Back to Journals » Journal of Inflammation Research » Volume 18
M-CSF Protects Against Ulcerative Colitis via Aconitate: Mendelian Randomization and Experimental Evidence
Authors Zhang Y ![]() , Huang L, Yue N, Mai Z, Kong C, Tian C, Wei DR, Yao J
, Huang L, Yue N, Mai Z, Kong C, Tian C, Wei DR, Yao J ![]() , Wang L
, Wang L ![]() , Li D
, Li D ![]()
Received 23 March 2025
Accepted for publication 30 June 2025
Published 31 July 2025 Volume 2025:18 Pages 10313—10329
DOI https://doi.org/10.2147/JIR.S528072
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Nadia Andrea Andreani
Yuan Zhang,1,2,* Longbin Huang,3,* Ningning Yue,3,* Zhiliang Mai,3,* Chen Kong,3 Chengmei Tian,4 Dao-ru Wei,5 Jun Yao,1 Lisheng Wang,1 Defeng Li1
1Department of Gastroenterology, Shenzhen People’s Hospital (the Second Clinical Medical College, Jinan University; The First Affiliated Hospital, Southern University of Science and Technology), Shenzhen, Guangdong, People’s Republic of China; 2Department of Medical Administration, Huizhou Institute for Occupational Health, Huizhou, Guangdong, People’s Republic of China; 3Department of Gastroenterology, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University), Shenzhen, Guangdong, People’s Republic of China; 4Department of Emergency, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University; The First Affiliated Hospital, Southern University of Science and Technology), Shenzhen, Guangdong, People’s Republic of China; 5Department of Rehabilitation, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University; the First Affiliated Hospital, Southern University of Science and Technology), Shenzhen, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lisheng Wang, Department of Gastroenterology, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University; the First Affiliated Hospital, Southern University of Science and Technology), No. 1017, Dongmen North Road, Luohu District, Shenzhen, 518020, People’s Republic of China, Tel +86 755 25533018, Email [email protected] Defeng Li, Department of Gastroenterology, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University; the First Affiliated Hospital, Southern University of Science and Technology), No. 1017, Dongmen North Road, Luohu District, Shenzhen, 518020, People’s Republic of China, Tel +86 755 25533018, Email [email protected]
Background: Although cytokines have been implicated in the development of ulcerative colitis (UC), the potential mediating role of metabolite levels in this association remains unclear.
Methods: Utilizing data from genome-wide association studies (GWAS) encompassing 91 circulating cytokines, 1400 blood metabolites, and 178,689 UC cases, we performed a two-sample Mendelian randomization (MR) analysis to investigate the effect of metabolites mediated cytokines on the development of UC. A two-step MR analysis was conducted to quantitatively evaluate the mediation effect. Additionally, dextran sodium sulfate (DSS)-induced colitis mice were used to further confirm our results.
Results: Mendelian randomization (MR) analysis indicated that macrophage colony-stimulating factor (M-CSF) had a causal and positive relationship with aconitate (OR: 1.10, 95% CI: 1.00– 1.20, p = 0.043, IVW beta 1 = 0.095). Moreover, MR analysis revealed that high level of aconitate were associated with reduced risk of UC (OR: 0.44, 95% CI: 0.24– 0.80, p = 0.008, IVW beta 2 = − 0.818). In addition, MR analysis showed M-CSF had an inverse correlation with the disease onset of UC (OR: 0.31, 95% CI: 0.15– 0.80, p = 0.002, IVW beta all = − 1.16). Furthermore, the mediation effect of aconitate mediated M-CSF on the risk of UC was − 0.0777 (95% CI: − 0.154 to − 0.0018, p = 0.045), accounting for 6.69% of the total effect, and indicating a modest contribution to the protective effect of M-CSF against UC. Subsequently, as an in vivo validation model, DSS-induced colitis was employed to demonstrate that M-CSF treatment significantly ameliorated weight loss, disease activity index (DAI) scores, colon shortening, and histological damage. Additionally, M-CSF treatment also significantly reduced M1 macrophage infiltration, elevated levels of aconitate as well as itaconate, and decreased the levels of pro-inflammatory cytokines in colitis. These results demonstrated that aconitate inhibited the expression of pro-inflammatory cytokines through its enzymatic conversion into the immunometabolite itaconate by aconitate decarboxylase 1 (Acod1), and downregulated the levels of M1 macrophages, thereby ameliorating colitis.
Conclusion: These findings suggest that M-CSF is an important anti-inflammatory cytokine in UC, which may be a promising therapeutic target in the treatment of UC.
Keywords: ulcerative colitis, Mendelian randomization, macrophage colony-stimulating factor, aconitate
Graphical Abstract:

Introduction
Ulcerative colitis (UC) is an immune-mediated chronic inflammation disease, characterized by chronic damage to the colonic epithelial mucosa resulting in a remitting and relapsing course.1 UC is a multifactorial disease involving environmental factors, genetic factors, dysregulated immune response, epithelial barrier defects, and inappropriate production of chemokines and proinflammatory cytokines.2,3 The treatment options for UC primarily rely on targeted biologic therapies, such as Janus kinase (JAK) signaling pathway inhibitors (eg, tofacitinib), anti-tumor necrosis factor (anti-TNF) antibodies, and non-targeted therapies, including 5-aminosalicylates, immunosuppressive agents, and glucocorticoids.4 However, anti-TNF agents, while effective in reducing inflammation, face limitations like immunogenicity and variable response rates.5,6 JAK inhibitors, though rapid-acting and oral, carry safety concerns (eg, pregnancy contraindications) and heterogeneous efficacy due to pathway redundancy.7,8 The toxicity, adverse events and intolerance associated with these treatments prevent the patients with UC from achieving remission.9 As a result, there is an urgency to identify novel therapeutic targets.
Cytokines, as circulating proteins, play a pivotal role in orchestrating inflammatory responses within the colonic microenvironment. Aberrant inflammatory responses are hallmarks of UC.10 The regulation of the intestinal immune system is contingent upon cytokines.11,12 Emerging evidence suggests that cytokine-mediated immune activation dynamically interacts with blood metabolite profiles, particularly through microbial-host co-metabolites.13 Metabolites, as intermediate or end products of metabolic processes, are modulated by cytokines, gut microbial factors, genetic factors, and dietary factors. Furthermore, their alterations have been implicated in the pathogenesis of UC. For instance, reduced levels of short-chain fatty acids (SCFAs), particularly butyrate, impair colonic epithelial barrier function and exacerbate mucosal inflammation in UC by disrupting energy metabolism and immune homeostasis.14 However, no studies have systematically explored the causal relationship network between cytokines and metabolites in UC.
MR is an emerging technique in medical research, which has been used to assess causal relationships between specific exposures and outcomes by using genetic variations as instrumental variables (IVs).15 The principle underlying MR bears resemblance to a randomized clinical trial, whereby genetic variation is randomly allocated during meiosis and conception. This approach is frequently regarded as the gold standard for establishing causality, particularly due to its capacity to circumvent the confounding effects and reverse causation issues that are commonly observed in traditional observational studies.16 This study aimed to ascertain the causal relationship between cytokines, metabolites, and UC by employing bidirectional two-sample, two-step MR analysis. We found that M-CSF had a causal and positive relationship with aconitate, and the level of aconitate were negatively associated with UC development. Research indicates that M-CSF maintains mucosal homeostasis by supporting the survival and functional plasticity of resident macrophages, which coordinate immune surveillance and tolerance to commensal microbiota.17,18 Aconitate, an intermediate of the tricarboxylic acid (TCA) cycle and precursor of the anti-inflammatory metabolite itaconate, may play a regulatory role in immune responses relevant to UC. Given the limitations of MR in fully elucidating biological mechanisms, coupled with the fact that although M-CSF has been implicated in modulating inflammation, its precise role in UC remains unclear and potentially context-dependent, we further validated the findings in a murine model of colitis. We demonstrated that M-CSF significantly reduced M1 macrophage infiltration and effectively elevated levels of aconitate as well as itaconate, thereby improving DSS-induced colitis.
