Back to Journals » Journal of Inflammation Research » Volume 16
Lysosome-Related Diagnostic Biomarkers for Pediatric Sepsis Integrated by Machine Learning
Received 25 August 2023
Accepted for publication 21 November 2023
Published 24 November 2023 Volume 2023:16 Pages 5575—5583
DOI https://doi.org/10.2147/JIR.S437110
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Yang Yang,1 Genhao Zhang2
1Department of Nuclear Medicine, Henan Provincial People’s Hospital, People’s Hospital of Zhengzhou University, People’s Hospital of Henan University, Zhengzhou, Henan, 450003, People’s Republic of China; 2Department of Blood Transfusion, Zhengzhou University First Affiliated Hospital, Zhengzhou, Henan, 450052, People’s Republic of China
Correspondence: Genhao Zhang, Email [email protected]
Background: There is currently no biomarker that can reliably identify sepsis, despite recent scientific advancements. We systematically evaluated the value of lysosomal genes for the diagnosis of pediatric sepsis.
Methods: Three datasets (GSE13904, GSE26378, and GSE26440) were obtained from the gene expression omnibus (GEO) database. LASSO regression analysis and random forest analysis were employed for screening pivotal genes to construct a diagnostic model between the differentially expressed genes (DEGs) and lysosomal genes. The efficacy of the diagnostic model for pediatric sepsis identification in the three datasets was validated through receiver operating characteristic curve (ROC) analysis. Furthermore, a total of 30 normal samples and 35 pediatric sepsis samples were gathered to detect the expression levels of crucial genes and assess the diagnostic model’s efficacy in diagnosing pediatric sepsis in real clinical samples through real-time quantitative PCR (qRT-PCR).
Results: Among the 83 differentially expressed genes (DEGs) related to lysosomes, four key genes (STOM, VNN1, SORT1, and RETN) were identified to develop a diagnostic model for pediatric sepsis. The expression levels of these four key genes were consistently higher in the sepsis group compared to the normal group across all three cohorts. The diagnostic model exhibited excellent diagnostic performance, as evidenced by area under the curve (AUC) values of 1, 0.971, and 0.989. Notably, the diagnostic model also demonstrated strong diagnostic ability with an AUC of 0.917 when applied to the 65 clinical samples, surpassing the efficacy of conventional inflammatory indicators such as procalcitonin (PCT), white blood cell (WBC) count, C-reactive protein (CRP), and neutrophil percentage (NEU%).
Conclusion: A four-gene diagnostic model of lysosomal function was devised and validated, aiming to accurately detect pediatric sepsis cases and propose potential target genes for lysosomal intervention in affected children.
Keywords: pediatric sepsis, diagnostic marker, ROC curves, lysosomal, GSEA
Introduction
The prevalence of pediatric sepsis, with both identified and unidentified causes, poses a significant threat to the well-being and survival of approximately 3 million children annually.1 Despite the potential for antibiotic overuse, empirical antibiotic therapy has demonstrated effectiveness in managing infection, stabilizing hemodynamics, and modulating the septic response.2 Regrettably, pediatric sepsis exhibits substantial heterogeneity throughout the entire pathobiological process, thereby posing significant challenges for clinicians in making appropriate decisions, such as early-stage antibiotic administration and organic supportive treatment, as well as risk assessment and discontinuation of antibiotic use in late-stage sepsis.3 Hence, to proficiently diagnose, treat, and prevent pediatric sepsis, it is imperative to actively explore biomarkers that can precisely ascertain pediatric sepsis.
Lysosomes, which are membrane-bound organelles responsible for intracellular macromolecule degradation, play a crucial role as signaling centers for calcium regulation and nutrient response control. Therefore, preserving the integrity and functionality of lysosomes is vital for maintaining cellular homeostasis.4 Lysosomes are known to have significant involvement in inflammation, antigen presentation, and the preservation of long-lived immune cells.5 Various types of stress can trigger an elevation in lysosomal membrane permeability, resulting in the discharge of intracellular components from the lysosomes into the cytoplasm and ultimately leading to cell death that is dependent on lysosomes.6 The excessive activation of the NLRP3 inflammasome can induce lysosomal cleavage, which plays a role in the regulation of inflammation during sepsis.7 Neutrophil extracellular traps (NETs) can promote cathepsin B (CatB) release from ruptured lysosomes by inducing thermal apoptosis in macrophages (Mϕ) and thus modulating the inflammatory response in sepsis.8 The lysosomal membrane-tethered Ragulator complex can participate in the progression of inflammation-associated diseases by activating intracellular NLRP3 inflammasome.9 In an acidic lysosomal environment, HMGB1 can induce leakage of LPS into the cytoplasm causing acute lung injury and death in children.10 The effects of morphine on lysosomal processes, such as production, acidification, and mitochondrial-lysosomal fusion, result in a decrease in the activation of mitochondrial autophagy in microglia, which finally promotes infectious shock.11 Melatonin with anti-oxidative stress and autophagy lysosomal modulation reduces the occurrence of sepsis-associated multiorgan dysfunction.12 α-MOFs can release calcium and zinc ions into monocyte/macrophage lysosomes to promote bacterial degradation and effectively improve the survival of septic patients.13 Therefore, targeting lysosomes is a potential therapeutic strategy to modulate inflammation and metabolism in various conditions.
