF-1 phosphorylation through the Neuropilin-1 (Nrp1) and PlexinA1/PlexinA4 receptors.21 When Nrp1 levels are downregulated in the hypoxic environment, Sema3A continues to regulate TAMs in an Nrp1-independent manner, by eliciting PlexinA1/PlexinA4-mediated stop signals, which retain TAM inside the hypoxic niche. Interestingly, in a mouse model, where macrophages had a genetic deletion of Nrp1, it was shown that TAMs were retained in normoxic tumor sites, and this resulted in the prevention of tumor growth and metastasis.21
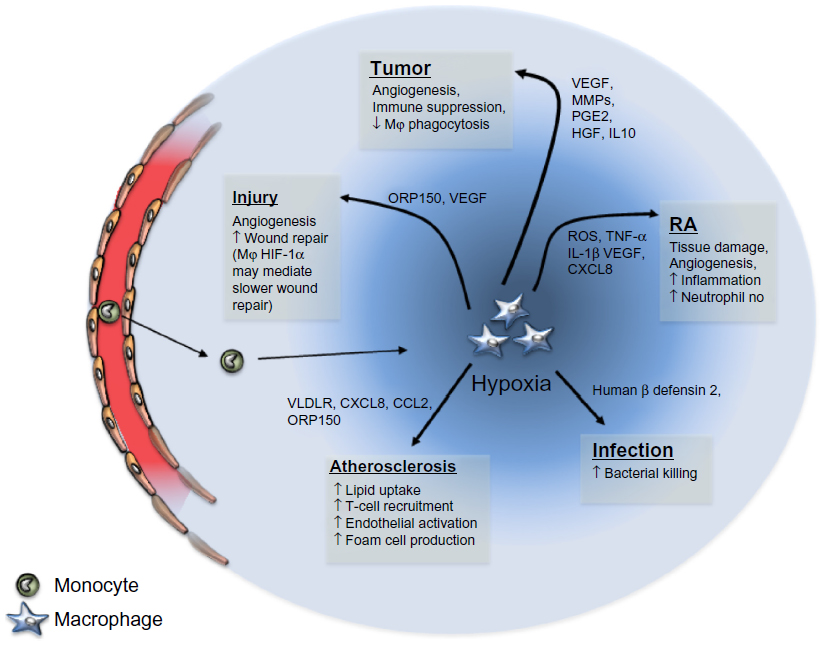 | Figure 2 Schematic representation of the role of macrophages following exposure to hypoxia in varying disease states. |
Since macrophages are phagocytes, they may also be attracted to hypoxic, perinecrotic areas via the trail of necrotic debris emanating from dead cells. Necrotic cells release high-mobility group box 1 (HMGB1) at sites of chronic tumor hypoxia, that has been shown to alter the phenotype of macrophages,22 although it has yet to be shown that tumor-derived necrotic debris can mediate monocyte recruitment to tumors. Once monocytes/macrophages are recruited, the presence of hypoxia has also been implicated in their retention in these areas. Hypoxic macrophages upregulate mitogen-activated protein kinase phosphatase (MKP-1)23 that, in turn, dephosphorylates the signaling enzymes MEK, ERK1/2, and p38 MAPK. This effectively prevents macrophages from responding to a multitude of chemotactic factors, including VEGF and chemokine (C-C motif) ligand (CCL) 2, that function through this signaling mechanism.24 Additionally, hypoxia reduces the expression of the chemokine receptors CCR2 and CCR5, further preventing macrophages from responding to chemotactic signals outside the hypoxic environment.25,26
Once within the hypoxic tumor microenvironment, macrophages are induced to undergo a phenotype change toward what has been termed an M2, or alternatively activated state.27 Exposure to hypoxia induces the build-up of the transcription factors HIF-1α and HIF-2α. These transcription factors bind to hypoxia response elements in a number of genes, increasing their transcription. Notably, in response to hypoxia, macrophages have been shown to induce such proangiogenic molecules as VEGF, fibroblast growth factor-(FGF) 2, CXCL8, cyclooxygenase-2, hepatocyte growth factor (HGF), and matrix metalloproteinase- (MMP) 12.2,18 There is now emerging data to show that tumor hypoxia also establishes an immunosuppressive macrophage phenotype. The expression of such immunosuppressive factors as prostaglandin E2 (PGE2)28 and interleukin (IL) 1029 are increased in hypoxia, and their presence within the tumor microenvironment can downregulate the tumoricidal abilities of TAMs. In addition, PGE2 and IL-10 inhibit the functions of cytotoxic CD8+ T lymphocytes and other effector cells of the immune system,30 which, combined with hypoxic inhibition of macrophage phagocytosis and antigen presentation, suppresses the triggering of an adaptive immune response toward tumor cells. Moreover, Doedens et al recently reported hypoxia- and HIF-1-dependent in vitro suppression of T-cell proliferation by macrophages.31
The hypoxia-induced skewing of macrophages toward an M2 state may not be entirely under the control of HIF-1α. Interestingly, Werno et al demonstrated that HIF-1α-deficient macrophages exposed to hypoxia were more M2-polarized, as assessed by their lower cytotoxicity toward tumor cells and the lack of a response to lipopolysaccharide (LPS) stimulation. In addition, the proangiogenic functions of these macrophages were decreased, compared to wild-type macrophages.32 Taken together, these data show a clear role for hypoxia in the promotion of tumors, by encouraging macrophages to support many aspects of tumor development.
Rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic systemic autoimmune disease characterized by a symmetrical, inflammatory arthropathy that results in destruction of bone and cartilage, leading to joint stiffness, significant pain, and loss of functional capacity.33 Although the pathogenesis of RA is still not completely understood and treatment is non-curative, studies suggest that genetic and environmental factors can trigger joint inflammation.34–36
An early event in inflammatory arthritic joints is hypoxia-driven angiogenesis, where new blood vessels invade the synovial tissue, resulting in persistent infiltration of leukocytes into the synovial fluid and the dramatically increased proliferation of synovial fibroblasts.33 Although these new blood vessels deliver oxygen to the inflammatory cell mass, the vascular network is dysfunctional, so the synovium remains a markedly hypoxic environment.37 Hypoxia and angiogenesis are now recognized as seminal events in the perpetuation of joint destruction in RA.38–41
The presence of hypoxia in the human RA joint was initially confirmed using microelectrodes that measured oxygen tension indirectly in the synovial fluid.42,43 More recent studies using a combined oxygen and temperature probe in patients with inflammatory arthritis showed that low synovial pO2 levels (<20 mmHg) significantly correlated with joint inflammation,44 lipid peroxidation, secretion of angiogenic factors, and the presence of T-cells and macrophages in the sublining layer of the synovium.45 Animal models of RA also display pO2 levels as low as 10 mmHg, and extensive hypoxia has been shown in the synovium, pannus, bone marrow, and articular cartilage chondrocytes in the arthritic joints of rats in in vivo models of the disease.38,46 Repetitive cycles of hypoxia and reoxygenation, together with oxidants produced by macrophages and neutrophils, lead to the production of reactive oxygen species that result in significant tissue damage in RA joints.47,48 Hypoxia also aggravates the synovial inflammatory lesion by increasing local production of such enzymes as COX-2 and MMPs by synoviocytes, monocytes/macrophages,49 and fibroblast-like synoviocytes.50,51 COX-2 catalyzes the conversion of arachidonic acid, which is cleaved from membrane phospholipids, to form proinflammatory eicosanoids such as PGE2 and prostacyclin I2 (PGI2). COX-2 expression is highly elevated in the RA synovium and has a direct relation to the degree of inflammation in synovial tissue.52 PGE2 is involved in the erosion of cartilage and juxta-articular bone,53 stimulates the production of MMPs,54 and contributes to angiogenesis,55 all of which are important mediators of connective tissue degradation in the synovium.
A large proportion of macrophages have been shown to accumulate in the RA synovium, with most residing in the inflamed lining layer. In fact, Hollander et al reported that >90% of the cells in the RA synovial lining stained positive for the pan-macrophage marker CD68.56 Immunohistochemical studies have revealed that both HIF-1α and HIF-2α are overexpressed in the synovial lining of patients with RA, compared to healthy joints.57 Importantly, Hollander et al found that the CD68+ cells in RA joints also expressed HIF-1α and were not detected in the synovium of normal or osteoarthritic (nonhypoxic) joints,56 suggesting that hypoxic, HIF-1α-activated macrophages are overrepresented in RA joints. In addition, Cramer et al showed that when arthritis was induced in the joints of mice bearing a targeted deletion of the HIF-1α gene in myeloid cells (macrophages and neutrophils), the mice displayed a marked reduction in joint swelling and reduced synovial infiltration by macrophages, strongly implicating the hypoxic-induced responses of these cells in RA pathology.58 A number of known hypoxia-regulated genes/proteins are also known to be expressed by macrophages in RA joints, including CXCL12,59 CXCL8,60 VEGF,61 IL-1β, and tumor necrosis factor-(TNF) α,62 which potentiate the inflammatory response and stimulate angiogenesis in RA. IL-1β and TNF-α are essential proinflammatory cytokines required for the progression of RA, because they are capable of inducing other proinflammatory cytokines and activating MMPs in autocrine and paracrine fashions.63 Inhibitors of IL-1 and TNF-α cause a reduction in synovial inflammation, bone destruction, and macrophage infiltration in patients with RA.64,65 A critical role of TNF-α and IL-1 during RA pathogenesis was confirmed by the recent development of new drugs targeting these molecules, that has revolutionized patient treatment.66
These data provide clear evidence that activation of HIF-1α in macrophages and the subsequent expression of HIF-1α target genes in these cells is an important contributor to the pathogenesis of RA. Whether HIF-1α alone is sufficient to drive RA has not been demonstrated. HIF-2α, a protein closely related to HIF-1α, is also expressed in the RA synovium.57 However, to date, there is little information on the role of HIF-2α-expressing macrophages in hypoxic joints of RA patients.
Precisely how hypoxia regulates the activity HIF-1α and HIF-2α transcription factors in the RA joint has yet to be determined. In view of the abundance of macrophages in the synovia of RA joints, and their increased activity of HIFs and subsequent expression of various proinflammatory HIF-target genes, it would be interesting to see whether inflammation in RA joints would be dampened by suppressing the expression/function of macrophage HIF-1α or HIF-2α. This could be achieved by injecting the synovial fluid of RA joints with a vector that bears short hairpin RNA (shRNA) for HIFs. More recently, highly selective antagonists have been developed that suppress HIF-1α and HIF-2α in vitro and in vivo, reducing the expression of HIF-related gene targets in tumor cells67,68 and inhibiting tumor angiogenesis;66,69 so, these could also be applied to RA. Such chemical tools provide a potential therapeutic opportunity to intervene in HIF-driven RA.