Materials and Methods
Methods
The three fundamental assumptions for MR analysis are relevance, independence, and exclusion restriction. These assumptions ensure that the genetic variants, as instrumental variables (IVs), are robust and unbiased.19 The first assumption, relevance, indicates that the genetic variants chosen must be linked to the risk factors being studied. The second assumption, independence, requires that these genetic variants are not influenced by any confounding factors. The third assumption, exclusion restriction, states that the genetic variants should affect the outcome only through the specific risk factors.
Mediation analysis typically estimates three parameters, which can also be referred to effects. The first is the total effect, which represents the effect of the exposure on the outcomes. The second is the direct effect, which represents the remaining impact of the exposure on the outcome and operates through pathways other than the specified mediator or set of mediators. The third is the indirect effect, also known as the mediating effect, which represents the path from the exposure to the outcome that operates through the mediators.20,21 Following standard causal mediation frameworks,22 mediation analysis requires two prerequisites: (1) a significant causal effect of the exposure on the mediator (p < 0.05), and (2) a significant causal effect of the mediator on the outcome (p< 0.05); Metabolites violating either assumption were excluded from mediation models.
The procedures were as follows. Firstly, a two-sample bidirectional MR analysis was employed to ascertain the causal relationship between cytokines and UC. This analysis identified cytokines highly associated with UC risk (referred to as single nucleotide polymorphism 1, SNP1) and yielded a total effect estimate. Next, we screened metabolites that demonstrated a correlation with UC, referred to as SNP2 (Figure 1A). Subsequently, a two-step MR approach was employed for mediation analysis. In the initial stage of the analysis, a two-sample MR was conducted to examine the relationship between the selected cytokine-associated SNP1 and SNP2, resulting in the extraction of beta 1. In the second stage of the analysis, a two-sample MR was conducted between the chosen metabolites SNP2 and UC, resulting in beta 2. The study design is illustrated in Figure 1B.
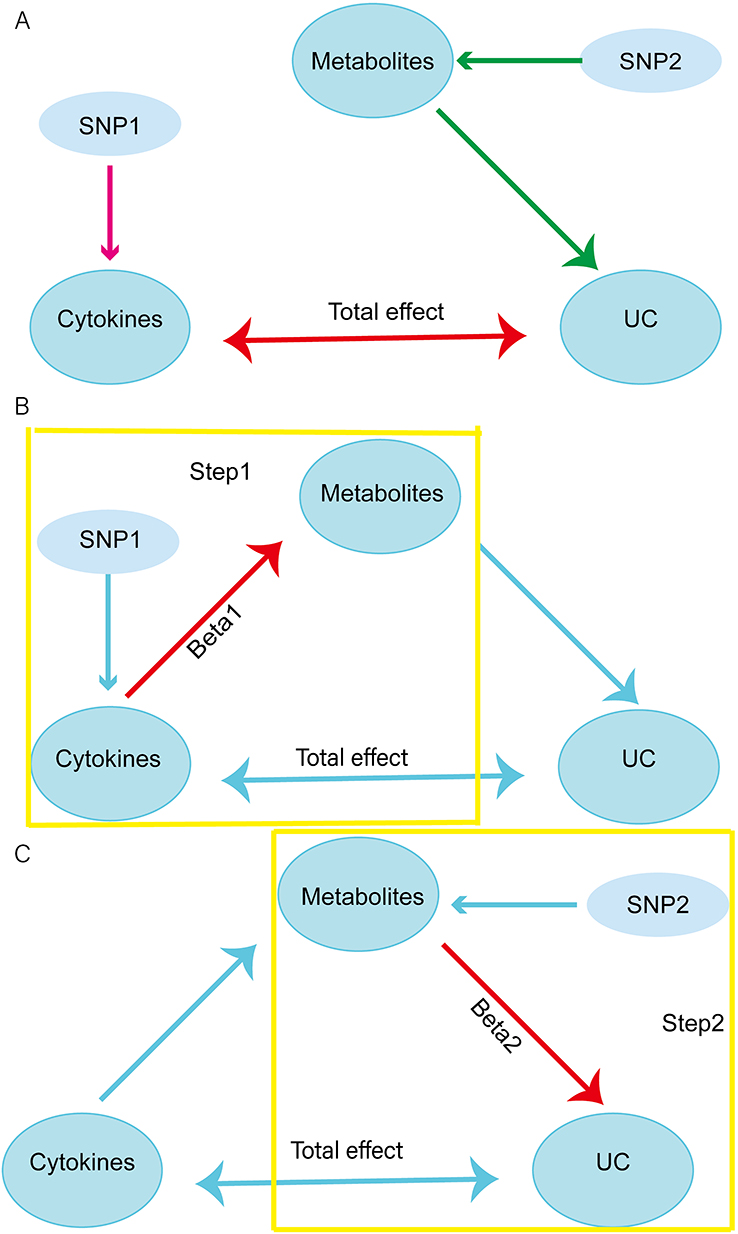 |
Figure 1 Schematic representation of a two-step MR analysis between cytokines and UC mediated by plasma metabolites. (A) Screening of UC-associated cytokines (SNP1) and metabolites (SNP2). (B) The first-stage MR analysis estimates the causal effect of cytokines on metabolites. (C) The second-stage MR analysis estimates the causal effect of metabolites on UC risk. |
Data Source
This dataset was derived from a GWAS investigation conducted by Zhao et al, which encompassed the analysis of 91 circulating cytokines.10 In their research publication, the authors conducted protein quantitative trait locus (p-QTL) mapping in a cohort comprising 14,824 individuals of European ancestry. The primary aim was to identify genetic variants associated with circulating cytokines. This was done by collating p-QTL information within ±1 megabase (Mb) of 91 candidate genes associated with these proteins. The GWAS data for metabolites are sourced from a study published in 2023 by Chen et al.23 They performed a GWAS on 8299 individuals from the Canadian Longitudinal Study on Aging (CLSA) cohort, using 1400 blood metabolites. The GWAS summary statistics for UC are accessible for download from https://gwas.mrcieu.ac.uk/. We selected data for ebi-a-GCST90018713. This dataset includes 178,689 cases (N case = 314; N control = 178,375) of European ancestry with UC and 12,454,670 SNPs.
This analysis used de-identified, publicly available summary-level GWAS data. According to Article 32 of China’s Measures for Ethical Review (2023), this research qualifies for exemption from institutional ethics approval as it involved no individual identifiers.
Two-Step MR Analysis
Selection Criteria for IVs
The criteria for the selection of the IVs are as follows. (1) We conducted a correlation analysis, selecting independent genetic variants at a genome-wide significant level (p < 1×10−5), and selected SNPs strongly associated with exposure as IVs.24,25 (2) The SNPs were then filtered to remove linkage disequilibrium (LD), using data from the European samples of the 1000 Genomes Project (r2 = 0.001, clumping window = 10,000 kb). (3) The F-statistic was used to assess the strength of the IVs. We excluded genetic variants with an F-statistic <10, thereby reducing the bias introduced by weak IVs.
Outcome Variable Selection Criteria
The outcome variable was obtained by matching the corresponding instrumental variable from the abovementioned databases.
Main Analysis Methods
In the two-sample MR analysis, MR-Egger, Weighted Median (WM), Inverse Variance Weighted (IVW), Simple Mode and Weighted Mode were used to assess causality. However, the IVW method is the primary MR analysis technique providing estimates of the causal effect.26 The WM and MR-Egger regression methods were performed to assess potential violations of the MR assumptions.27
Sensitivity Analysis
The sensitivity analysis included heterogeneity and pleiotropy test based on three perspectives: (a) Cochran’s Q test assessed heterogeneity across IVs, p < 0.05 suggests potential pleiotropy. (b) Egger regression examined directional pleiotropy (systematic bias) across genetic variants by testing whether the intercept term significantly deviates from zero, a non-zero intercept (p < 0.05) indicated systematic pleiotropic effects.28 (c) The MR pleiotropy residual sum and outlier (MR-PRESSO) identified and corrected outlier IVs contributing to pleiotropy by comparing observed and expected causal estimates before and after outlier removal.
