Back to Journals » Drug Design, Development and Therapy » Volume 14
Lysosomal Acid Lipase Deficiency: Therapeutic Options
Authors Pastores GM, Hughes DA
Received 5 December 2019
Accepted for publication 16 January 2020
Published 11 February 2020 Volume 2020:14 Pages 591—601
DOI https://doi.org/10.2147/DDDT.S149264
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Gregory M Pastores,1 Derralynn A Hughes2
1Department of Medicine (Genetics)/National Centre for Inherited Metabolic Disorders, Mater Misericordiae University Hospital and University College Dublin, Dublin, Ireland; 2Royal Free London NHS Foundation Trust, University College London, London NW3 2QG, UK
Correspondence: Gregory M Pastores
National Centre for Inherited Metabolic Disorders, Mater Misericordiae University Hospital and University College Dublin, Dublin, Ireland
Tel +353 1 803 4878
Fax +353 1 803 4876
Email [email protected]
Abstract: Lysosomal acid lipase (LAL) deficiency is a metabolic (storage) disorder, encompassing a severe (Wolman disease) and attenuated (Cholesterol ester storage disease) subtype; both inherited as autosomal recessive traits. Cardinal clinical features include the combination of hepatic dysfunction and dyslipidemia, as a consequence of cholesteryl esters and triglyceride accumulation, predominately in the liver and vascular and reticuloendothelial system. Significant morbidity can arise, due to liver failure and/or atherosclerosis; in part related to the severity of the underlying gene defect and corresponding enzyme deficiency. Diagnosis is based on demonstration of decreased LAL enzyme activity, complemented by analysis of the cognate gene defects. Therapeutic options include dietary manipulation and the use of lipid-lowering drugs. Sebelipase alfa, a recombinant enzyme replacement therapy, has garnered regulatory approval, following demonstration of improvements in disease-relevant markers and clinical benefit in clinical trials, which included increased survival in the most severe cases.
Keywords: atherosclerosis, dyslipidemia, enzyme replacement therapy, hepatomegaly, lipid-lowering medications, lysosomal acid lipase deficiency, lysosomal storage disease
Introduction
Lysosomal acid lipase (LAL) deficiency is an autosomal recessive disorder of metabolism caused by defects of the LIPA gene, resulting in disruption of intralysosomal degradation of cholesteryl esters and triglycerides and accumulation of relevant substrates. Clinical pathology primarily involves the liver and the vascular and reticuloendothelial systems. Causal gene defects are heterogeneous, with over 100 LIPA mutations having been identified in patients with LAL deficiency.1
Hepatomegaly associated with liver dysfunction and an abnormal lipid profile (elevated low-density lipoprotein [LDL] cholesterol and triglyceride levels, decreased high-density lipoprotein [HDL] cholesterol) are characteristic clinical features. The dyslipidemia has been attributed to the inability to release free (unesterified) cholesterol from lysosomes, leading to a cascade of downstream intracellular events and an associated atherogenic lipid profile. Liver biopsy demonstrates micro- or macro-vesicular steatosis involving Kupffer cells and hepatocytes, accompanied by fibrosis and cirrhosis as the pathology progresses. Chronic (untreated) dyslipidemia may lead to accelerated atherosclerosis and a high risk of cardio- (i.e., myocardial infarction, coronary heart disease) and cerebrovascular (stroke) complications.
Disease severity is related to the presence of residual LAL enzyme activity and indicated by the age of onset (Figure 1). In the rapidly progressive form (Wolman disease, WD), the median age at initial presentation is in the first month of life, with a median age of death in untreated patients at 3.7 months.2 The eponymous designation is an acknowledgment of an early case reported by Moshe Wolman. In addition to hepatosplenomegaly, adrenal calcification is a characteristic feature of WD.
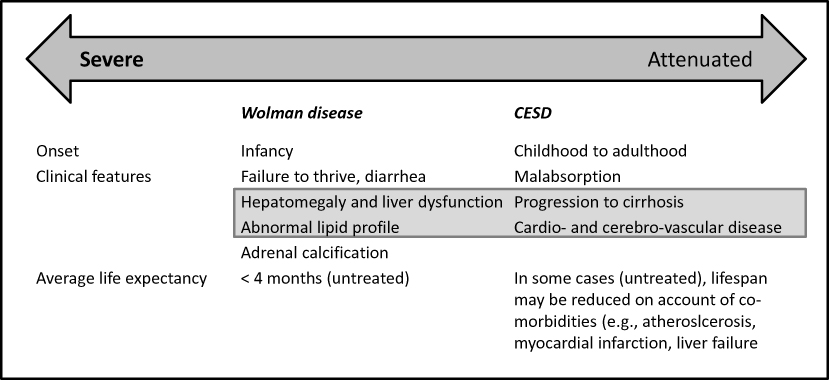 |
Figure 1 LAL deficieincy: clinical manifestations*. Note: *Although historically delineated into a severe Wolman disease and attenuated (mild) clinical subtype AKA Cholesterol ester storage disease (CESD), it is recognized that these clinical entities represent the spectrum of clinical manifestations, caused by deficiency of lysosomal lipase. NB. Clinical features listed within gray box are major signs common between severe and attenuated disease variants; with heterogeneity in disease severity and progression in the latter subtype. |
In two unrelated infants the diagnosis of hemophagocytic lymphohistiocytosis (HLH) was initially considered; with WD established as a diagnosis following progression to cholestasis and liver failure.3 Hemophagocytic lymphohistiocytosis (HLH) is a potentially lethal condition resulting from macrophage activation; it can arise as inherited or be acquired; secondary to infections, immune deficiency, and malignancy. It has been hypothesized that cholesteryl ester-induced inflammasome activation in cells of monocytic lineage may play a role, leading to secondary HLH or a phenocopy of LAL deficiency.4 However, it is notable that the pattern of pathological disruption as a result of macrophage activation in HLH is similar to another reticuloendothelial storage disorder, namely Gaucher disease. Studies in the LAL gene-knockout mouse model indicate critical roles of LAL in the development, homeostasis, and function of T cells, defects of which have subsequently been shown to be corrected by myeloid-specific expression of human lysosomal acid lipase.5 Interestingly, a similar effect was achieved by blocking stat3 and NF-κB p65 signaling using small-molecule inhibitors in myeloid-derived suppressor cells; pointing to putative molecular pathways.6
Diagnosis and Differential Diagnosis
The attenuated phenotype, cholesteryl ester storage disease (CESD), is often heralded by an abnormal lipid profile, liver function abnormalities or gallstones as incidental findings. Manifestations are evident in childhood or later.2 Diagnosis may be delayed; especially in the absence of a family history (e.g., affected sibling). In some patients delayed initiation of therapy can lead to significant hepatic dysfunction with cirrhosis and resultant co-morbidity (e.g., esophageal varices and upper gastrointestinal bleeding, liver failure).7
Of note, LAL enzyme activity does not fully predict disease severity, and assay results should therefore not be used to assign disease subtype. In addition, there is minimal information on the prognostic value of genotype, although homozygosity for null alleles where enzyme activity is undetectable tends to correlate with severe disease; whilst mutations associated with residual enzyme activity may have a mitigating effect on clinical expression.