In this study, we integrated multiple high-throughput sequencing datasets of pediatric sepsis to develop a diagnostic model focusing on lysosomal function, which effectively identifies cases of pediatric sepsis. The reliability of this diagnostic model was subsequently verified using a cohort comprising 30 healthy individuals and 35 children diagnosed with sepsis, based on peripheral blood samples. It is anticipated that this diagnostic model will offer healthcare practitioners novel insights into the diagnosis and treatment of pediatric sepsis.
Materials and Methods
Acquisition of Pediatric Sepsis Cohorts and Lysosomal Genes
We designed the current study according to the Transparent Reporting of Individual Prognostic or Diagnostic Multivariate Predictive Models (TRIPOD). Three datasets (GSE13904, GSE26378, and GSE26440) comprising whole blood gene expression profiles of children with sepsis and healthy controls were acquired from the gene expression omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). The normalization of each dataset was performed using the “normalizeBetweenArrays” function of the R “Limma” package. The batch effects between the three datasets were corrected using the Combat function from the “SVA” R package. GSE26378 (21 Normal vs 82 Pediatric sepsis) served as the training set for subsequent analysis, while GSE26440 (32 Normal vs 98 Pediatric sepsis) and GSE13904 (18 Normal vs 106 Pediatric sepsis) were utilized as validation sets. In addition, a comprehensive set of 894 genes associated with lysosomes was obtained from the Gene Ontology (GO) database (https://geneontology.org/) and the Gene Set Enrichment Analysis (GSEA) database (http://www.gsea-msigdb.org/).
Screening Differentially Expressed Genes (DEGs)
DEGs were identified between pediatric sepsis and healthy samples in the training dataset, using adjusted P-values less than 0.05 and |logFC| > 1 as the criteria. The STRING database (http://string-db.org) was utilized to conduct protein-protein interaction (PPI) network analysis based on these DEGs. Subsequently, gene set enrichment analysis (GSEA)14 was employed to identify signaling pathways enriched with the DEGs.
Lysosomal-Related Diagnostic Biomarkers Developing
The overlapping genes of DEGs and lysosomal genes in pediatric sepsis were subjected to further analysis in the GSE26378 dataset. Initially, a least absolute shrinkage and selection operator (LASSO) regression analysis was conducted using the R package “glmnet” to select the optimal parameters through 10-fold cross-validation. Subsequently, a random forest analysis was performed using the R package “randomForest” to filter the optimal parameters. The associated out-of-pocket (OOB) data were utilized to determine each decision tree’s out-of-pocket data error. ErrorOOB1 might be used to represent the error value. To determine the out-of-pocket data error errorOOB2, the noise was added at random to the out-of-pocket data I. The relevance of out-of-pocket data I (score(I)) was finally calculated using the formula 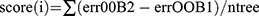 , where ntree is the number of decision trees. The number of decision trees to be utilized was chosen to be between 100 and 1000, and the default values were used for all other parameters for ordering feature significance. Moreover, the shared genes in the two machine learning algorithms were selected for logistic regression analysis to develop a diagnostic model for pediatric sepsis. Subsequently, the validity of the diagnostic model was evaluated using receiver operating characteristic curve (ROC) analysis in both the training and testing datasets and the area under the curve (AUC) was calculated to assess the predictive performance of the algorithm.
, where ntree is the number of decision trees. The number of decision trees to be utilized was chosen to be between 100 and 1000, and the default values were used for all other parameters for ordering feature significance. Moreover, the shared genes in the two machine learning algorithms were selected for logistic regression analysis to develop a diagnostic model for pediatric sepsis. Subsequently, the validity of the diagnostic model was evaluated using receiver operating characteristic curve (ROC) analysis in both the training and testing datasets and the area under the curve (AUC) was calculated to assess the predictive performance of the algorithm.