Visualization of Results
In order to facilitate the interpretation of the results of the MR analysis, a series of visual representations were developed.29 A leave-one-out analysis was conducted, whereby one SNP was removed at a time to assess the overall robustness of the analysis. The scatter plots demonstrated the influence of each single nucleotide polymorphism on the exposure and outcome variables, thereby capturing their combined effect. The use of forest plots, employing the Wald ratio method, facilitated the clarification of the individual IVs contribution to the overall causal estimate. The utilization of funnel plots facilitated the visualization of potential bias in the selection of SNPs.
Bidirectional MR Analysis
Reverse MR analysis demonstrated no evidence of reverse causation from UC to cytokines. This approach ensured that the analysis strictly followed a unidirectional causal pathway from exposure to outcome. The remaining analytical procedures were analogous to those previously outlined for the forward MR analysis. All analyses were performed in R, version 4.3.3, an open-access software platform (https://www.r-project.org/). The R package “TwoSampleMR” and the R/Bioconductor packages “gwasglue” and “VariantAnnotation” were utilized for these analyses.
Mediation Analysis
Moreover, a two-step MR analysis was conducted to investigate whether metabolites act as mediators in the causal relationship between cytokines and UC. Subsequently, the overall effect was divided into a mediation effect and a direct effect. The direct effect was calculated as: Direct effect = Total effect - Mediating effect.30
The mediation effect of the cytokines on UC was decomposed into two distinct components: the causal effect of the exposure on the mediator, represented by the parameter beta 1, and the causal effect of the mediator on the outcome, represented by the parameter beta 2. The following formula was employed to calculate the mediating effect: Mediating effect = beta 1×beta 221 (Figure 2).
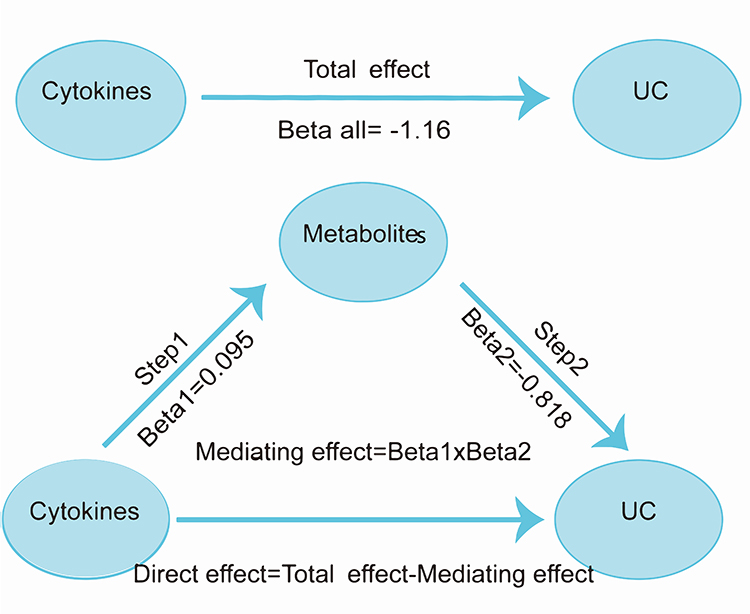 |
Figure 2 The steps and results of mediation analysis. |
Experimental Validation
Mice
C57BL/6 male mice were purchased from Biotechnology Co., Ltd, Beijing, China. All mice were maintained in a pathogen-free setting and were subjected to a 12-hour alternating light/dark regimen. At 8 weeks of age, they were used for experiments according to the guidelines and regulations of the Animal Care Committee of Shenzhen People’s Hospital, Shenzhen, China. Adherence to all institutional and national protocols regarding animal care and utilization was strictly maintained.
Cell Culture
The mouse macrophage cell line RAW264.7 was obtained from American Type Culture Collection (ATCC) and maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, USA) with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS), penicillin (100 U/mL), and streptomycin (100 mg/mL) in an incubator with 5% CO2 at 37°C. RAW264.7 cells were seeded in a 6-well culture plate at a density of 2×106 cells/mL and stimulated with 1 μ g /mL lipopolysaccharide (LPS from Escherichia coli serotype 055:B5, Sigma-Aldrich, USA) for 24 h to induce M1 macrophage polarization. To evaluate the effect of the M-CSF on macrophage polarization, cells were incubated with M-CSF, for 24 h.
Model Establishment
The mice of the DSS group were administered 3% DSS (MW 36,000 to 50,000 k Da, MP Biomedical) dissolved in drinking water, while the control group received the drinking water without DSS. The DSS + M-CSF group received a daily intraperitoneal injection of 50 ng of recombinant mouse M-CSF (Pepro Tech). During the experimental period, the status of the body weight, diarrhea, and bleeding was monitored daily. After 7 days, mice were sacrificed, and blood samples as well as colorectal tissues were collected.
Hematoxylin–Eosin (H&E) Staining
Colon tissues were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at 4-μm thickness. Sections were stained with hematoxylin, differentiated in acid alcohol, counterstained with eosin, then dehydrated, cleared, and mounted for microscopic examination. Histological scoring was performed according to previously described.31
Metabolomic LC–MS/MS Analysis
The samples were processed using methanol and 3-NPH (Shanghai Aladdin Biochemical Technology Co., Ltd). Metabolomic liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was performed using a liquid chromatography system (Waters Acquity UPLC) coupled to a quadrupole time-of-flight instrument (AB SCIEX 5500 Qtrap-MS). The chromatographic separation was carried out on an Acquity UPLC HSS T3 column with a gradient elution program, and the mass spectrometry was conducted under electrospray ionization (ESI) conditions for the detection of aconitate and itaconate. Data were processed using Multi Quant software for integration.
Quantitative Real-Time Polymerase Chain Reaction (qPCR)
Colon samples were subjected to RNA extraction using the TRIzol Reagent (Thermo, USA). RNA concentration was measured using the NanoDrop 2000 (Thermo Scientific, Massachusetts, USA). Reverse transcription was performed using the PrimeScript RT Master Mix (Takara).32 The qPCR amplification was carried out in triplicate using SYBR Green master mix (Applied Biosystems) with the following conditions: initial denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min, with a final melt curve analysis from 65°C to 95°C to verify primer specificity.33,34 Two validated internal control genes (GAPDH and proactivator saposin precursor (PSAP)) were included in all reactions to normalize target gene expression levels.35 The geometric mean of their threshold cycle (Ct) values was used for normalization according to the comparative Ct method.36 Primers were designed using Primer-BLAST (National Center for Biotechnology Information) with the following criteria: amplicon length 80–150 bp, guanine-cytosine (GC) content 40–60%, and absence of secondary structures. Sequences were listed in Table S1.
Enzyme-Linked Immunosorbent Assay (ELISA)
The concentrations of IL-6, IL-17A, and IL-10 in serum samples were quantified using commercial ELISA kits, following the manufacturer’s protocols for optimized capture ELISAs.37 Briefly, samples were incubated in pre-coated microplates, washed with phosphate-buffered saline (PBS) buffer, and incubated with biotin-labeled detection antibodies. After adding streptavidin-HRP conjugate and 3,3′,5,5′-tetramethylbenzidine (TMB) substrate, the colorimetric reaction was stopped with sulfuric acid solution, and absorbance was measured at 450 nm using a microplate reader. Standard curves derived from recombinant cytokines were employed to calculate cytokine concentrations, with each sample analyzed in duplicate to ensure reproducibility. This method has been validated for quantifying inflammatory cytokines as described in previous studies.38,39
Flow Cytometry Analysis
For flow cytometry analysis, cells were first blocked with appropriate serum to prevent non-specific antibody binding. Subsequently, the mouse macrophage cell line RAW264.7 was stained with fluorochrome-conjugated antibodies specific to surface markers that define M1 (CD86 (BioLegend, 105011)) and M2 (CD206 (BioLegend, 141717)) phenotype. Gating strategy: Viable cells were identified using FSC-A vs SSC-A scatter plots to exclude debris and cell aggregates. Doublets were subsequently excluded through FSC-H vs FSC-A analysis.40,41 For RAW264.7 characterization, positive thresholds for CD86 (M1) and CD206 (M2) were established using unstained controls and fluorescence-minus-one (FMO) controls.42,43 Instrument settings: FITC (CD86) and PE (CD206) fluorochromes were excited by a 488 nm laser, with detection through 530/30 nm and 575/26 nm filters respectively.44 Following staining, cells were acquired on a flow cytometer, with ≥10,000 cellular events collected per sample to ensure statistical power.45 Data analysis was assessed using FlowJo (TreeStar, USA).