LAL deficiency has an estimated prevalence ranging from 1:40,000 to 1:300,000. A recent UK-based study among a cohort (n-1825 patients) with dyslipidaemia and elevated transaminases failed to identify a case of LAL deficiency.8 Liver biopsies from six of the study patients enabled DNA extraction from four cases; two of these were found to be homozygous for the LAL c.46A>C;p.Thr16Pro variant in exon 2 of uncertain significance.8 These observations highlight caution when making a diagnosis, based on molecular genetic analysis alone.
Diagnostic delays are frequent; unless there is relevant family history and as with other rare diseases are best explained by the association of non-specific and highly prevalent clinical manifestations. This has prompted a search for biomarkers that can be used for screening, amongst high-risk population.9
Presenting features such as hepatomegaly and evidence of storage (foam cells) can be seen in other relatively more common lysosomal storage disorders (LSD) such as Gaucher disease (GD) and Niemann-Pick disease (NPD) types B and C; thus if the strategy is single-gene testing with analysis of enzyme activity, evaluation for LAL deficiency would be appropriate when these conditions are ruled out. However, increasingly gene panels for associated diagnoses are being employed and may allow diagnosis of LAL deficiency even when clinical awareness is low. Of note, primary central nervous system (CNS) involvement, as encountered in neuropathic GD and NPD types A and C, is not a feature of LAL deficiency. Moreover, thrombocytopenia - a major feature of GD and NPD – is less frequently seen in LAL deficiency, although this is not necessarily an exclusionary finding. A biomarker (ie, chitotriosidase), found to be elevated in GD and used in screening tests for this disorder, is also increased, although usually to a lesser extent, in LAL deficiency and NPD.9 Most laboratories that offer testing for LSDs often perform multiplex screening, which would include measuring the enzymatic activity of β-glucosidase (GD), acid sphingomyelinase (NPD types A and B) and LAL. Including a novel biomarker, lysosphingomyelin-509, in screening tests for suspected cases of NPD type C has been suggested to potentially increase diagnostic yield.10,11 Other biomarkers, such as oxysterols (e.g., cholestane-3beta,5alpha,6beta-triol), have also been described to be abnormally elevated non-specifically in LAL deficiency, NPD type C, and cerebrotendinous xanthomatosis.12 The possibility oxysterol intermediates may have a contributory role in the clinical expression of these disorders has been raised, although more studies would be required to establish mechanistic links.13 As with LAL deficiency, these other LSDs (GD, NPD) are inherited in an autosomal recessive fashion.
LAL deficiency should be considered in cases wherein dyslipidemia (± liver dysfunction) prompts evaluation for suspected familial hypercholesterolemia, non-alcoholic fatty liver disease (NAFLD) or non-alcoholic steatohepatitis (NASH), and/or metabolic syndrome (i.e., a combination of features including abnormal lipid profile, elevated blood glucose, hypertension, excess waist circumference fat). Of interest, screening by testing peripheral dried blood spot (DBS) samples in a high-risk cohort of 81 patients with a diagnosis of NAFLD, with 78 matched control patients with HCV-related liver disease, revealed LAL activity to be significantly reduced in this population, particularly evident in those with pre-cirrhotic disease.14 Furthermore, LAL activity correlated with platelet and white blood cell count, which suggested the presence of portal hypertension-associated cytopenias may be a confounder in the accurate determination of enzyme activity.14 This is an important consideration in screening studies of relevant patient populations, wherein testing involves measuring LAL activity on DBS cards. It has further been reported that reduced LAL activity may be observed in patients with severe liver disease and may be a contributory factor in pathogenesis.15 The latter observation underscores the need to differentiate primary from secondary LAL deficiency, so patients are treated appropriately.
Management
Until recently, treatment for LAL deficiency was primarily symptomatic and involved a combination of dietary manipulation and the use of lipid-lowering drugs.16 Several procedures could also be performed in those with advanced disease; these measures included splenectomy, ligation of esophageal varices, and liver transplantation, in an effort to assuage symptoms or deal with end-organ failure; hematopoietic stem cell transplantation had also been undertaken to halt disease progression.
Hematopoietic Stem Cell/Bone Marrow Transplantation
Prior to the availability of enzyme replacement therapy (ERT), hematopoietic stem cell transplantation (HSCT) had been undertaken in several patients with WD. A single-center report by Tolar et al provides outcome data on two affected patients subjected to HSCT.17 In one child, transplanted at 19 weeks of age, disease course was complicated by sinusoidal obstruction syndrome (SOS) on 19 days post-procedure (PP); the child subsequently died (day 67 PP) from a combination of problems: multi-organ (i.e., liver, kidney and lung) failure, coagulopathy, and sepsis. The other child, transplanted at a later age (19 months), developed sepsis and liver failure on day 14 PP and died at age 27 months.18
At the time of the above report (2009), two additional WD patients were noted as long-term survivors; outcome described the resolution of diarrhea within weeks after engraftment, improvement of hepatosplenomegaly and normalization of liver function.18 The elder patient, age 11 years at follow-up, had normal adaptive functions; however, mild to moderate neurocognitive deficiencies were evident, felt to be secondary to the treatment course and other ensuing medical problems. The younger patient, at age 4 years had age-appropriate neurodevelopmental and adaptive abilities.