Atherosclerosis
Atherogenesis is most commonly thought to occur at sites of vessel wall injury, where it is heralded by endothelial cell dysfunction. This results in lipid accumulation and inflammatory cell infiltration, including monocyte recruitment into the arterial intima.70 These monocytes differentiate into macrophages that ingest such lipoproteins as oxidized low-density lipoprotein (LDL) and native LDL, resulting in cells laden with cytoplasmic lipid vacuoles, that are often termed “foam” cells.71 Accumulating “foam” cells form the fatty streak characteristic of early atherosclerosis and remain present in the mature atherosclerotic plaque.72 As the plaque develops, the arterial wall increases in volume, resulting in reduced oxygen diffusion and subsequent hypoxia. Bjornheden et al demonstrated arterial hypoxia at distances of 200–300 μm from the endothelial surface of atherosclerotic plaques in rabbits, and observed large numbers of “foam” cells within these regions, indicating these macrophages experience hypoxia in atherosclerosis.73
Hypoxia significantly contributes to lipid metabolism in human macrophages in vitro. Bostrom et al demonstrated that human macrophages were capable of increasing triglyceride content in response to hypoxia, while Parathath et al observed an increase in cholesterol content within macrophages.74,75 In vitro, hypoxia upregulates macrophage expression of 15-lipooxygenase-2 leading to increased LDL oxidation.76 15-lipooxygenase-2 expression also lead to increased expression of CXCL10 and CCL2, resulting in the increased T-cell recruitment observed in atherosclerosis.77 Very low-density lipoprotein (VLDL) receptors are upregulated on macrophages experiencing hypoxia, and are also known to be upregulated on plaque macrophages in vivo,78 suggesting a correlation between hypoxia and plaque macrophage expression of VLDL receptors. The VLDL receptor mediates the uptake of such lipids as β-VLDL, chylomicron remnant, and lipoprotein Lp(α), suggesting hypoxia also affects lipid metabolism through VLDL receptor-derived pathways.78–80
T lymphocyte recruitment, along with endothelial cell and smooth muscle migration and proliferation, is vital for the progression of atherosclerosis. CXCL8 is chemotactic for T lymphocytes,81 and both chemotactic and mitogenic for endothelial82 and smooth muscle cells.83 Human foam cells taken from atherosclerotic plaques release increased levels of CXCL8, compared to cultured monocytes and macrophages,84 while CXCR2-deficient mice demonstrated reduced atherosclerosis compared to wild-type mice.85 These findings suggest CXCL8 aids atherosclerotic plaque development. Hypoxia itself has been shown to upregulate macrophage CXCL8 gene expression and foam cell production of CXCL8,86,87 suggesting hypoxia prompts increased CXCL8 secretion from foam cells, leading to atherosclerotic development via interactions with T lymphocytes, endothelial cells, and smooth muscle.
Foam cells secrete a wide variety of other proteins in response to hypoxia. Foam cells from human atherosclerotic lesions have shown oxygen-regulated protein (ORP) 150 expression in response to hypoxia and the presence of oxidized LDL.88 ORP150 expression by macrophages has been linked to subsequent increased VEGF expression and angiogenesis,67 and it has been suggested that ORP150 plays an important role in preventing hypoxia-induced apoptotic cell death.90 This, combined with its expression in areas of high oxidized LDL concentration, suggests that this factor has a protective role against environmental stressors in the atherosclerotic milieu. Macrophage inhibitory factor (MIF) is a protein that is highly expressed by human foam cells, and its expression is strongly correlated to neovascularization and other signs of atherosclerotic plaque instability.91 Burger-Kentischer et al found treatment with a MIF antibody in an atherosclerotic-prone murine model resulted in reduction of a number of inflammatory markers typically associated with atherosclerosis, suggesting that MIF had a mediating effect on intimal inflammation.92 In addition, MIF blockade in murine models has resulted in plaque regression and reduced levels of T lymphocytes and macrophages.93 In addition, VEGF has been shown to be expressed in significantly greater amounts in hypoxic foam cells than nonhypoxic foam cells in vivo.75 Other factors known to be induced by hypoxia are also expressed by foam cells, such as platelet-derived growth factor (PDGF),94 TNF-α, IL-1,95 CCL2,96 MMP-1,97 MMP-7,98 though it is not yet directly known if foam cells vary their expression of these factor in a hypoxia-dependent manner.
One of the complications of atherosclerosis is myocardial infarction (MI), which results from plaque rupture, creating microemboli that can occlude blood vessels.99 If this occurs in coronary arteries, it can cause chronic hypoxia in the myocardium. Regions of myocardial hypoxia have been demonstrated in canine models of MI, and macrophages have been found in significantly greater quantities in human myocardium from ischemic heart disease patients than in controls.100,101 Macrophages recruited to the damaged tissue may be beneficial in heart recovery. Researchers have demonstrated that, after MI, these recruited macrophages express high levels of PR-39 (originally identified as an antibacterial peptide).102 This research group went on to demonstrate that PR-39 had a proangiogenic effect on mice myocardium via inhibition of HIF-1α degradation, resulting in increased HIF-1α levels and, consequently, growth factor expression.103 Evidence that elevated levels of PR-39 may be beneficial in MI has been shown with the use of recombinant adeno-associated virus, engineered to overexpress this gene. Use of this virus in a porcine model of MI resulted in decreased infarct areas in MI, creating the tantalizing possibility of utilizing PR-39 as a potential therapeutic agent in the future.104
Wound healing
Ischemia is a common event in wounds that often leads to local tissue hypoxia. Wounds compromising the epidermis and entering the underlying connective tissue disrupt the local vasculature, leading to localized bleeding, blood vessel vasoconstriction, and clot formation. Hypoxia within the wound can then develop, due to both a reduction in local perfusion and increased oxygen demand of the cells within the healing tissue.105 Oxygen tension measurements in rodent models decreased from 150 mmHg to 20–30 mmHg immediately after injury and, in some instances, these oxygen tensions decreased further to 5–7 mmHg 3–5 days post-injury.106 This acute hypoxia is thought to be essential for tissue repair, by activating platelet aggregation and promoting release of such growth factors and cytokines as VEGF and TNF-α.107–109 In addition to the typical laceration wound, hypoxia is also observed in burn wounds. Xing et al studied the expression of HIF-1α along with the hypoxic marker pimonidazole, in order to examine the spatial and temporal course of hypoxia in a murine burn model. They found that the leading edge of the healing burn was hypoxic and that this level of hypoxia correlated positively with increased expression of HIF-1α, as well as the proangiogenic factors VEGF and CXCL12.108