Statistical Analysis
Comparisons of multiple experimental groups were performed using one-way or two-way analysis of variance (ANOVA). A t-test was calculated to compare the means of the two groups. Data are presented as means ± SEM. The P <0.05*P < 0.01**P < 0.001***P < 0.0001**** were considered statistically significant, and ns was not significant.
Results
Selection of Exposure Variable: Bidirectional MR Analysis Between Cytokines to UC
We excluded SNPs that had both an LD with palindromic structure and prior association with the pathway. The IVW methods indicated a causal association of M-CSF (IVW, odds ratio (OR): 0.31,95% confidence interval (CI): 0.15–0.65, p = 0.002), interleukin-10 (IL-10) receptor subunit alpha (IVW, OR: 1.80, 95% CI: 1.14–2.83, p = 0.012), interleukin-6 (IL-6) (IVW, OR:0.42, 95% CI: 0.20–0.88, p = 0.022), SIR2-like protein 2 (IVW, OR:2.44, 95% CI: 1.04–5.76, p = 0.041), and urokinase-type plasminogen activator (IVW, OR: 1.71, 95% CI: 1.07–2.72, p = 0.024) with UC (Table S2). Reverse MR analysis indicated no reverse causal relationship between M-CSF (IVW, OR: 1.00, 95% CI: 0.99–1.00, p = 0.97), IL-10 receptor subunit alpha (IVW, OR: 1.00, 95% CI: 0.99–1.00, p = 0.99), IL-6 (IVW, OR:1.00, 95% CI: 0.99–1.00, p = 0.58), SIR2-like protein 2 (IVW, OR:1.00, 95% CI: 0.99–1.00, p = 0.64) and UC (Table S3). Finally, M-CSF, IL-10 receptor subunit alpha, IL-6 and SIR2-like protein 2 were selected as the exposure variable (SNP1).
Selection of Mediator Variable: MR Analysis Between Metabolites and UC
We performed MR analysis on SNPs representing 1400 metabolites in the CLSA cohort. We identified that there were ten metabolites with a causal association with UC, including Aconitate (IVW, OR: 0.44, 95% CI: 0.24–0.80, p = 0.008), 4-hydroxycoumarin (IVW, OR: 2.06, 95% CI: 1.20–3.56, p = 0.010), Alpha-ketoglutaramate (IVW, OR: 0.58, 95% CI: 0.39–0.88, p = 0.008), Sphingomyelin (IVW, OR: 0.27, 95% CI: 0.11–0.64, p = 0.003), Cortolone glucuronide (IVW, OR: 2.28, 95% CI: 1.22–4.25, p = 0.009), 6-bromotryptophan (IVW, OR: 0.45, 95% CI: 0.26–0.79, p = 0.005), Hydroxy-N6, N6, N6-trimethyllysine (IVW, OR: 0.37, 95% CI: 0.19–0.69, p = 0.002), Anthranilate (IVW, OR: 1.87, 95% CI: 1.78–2.96, p = 0.008), Cholesterol to cortisol ratio (IVW, OR: 3.48, 95% CI: 1.69–7.19, p = 0.0007) and Benzoate to linoleoyl-arachidonoyl-glycerol ratio (IVW, OR: 1.48, 95% CI: 1.16–1.89, p = 0.002) (Table S4).
Results of Mediation Analysis
Using IVW regression, we identified a unidirectional causal effect of M-CSF on aconitate levels (beta 1 = 0.095, OR: 1.10, 95% CI: 1.00–1.20, p = 0.043) (Table S5), showing aconitate as a significant mediator associated M-CSF with UC. Other UC-associated metabolites were excluded from mediation analysis because they violated the fundamental assumptions of causal mediation, specifically due to the absence of a significant causal effect of M-CSF on these metabolites (p > 0.05).
Further MR analysis demonstrated that aconitate exerted a protective effect against UC (beta 2 = −0.818, OR: 0.44, 95% CI: 0.24–0.80, p = 0.008) (Table S6). Following the identification of M-CSF as the cytokine of interest, the causal relationship between M-CSF and UC was investigated (beta all = −1.16, OR: 0.31, 95% CI: 0.15–0.80, p = 0.002) (Table S7).
Our analysis identified a single mediator, aconitate, which mediated the effect of M-CSF on the risk of developing UC. The mediation effect and proportion were calculated using the delta method. The results indicated that the mediation effect of aconitate was −0.0777 (95% CI: −0.154 to −0.0018, p = 0.045), accounting for 6.69% of the total effect (Table 1).
 |
Table 1 Mediation Effect and Proportion of Aconitate in the Pathway Linking Macrophage Colony-Stimulating Factor (M-CSF) to Ulcerative Colitis (UC) Risk |
Results of Sensitivity Analysis and Visualization
All selected SNPs exhibited F-statistics above the critical threshold of 10, indicating minimal risk of weak instrument bias for these IVs (Table S8).46 The r² value of 0–1 indicated no LD between the two SNPs, which was consistent with random genetic distribution.47,48 To evaluate the reliability of our findings, sensitivity analyses were conducted using MR-Egger regression and Cochran’s Q test.49 The p-values of Cochran’s Q test were all greater than 0.05; the intercepts of Egger regression were approximately zero, indicating no significant heterogeneity.50 Following the removal of outlier SNPs using the multi-sample MR-PRESSO approach test, p-values were all greater than 0.05. The findings confirmed the alignment of our analysis with the three core postulates inherent to MR methodology (Table 2).
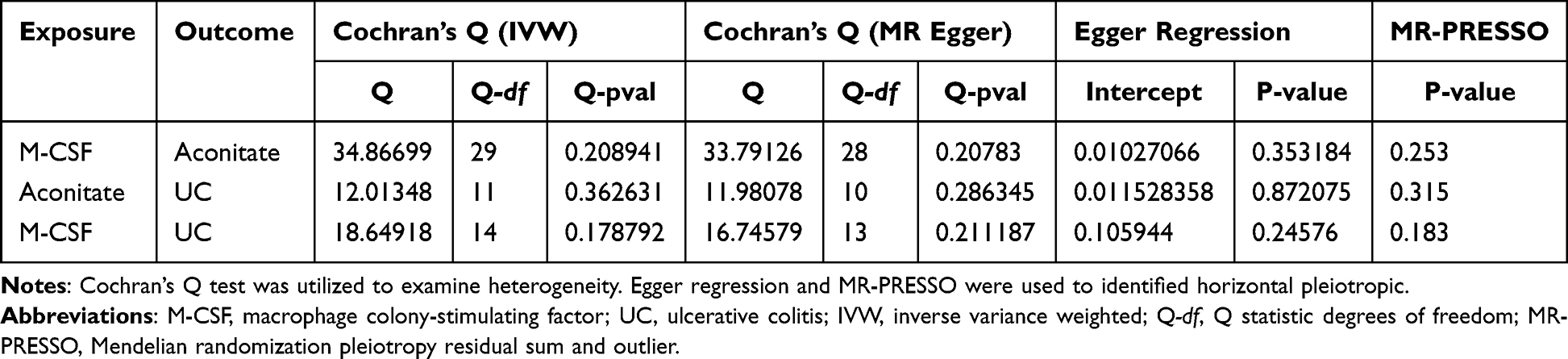 |
Table 2 The Results of Sensitivity Analysis |
Furthermore, the results of the MR analysis were visualized. The scatter plots demonstrate the trends in effects observed when utilizing different parameter estimation methods (Figure 3). A leave-one-out analysis was conducted to evaluate the influence of each SNP on the overall causal estimate (Figure 4). The funnel plots demonstrate the outcomes of the heterogeneity assessment, using both the IVW and MR-Egger approaches (Figure 5). The forest plots provide a visual illustration of the strength of association between each SNP and the outcome variable (Figure 6).