RNA Extraction and qRT-PCR Analysis
This study included a total of 35 pediatric sepsis cases that met the diagnostic criteria for sepsis,2,15 as well as 30 normal children, between January 1, 2023, and May 30, 2023. The confirmation of pediatric sepsis samples was based on blood culture results. The basic clinical characteristics of both groups can be found in Table S1. The sepsis group exhibited higher levels of procalcitonin (PCT) and neutrophil percentage (NEU%) compared to the normal group. However, there were no statistically significant differences observed in terms of age, gender, white blood cell (WBC) count, and C-reactive protein (CRP) level. Three milliliters of whole blood were collected from each child after fasting. Peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Paque isolate. Total RNA was extracted from PBMCs and mRNA levels of diagnostic markers were detected using qRT-PCR. The normalization was done using β-ACTIN.16 The primer sequences can be found in Table S2.
Evaluation of Immune Cell Infiltration
Given the significant role of immune cells in the pathogenesis of pediatric sepsis,17 the CIBERSORT18 database was utilized to assess the infiltration of diverse immune cell types. This analysis aimed to elucidate the immune cell types that strongly correlate with model scores in different cohorts of children with sepsis.
Statistical Analysis
Categorical data were analyzed using the Fisher exact test or the chi-square test, while quantitative variables were assessed using the independent samples t-test. All p-values were considered statistically significant at a significance level of P<0.05, with a two-tailed approach.
Results
Screening Lysosomal-Related DEGs
In the GSE26378 dataset (Figure 1A), a total of 468 upregulated DEGs and 271 downregulated DEGs were identified. Subsequently, 83 lysosomal-related DEGs were screened (Figure 1B). To visualize the interrelationships between different proteins and suggest potential mechanisms by which the proteins function, a PPI network was constructed using these lysosome-associated DEGs in pediatric sepsis (Figure 1C). The GSEA analysis findings indicated that these lysosome-associated DEGs were primarily associated with neutrophil deregulation, lysosomal, and immune effector processes (Figure 1D).
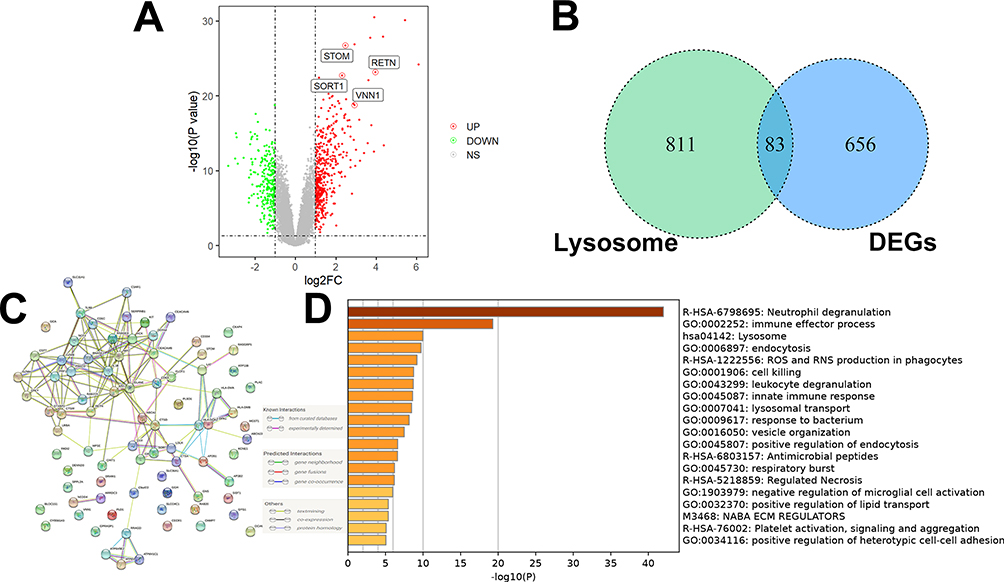 |
Figure 1 Screening lysosomal-related DEGs in the GSE26378 dataset. (A) A total of 7468 upregulated DEGs and 271 downregulated DEGs were identified. (B) 83 lysosomal-related DEGs were screened. (C) PPI network analysis. (D) The GSEA analysis findings indicated that these lysosome-associated DEGs were primarily associated with neutrophil deregulation, lysosomal, and immune effector processes. |
Lysosomal-Related Diagnostic Markers Construction
The LASSO regression analysis yielded a selection of the top 13 parameters (Figure 2A), while the random forest analysis also identified 13 optimal parameters (Figure 2B). From these, a total of four genes were found to be common to both analyses and were subsequently utilized in the development of a diagnostic model for pediatric sepsis (Figure 2C). 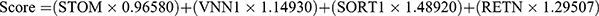 . Subsequently, ROC analysis was conducted on both the training and validation cohorts to evaluate the efficacy of the diagnostic model. As depicted in Figure 2D, the expression levels of all four genes implicated in the diagnostic model were consistently higher in the sepsis group compared to the normal group across all three cohorts. This trend was also observed in the diagnostic model scores (Figure 2E). Notably, the diagnostic model exhibited commendable diagnostic capability, as evidenced by AUC values of 1 in the GSE26378 cohort, 0.989 in the GSE26440 cohort, and 0.971 in the GSE13904 cohort, respectively (Figure 2F).