Inflammatory cell infiltration of wounds has long been accepted. Neutrophils are the first cells to be recruited to wound sites, and these are followed by infiltration of monocytes that quickly differentiate into macrophages. Later, lymphocytes enter the wounds and, eventually, inflammatory resolution and matrix remodeling occurs.110 The importance of macrophages in wound healing was clearly demonstrated by macrophage depletion studies. Wounded transgenic mice devoid of macrophages showed an increased expression of TNF-α but decreased levels of VEGF and tumor growth factor (TGF)-β at the injured site. Interestingly, the altered expressions of these molecules were associated with a reduced rate of reepithelialization, impaired collagen deposition, angiogenesis, and decreased cell proliferation, suggesting that macrophages are important for wound closure and healing.111 Furthermore, application of macrophages into experimentally induced cutaneous wounds was shown to accelerate wound repair.112 Although a clear role for hypoxia and macrophages in wound healing is evident, the precise role of hypoxia-induced macrophages in this process is less understood. Macrophages at the site of tissue injury have been shown to upregulate and express many factors that are typically secreted by hypoxic macrophages, including VEGF,113 PDGF,114 MMP-1,115 TNF-α, IL-1,116 and FGF-2.117 More specifically, the increased expression of these molecules in hypoxic macrophages is a direct result of HIF-1α and HIF-2α activation.2 The importance of hypoxic macrophages in wounds has been suggested by Ozawa et al, who, using immunohistochemical staining of human wound granular tissue, found that CD68-positive macrophages coexpressed the proangiogenic factors VEGF and ORP150 at these sites. Overexpression of ORP150 by macrophages in a murine wound model increased the expression of VEGF by these cells, that accelerated neovascularization and wound healing.90 Additional in vitro experiments using cultured macrophages under hypoxic and normoxic conditions have revealed a mechanism for the coexpression of these two factors. ORP150 is an endoplasmic reticulum chaperone that is encoded by a hypoxia-responsive gene. In hypoxic cultures, increased expression of ORP150 promoted secretion of VEGF by macrophages, while its inhibition caused retention of VEGF within the endoplasmic reticulum, thereby reducing the secretion of this angiogenic growth factor. This suggests that hypoxia-induced expression of ORP150 is important for the secretion of VEGF by hypoxic macrophages within healing wounds.90 Similarly, in burn wounds, macrophages have been found in the deep, hypoxic regions of the injury, where they express VEGF.109
Taken together, these data suggest that macrophages experiencing hypoxia within wounds drive a proangiogenic phenotype, with the expression of VEGF and other proangiogenic factors to revascularize the wound site. However, there are still a limited number of publications that have examined the presence of macrophages in hypoxic wound areas, and, more importantly, the effect that hypoxia-driven macrophage gene products have on would healing remains speculative. Indeed, contradicting data regarding the role of hypoxic macrophages following tissue injury have been published. Owings et al showed that HIF-1α expression in myeloid cells infiltrating wounds mediated a slower rate of wound repair. In their experiments, myeloid-specific HIF-1α knockout mice showed a more rapid rate of wound closure when compared to wild-type controls, and this was not related to an alteration in angiogenesis, cell proliferation, or the number of recruited neutrophils or macrophages at the wound site.118
Bacterial infection
Hypoxia is also a feature of tissues experiencing bacterial infection.119 This hypoxic state is due to increased oxygen consumption by the proliferation of bacteria, the accumulation of phagocytes at the infected site, and/or the vasoconstriction of vessels in the area, which could impede delivery of oxygen to the site of inflammation. Therefore, macrophages are likely to experience hypoxia at sites of bacterial infection. However, there is currently little in vivo evidence that this directly alters/regulates macrophage antibacterial functions. Johnson’s group used transgenic mice with a targeted HIF-1α knockout in myeloid cells to demonstrate the importance of HIF-1α in the antibacterial functions of macrophages, in vitro and in vivo, under both normoxic and hypoxic conditions.58 It has been shown in vitro that macrophage behavior is altered by hypoxia in bacterial infections. Anand et al observed an increase in phagocytosis of Escherichia coli by murine macrophages in a p38- and HIF-1α-dependent manner.120 Curiously, a contradictory observation has also been made; Shirato et al found hypoxia resulted in HIF-1α expression that was correlated with macrophage scavenger receptor 1 suppression and subsequent reduced phagocytic capacity on exposure to Listeria monocytogenes.121 Hypoxia has also been observed changing the expression of factors by macrophages during bacterial infection. Nickel et al observed expression of human β defensin 2 and upregulation of the vitamin D-dependent antimicrobial pathway, exclusively in response to hypoxia in Mycobacterium tuberculosis infections. This, in turn, resulted in decreased intracellular growth of M. tuberculosis.122
It should be noted that HIF-1α levels can be upregulated by rat and mouse macrophage cell lines in the absence of hypoxia by LPS (which is present in bacterial cell walls).123,124 Furthermore, mouse macrophages rapidly upregulate their expression of HIF-1α when exposed to Gram-positive or Gram-negative bacteria.124 Together, these findings indicate that HIF-1-mediated mechanisms play an important part in macrophage responses to bacterial infections and that these do not necessarily require activation by hypoxia.
Conclusion
It is clear from the evidence cited above that macrophages accumulate in areas of severe hypoxia in various inflamed sites, where they respond to microenvironmental cues and contribute to hypoxia by consuming oxygen themselves. This raises the question of whether it might be possible to exploit our knowledge of how macrophages are recruited/trapped in these hypoxic/necrotic areas, to develop new treatment strategies devised at targeting these pathways. One such treatment may also be able to exploit the natural propensity of macrophages to home to hypoxic areas. We previously described how macrophages could be isolated from patients, treated ex vivo to carry hypoxically-activated therapeutic agents, and then reintroduced back into the body, systemically.15 In addition, we recently demonstrated that the administration of macrophages “armed” with oncolytic virus 48 hours after treatment with the chemotherapeutic agent docetaxel, or following tumor irradiation, abolished the subsequent post-therapy regrowth of primary prostate tumors in mice.125 Alternatively, new therapies involving the use of peptides or small molecule antagonists to inhibit macrophage receptors for specific chemoattractants produced by hypoxic areas of tumors may help to reduce macrophage accumulation in these sites, while leaving macrophage functions in healthy tissues unaffected. This would then remove their proangiogenic contribution under hypoxia and reduce the net proangiogenic activity of cells in such areas.