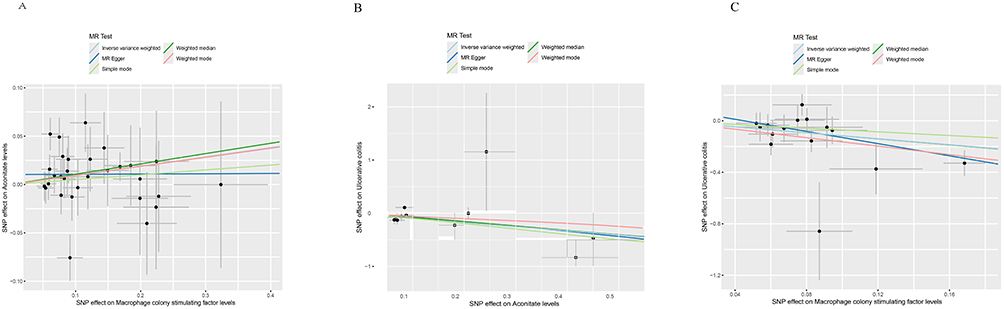 |
Figure 3 Scatter plots of Mendelian randomization analyses. The y-axis represents the causal effect of IVs on the outcome, while the x-axis represents the effect on the exposure. The slope illustrates the effect of exposure on the outcome. (A) The trends of effects between M-CSF and aconitate. (B) The trends of effects between Aconitate and UC. (C) The trends of effects between M-CSF and UC. |
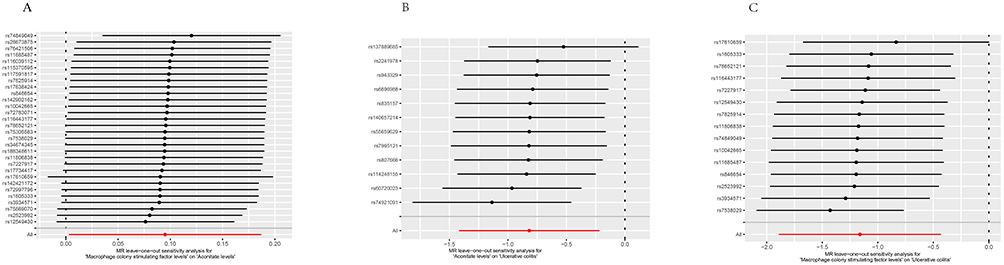 |
Figure 4 Analysis diagram of leave-one-out method. The y-axis represents the removed SNPs, and the x-axis represents the effect of the remaining SNPs on the outcome. (A)The stability of SNPs between M-CSF and Aconitate. (B)The stability of SNPs between aconitate and UC. (C)The stability of SNPs between M-CSF and UC. |
 |
Figure 5 Funnel plots. Symmetry indicates the absence of heterogeneity in IVs. (A)The heterogeneity of SNPs between M-CSF and aconitate. (B)The heterogeneity of SNPs between Aconitate and UC. (C)The heterogeneity of SNPs between M-CSF and UC. |
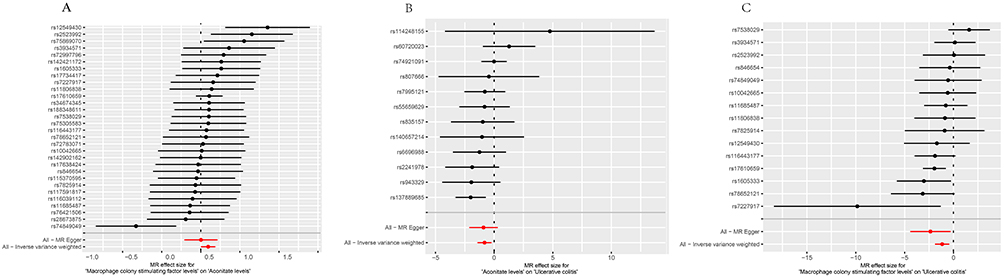 |
Figure 6 Forest plots. The y-axis represents individual SNPs, while the x-axis represents the effect of each SNP on exposure. (A)The association between each SNP and the outcome between M-CSF and Aconitate. (B)The association between each SNP and the outcome between aconitate and UC. (C)The association between each SNP and the outcome between M-CSF and UC. |
M-CSF Alleviate DSS-Induced Colitis
Subsequently, we investigated whether M-CSF mitigated the experimental colitis. C57BL/6 mice were randomized into three groups: control group, DSS group, and M-CSF + DSS group. The mice of the DSS group were administered with 3% DSS dissolved in drinking water, while the M-CSF group received a daily intraperitoneal injection of 50 ng of recombinant mouse M-CSF (Figure 7A). M-CSF treatment significantly reduced the DSS-induced body weight loss and markedly decreased DAI compared with the DSS group (Figure 7B - C). As expected, shortening of the colon was significantly attenuated in DSS-induced colitis (Figure 7D -E). In addition, histological analyses showed that reduced pathological damage and inflammatory cell infiltration were observed after M-CSF administration (Figure 7F -G).
 |
Figure 7 M-CSF alleviate the DSS-induced colitis. (A) Schematic illustration for the administration regimen of DSS-induced M-CSF depletion colitis mice. (B) Daily body weight over 7 days. (C) Disease activity index (DAI) scores. (D) Photographs of colons. (E) Colon length. (F)Hematoxylin-eosin (H&E) staining. (G) Histological scores. *p<0.05, **p <0.01, ***p <0.001, ****p<0.0001. |
Impact of M-CSF on Colonic Metabolites
As expected, qRT-PCR and ELISA results showed a significant increase in the levels of IL-6, IL-17A in the colon tissue when comparing the DSS group to the control group, whereas the expression of IL-10 was dramatically decreased in the DSS group (Figure 8A–F). Nevertheless, M-CSF administration mitigated the DSS-induced increases in IL-6 and IL-17A, as well as the decrease in IL-10 (Figure 8A–F). Moreover, metabolomics analysis revealed aconitate, itaconate and Acod1, was significantly reduced in the DSS group compared to the healthy control group. However, M-CSF treatment effectively alleviated the decrease of aconitate, itaconate and Acod1 in the DSS group (Figure 8G–I).
 |
Figure 8 Impact of M-CSF on the metabolites in the DSS-induced colitis model. (A–C) The mRNA expression profiles of IL-6, IL-17A and IL-10 in colonic tissues after M-CSF administration. (D–F) The concentrations of IL-6, IL-17A and IL-10 in colonic tissues quantified by the ELISA method. (G–I) The levels of aconitate, itaconate, and acod1 in colonic tissues after M-CSF treatment. (J) Flow cytometry analysis of CD86+ cells. (K) Percentage of CD86+ cells as determined by flow cytometry. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns=not significant. |
Furthermore, flow cytometry analysis revealed that lipopolysaccharide (LPS) treatment significantly increased the percentage of CD86+ RAW264.7 cells compared to untreated controls, indicating enhanced M1 macrophage polarization. This LPS-induced effect was markedly attenuated by M-CSF co-treatment, as evidenced by reduced CD86+ cell population (Figure 8J -K).