. Subsequently, ROC analysis was conducted on both the training and validation cohorts to evaluate the efficacy of the diagnostic model. As depicted in Figure 2D, the expression levels of all four genes implicated in the diagnostic model were consistently higher in the sepsis group compared to the normal group across all three cohorts. This trend was also observed in the diagnostic model scores (Figure 2E). Notably, the diagnostic model exhibited commendable diagnostic capability, as evidenced by AUC values of 1 in the GSE26378 cohort, 0.989 in the GSE26440 cohort, and 0.971 in the GSE13904 cohort, respectively (Figure 2F).
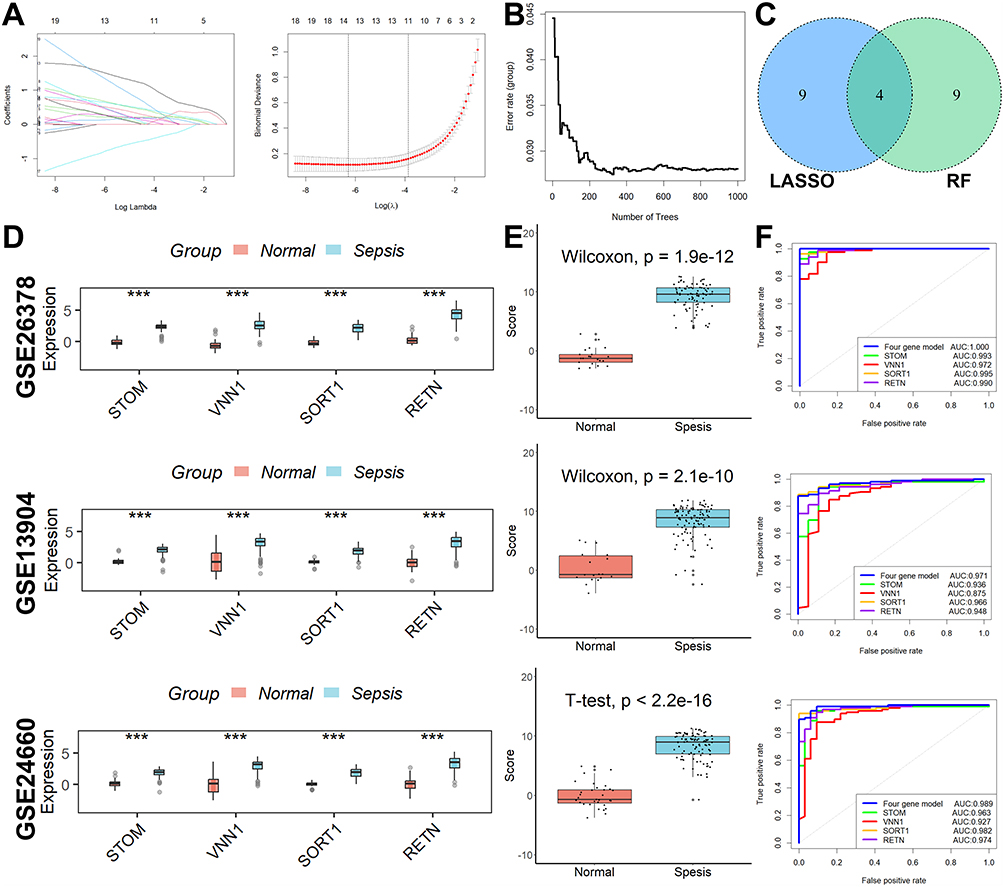 |
Figure 2 Lysosomal-related diagnostic markers construction and validation. (A) The LASSO regression analysis yielded a selection of the top 13 parameters. (B) The random forest analysis also identified 13 optimal parameters. (C) Four overlapped genes. (D) The expression of all four genes involved in the diagnostic model was higher in the sepsis group than in the normal group in all three cohorts. (E) The diagnostic model scores were significantly elevated in the sepsis group compared to the normal group in all three cohorts. (F) The diagnostic model showed good diagnostic ability. ***p < 0.001. |
Performance Analysis of the Diagnostic Markers in a Clinical Cohort
The bacterial strains responsible for sepsis in children are depicted in Figure 3A. Following the findings from the aforementioned bioinformatics analysis, the expression levels of all four genes implicated in the diagnostic model were significantly elevated in the sepsis group compared to the normal group (Figure 3B), along with the diagnostic model scores (Figure 3C). The diagnostic model exhibited commendable diagnostic efficacy, as evidenced by the area under the curve (AUC) values of 0.917 in the 65 clinical samples (Figure 3D). Furthermore, the diagnostic model demonstrated superior effectiveness in comparison to conventional inflammatory markers including PCT, CRP, WBC, and NEU% (Figure 3E). Additionally, we assessed the diagnostic model’s ability to differentiate between bacterial and non-bacterial sepsis and observed it to possess a moderate level of competence (Figure 3F).