Acknowledgments
The authors gratefully acknowledge the grant support of the following UK funding bodies for their work on macrophages responses to hypoxia. Simon Tazzyman: Yorkshire Cancer Research, Cancer Research UK; Craig Murdoch: BBSRC, Breast Cancer Campaign; Munitta Muthana: Yorkshire Cancer Research, Prostate cancer Charity, MRC.
Disclosure
The authors report no conflicts of interest in this work.
References
Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. | |
Fang HY, Hughes R, Murdoch C, et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood. 2009;114(4):844–859. | |
Burke B, Giannoudis A, Corke KP, et al. Hypoxia-induced gene expression in human macrophages: implications for ischemic tissues and hypoxia-regulated gene therapy. Am J Pathol. 2003;163(4):1233–1243. | |
Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. | |
Yu F, White SB, Zhao Q, Lee FS. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci U S A. 2001;98(17):9630–9635. | |
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16(12):1466–1471. | |
Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295(5556):858–861. | |
Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol. 2002;64(5–6):993–998. | |
Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66(2):605–612. | |
Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49(23):6449–6465. | |
Vaupel P, Kelleher DK, Hockel M. Oxygen status of malignant tumors: pathogenesis of hypoxia and significance for tumor therapy. Semin Oncol. 2001;28(2 Suppl 8):29–35. | |
Kennedy AS, Raleigh JA, Perez GM, et al. Proliferation and hypoxia in human squamous cell carcinoma of the cervix: first report of combined immunohistochemical assays. Int J Radiat Oncol Biol Phys. 1997;37(4):897–905. | |
Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618–631. | |
Coffelt SB, Chen YY, Muthana M, et al. Angiopoietin 2 stimulates TIE2-expressing monocytes to suppress T cell activation and to promote regulatory T cell expansion. J Immunol. 2011;186(7):4183–4190. | |
Muthana M, Giannoudis A, Scott SD, et al. Use of macrophages to target therapeutic adenovirus to human prostate tumors. Cancer Res. 2011;71(5):1805–1815. | |
Bingle L, Lewis CE, Corke KP, Reed MW, Brown NJ. Macrophages promote angiogenesis in human breast tumour spheroids in vivo. Br J Cancer. 2006;94(1):101–107. | |
Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104(8):2224–2234. | |
Lewis JS, Landers RJ, Underwood JC, Harris AL, Lewis CE. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J.Pathol. 2000;192(2):150–158. | |
Wang SC, Hong JH, Hsueh C, Chiang CS. Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Lab invest. 2012;92(1):151–162. | |
Schioppa T, Uranchimeg B, Saccani A, et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Medicine. 2003;198(9):1391–1402. | |
Casazza A, Laoui D, Wenes M, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer cell. 2013;24(6):695–709. | |
Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta. 2010;1799(1–2):101–113. | |
Grimshaw MJ, Balkwill FR. Inhibition of monocyte and macrophage chemotaxis by hypoxia and inflammation – a potential mechanism. Eur J Immunol. 2001;31(2):480–489. | |
Ashida N, Arai H, Yamasaki M, Kita T. Differential signaling for MCP-1-dependent integrin activation and chemotaxis. Ann N Y Acad Sci. 2001;947:387–389. | |
Sica A, Saccani A, Bottazzi B, et al. Defective expression of the monocyte chemotactic protein –1 receptor CCR2 in macrophages associated with human ovarian carcinoma. J Immunol. 2000;164(2):733–738. | |
Bosco MC, Reffo G, Puppo M, Varesio L. Hypoxia inhibits the expression of the CCR5 chemokine receptor in macrophages. Cell Immunol. 2004;228(1):1–7. | |
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. | |
Ertel W, Singh G, Morrison MH, Ayala A, Chaudry IH. Chemically induced hypotension increases PGE2 release and depresses macrophage antigen presentation. Am J Physiol. 1993;264(4 Pt 2):R655–R660. | |
Murata Y, Ohteki T, Koyasu S, Hamuro J. IFN-gamma and pro-inflammatory cytokine production by antigen-presenting cells is dictated by intracellular thiol redox status regulated by oxygen tension. Eur J Immunol. 2002;32(10):2866–2873. | |
Elgert KD, Alleva DG, Mullins DW. Tumor-induced immune dysfunction: the macrophage connection. J Leukoc Biol. 1998;64(3):275–290. | |
Doedens AL, Stockmann C, Rubinstein MP, et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer. October 1, 2010;70(19):7465–7475. | |
Werno C, Menrad H, Weigert A, et al. Knockout of HIF-1alpha in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis. 2010;31(10):1863–1872. | |
Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423(6937):356–361. | |
Sweeney SE, Firestein GS. Rheumatoid arthritis: regulation of synovial inflammation. Int J Biochem Cell Biol. 2004;36(3):372–378. | |
Tak PP, Zvaifler NJ, Green DR, Firestein GS. Rheumatoid arthritis and p53: how oxidative stress might alter the course of inflammatory diseases. Immunol Today. 2000;21(2):78–82. | |
Karouzakis E, Neidhart M, Gay RE, Gay S. Molecular and cellular basis of rheumatoid joint destruction. Immunol Lett. 2006;106(1):8–13. | |
Taylor PC, Sivakumar B. Hypoxia and angiogenesis in rheumatoid arthritis. Curr Opin Rheumatol. 2005;17(3):293–298. | |