Discussion
In this study, bidirectional two-step MR analysis was employed to investigate whether specific cytokines associated with UC pathogenesis through the modulation of metabolites. Our findings indicated that M-CSF had a causal and positive relationship with aconitate. Moreover, the level of aconitate was associated with reduced risk of UC. In addition, M-CSF had an inverse causal relationship with the disease onset of UC. Furthermore, the mediation analysis suggested that M-CSF exerted regulatory effect on the pathogenesis of UC through regulating the expression of aconitate. It was found that M-CSF treatment could effectively alleviate DSS-induced colitis in mice. These findings demonstrate that M-CSF promoted the secretion of aconitate, itaconate and Acod1, subsequently inhibiting the expressions of pro-inflammatory cytokines and downregulating the levels of M1 macrophage, thereby ameliorating colitis.
Our findings demonstrated that M-CSF acts as a protective cytokine against UC. Several studies have demonstrated that M-CSF binds to its cognate receptor colony-stimulating factor 1 receptor (CSF-1R), and plays a crucial role in maintaining intestinal homeostasis and modulating inflammatory responses by modulating macrophage differentiation and function.51,52 In DSS-induced colitis, M-CSF/CSF-1R signaling promotes release of reparative factors (eg, Insulin-like Growth Factor 1, Vascular Endothelial Growth Factor) from macrophages, thereby accelerating epithelial repair and ulcer healing.53,54 Huynh et al also discovered that mice lacking the M-CSF/CSF-1R exhibited reduced numbers of proliferating epithelial progenitor cells and lamina propria macrophages.55 Notably, our study extends prior knowledge by demonstrating that M-CSF treatment can decrease the population of M1 macrophage in DSS-induced colitis.
M-CSF primes macrophages by promoting oxidative phosphorylation (OXPHOS) and enhancing TCA cycle activity.56,57 In these M-CSF-primed macrophages, inflammatory stress alters TCA cycle flux, impacting Acod1 (encoded by immune-responsive gene 1, Irg1), the enzyme responsible for converting aconitate to itaconate.58 The resulting itaconate production establishes the Irg1-Acod1 axis as a central regulatory node in immunometabolic reprogramming, enabling an anti-inflammatory phenotype.59,60 Mechanistically, itaconate promotes inflammation resolution through alkylation of Keap1, which activates the Nrf2 pathway and its downstream antioxidant response, thereby suppressing oxidative stress and inflammatory signaling.61,62 Moreover, Acod1 deficiency has been observed to intensify the DSS-induced colitis and result in substantial increases in inflammatory cytokine and chemokine levels.63 The deficiency of itaconate in Acod1-/- mice contributed to the initiation and progression of intestinal inflammation through mechanisms involving impaired immune homeostasis, notably via dysregulated intestinal tuft cell-mediated mucosal immunity and compromised mucosal barrier function.64 In this study, M-CSF treatment significantly elevated the levels of aconitate, itaconate and Acod1 of colonic tissues in DSS-treated colitis.
While our data support a protective role of M-CSF in UC, its context-dependent actions necessitate cautious interpretation. Jiarui et al65 reported opposing results wherein genetically elevated M-CSF increased inflammatory bowel disease (IBD) risk, potentially due to population stratification (eg, ethnic differences in GWAS cohorts) and unaddressed pleiotropy. Importantly, M-CSF exhibits microenvironment-specific duality: in the spinal cord, astrocyte-derived M-CSF exacerbates neuroinflammation via microglial CSF-1R activation,66 whereas intestinal M-CSF promotes barrier repair through IL-22-dependent epithelial regeneration.54,67 Notably, therapeutically administered M-CSF in our high-dose DSS model likely engaged compensatory metabolic reprogramming, contrasting with pro-inflammatory outcomes in low-dose acute models.68
Despite these contextual complexities, our findings highlight the therapeutic potential of M-CSF in intestinal inflammation, supported by three key strengths: firstly, MR analysis mitigated confounding effects and reverse causality by leveraging genetic instruments, overcoming limitations of traditional observational studies. Secondly, the methodology employed was capable of surmounting the constraints intrinsic to traditional observational studies.69 Thirdly, experimental validation confirmed M-CSF efficacy in alleviating colitis in vivo and in vitro. Despite these strengths, several limitations warrant cautious interpretation. First, the modest mediation proportion of aconitate (6.69%) implies contributions from unmeasured pathways, such as alternative TCA cycle metabolites, cytokine networks (eg, IL-6, IL-10 receptor subunit alpha), or direct immunoregulatory actions of M-CSF on macrophage subsets. Furthermore, the lack of interventional studies with exogenous aconitate leaves unresolved whether M-CSF acts through this metabolite alone or via unidentified intermediaries, potentially confounding mechanistic interpretations. Finally, while the DSS model replicates acute colitis, it poorly recapitulates chronic UC progression, and the long-term safety of M-CSF therapy remains unaddressed.
To address these challenges and refine therapeutic strategies, future studies should prioritize the following: First, given that additional pathways or metabolites may contribute to its overall effect, exploration of alternative mediators or downstream targets is essential to fully elucidate the mechanism of action of M-CSF. Second, translating M-CSF-targeted therapies will require careful consideration of its dual pro-inflammatory and tissue-repair functions; specifically, delineating the spatiotemporal roles of M-CSF in colitis will be critical to optimize therapeutic windows and reduce off-target effects.
In conclusion, our findings demonstrated that M-CSF treatment significantly attenuated UC by reducing M1 macrophage infiltration, elevating aconitate/itaconate levels, and suppressing pro-inflammatory cytokines, as evidenced through MR analysis and experimental validation. Although aconitate partially mediated this protective effect (proportion mediated = 6.69%), the majority of M-CSF’s benefits may involve broader immunoregulatory mechanisms. While the current UC model highlights M-CSF’s therapeutic potential, two critical limitations warrant attention: First, the context-specific roles of M-CSF (eg, tissue microenvironment variations) require systematic evaluation. Second, despite the DSS model replicates acute colitis, the long-term safety and translational relevance of M-CSF-targeted interventions must be validated in human studies.
Abbreviation
UC, ulcerative colitis; GWAS, genome-wide association studies; MR, Mendelian randomization; DSS, dextran sodium sulfate; M-CSF, macrophage colony-stimulating factor; DAI, disease activity index; Acod1, aconitate decarboxylase 1; JAK, Janus Kinase; anti-TNF, anti-tumor necrosis factor; SCFAs, short-chain fatty acids; IVs, instrumental variables; TCA cycle, tricarboxylic acid cycle; SNP, single nucleotide polymorphism; p-QTL, protein quantitative trait locus; Mb, megabase; CLSA, Canadian Longitudinal Study on Aging; LD, linkage disequilibrium; WM, Weighted Median; IVW, Inverse Variance Weighted; MR-PRESSO, Mendelian randomization pleiotropy residual sum and outlier; ATCC, American Type Culture Collection; DMEM, Dulbecco’s Modified Eagle Medium; ESI, electrospray ionization; PSAP, proactivator saposin precursor; H&E, Hematoxylin–eosin; LC–MS/MS, Liquid Chromatography–Tandem Mass Spectrometry; qPCR, Quantitative Real-Time Polymerase Chain Reaction; Ct, threshold cycle; GC, guanine-cytosine; ELISA, Enzyme-linked immunosorbent assay; PBS, phosphate-buffered saline; TMB, 3,3′,5,5′-tetramethylbenzidine; FMO, fluorescence-minus-one; ANOVA, analysis of variance; OR, odds ratio; CI, confidence interval; IL-6, interleukin-6; IL-10, interleukin-10; IL-17A, interleukin-17A; LPS, Lipopolysaccharide; CSF-1R, Colony-Stimulating Factor 1 Receptor; OXPHOS, oxidative phosphorylation; IBD, inflammatory bowel disease; IL-22, interleukin-22.
Data Sharing Statement
All data are available in the main text and supplementary materials. Additional data related to this paper may be requested from the authors.
Ethics Statement
Human data: The genome-wide association study (GWAS) data used in this Mendelian randomization analysis were obtained from public, de-identified summary-level databases. In accordance with Article 32 of the Measures for the Ethical Review of Life Science and Medical Research Involving Human Subjects (National Health Commission of China, effective February 18, 2023), this component of the research is exempt from institutional ethics review approval because:
(i) It utilizes legally accessible public data without individual identifiers;
(ii) The analytical methodology poses no risk to personal rights or public interests.