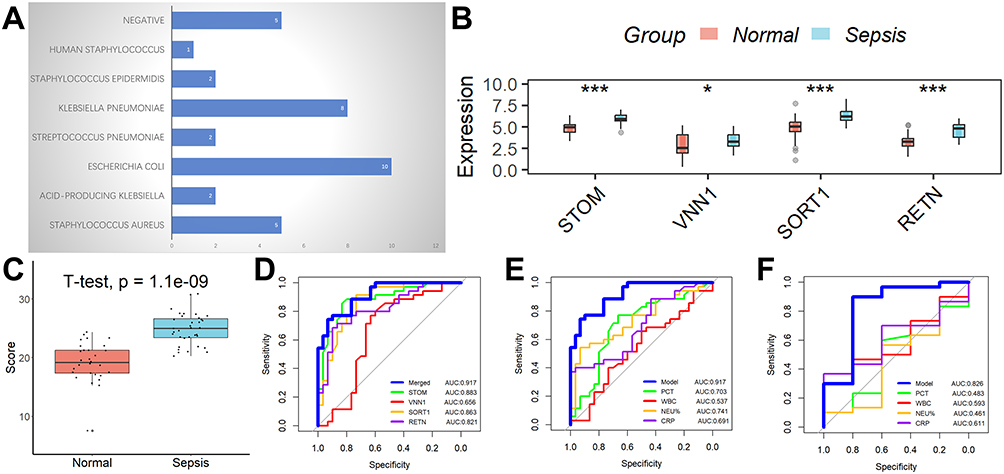 |
Figure 3 Performance analysis of the diagnostic markers in a clinical cohort. (A) The types of bacteria infecting children with sepsis. (B) The expression of all four genes involved in the diagnostic model was higher in the sepsis group than in the normal group. (C) The diagnostic model scores were significantly elevated in the sepsis group compared to the normal group. (D) The diagnostic model showed good diagnostic ability in the 65 clinical samples. (E) The diagnostic model showed better efficacy compared to conventional inflammatory indicators such as PCT, CRP, WBC, and NEU%. (F) The value of this diagnostic model in distinguishing bacterial from non-bacterial sepsis. *p < 0.05; ***p < 0.001. |
Immune Cell Infiltration Analysis
The infiltration of various immune cell types in different categories of pediatric sepsis was evaluated using the CIBERSORT database. Subsequently, the diagnostic scores showed significant correlations with various immune cell types, namely CD8 T cell, T. cell. gamma. delta, monocyte, macrophage M0, resting dendritic cell, activation dendritic cell, and neutrophil in the GSE26378 cohort (Figure 4A), and B. cell. naive, CD8 T cell, follicular helper T cell, activation NK cell, macrophage M1, resting dendritic cell, and neutrophil in the GSE13904 cohort (Figure 4B), and of T. cells. follicular. helper, dendritic cells. resting and neutrophils in the GSE26440 cohort (Figure 4C). Resting dendritic cells and neutrophils exhibited a correlation with model scores across all three datasets (Figure 4D). Due to the limited presence of resting dendritic cells in peripheral blood, we exclusively confirmed the correlation between neutrophils and model scores in clinical samples, yielding consistent results with the aforementioned analysis (Figure 4E). By integrating the findings, it can be inferred that the shared presence of neutrophils potentially serves as the underlying mechanism through which the diagnostic model gene influences the onset of pediatric sepsis.