A more recent publication describes two brothers with the WD phenotype subjected to HSCT; unfortunately, both subsequently died from liver-related complications.18 The older sib, transplanted at 127 days of life, succumbed at age 6 years; whilst the younger sib who underwent HSCT at the age of 8 months developed multi-organ failure and died at day 21 PP. Incidentally, the paper by Yanir et al provides a summary of transplanted cases reported by 2013; altogether 10 subjects, including the cases reported by Tolar et al18.
Liver Transplantation
As with HSCT, liver transplantation had been undertaken in patients with LAL deficiency prior to the availability of disease-specific enzyme therapy. On the whole, the outcome for the severe phenotype has been dismal. For instance, a report on two sisters, both diagnosed in infancy, describes progressive deterioration despite prior splenectomy and liver transplantation.19 One sister survived for 5 years postliver transplant, the course of which had been complicated by intermittent, acute rejection. Examination of the liver allograft revealed features characteristic of LAL deficiency, including progressive, microvesicular steatosis, foamy macrophage aggregates, and vacuolated Kupffer cells, associated with advanced fibrosis and micronodular cirrhosis.19 Recurrence of disease in the donor liver was attributed to infiltration by host-derived cells of monocyte-derived macrophages, which remained deficient in LAL enzyme activity.19
Additionally, the above report by Bernstein et al provides the outcome of 18 LAL deficiency cases-postliver transplant; noted were disease progression in 11 (61%) patients and death in six (33%).19 These observations demonstrate both the inadequacy of liver transplantation alone as a therapeutic approach and procedure-related morbidity.
There are no reported cases of patients with LAL deficiency subjected to both HSCT and liver transplantation. Whether outcomes following transplant can be modified either by pretreatment with enzyme replacement or a combination strategy has not been examined. Obviously, due consideration must be given to risk: benefit, and early diagnosis with timely initiation of ERT will likely be prioritized.
Enzyme Replacement Therapy
Proof of concept for ERT as a therapeutic option was demonstrated through outcomes observed in the null mouse model, generated through a targeted disruption of the LAL locus and reported as mimicking the human LAL deficiency phenotype.20 Mannose-terminated human LAL (1.5 U/dose) expressed in Pichia pastoris (phLAL), given via tail vein injections to two-month-old LAL null mice once every 3 days for 30 days (total=10 doses), led to near-complete resolution of the hepatic discoloration; associated with a 36% decrease in hepatic weight, compared to PBS(sham)-treated LAL null mice.21 Examination of tissues obtained from phLAL-treated mice revealed reductions in macrophage lipid accumulation. Additionally, triglycerides and cholesteryl levels decreased; by approximately 50%, 69% and 50% in the liver, spleen and small intestine, respectively.21 Similar findings were obtained following the administration of an alternative plant-expressed enzyme formulation, generated using the GENEWARE expression system in Nicotiana benthamiana (tobacco) cells.22 Further observations included the detection of anti-hLAL antibodies, but there were no adverse events observed in LAL null mice.22
The outcome of subsequent open-label clinical trials involving recombinant human LAL (sebelipase alfa) is described in relation to publication dates. Of note, the majority of patients (56/84, 67%) enrolled in the sebelipase alfa clinical trials (LAL-CL01/LAL-CL04 and LAL-CL03, see Table 1) were children (from 1 month up to age 18 years).
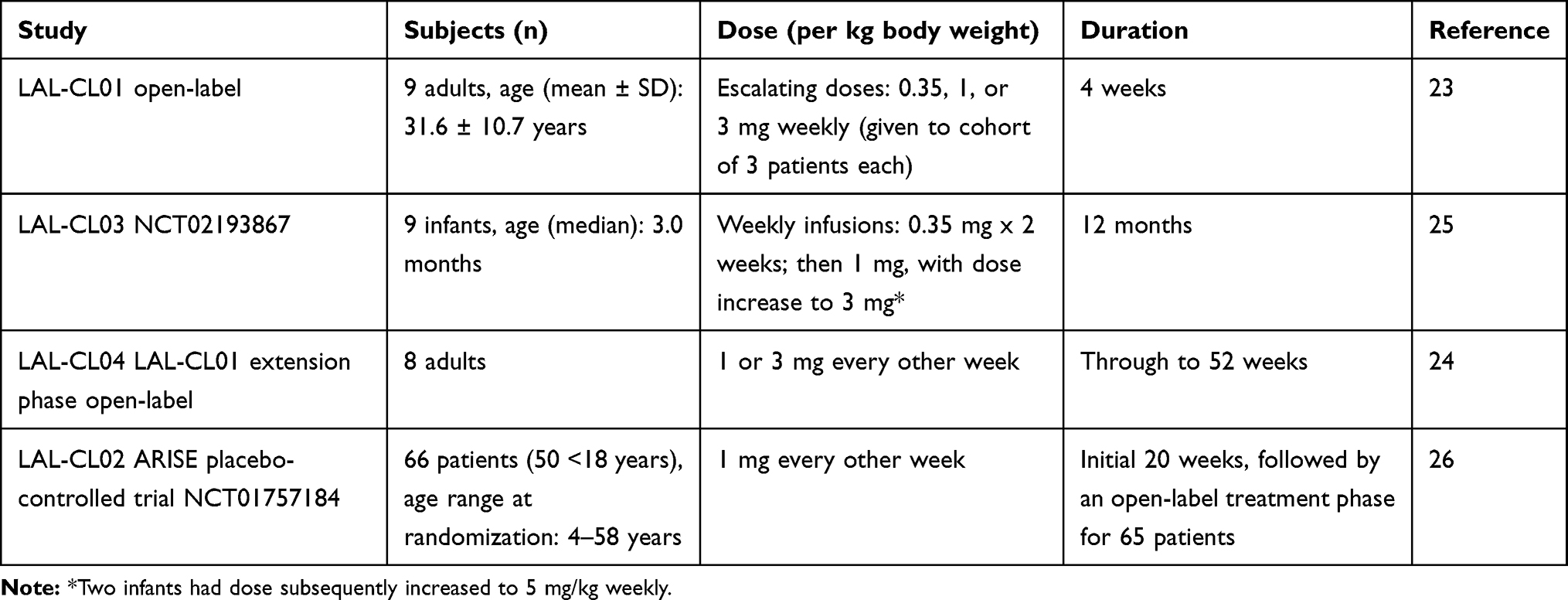 |
Table 1 Clinical Studies Involving Sebalipase Alfa |
A Phase 1/2 open-label study (aka LAL-CL01) enrolled nine adult patients; grouped into 3 cohorts, each of which received an escalating dose of sebelipase alfa (0.35, 1, or 3 mg/kg/body weight) given weekly for 12 weeks.23 All patients (mean age 31.6 ± 10.7 years; range 19 to 45 years) were Caucasian; predominantly males (n-6). Sebelipase alfa was well tolerated, with adverse events that were mild and deemed unrelated to study drug. No antidrug antibodies were detected, although the period of observation was short.