Animal experiments: All mouse procedures were reviewed and approved by the Ethics Committee of Shenzhen People’s Hospital (Approval No.: 2024-085-01; Date: February 19, 2024). The study strictly adhered to the ARRIVE Guidelines 2.0 and the NIH Guide for the Care and Use of Laboratory Animals (8th edition).
Acknowledgments
This work was supported by the Science and Technology Innovation Committee of Shenzhen (JCYJ20210324113802006, JCYJ2022053015180024 and JCYJ20210324113613035).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
All authors declare no conflicts of interest in this study.
References
1. Rubin DT, Ananthakrishnan AN, Siegel CA, et al. ACG clinical guideline: ulcerative colitis in adults. Am J Gastroenterol. 2019;114(3):384–413. doi:10.14309/ajg.0000000000000152
2. Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390(10114):2769–2778. doi:10.1016/s0140-6736(17)32448-0
3. Yilmaz B, Juillerat P, Øyås O, et al. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat Med. 2019;25:323–336. doi:10.1038/s41591-018-0308-z
4. Lu Q, Yang MF, Liang YJ, et al. Immunology of inflammatory bowel disease: molecular mechanisms and therapeutics. J Inflamm Res. 2022;15:1825–1844. doi:10.2147/jir.S353038
5. Adimadhyam S, Lewis JD, Simon AL, et al. Real-world evidence comparing tofacitinib and vedolizumab in anti-TNF-experienced patients with ulcerative colitis. Inflamm Bowel Dis. 2024;30:554–562. doi:10.1093/ibd/izad115
6. Anjie SI, Hulshoff MS, D’Haens G. Efficacious dosing regimens for anti-TNF therapies in inflammatory bowel disease: where do we stand? Expert Opin Biol Ther. 2023;23:341–351. doi:10.1080/14712598.2023.2198086
7. Mitrova K, Julsgaard M, Augustijns P, et al. Tofacitinib in pregnancy: assessing pregnancy and infant outcomes, cord blood, and breast milk concentrations. Clin Gastroenterol Hepatol. 2025;23:163–165.e3. doi:10.1016/j.cgh.2024.01.019
8. Veltkamp SHC, Voorneveld PW. The cell-specific effects of JAK1 inhibitors in ulcerative colitis. J Clin Med. 2025;14. doi:10.3390/jcm14020608
9. Baumgart DC, Le Berre C. Newer biologic and small-molecule therapies for inflammatory bowel disease. N Engl J Med. 2021;385(14):1302–1315. doi:10.1056/NEJMra1907607
10. Zhao JH, Stacey D, Eriksson N, et al. Genetics of circulating inflammatory proteins identifies drivers of immune-mediated disease risk and therapeutic targets. Nat Immunol. 2023;24(9):1540–1551. doi:10.1038/s41590-023-01588-w
11. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14(5):329–342. doi:10.1038/nri3661
12. Abraham C, Abreu MT, Turner JR. Pattern recognition receptor signaling and cytokine networks in microbial defenses and regulation of intestinal barriers: implications for inflammatory bowel disease. Gastroenterology. 2022;162:1602–1616.e6. doi:10.1053/j.gastro.2021.12.288
13. Aonghus L, Harry S. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17. doi:10.1038/s41575-019-0258-z
14. Zhao H, Zhou Y, Xu J, et al. Short-chain fatty acid-producing bacterial strains attenuate experimental ulcerative colitis by promoting M2 macrophage polarization via JAK/STAT3/FOXO3 axis inactivation. J Transl Med. 2024;22:369. doi:10.1186/s12967-024-05122-w
15. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23:R89–98. doi:10.1093/hmg/ddu328
16. Hemani G, Zheng J, Elsworth B, et al. The MR-base platform supports systematic causal inference across the human phenome. Elife. 2018;7. doi:10.7554/eLife.34408
17. Shang Q, Zhang P, Lei X, et al. Insights into CSF-1/CSF-1R signaling: the role of macrophage in radiotherapy. Front Immunol. 2025;16:1530890. doi:10.3389/fimmu.2025.1530890
18. Petrina M, Alothaimeen T, Bouzeineddine NZ, et al. Granulocyte macrophage colony stimulating factor exerts dominant effects over macrophage colony stimulating factor during macrophage differentiation in vitro to induce an inflammatory phenotype. Inflamm Res. 2024;73:253–262. doi:10.1007/s00011-023-01834-9
19. Connor AE, Amit VK, Sekar K. Mendelian Randomization. JAMA. 2017;318. doi:10.1001/jama.2017.17219
20. Tyler JV. Mediation analysis: a practitioner’s guide. Annu Rev Public Health. 2015;37. doi:10.1146/annurev-publhealth-032315-021402
21. Alice RC, Eleanor S, Gemma H, et al. Mendelian randomisation for mediation analysis: current methods and challenges for implementation. Eur J Epidemiol. 2021;36. doi:10.1007/s10654-021-00757-1
22. Coscia C, Molina-Montes E, Benitez R, et al. New proposal to address mediation analysis interrogations by using genetic variants as instrumental variables. Genet Epidemiol. 2023;47:287–300. doi:10.1002/gepi.22519
23. Yiheng C, Tianyuan L, Ulrika P-K, et al. Genomic atlas of the plasma metabolome prioritizes metabolites implicated in human diseases. Nat Genet. 2023:55. doi:10.1038/s41588-022-01270-1
24. Bin L, Ding Y, Hong Y, et al. Assessing the relationship between gut microbiota and irritable bowel syndrome: a two-sample Mendelian randomization analysis. BMC Gastroenterol. 2023;23:23. doi:10.1186/s12876-023-02791-7
25. Alexander K, Carolina M-G, Rodrigo B, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021:53. doi:10.1038/s41588-020-00763-1
26. Lawlor DA, Harbord RM, Sterne JA, et al. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–1163. doi:10.1002/sim.3034
27. Bowden J, Holmes MV. Meta-analysis and Mendelian randomization: a review. Res Synth Methods. 2019;10:486–496. doi:10.1002/jrsm.1346
28. Jack B, George DS, Stephen B. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44. doi:10.1093/ije/dyv080
29. Veronika WS, Rebecca CR, Benjamin ARW, et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomization: the STROBE-MR statement. JAMA. 2021:326. doi:10.1001/jama.2021.18236
30. David PM, Chondra ML, Jeanne MH, et al. A comparison of methods to test mediation and other intervening variable effects. Psychol Methods. 2002:7. doi:10.1037/1082-989x.7.1.83
31. Lee Y, Sugihara K, Gillilland MG, et al. Hyaluronic acid-bilirubin nanomedicine for targeted modulation of dysregulated intestinal barrier, microbiome and immune responses in colitis. Nat Mater. 2020;19:118–126. doi:10.1038/s41563-019-0462-9
32. Min-Zheng Z, Hao-Ming X, Yu-Jie L, et al. Edible exosome-like nanoparticles from portulaca oleracea L mitigate DSS-induced colitis via facilitating double-positive CD4(+)CD8(+)T cells expansion. J Nanobiotechnology. 2023;21:21. doi:10.1186/s12951-023-02065-0
33. Lima A, Franca A, Muzny CA, et al. DNA extraction leads to bias in bacterial quantification by qPCR. Appl Microbiol Biotechnol. 2022;106(24):7993–8006. doi:10.1007/s00253-022-12276-4
34. Damgaard MV, Treebak JT. Protocol for qPCR analysis that corrects for cDNA amplification efficiency. STAR Protoc. 2022;3:101515. doi:10.1016/j.xpro.2022.101515
35. Oh D, De Spiegelaere W, Nauwynck HJ. Selection and validation of reference genes for RT-qPCR normalization of porcine alveolar macrophages (PAMs) for PRRSV studies. Sci Rep. 2023;13:8840. doi:10.1038/s41598-023-35873-3