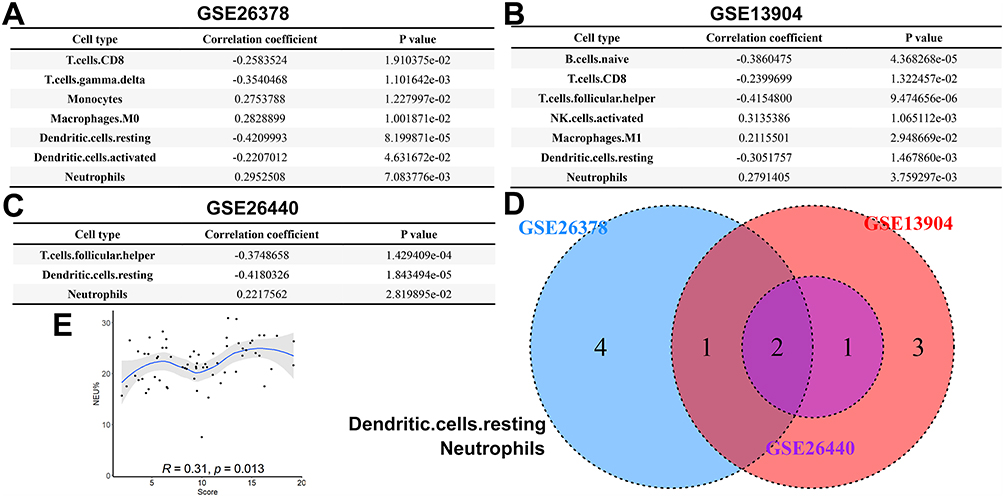 |
Figure 4 Immune cell infiltration analysis in pediatric sepsis. (A) Cell types associated with model scores in the GSE26378 cohort. (B) Cell types associated with model scores in the GSE13904 cohort. (C) Cell types associated with model scores in the GSE26440 cohort. (D) Veen plot of the correlation analysis in the three cohorts. (E) The correlation between neutrophils and model scores in clinical samples. |
Functional Enrichment Analysis
We divided the pediatric sepsis samples into high and low groups according to the diagnostic scores and analyzed the differential genes between the two groups for functional enrichment. As shown in Figure 5A, GO analysis showed that the differential genes were mainly enriched in ribosomal subunits and biological processes related to RNA production and metabolism, whereas KEGG analysis showed that the differential genes were mainly enriched in lysosomal, ribosomal, and complement-related biological processes (Figure 5B).
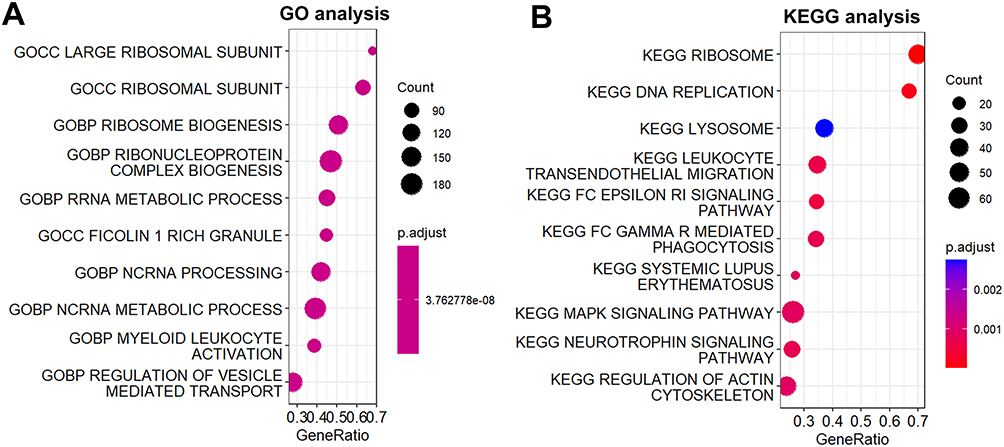 |
Figure 5 Functional enrichment analysis. (A) Go analysis. (B) KEGG analysis. |
Discussion
Pediatric sepsis, characterized by a dysregulated immune response to pathogens, is a condition of significant morbidity and mortality worldwide.13 The timely identification and management of pediatric sepsis pose considerable challenges due to the frequently variable clinical manifestations it presents. The aforementioned biomarkers, namely CRP and PCT, are widely accessible in the majority of pediatric institutions and continue to serve as valuable clinical tools for diagnosing pediatric sepsis.19 Nonetheless, their sensitivity and specificity are relatively limited, necessitating the identification of novel biomarkers capable of promptly and accurately diagnosing pediatric sepsis in real-world medical settings. The diagnostic biomarkers of lysosomes that have been developed in our present study exhibit considerable potential in addressing the aforementioned clinical challenges. This diagnostic approach surpasses conventional biomarker diagnostics and demonstrates enhanced precision in capturing the intricate dynamics of the human immune system.