Patients who completed study participation in LAL-CL01 were eligible to enroll in an extension phase (LAL-CL04), which lasted through to week 52.24 In these adults-only study, patients already on lipid-lowering drugs were maintained on their regimen at a stable dose.
Liver transaminase levels decreased in treated patients in the LAL-CL01 study and increased between studies when treatment was interrupted (i.e., while awaiting enrolment into LAL-CL04). In seven patients on sebelipase alfa treatment, the mean (± standard deviation) percent decreases for alanine aminotransaminase (ALT) and aspartate aminotransferase (AST) at week 12 (compared to the baseline values in LAL-CL01) were −46 ± 21 U/L (−52%) and −21 ± 14 U/L (−36%), respectively (p ≤ 0.05).23 Through week 12 (i.e., 24 weeks of therapy), these patients also showed mean decreases from baseline in total cholesterol of −44 ± 41 mg/dL (−22%; p = 0.047), LDL cholesterol of −29 ± 31 mg/dL (−27%; p = 0.078), and triglycerides of −50 ± 38 mg/dL (−28%, p = 0.016), with an increase in HDL cholesterol of 5 mg/dL (15%; p = 0.016).23
Of note, in the extension phase (LAL-CL04), sebelipase alfa was given at 1mg/kg or 3mg/kg every-other-week; 8/9 adult subjects (mean age at first dose=31±11 years) enrolled. At week 52, the mean ALT and AST levels for the group were within normal limits; representing a mean change from baseline of −58% and −40%, respectively.24 Also noted was a mean reduction in liver volume (assessed by magnetic resonance imaging) and in hepatic proton density fat fraction by −12% and −55%, respectively.24 Moreover, the mean changes for LDL cholesterol, total cholesterol, triglyceride and HDL cholesterol were observed by −60%, −39%, −36% and +29%, respectively.24 These observations were interpreted as consistent with a decrease in disease burden.
Adverse events in these adult trials were mainly mild and deemed unrelated to sebelipase alfa.23,24 Infusion-related reactions were uncommon, although three events of moderate severity were reported in two subjects; in one patient this was suggestive of a hypersensitivity-like reaction, but this was reported as not confirmed on additional testing, and the subject continued treatment with sebelipase alfa. Of samples tested, no anti-drug antibodies have been detected.23
Study (LAL-CL03) evaluated the safety and efficacy of ERT with sebelipase alfa in infants (n-9) with LAL deficiency, who demonstrated growth failure or other evidence of rapidly progressive disease with onset before 6 months of age.25 The median age at baseline was 3.0 months (range 1.1–5.8 months). Patients received sebelipase alfa at 0.35 mg/kg once weekly for the first 2 weeks and then increased to 1 mg/kg once weekly. Dose escalation to 3 mg/kg once weekly was allowed (based on clinical judgment); this change in dose occurred as early as 1 month and up to 20 months after starting treatment at the 1 mg/kg dose. A further dose escalation to 5 mg/kg once weekly was also permitted; subsequently undertaken for two patients, one with neutralizing antibodies and less than expected improvements in growth (compared with other infants in the study) and in another patient due to persistence of elevated liver transaminase levels, hypoalbuminemia, and both poor weight gain.25 During this trial, there were three deaths in the first few months of life; two cases because of advanced disease, and a third following complications related to abdominal paracentesis that was not a protocol-specified undertaking.25
Survival to 12 months of age was noted in 6/9 (67%) treated infants, compared with 0% for a historical control group (n = 21).25 These six children exhibited improvements in weight-for-age; additional findings included reductions in both hepatosplenomegaly and markers of liver dysfunction (ALT, AST), as noted in the adult trials. Also, there were improvements in anemia and gastrointestinal symptoms. Unfortunately, one of these patients subsequently died at 15 months of age attributed to cardiac complications, deemed unlikely related to study drug.25 Post-mortem studies revealed evidence of early hepatic cirrhosis, xanthomatous changes in the lungs and intestine, and widespread lipid vacuoles and lipid-laden macrophages in the spleen and bone marrow.25
All five remaining survivors (56%) from the pediatric study (LAL-CL03) lived to age ≥24 months; on sebelipase alfa treatment, they displayed marked improvement in liver function and overall growth.25 Serious adverse events (i.e., infusion-related reactions [IRR]: tachycardia, pallor, chills, and pyrexia) were reported in one infant. Most of the other IRRs were deemed mild and no serious adverse events, in line with enzyme therapy infusion experience for other LSDs.