Etherington PJ, Winlove P, Taylor P, Paleolog E, Miotla JM. VEGF release is associated with reduced oxygen tensions in experimental inflammatory arthritis. Clin Exp Rheumatol. 2002;20(6):799–805. | |
Koch AE. Angiogenesis as a target in rheumatoid arthritis. Ann Rheum Dis. 2003;62 Suppl 2:ii60–ii67. | |
Distler JH, Wenger RH, Gassmann M, et al. Physiologic responses to hypoxia and implications for hypoxia-inducible factors in the pathogenesis of rheumatoid arthritis. Arthritis Rheum. 2004;50(1):10–23. | |
Szekanecz Z, Koch AE. Vasculogenesis in rheumatoid arthritis. Arthritis Res Ther. 2010;12(2):110. | |
Lund-Olesen K. Oxygen tension in synovial fluids. Arthritis Rheum. 1970;13(6):769–776. | |
Treuhaft PS, DJ MC. Synovial fluid pH, lactate, oxygen and carbon dioxide partial pressure in various joint diseases. Arthritis Rheum. 1971;14(4):475–484. | |
Ng CT, Biniecka M, Kennedy A, et al. Synovial tissue hypoxia and inflammation in vivo. Ann Rheum Dis. 2010;69(7):1389–1395. | |
Biniecka M, Kennedy A, Fearon U, Ng CT, Veale DJ, O’Sullivan JN. Oxidative damage in synovial tissue is associated with in vivo hypoxic status in the arthritic joint. Ann Rheum Dis. 2010;69(6):1172–1178. | |
Peters CL, Morris CJ, Mapp PI, Blake DR, Lewis CE, Winrow VR. The transcription factors hypoxia-inducible factor 1alpha and Ets-1 colocalize in the hypoxic synovium of inflamed joints in adjuvant-induced arthritis. Arthritis Rheum. 2004;50(1):291–296. | |
Woods JM, Mogollon A, Amin MA, Martinez RJ, Koch AE. The role of COX-2 in angiogenesis and rheumatoid arthritis. Exp Mol Pathol. 2003;74(3):282–290. | |
Mirshafiey A, Mohsenzadegan M. The role of reactive oxygen species in immunopathogenesis of rheumatoid arthritis. Iran J Allergy Asthma Immunol. 2008;7(4):195–202. | |
Demasi M, Cleland LG, Cook-Johnson RJ, Caughey GE, James MJ. Effects of hypoxia on monocyte inflammatory mediator production: Dissociation between changes in cyclooxygenase-2 expression and eicosanoid synthesis. J Biol Chem. 2003;278(40):38607–38616. | |
Demasi M, Cleland LG, Cook-Johnson RJ, James MJ. Effects of hypoxia on the expression and activity of cyclooxygenase 2 in fibroblast-like synoviocytes: interactions with monocyte-derived soluble mediators. Arthritis Rheum. 2004;50(8):2441–2449. | |
Lee YH, Choi SJ, Kim A, Kim CH, Ji JD, Song GG. Expression of cyclooxygenase-1 and -2 in rheumatoid arthritis synovium. J Korean Med Sci. 2000;15(1):88–92. | |
Siegle I, Klein T, Backman JT, Saal JG, Nusing RM, Fritz P. Expression of cyclooxygenase 1 and cyclooxygenase 2 in human synovial tissue: differential elevation of cyclooxygenase 2 in inflammatory joint diseases. Arthritis Rheum. 1998;41(1):122–129. | |
Robinson DR, Tashjian AH Jr, Levine L. Prostaglandin-stimulated bone resorption by rheumatoid synovia. A possible mechanism for bone destruction in rheumatoid arthritis. J Clin Invest. 1975;56(5):1181–1188. | |
Mehindate K, al-Daccak R, Dayer JM, et al. Superantigen-induced collagenase gene expression in human IFN-gamma-treated fibroblast-like synoviocytes involves prostaglandin E2. Evidence for a role of cyclooxygenase-2 and cytosolic phospholipase A2. J Immunol. 1995;155(7):3570–3577. | |
Ben-Av P, Crofford LJ, Wilder RL, Hla T. Induction of vascular endothelial growth factor expression in synovial fibroblasts by prostaglandin E and interleukin-1: a potential mechanism for inflammatory angiogenesis. FEBS Lett. 1995;372(1):83–87. | |
Hollander AP, Corke KP, Freemont AJ, Lewis CE. Expression of hypoxia-inducible factor 1alpha by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint. Arthritis Rheum. 2001;44(7):1540–1544. | |
Giatromanolaki A, Sivridis E, Maltezos E, et al. Upregulated hypoxia inducible factor-1 alpha and -2 alpha pathway in rheumatoid arthritis and osteoarthritis. Arthritis Res Ther. 2003;5(4):R193–R201. | |
Cramer T, Yamanishi Y, Clausen BE, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–657. | |
Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El-Gabalawy H. Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum. 2002;46(10):2587–2597. | |
Rodenburg RJ, van Den Hoogen FH, Barrera P, van Venrooij WJ, van De Putte LB. Superinduction of interleukin 8 mRNA in activated monocyte derived macrophages from rheumatoid arthritis patients. Ann Rheum Dis. 1999;58(10):648–652. | |
Ikeda M, Hosoda Y, Hirose S, Okada Y, Ikeda E. Expression of vascular endothelial growth factor isoforms and their receptors Flt-1, KDR, and neuropilin-1 in synovial tissues of rheumatoid arthritis. J Pathol. 2000;191(4):426–433. | |
MacNaul KL, Hutchinson NI, Parsons JN, Bayne EK, Tocci MJ. Analysis of IL-1 and TNF-alpha gene expression in human rheumatoid synoviocytes and normal monocytes by in situ hybridization. J Immunol. 1990;145(12):4154–4166. | |
Ma Y, Pope RM. The role of macrophages in rheumatoid arthritis. Curr Pharm Design. 2005;11(5):569–580. | |
Nuki G, Bresnihan B, Bear MB, McCabe D, European Group Of Clinical I. Long-term safety and maintenance of clinical improvement following treatment with anakinra (recombinant human interleukin-1 receptor antagonist) in patients with rheumatoid arthritis: extension phase of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46(11):2838–2846. | |