36. Zhang P, Chen S, Chen S, et al. Selection and validation of qRT-PCR internal reference genes to study flower color formation in camellia impressinervis. Int J Mol Sci. 2024;25. doi:10.3390/ijms25053029
37. Krzysica P, Verhoog L, de Vries S, et al. Optimization of capture ELISAs for chicken cytokines using commercially available antibodies. Animals. 2022;13:12. doi:10.3390/ani12213040
38. Zhang B, Zeng M, Zhang Q, et al. Ephedrae Herba polysaccharides inhibit the inflammation of ovalbumin induced asthma by regulating Th1/Th2 and Th17/Treg cell immune imbalance. Mol Immunol. 2022;152:14–26. doi:10.1016/j.molimm.2022.09.009
39. Oyegue-Liabagui SL, Mbani Mpega Ntigui CN, Ada Mengome MF, et al. Cytokine response in asymptomatic and symptomatic Plasmodium falciparum infections in children in a rural area of south-eastern Gabon. PLoS One. 2023;18(2):e0280818. doi:10.1371/journal.pone.0280818
40. Chauhan A, Pal A, Sachdeva M, et al. A FACS-based novel isolation technique identifies heterogeneous CTCs in oral squamous cell carcinoma. Front Oncol. 2024;14:1269211. doi:10.3389/fonc.2024.1269211
41. Rico LG, Bardina J, Bistue-Rovira A, et al. Accurate identification of cell doublet profiles: comparison of light scattering with fluorescence measurement techniques. Cytometry A. 2023;103:447–454. doi:10.1002/cyto.a.24690
42. Wang Y, Zhou Y, Liu J, et al. Temporal and spatial expression of Phosphodiesterase-4B after sciatic nerve compression in rats and its mechanism of action on sciatic nerve repair. Neurochem Int. 2025;185:105940. doi:10.1016/j.neuint.2025.105940
43. Zhang L, Nie F, Zhao J, et al. PGRN is involved in macrophage M2 polarization regulation through TNFR2 in periodontitis. J Transl Med. 2024;22:407. doi:10.1186/s12967-024-05214-7
44. Song Y, Lee Y. Brief guide to flow cytometry. Mol Cells. 2024;47:100129. doi:10.1016/j.mocell.2024.100129
45. Ullas S, Sinclair C. Applications of flow cytometry in drug discovery and translational research. Int J Mol Sci. 2024;25:3851. doi:10.3390/ijms25073851
46. Rasooly D, Peloso GM. Two-sample multivariable Mendelian randomization analysis using R. Curr Protoc. 2021;1:e335. doi:10.1002/cpz1.335
47. VanLiere JM, Rosenberg NA. Mathematical properties of the r2 measure of linkage disequilibrium. Theor PopulBiol. 2008;74:130–137. doi:10.1016/j.tpb.2008.05.006
48. Gerard D. Scalable bias-corrected linkage disequilibrium estimation under genotype uncertainty. Heredity. 2021;127:357–362. doi:10.1038/s41437-021-00462-5
49. Yang K, Li J, Hui X, et al. assessing the causal relationship between metabolic biomarkers and coronary artery disease by Mendelian randomization studies. Sci Rep. 2024;14(1):19034. doi:10.1038/s41598-024-69879-2
50. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32:377–389. doi:10.1007/s10654-017-0255-x
51. Monteleone G, Franzè E, Troncone E, Maresca C, Marafini I. Interleukin-34 mediates cross-talk between stromal cells and immune cells in the gut. Front Immunol. 2022;13:873332. PMID: 35529879; PMCID: PMC9073079. doi:10.3389/fimmu.2022.873332
52. Zwicker S, Martinez GL, Bosma M, et al. Interleukin 34: a new modulator of human and experimental inflammatory bowel disease. Clin Sci. 2015;129(3):281–290. doi:10.1042/CS20150176
53. Hernandez-Espinosa DR, Medina-Ruiz GI, Scrabis MG, et al. Proinflammatory microglial activation impairs in vitro cortical tissue repair via zinc-dependent ADAM17 cleavage of the CSF-1CSF −1 receptor. J Neurochem. 2025;169:e16239. doi:10.1111/jnc.16239
54. Brown ND, Vomhof-DeKrey EE, Aoki H. Focal adhesion kinase and colony stimulating factors: intestinal homeostasis and innate immunity crosstalk. Cells. 2024;14:13. doi:10.3390/cells13141178
55. Duy H, Dilara A, Jordane M, et al. CSF-1 receptor-dependent colon development, homeostasis and inflammatory stress response. PLoS One. 2013:8. doi:10.1371/journal.pone.0056951
56. Wculek SK, Dunphy G, Heras-Murillo I, et al. Metabolism of tissue macrophages in homeostasis and pathology. Cell Mol Immunol. 2022;19:384–408. doi:10.1038/s41423-021-00791-9
57. Qianyue Z, Qiaoling S, Shan L, et al. Integrated transcriptomic and metabolomic analysis reveals the metabolic programming of GM-CSF- and M-CSF- differentiated mouse macrophages. Front Immunol. 2023:14. doi:10.3389/fimmu.2023.1230772
58. MacLean A, Legendre F, Appanna VD. The tricarboxylic acid (TCA) cycle: a malleable metabolic network to counter cellular stress. Crit Rev Biochem Mol Biol. 2023;58:81–97. doi:10.1080/10409238.2023.2201945
59. Zhang MN, Xie R, Wang HG, et al. Cepharanthine alleviates DSS-induced ulcerative colitis via regulating aconitate decarboxylase 1 expression and macrophage infiltration. Molecules. 2023;29:28. doi:10.3390/molecules28031060
60. Kang H, Liu T, Wang Y, et al. Neutrophil-macrophage communication via extracellular vesicle transfer promotes itaconate accumulation and ameliorates cytokine storm syndrome. Cell Mol Immunol. 2024;21:689–706. doi:10.1038/s41423-024-01174-6
61. Zhanyang Q, Mingjie X, Tianyu Z, et al. ACOD1, rather than itaconate, facilitates p62-mediated activation of Nrf2 in microglia post spinal cord contusion. Clin Transl Med. 2024:14. doi:10.1002/ctm2.1661
62. Jean-Philippe A, Max Z, Maria F, et al. Metabolic rewiring promotes anti-inflammatory effects of glucocorticoids. Nature. 2024:629. doi:10.1038/s41586-024-07282-7
63. Ho Won K, A-Reum Y, Ji Won L, et al. Aconitate decarboxylase 1 deficiency exacerbates mouse colitis induced by dextran sodium sulfate. Int J Mol Sci. 2022. doi:10.3390/ijms23084392
64. Ting Z, Yuko H, Matthew KW. Enteric bacterial infection stimulates remodelling of bile metabolites to promote intestinal homeostasis. Nat Microbiol. 2024;9. doi:10.1038/s41564-024-01862-z
65. Mi J, Wu X, Bai X, et al. ST2 and CSF-1as potential druggable targets of inflammatory bowel diseases: results from two-sample Mendelian randomization study. Clin Transl Sci. 2023;16:236–245. doi:10.1111/cts.13442
66. Wu K, Shao S, Dong YT, et al. Spinal astrocyte-derived M-CSF mediates microglial reaction and drives visceral hypersensitivity following DSS-induced colitis. Neuropharmacology. 2025;270:110373. doi:10.1016/j.neuropharm.2025.110373
67. Carnevale S, Ponzetta A, Rigatelli A, et al. Neutrophils mediate protection against colitis and carcinogenesis by controlling bacterial invasion and IL22 production by gammadelta T cells. Cancer Immunol Res. 2024;12:413–426. doi:10.1158/2326-6066.Cir-23-0295
68. Diane M, James C, Daniel L, et al. Blockade of colony stimulating factor-1 (CSF-I) leads to inhibition of DSS-induced colitis. Inflamm Bowel Dis. 2007:13. doi:10.1002/ibd.20055
69. Frederick JB, Xiang Z. Statistical methods for Mendelian randomization in genome-wide association studies: a review. Comput Struct Biotechnol J. 2022;20. doi:10.1016/j.csbj.2022.05.015
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.