Independent of the above open-label (pediatric and adult) studies, there was a multicentre, randomized, double-blind, placebo-controlled trial (LAL-CL02, aka ARISE ClinicalTrials.gov number, NCT01757184) involving 66 patients (76% [50] of whom were ≤18 years of age).26 Patients who had undergone transplantation or had severe liver disease (defined as Child–Pugh class C\) were excluded from enrolment.26 Age range at randomization was 4–58 years old. Sebelipase alfa was given intravenously (at a dose of 1 mg/kg of body weight every other week) for the initial 20 weeks; a comparator placebo arm enrolled 30 patients. This trial was followed by an open-label treatment phase for 65 patients; four (6%) of whom had their dose increased to 3 mg/kg once every other week (based on clinical response; in line with the approach taken in other sebelipase alfa studies).26
Baseline disease burden was deemed “substantial” in the majority of the LAL-CL02 study subjects; for instance, LDL cholesterol levels were very high (≥190 mg per deciliter) in 38 (58%) patients; of these 9 (24%) were on maintenance (stable) doses of lipid-lowering drugs.26 Additional findings noted for this “substantially” affected cohort included: hepatomegaly (67% had a liver volume ≥1.25 times the normal volume) and splenomegaly (21% spleen volume ≥4 times the normal volume). Liver-biopsy samples, obtained at baseline for 32 (48%) patients, revealed fibrosis (Ishak score, ≥1, on a scale from 0 to 6, with higher scores indicating a greater degree of fibrosis); 15 (47%) of these had bridging fibrosis (Ishak score, 3 or 4), whilst cirrhosis (Ishak score, 5 or 6) was noted in 10 (31%); the mean age for this later severe subgroup was 12 years.26
The endpoints assessed in the LAL-CL02 (ARISE) study were: Primary-normalization of ALT; Secondary-normalization of AST; decreases in LDL-/non-HDL cholesterol, triglyceride levels and liver fat content (assessed by multi-echo gradient echo magnetic resonance imaging, MEGE-MRI); increase in HDL cholesterol; and improvement in hepatic steatosis (based on morphometry).26
At the end of the placebo-controlled phase (20 weeks), the ALT level was normal in 11/36 (31%) treated patients, as opposed to 2/30 (7%) in the placebo group (p=0.03); reflecting a mean change from baseline of −58 U per liter versus −7 U per liter (P<0.001), respectively.26 There were also improvements in the secondary endpoints of lipid profile and a reduction in hepatic fat content (P<0.001 for all comparisons, except p=0.04 for triglycerides). Paired liver biopsies (at baseline and week 20) were available in 26 patients (39% of enrolled subjects); in 10/16 (63%) patients receiving sebelipase alfa there was improvement in hepatic steatosis (≥5% reduction), as opposed to 4/10 (40%) subjects on placebo; although this was not a statistically significant difference.26 The number of patients who experienced adverse events was similar in study drug and placebo groups; most episodes were deemed mild and considered by the investigator to be unrelated to study drug.
As with other studies, atherogenic biomarkers were evaluated for the patients enrolled in the ARISE study (reported separately), which included some subjects on lipid-lowering drugs (LLD, 26 of 61 evaluable subjects, 43%).27 Baseline values for LDL-C, LDL-P number (mean total concentration was 2151 nmol/L), and apoB were elevated, while HDL-C and apoA1 were low; a pattern consistent with cholesterol dysregulation. Treatment with sebelipase alfa for 20 weeks significantly improved altered lipid profiles, irrespective of lipid-lowering medication usage; the improvements were sustained for the period of observation (up to 52 weeks).27 The reduction in LDL-C with treatment was associated with a reduction in the LDL-P number. For those on sebalipase alfa from the start, at the 20-weeks timepoint mean (SD) percentage change from baseline in the LDL-P number for patients using LLD or not was −34.2% (−13.6) and −21.7% (−20.6), respectively; both of these results were statistically significantly different from values noted for the cohort on placebo. At study week 52, the corresponding LDL-P changes were −43.0% (11.8) and −43.8% (16.2) for these patients on sebalipase alfa from the start.27
Hypersensitivity and Immunogenicity
In the clinical studies (and as noted in the SPC, EMEA summary of product characteristics), infusion-related reactions were noted as consistent with anaphylaxis (in 3/106, 3%) or hypersensitivity (21/106, 20%).28 The events were observed in both infants and older subjects.24,26 In these treated patients, the majority of reactions occurred during or within 4 hrs of the completion of the infusion. As with other enzyme therapies for LSDs, premedication and/or slower rate of infusion enabled the administration of further treatments.
With respect to seroconversion (ie, antibody formation), in the LAL-CL02 study, 5 of 35 (14%) evaluable children and adults who were given sebelipase alfa (at 1 mg/kg once every other week) during the 20-week double-blind period of the study developed anti-drug antibodies (ADA).26,28 While in the LAL-CL03 study, 4 of 7 (57%) evaluable infants developed ADA during treatment with sebelipase alfa given once weekly.25,28 At the time of initial ADA positivity, three of these infants were receiving a dose of 1 mg/kg and the fourth patient was on 3 mg/kg.
Most patients who developed ADA did so within the first 3 months of exposure. ADA titers decreased to undetectable levels during continued treatment in the majority of treated patients. Two of the patients in the LAL-CL03 study were determined to be positive for ADA that inhibited in vitro enzyme activity and cellular uptake of the enzyme, which was not observed in the patients enrolled in the LAL-CL02 study who had seroconverted.25,27
There was no determination of an association between the development of ADA to sebelipase alfa and impact of treatment or the occurrence of adverse events.
Adjunctive Therapies-Lipid-Lowering Drugs
There are several classes of lipid (cholesterol)-reducing drugs (including HMG-CoA reductase inhibitors, bile acid sequestrants, fibric acid derivatives, and plant stanols). In patients with hypercholesterolemia, a statin is considered by most as an established first-line therapy.29 Other drug classes are used to augment statin effects on LDL cholesterol, as a substitute for statins (when contraindicated or cannot be used), or to treat non-LDL cholesterol disorders (primarily hypertriglyceridemia).
Apart from the mechanism(s) of action relating to regulation of blood lipids, the benefit seen with lipid-lowering drugs (LLD) may include regression of atherosclerosis, plaque stabilization, reversal of endothelial dysfunction, inhibition of inflammatory pathways, and decreased thrombogenicity.30 At present, it is not clear whether these putative processes are relevant disease pathways contributing to morbidity in LAL deficiency. Anyhow, prior to the availability of ERT, LLDs had been prescribed to LAL deficient patients; primarily those with an attenuated (CESD) phenotype.31,32 These reports suggest the patients showed continued progression to liver cirrhosis; although disease stage at initiation of lipid-lowering drugs may have been a factor.
A recent report describes the use of ezetimibe, a drug that inhibits the gastrointestinal absorption of cholesterol; mediated through binding with the Niemann-Pick C1-Like 1 (NPC1L1) protein. Thus, unlike other classes of LLDs, ezetimibe does not inhibit hepatic cholesterol synthesis or increase its excretion in bile.33
The aim for prescribing ezetimibe in patients with LAL deficiency would be to promote an increase in the circulatory clearance of cholesterol and a reduction of hepatic cholesterol deposits. Three patients with CESD, not on enzyme therapy, had been treated with ezetimibe for 9–10 years; this was supplemented with low dose atorvastatin in two patients during the last 6 years of observation.34 All patients showed significant changes in their lipid profile, including a reduction of ALT, cholesterol and triglyceride. Furthermore, there was no demonstrable progression of liver fibrosis.34
Although the use of lipid-lowering agents may be indicated to achieve an optimal outcome in patients with CESD, there may be other mechanisms of disease beyond those attributable to disruption of lipid metabolism associated with LAL deficiency that may be drivers of morbidity. Thus, close monitoring of patients on lipid-lowering agents will be necessary, so sebalipase alfa therapy can be initiated, if deemed appropriate.