The integration of big data, machine learning, and artificial intelligence presents an opportunity for the development of improved sepsis identification tools, which can effectively support decision-making in pediatric sepsis cases and facilitate the implementation of precision-based therapeutic approaches.2 By employing machine learning techniques for the identification of crucial genes and conducting a comprehensive analysis of high-throughput sequencing data derived from multiple instances of pediatric sepsis, we have successfully developed a diagnostic model in this present study. This model exhibits the capability to effectively screen for pediatric sepsis, as evidenced by its accurate discrimination between normal cases and instances of pediatric sepsis, both within the clinical peripheral blood samples we collected and in publicly available datasets of pediatric sepsis. Additionally, an examination of the correlation between the aforementioned diagnostic model and immune cell infiltration revealed a strong association. This can be attributed to the substantial involvement of cellular immune regulation, which encompasses immune cells, in the pathogenesis of pediatric sepsis. Monocytes and macrophages play an important role in inflammation and after pathogen attack.20 Monocytes transform sepsis from being cells with powerful positive immunomodulatory capacity, like Pha of bacteria, to defective cells that are unable to mount a successful defense against infection.13 A possible increased lysosomal load during sepsis may result from a suppression of bacterial absorption and lysosomal degradation by monocytes or macrophages.21 Bacteria hiding in lysosomes use dysfunctional monocytes or macrophages as hosts to avoid normal immune cell activity and cytokines.13 In addition, phagocytosis is closely related to lysosomes in the pathogenesis of inflammatory disease.8 Phagocytosis and lysosomal trafficking are necessary for the inflammatory cytokine response to the Bacillus anthracis peptidoglycan.22 Treatment for sepsis may be hampered by dysfunctional monocytes/macrophages, which may even help germs evade immune clearance.13 Consequently, these findings have the potential to enhance our comprehension of the underlying molecular immunological mechanisms that precipitate the onset of pediatric sepsis.
Despite the utilization of diverse high-throughput sequencing data from pediatric sepsis and the application of machine learning techniques to identify characteristic genes, our study presents distinct advantages compared to previous investigations.17,23,24 Firstly, our diagnostic model exhibits enhanced diagnostic efficacy, as evidenced by its successful application to clinical samples and subsequent validation using three publicly available datasets. Secondly, our developed diagnostic model demonstrates effectiveness in distinguishing between bacterial and non-bacterial etiologies of pediatric sepsis (AUC=0.82).
There are several limitations to our study. Firstly, the lack of mortality data in the three publicly accessible pediatric sepsis datasets hinders the assessment of the predictive efficacy of our developed model. Secondly, the potential influence of co-morbid conditions in the pediatric sepsis patients included in our research may impact the accuracy of our model in predicting outcomes. Finally, despite the satisfactory performance of the model we constructed in analyzing the 65 clinical samples collected, the limited sample size and the single-center nature of the study necessitate the conduction of additional multicenter investigations with larger sample sizes to validate and enhance the diagnostic efficacy of the model.
Conclusions
We have successfully identified a diagnostic model consisting of four genes associated with lysosomes, which exhibits a high level of reliability in identifying pediatric sepsis. This model presents potential candidate genes for diagnostic testing using peripheral blood samples in pediatric sepsis patients, and also offers novel perspectives for the development of lysosome-targeted therapies in this specific patient population.
Abbreviation
GEO, gene expression omnibus; ROC, Receiver operating characteristic curve; DEGs, differentially expressed genes; NETs, Neutrophil extracellular traps (NETs); CatB, cathepsin B; GSEA, gene set enrichment analysis; LASSO, Least absolute shrink and selection operator; RF, random forests; AUC, area under the curve; qRT-PCR, quantitative real-time PCR; PCT, procalcitonin; NEU%, neutrophil percentage; WBC, white blood cell; CRP, C-reactive protein; PBMCs, peripheral blood mononuclear cells; TRIPOD, Transparent Reporting of a multivariable prediction model for Individual Prognosis Or Diagnosis.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
This study was supported by the Ethics Committees of The First Affiliated Hospital of Zhengzhou University (2023-KY-0932-002). Written informed consent was obtained from parents or legal guardians of all patients and healthy controls. All methods were performed following the relevant guidelines and regulations. The manuscript is consistent with the Declaration of Helsinki.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Henan Provincial Health and Health Commission Joint Construction Project (LHGJ20230209).
Disclosure
All authors declare no conflicts of interest in this work.