De Rycke L, Baeten D, Foell D, et al. Differential expression and response to anti-TNFalpha treatment of infiltrating versus resident tissue macrophage subsets in autoimmune arthritis. J Pathol. 2005;206(1):17–27. | |
Olsen NJ, Stein CM. New drugs for rheumatoid arthritis. N Engl J Med. 2004;350(21):2167–2179. | |
Scheuermann TH, Li Q, Ma HW, et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat Chem Biol. 2013;9(4):271–276. | |
Rogers JL, Bayeh L, Scheuermann TH, et al. Development of inhibitors of the PAS-B domain of the HIF-2alpha transcription factor. J Med Chem. 2013;56(4):1739–1747. | |
Schwartz DL, Bankson JA, Lemos R Jr, et al. Radiosensitization and stromal imaging response correlates for the HIF-1 inhibitor PX-478 given with or without chemotherapy in pancreatic cancer. Mol Cancer Ther. 2010;9(7):2057–2067. | |
Vanepps JS, Vorp DA. Mechano-pathobiology of atherogenesis: a review. J Surg Res. 2007;142(1):202–217. | |
Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145(3):341–355. | |
Manduteanu I, Simionescu M. Inflammation in atherosclerosis: a cause or a result of vascular disorders: J Cell Mol Med. 2012;16(9):1978–1990. | |
Bjornheden T, Levin M, Evaldsson M, Wiklund O. Evidence of hypoxic areas within the arterial wall in vivo. Arterioscler Thromb Vasc Biol. 1999;19(4):870–876. | |
Bostrom P, Magnusson B, Svensson PA, et al. Hypoxia converts human macrophages into triglyceride-loaded foam cells. Arterioscler Thromb Vasc Biol. 2006;26(8):1871–1876. | |
Parathath S, Yang Y, Mick S, Fisher EA. Hypoxia in murine atherosclerotic plaques and its adverse effects on macrophages. Trends Cardiovasc Med. 2013;23(3):80–84. | |
Rydberg EK, Krettek A, Ullstrom C, et al. Hypoxia increases LDL oxidation and expression of 15-lipoxygenase-2 in human macrophages. Arterioscler Thromb Vasc Biol. 2004;24(11):2040–2045. | |
Danielsson KN, Rydberg EK, Ingelsten M, et al. 15-Lipoxygenase-2 expression in human macrophages induces chemokine secretion and T cell migration. Atherosclerosis. 2008;199(1):34–40. | |
Nakazato K, Ishibashi T, Nagata K, et al. Expression of very low density lipoprotein receptor mRNA in circulating human monocytes: its up-regulation by hypoxia. Atherosclerosis. 2001;155(2):439–444. | |
Niemeier A, Gafvels M, Heeren J, Meyer N, Angelin B, Beisiegel U. VLDL receptor mediates the uptake of human chylomicron remnants in vitro. J Lipid Res. 1996;37(8):1733–1742. | |
Argraves KM, Kozarsky KF, Fallon JT, Harpel PC, Strickland DK. The atherogenic lipoprotein Lp(a) is internalized and degraded in a process mediated by the VLDL receptor. J Clin Invest. 1997;100(9):2170–2181. | |
Larsen CG, Anderson AO, Appella E, Oppenheim JJ, Matsushima K. The neutrophil-activating protein (NAP-1) is also chemotactic for T lymphocytes. Science. 1989;243(4897):1464–1466. | |
Koch AE, Polverini PJ, Kunkel SL, et al. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 1992;258(5089):1798–1801. | |
Yue TL, Wang X, Sung CP, et al. Interleukin-8. A mitogen and chemoattractant for vascular smooth muscle cells. Circ Res. 1994;75(1):1–7. | |
Liu Y, Hulten LM, Wiklund O. Macrophages isolated from human atherosclerotic plaques produce IL-8, and oxysterols may have a regulatory function for IL-8 production. Arterioscler Thromb Vasc Biol. 1997;17(2):317–323. | |
Boisvert WA, Curtiss LK, Terkeltaub RA. Interleukin-8 and its receptor CXCR2 in atherosclerosis. Immunol Res. 2000;21(2–3):129–137. | |
White JR, Harris RA, Lee SR, et al. Genetic amplification of the transcriptional response to hypoxia as a novel means of identifying regulators of angiogenesis. Genomics. 2004;83(1):1–8. | |
Rydberg EK, Salomonsson L, Hulten LM, et al. Hypoxia increases 25-hydroxycholesterol-induced interleukin-8 protein secretion in human macrophages. Atherosclerosis. 2003;170(2):245–252. | |
Tsukamoto Y, Kuwabara K, Hirota S, et al. 150-kD oxygen-regulated protein is expressed in human atherosclerotic plaques and allows mononuclear phagocytes to withstand cellular stress on exposure to hypoxia and modified low density lipoprotein. J Clin Invest. 1996;98(8):1930–1941. | |
Ozawa K, Kondo T, Hori O, et al. Expression of the oxygen-regulated protein ORP150 accelerates wound healing by modulating intracellular VEGF transport. J Clin Invest. 2001;108(1):41–50. | |
Ozawa K, Kuwabara K, Tamatani M, et al. 150-kDa oxygen-regulated protein (ORP150) suppresses hypoxia-induced apoptotic cell death. J Biol Chem. 1999;274(10):6397–6404. | |
Schmeisser A, Marquetant R, Illmer T, et al. The expression of macrophage migration inhibitory factor 1alpha (MIF 1alpha) in human atherosclerotic plaques is induced by different proatherogenic stimuli and associated with plaque instability. Atherosclerosis. 2005;178(1):83–94. | |
Burger-Kentischer A, Gobel H, Kleemann R, et al. Reduction of the aortic inflammatory response in spontaneous atherosclerosis by blockade of macrophage migration inhibitory factor (MIF). Atherosclerosis. 2006;184(1):28–38. | |
Bernhagen J, Krohn R, Lue H, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13(5):587–596. | |
Ross R, Masuda J, Raines EW, et al. Localization of PDGF-B protein in macrophages in all phases of atherogenesis. Science. 1990;248(4958):1009–1012. | |