Current Perspectives
Lysosomal acid lipase (LAL) deficiency in its most severe form (i.e., Wolman disease) is a life-limiting illness; unlike Hurler syndrome (mucopolysaccharidosis type I) disease course in LAL deficiency has not been significantly impacted by hematopoietic stem cell transplantation.35 As noted, this lack of improvement following HSCT is likely due to continued disease process in affected tissues not derived from hematopoietic elements. Moreover, patients with an attenuated phenotype (i.e., Cholesteryl-ester storage disease) continue to progress to liver cirrhosis, albeit at varying rates, despite the use of lipid-lowering drugs (LLD) to modify abnormal lipid profile, a contributor to morbidity in LAL deficiency. However, the majority of this later group of patients were on various drug classes; it is possible a more favorable outcome with newer LLDs may be discerned, but this would require additional long-term studies.
The outcome of enzyme replacement therapy with sebelipase alfa points to a more beneficial prognosis among treated cases. To summarize: Increased survival has been observed in treated infants with Wolman disease, and significant improvements in liver function with reduction in hepatic fat content have been observed in both treated children and adults. Moreover, changes favoring a less atherogenic lipid profile were observed. These findings suggest a reduction in the risk of hepatic fibrosis and progression to cirrhosis among treated patients with LAL deficiency can be anticipated; perhaps also ultimately leading to a reduction in the incidence of cardio- and cerebrovascular-related events. Determinants of outcome, including the impact of pretreatment disease severity, remain to be more fully delineated. Ultimately, long-term studies will clarify the scope of clinical benefit for sebelipase alfa in patients with LAL deficiency. Anyhow, sebelipase alfa treatment will likely become the standard of care in jurisdictions where available, given the procedural morbidity and mortality risks associated with HSCT. In these severe cases, it would be deemed appropriate to initiate therapy at diagnosis. Initiation of therapy in attenuated cases of LAL deficiency necessitates local guidelines, given the heterogeneity not only in terms of clinical presentation, but also with rates of disease progression. Regardless, in patients with dyslipidemia, it would be appropriate to consider dietary advice and the use of lipid-lowering agents prior to and following diagnostic confirmation, which unfortunately appears to be significantly delayed in a significant proportion of cases. Involvement of lipid and liver specialty teams, in drawing a multidisciplinary consensus regarding follow-up would be ideal. A limited number of reports have described findings on liver biopsy, and noninvasive tests, such as FibroScan and FibroTest in assessment of patients with LAL deficiency;36 and there continues to be debate regarding how findings from these investigations should be used to inform decisions in relation to enzyme replacement therapy. Consensus recommendations for the initial assessment and ongoing monitoring of children and adults with LAL deficiency have recently been published.37
Enzyme replacement therapy (ERT)-based on the administration of sebelipase alfa – for LAL deficiency adds to the growing list of recombinant proteins currently available for infusion in patients with a lysosomal storage disorder (LSD). As with other ERTs, it has been shown to be relatively safe and efficacious across a range of phenotypes, from the severe Wollman to the attenuated (CESD) clinical forms. Although there were >100 patients treated during the clinical trial phase, with additional experience drawn from those initiated on therapy in certain jurisdictions where treatment was made available, additional studies will be required to ascertain whether the changes in the natural course of disease are full or may in some cases result in emergent phenotypes that were not a previously recognized feature of LAL deficiency, given historically limited survival; particularly in patients with Wollman disease. The experience with other LSDs may be instructive in this regard. For instance, in Gaucher disease, one of the earliest LSDs for which enzyme replacement therapy (ERT) became available, it has emerged affected individuals and their carrier relatives have an increased risk for Parkinson disease (compared with the general population), and that amongst affected patients this risk does not appear to be mitigated by ERT.38
Perhaps a more relevant situation is the experience relating to follow-up of patients with infantile Pompe disease (including only those with survival to age ≥5 years) on ERT (age ≤6 months at treatment initiation with alglucosidase alfa). Long-term survivors (n-11, median age of 8.0 years; range: 5.4–12.0 years) exhibited sustained improvements in cardiac structure and function, and in gross motor function.39 However, some patients displayed a variety of issues including residual muscle weakness, hearing loss, risk for arrhythmias, hypernasal speech, dysphagia with risk for aspiration, and osteopenia.39 Of note, these were children – all of whom were cross-reactive immunologic material (CRIM) positive – had low or undetectable anti-alglucosidase alfa antibody titers. It is known that patients with infantile onsetdisease and null alleles and who are CRIMnegative deteriorate despite ERT when not given immunomodulatory agents concomitantly; indeed, outcome may remain unfavorable in some cases despite immunomodulation.40,41
With respect to patients with the attenuated variant - late-onset Pompe disease (LOPD) - a systematic review of outcomes revealed a nearly five-fold lower mortality rate in ERT-treated patients, compared to those who were untreated (rate ratio: 0.21; 95% credible interval: 0.11, 0.41).42 In this report, 22 publications pertaining to 19 studies/trials (with an average duration of 45.7 months) were examined, encompassing 438 patients.43 On average, treated patients showed a rapid improvement, with an increase of 1.4% FVC after 2 months; however, this was followed by a slow regression back to baseline over a three-year period. In the 6-min walk test, the largest improvement was observed over the first 20 months in treated patients; approximately 50 m increase in performance over baseline, with stabilization in the following years. As deterioration would have been anticipated, stabilization could be deemed a measure of disease control. A more recent report notes that among treated patients with LOPD followed for an extended period, extra-muscular features have been found; findings not previously within the spectrum of disease manifestation.43 A potential explanation for these observations may be that tissues that retain pathology (whether persistent storage material and/or secondary fibrotic changes) are “sanctuaries,” that is, sites not accessible to intravenously infused enzyme. It may also be disease at a certain stage may be “beyond storage,” a consequence of concomitant or downstream phenomenon, not fully reversible. The relevance of these findings to treatment with ERT in LAL deficiency remains to be determined; hopefully, these issues will be addressed by ongoing data collection through registries. As with other genetic lipid disorders, there is great value in a registry program to capture ongoing outcomes and enable the identification of deficiencies in current diagnostic and management practices.44
As evident from the experience of treatment of LSDs with recombinant enzyme infusions, early diagnosis is critical as burden of disease when therapy is initiated can limit clinical response. This is evident in the experience with patients with Fabry disease on ERT; for instance, it has been shown that those with cardiomyopathy associated with myocardial fibrosis may continue to suffer from or be at risk for serious cardiac related events, including sudden cardiac death.45
The outcome of clinical trials and early experience with enzyme therapy for LAL deficiency indicate a favorable impact on disease course; both safety and efficacy data suggest a significant proportion of patients will benefit from therapy. Although there are patients who develop neutralizing antibodies against the enzyme, desensitization measures can be introduced as a mitigating approach.46 It is also evident there are aspects of the condition which can be modified by other approaches such as HSCT and the use of lipid-lowering drugs. Although the experience with these other measures, prior to introduction of sebalipase alfa, has revealed their limitations, it is possible a combination of options or a multi-modality approach may enable a better or more optimal outcome. However, there will need to be a careful evaluation of the overall risk to benefit ratio. As with other ERTs for LSDs, cost remains a major challenge, particularly for countries with constrained healthcare resources.47
Disclosure
Gregory M Pastores reports personal fees from Alexion, for participation in an advisory borad meeting. The authors report no other conflicts of interest in this work.