References
1. Qin Y, Caldino Bohn RI, Sriram A, et al. Refining empiric subgroups of pediatric sepsis using machine-learning techniques on observational data. Front Pediatr. 2023;11:1035576. doi:10.3389/fped.2023.1035576
2. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
3. Evans L, Rhodes A, Alhazzani W, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2021. Crit Care Med. 2021;49(11):e1063–e1143. doi:10.1097/CCM.0000000000005337
4. Wang F, Gómez-Sintes R, Boya P. Lysosomal membrane permeabilization and cell death. Traffic. 2018;19(12):918–931. doi:10.1111/tra.12613
5. Yambire KF, Rostosky C, Watanabe T, et al. Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. eLife. 2019;8. doi:10.7554/eLife.51031
6. Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27(50):6434–6451. doi:10.1038/onc.2008.310
7. Biasizzo M, Kopitar-Jerala N. Interplay between NLRP3 inflammasome and autophagy. Front Immunol. 2020;11:591803. doi:10.3389/fimmu.2020.591803
8. Chen L, Zhao Y, Lai D, et al. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis. 2018;9(6):597. doi:10.1038/s41419-018-0538-5
9. Tsujimoto K, Jo T, Nagira D, et al. The lysosomal ragulator complex activates NLRP3 inflammasome in vivo via HDAC6. EMBO J. 2023;42(1):e111389. doi:10.15252/embj.2022111389
10. Yang R, Zhang X. A potential new pathway for heparin treatment of sepsis-induced lung injury: inhibition of pulmonary endothelial cell pyroptosis by blocking hMGB1-LPS-induced caspase-11 activation. Front Cell Infect Microbiol. 2022;12:984835. doi:10.3389/fcimb.2022.984835
11. Peng J, Pan J, Wang H, Mo J, Lan L, Peng Y. Morphine-induced microglial immunosuppression via activation of insufficient mitophagy regulated by NLRX1. J Neuroinflammation. 2022;19(1):87. doi:10.1186/s12974-022-02453-7
12. Liu Y, Wang D, Li T, et al. Melatonin: a potential adjuvant therapy for septic myopathy. Biomed Pharmacother. 2023;158:114209. doi:10.1016/j.biopha.2022.114209
13. Zhao Q, Gong Z, Wang J, et al. A zinc- and calcium-rich lysosomal nanoreactor rescues monocyte/macrophage dysfunction under sepsis. Adv. Sci. 2023;10(6):e2205097. doi:10.1002/advs.202205097
14. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
15. Weiss SL, Peters MJ, Alhazzani W, et al. Surviving sepsis campaign international guidelines for the management of septic shock and sepsis-associated organ dysfunction in children. Intensive Care Med. 2020;46(Suppl 1):10–67. doi:10.1007/s00134-019-05878-6
16. Zhang G. Regulatory T-cells-related signature for identifying a prognostic subtype of hepatocellular carcinoma with an exhausted tumor microenvironment. Front Immunol. 2022;13:975762. doi:10.3389/fimmu.2022.975762
17. Fan J, Shi S, Qiu Y, Liu M, Shu Q. Analysis of signature genes and association with immune cells infiltration in pediatric septic shock. Front Immunol. 2022;13:1056750. doi:10.3389/fimmu.2022.1056750
18. Gentles AJ, Newman AM, Liu CL, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–945. doi:10.1038/nm.3909
19. Lim PPC, Bondarev DJ, Edwards AM, Hoyen CM, Macias CG. The evolving value of older biomarkers in the clinical diagnosis of pediatric sepsis. Pediatr Res. 2023;93(4):789–796. doi:10.1038/s41390-022-02190-w
20. Germic N, Frangez Z, Yousefi S, Simon HU. Regulation of the innate immune system by autophagy: monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. 2019;26(4):715–727. doi:10.1038/s41418-019-0297-6
21. Cai J, Li J, Zhou Y, et al. Staphylococcus aureus facilitates its survival in bovine macrophages by blocking autophagic flux. J Cell Mol Med. 2020;24(6):3460–3468. doi:10.1111/jcmm.15027
22. Iyer JK, Khurana T, Langer M, et al. Inflammatory cytokine response to Bacillus anthracis peptidoglycan requires phagocytosis and lysosomal trafficking. Infect Immun. 2010;78(6):2418–2428. doi:10.1128/IAI.00170-10
23. Wang X, Guo Z, Wang Z, et al. Diagnostic and predictive values of pyroptosis-related genes in sepsis. Front Immunol. 2023;14:1105399. doi:10.3389/fimmu.2023.1105399
24. Zhang WY, Chen ZH, An XX, et al. Analysis and validation of diagnostic biomarkers and immune cell infiltration characteristics in pediatric sepsis by integrating bioinformatics and machine learning. World J Pediatr. 2023;19(11):1094–1103. doi:10.1007/s12519-023-00717-7
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Predictive Value of a Diagnostic Five-Gene Biomarker for Pediatric Sepsis
Xiao Y, Zhang G
Journal of Inflammation Research 2024, 17:2063-2071
Published Date: 4 April 2024