Tipping PG, Hancock WW. Production of tumor necrosis factor and interleukin-1 by macrophages from human atheromatous plaques. Am J Pathol. 1993;142(6):1721–1728. | |
Yla-Herttuala S, Lipton BA, Rosenfeld ME, et al. Expression of monocyte chemoattractant protein 1 in macrophage-rich areas of human and rabbit atherosclerotic lesions. Proc Natl Acad Sci U S A. 1991;88(12):5252–5256. | |
Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94(6):2493–2503. | |
Abbas A, Aukrust P, Russell D, et al. Matrix metalloproteinase 7 is associated with symptomatic lesions and adverse events in patients with carotid atherosclerosis. PLoS One. 2014;9(1):e84935. | |
van der Wal AC, Becker AE, van der Loos CM, Tigges AJ, Das PK. Fibrous and lipid-rich atherosclerotic plaques are part of interchangeable morphologies related to inflammation: a concept. Coron Artery Dis. 1994;5(6):463–469. | |
Lewis JS, Herrero P, Sharp TL, et al. Delineation of hypoxia in canine myocardium using PET and copper(II)-diacetyl-bis(N(4)-methylthiosemicarbazone). J Nucl Med. 2002;43(11):1557–1569. | |
Azzawi M, Kan SW, Hillier V, Yonan N, Hutchinson IV, Hasleton PS. The distribution of cardiac macrophages in myocardial ischaemia and cardiomyopathy. Histopathology. 2005;46(3):314–319. | |
Li J, Brown LF, Laham RJ, Volk R, Simons M. Macrophage-dependent regulation of syndecan gene expression. Circ Res. 1997;81(5):785–796. | |
Li J, Post M, Volk R, et al. PR39, a peptide regulator of angiogenesis. Nat Med. 2000;6(1):49–55. | |
Sun L, Hao Y, Nie X, et al. Recombinant AAV-PR39-mediated hypoxia-inducible factor 1alpha gene expression attenuates myocardial infarction. Int J Mol Med. 2014;33(1):171–177. | |
Schreml S, Szeimies RM, Prantl L, Karrer S, Landthaler M, Babilas P. Oxygen in acute and chronic wound healing. Br J Dermatol. 2010;163(2):257–268. | |
Ninikoski J, Heughan C, Hunt TK. Oxygen and carbon dioxide tensions in experimental wounds. Surg Gynecol Obstet. 1971;133(6):1003–1007. | |
Gorlach A, Brandes RP, Bassus S, et al. Oxidative stress and expression of p22phox are involved in the up-regulation of tissue factor in vascular smooth muscle cells in response to activated platelets. FASEB J. 2000;14(11):1518–1528. | |
Xing D, Liu L, Marti GP, et al. Hypoxia and hypoxia-inducible factor in the burn wound. Wound Repair Regen. 2011;19(2):205–213. | |
Guida E, Stewart A. Influence of hypoxia and glucose deprivation on tumour necrosis factor-alpha and granulocyte-macrophage colony-stimulating factor expression in human cultured monocytes. Cell Physiol Biochem. 1998;8(1–2):75–88. | |
Ross R, Odland G. Human wound repair. II. Inflammatory cells, epithelial-mesenchymal interrelations, and fibrogenesis. J Cell Biol. 1968;39(1):152–168. | |
Mirza R, DiPietro LA, Koh TJ. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am J Pathol. 2009;175(6):2454–2462. | |
Danon D, Kowatch MA, Roth GS. Promotion of wound repair in old mice by local injection of macrophages. Proc Natl Acad Sci U S A. 1989;86(6):2018–2020. | |
Swift ME, Kleinman HK, DiPietro LA. Impaired wound repair and delayed angiogenesis in aged mice. Lab Invest. 1999;79(12):1479–1487. | |
Rappolee DA, Mark D, Banda MJ, Werb Z. Wound macrophages express TGF-alpha and other growth factors in vivo: analysis by mRNA phenotyping. Science. 1988;241(4866):708–712. | |
Gu Q, Wang D, Gao Y, et al. Expression of MMP1 in surgical and radiation-impaired wound healing and its effects on the healing process. J Environ Pathol Toxicol Oncol. 2002;21(1):71–78. | |
Fahey TJ 3rd, Sherry B, Tracey KJ, et al. Cytokine production in a model of wound healing: the appearance of MIP-1, MIP-2, cachectin/TNF and IL-1. Cytokine. 1990;2(2):92–99. | |
Kibe Y, Takenaka H, Kishimoto S. Spatial and temporal expression of basic fibroblast growth factor protein during wound healing of rat skin. Br J Dermatol. 2000;143(4):720–727. | |
Owings RA, Boerma M, Wang J, et al. Selective deficiency of HIF-1alpha in myeloid cells influences secondary intention wound healing in mouse skin. In Vivo. 2009;23(6):879–884. | |
Sawyer RG, Spengler MD, Adams RB, Pruett TL. The peritoneal environment during infection. The effect of monomicrobial and polymicrobial bacteria on pO2 and pH. Ann Surg. 1991;213(3):253–260. | |
Anand RJ, Gribar SC, Li J, et al. Hypoxia causes an increase in phagocytosis by macrophages in a HIF-1alpha-dependent manner. J Leukoc Biol. 2007;82(5):1257–1265. | |
Shirato K, Kizaki T, Sakurai T, et al. Hypoxia-inducible factor-1alpha suppresses the expression of macrophage scavenger receptor 1. Pflugers Arch. 2009;459(1):93–103. | |
Nickel D, Busch M, Mayer D, Hagemann B, Knoll V, Stenger S. Hypoxia triggers the expression of human beta defensin 2 and antimicrobial activity against Mycobacterium tuberculosis in human macrophages. J Immunol. 2012;188(8):4001–4007. | |
Blouin CC, Page EL, Soucy GM, Richard DE. Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia-inducible factor 1alpha. Blood. 2004;103(3):1124–1130. | |
Peyssonnaux C, Datta V, Cramer T, et al. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115(7):1806–1815. | |
Muthana M, Rodrigues S, Chen YY, et al. Macrophage delivery of an oncolytic virus abolishes tumor regrowth and metastasis after chemotherapy or irradiation. Cancer Res. 2013;73(2):490–495. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.