References
1. Pisciotta L, Tozzi G, Travaglini L, et al. Molecular and clinical characterization of a series of patients with childhood-onset lysosomal acid lipase deficiency. Retrospective investigations, follow-up and detection of two novel LIPA pathogenic variants. Atherosclerosis. 2017;265:124–132. doi:10.1016/j.atherosclerosis.2017.08.021
2. Pericleous M, Kelly C, Wang T, et al. Wolman’s disease and cholesteryl ester storage disorder: the phenotypic spectrum of lysosomal acid lipase deficiency. Lancet Gastroenterol Hepatol. 2017;2(9):670–679. doi:10.1016/S2468-1253(17)30052-3
3. Santos Silva E, Klaudel-Dreszler M, Bakuła A, et al. Early onset lysosomal acid lipase deficiency presenting as secondary hemophagocytic lymphohistiocytosis: two infants treated with sebelipase alfa. Clin Res Hepatol Gastroenterol. 2018;42(5):77–82. doi:10.1016/j.clinre.2018.03.012
4. Taurisano R, Maiorana A, De Benedetti F, et al. Wolman disease associated with hemophagocytic lymphohistiocytosis: attempts for an explanation. Eur J Pediatr. 2014;173(10):1391–1394. doi:10.1007/s00431-014-2338-y
5. Qu P, Du H, Wilkes DS, Yan C. Critical roles of lysosomal acid lipase in T cell development and function. Am J Pathol. 2009;174(3):944–956. doi:10.2353/ajpath.2009.080562
6. Qu P, Yan C, Blum JS, Kapur R, Du H. Myeloid-specific expression of human lysosomal acid lipase corrects malformation and malfunction of myeloid-derived suppressor cells in lal-/- mice. J Immunol. 2011;187(7):3854–3866. doi:10.4049/jimmunol.1003358
7. Canbay A, Müller MN, Philippou S, et al. Cholesteryl ester storage disease: fatal outcome without causal therapy in a female patient with the preventable sequelae of progressive liver disease after many years of mild symptoms. Am J Case Rep. 2018;19:577–581. doi:10.12659/AJCR.907755
8. Reynolds TM, Mewies C, Hamilton J, Wierzbicki AS, PATHFINDER Project Collaboration group. Identification of rare diseases by screening a population selected on the basis of routine pathology results-the PATHFINDER project: lysosomal acid lipase/cholesteryl ester storage disease substudy. J Clin Pathol. 2018;71(7):608–613. doi:10.1136/jclinpath-2017-204727
9. Barone R, Sotgiu S, Musumeci S. Plasma chitotriosidase in health and pathology. Clin Lab. 2007;53(5–6):321–333.
10. Geberhiwot T, Moro A, Dardis A, et al. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J Rare Dis. 2018;13(1):50. doi:10.1186/s13023-018-0785-7
11. Deodato F, Boenzi S, Taurisano R, et al. The impact of biomarkers analysis in the diagnosis of Niemann-Pick C disease and acid sphingomyelinase deficiency. Clin Chim Acta. 2018;486:387–394. doi:10.1016/j.cca.2018.08.039
12. Pajares S, Arias A, García-Villoria J, et al. Cholestane-3β,5α,6β-triol: high levels in Niemann-Pick type C, cerebrotendinous xanthomatosis, and lysosomal acid lipase deficiency. J Lipid Res. 2015;56(10):1926–1935. doi:10.1194/jlr.M060343
13. Griffiths WJ, Yutuc E, Abdel-Khalik J, et al. Metabolism of non-enzymatically derived oxysterols: clues from sterol metabolic disorders. Free Radic Biol Med. 2019;144:124–133. doi:10.1016/j.freeradbiomed.2019.04.020
14. Tovoli F, Napoli L, Negrini G, et al. A relative deficiency of lysosomal acid lipase activity characterizes non-alcoholic fatty liver disease. Int J Mol Sci. 2017;18(6):1134. doi:10.3390/ijms18061134
15. Baratta F, Pastori D, Ferro D, et al. Reduced lysosomal acid lipase activity: a new marker of liver disease severity across the clinical continuum of non-alcoholic fatty liver disease? World J Gastroenterol. 2019;25(30):4172–4180. doi:10.3748/wjg.v25.i30.4172
16. Camarena C, Aldamiz-Echevarria LJ, Polo B, et al. Update on lysosomal acid lipase deficiency: diagnosis, treatment and patient management. Med Clin (Barc). 2017;148(9):
17. Tolar J, Petryk A, Khan K, et al. Long-term metabolic, endocrine, and neuropsychological outcome of hematopoietic cell transplantation for Wolman disease. Bone Marrow Transplant. 2009;43(1):21–27. doi:10.1038/bmt.2008.273
18. Yanir A, Allatif MA, Weintraub M, Stepensky P. Unfavorable outcome of hematopoietic stem cell transplantation in two siblings with Wolman disease due to graft failure and hepatic complications. Mol Genet Metab. 2013;109(2):224–226. doi:10.1016/j.ymgme.2013.03.007
19. Bernstein DL, Lobritto S, Iuga A, et al. Lysosomal acid lipase deficiency allograft recurrence and liver failure - clinical outcomes of 18 liver transplantation patients. Mol Genet Metab. 2018;124(1):11–19. doi:10.1016/j.ymgme.2018.03.010
20. Du H, Duanmu M, Witte D, Grabowski GA. Targeted disruption of the mouse lysosomal acid lipase gene: long-term survival with massive cholesteryl ester and triglyceride storage. Hum Mol Genet. 1998;7(9):1347–1354. doi:10.1093/hmg/7.9.1347
21. Du H, Schiavi S, Levine M, et al. Enzyme therapy for lysosomal acid lipase deficiency in the mouse. Hum Mol Genet. 2001;10(16):1639–1648. doi:10.1093/hmg/10.16.1639
22. Du H, Cameron TL, Garger SJ, et al. Wolman disease/cholesteryl ester storage disease: efficacy of plant-produced human lysosomal acid lipase in mice. J Lipid Res. 2008;49(8):1646–1657. doi:10.1194/jlr.M700482-JLR200
23. Balwani M, Breen C, Enns GM, et al. Clinical effect and safety profile of recombinant human lysosomal acid lipase in patients with cholesteryl ester storage disease. Hepatology. 2013;58(3):950–957. doi:10.1002/hep.v58.3
24. Valayannopoulos V, Malinova V, Honzík T, et al. Sebelipase alfa over 52 weeks reduces serum transaminases, liver volume and improves serum lipids in patients with lysosomal acid lipase deficiency. J Hepatol. 2014;61(5):1135–1142. doi:10.1016/j.jhep.2014.06.022
25. Jones SA, Rojas-Caro S, Quinn AG, et al. Survival in infants treated with sebelipase Alfa for lysosomal acid lipase deficiency: an open-label, multicenter, dose-escalation study. Orphanet J Rare Dis. 2017;12(1):25. doi:10.1186/s13023-017-0587-3
26. Burton BK, Balwani M, Feillet F, et al. A Phase 3 trial of Sebelipase alfa in lysosomal acid lipase deficiency. N Engl J Med. 2015;373(11):1010–1020. doi:10.1056/NEJMoa1501365
27. Wilson DP, Friedman M, Marulkar S, Hamby T, Bruckert E. Sebelipase alfa improves atherogenic biomarkers in adults and children with lysosomal acid lipase deficiency. J Clin Lipidol. 2018;12(3):604–614. doi:10.1016/j.jacl.2018.02.020
28. Kanuma, INN-sebelipase alfa - European Medicines Agency - europa.eu Available from: www.ema.europa.eu/docs/en_GB/document_library/…/WC500192715.pdf.
29. Ahangari N, Ghayour Mobarhan M, Sahebkar A, et al. Molecular aspects of hypercholesterolemia treatment: current perspectives and hopes. Ann Med. 2018;50(4):303–311. doi:10.1080/07853890.2018.1457795
30. Geovanini GR, Libby P. Atherosclerosis and inflammation: overview and updates. Clin Sci (Lond). 2018;132(12):1243–1252. doi:10.1042/CS20180306
31. Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58:1230–1243. doi:10.1016/j.jhep.2013.02.014
32. Reiner Z, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency — an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235:21–30. doi:10.1016/j.atherosclerosis.2014.04.003
33. Savarese G, De Ferrari GM, Rosano GMC, Perrone-Filardi P. Safety and efficacy of ezetimibe: a meta-analysis. Int J Cardiol. 2015;201:247–252. doi:10.1016/j.ijcard.2015.08.103
34. Di Rocco M, Pisciotta L, Madeo A, et al. Long term substrate reduction therapy with ezetimibe alone or associated with statins in three adult patients with lysosomal acid lipase deficiency. Orphanet J Rare Dis. 2018;13(1):24. doi:10.1186/s13023-018-0768-8
35. Lum SH, Stepien KM, Ghosh A, et al. Long term survival and cardiopulmonary outcome in children with Hurler syndrome after haematopoietic stem cell transplantation. J Inherit Metab Dis. 2017;40(3):455–460. doi:10.1007/s10545-017-0034-6
36. Poinsot P, Collardeau Frachon S, Restier L, et al. Childhood/adult-onset lysosomal acid lipase deficiency: a serious metabolic and vascular phenotype beyond liver disease-four new pediatric cases. J Clin Lipidol. 2017;11(1):167–177. doi:10.1016/j.jacl.2016.11.008
37. Kohli R, Ratziu V, Fiel MI, et al. Initial assessment and ongoing monitoring of lysosomal acid lipase deficiency in children and adults: consensus recommendations from an international collaborative working group. Mol Genet Metab. 2019. doi:10.1016/j.ymgme.2019.11.004
38. Thaler A, Bregman N, Gurevich T, et al. Parkinson’s disease phenotype is influenced by the severity of the mutations in the GBA gene. Parkinsonism Relat Disord. 2018;55:45–49. doi:10.1016/j.parkreldis.2018.05.009
39. Prater SN, Banugaria SG, DeArmey SM, et al. The emerging phenotype of long-term survivors with infantile Pompe disease. Genet Med. 2012;14(9):800–810. doi:10.1038/gim.2012.44
40. Owens P, Wong M, Bhattacharya K, Ellaway C. Infantile-onset Pompe disease: a case series highlighting early clinical features, spectrum of disease severity and treatment response. J Paediatr Child Health. 2018;54(11):1255–1261. doi:10.1111/jpc.2018.54.issue-11
41. Poelman E, Hoogeveen-Westerveld M, Kroos-de Haan MA, et al. High sustained antibody titers in patients with classic infantile Pompe disease following immunomodulation at start of enzyme replacement therapy. J Pediatr. 2018;195:236–243. doi:10.1016/j.jpeds.2017.11.046
42. Schoser B, Stewart A, Kanters S, et al. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J Neurol. 2017;264(4):621–630. doi:10.1007/s00415-016-8219-8
43. Chan J, Desai AK, Kazi ZB, et al. The emerging phenotype of late-onset Pompe disease: a systematic literature review. Mol Genet Metab. 2017;120(3):163–172. doi:10.1016/j.ymgme.2016.12.004
44. Ng DM, Hooper AJ, Bellgard MI, Burnett JR. The role of patient registries for rare genetic lipid disorders. Curr Opin Lipidol. 2018;29(2):156–162. doi:10.1097/MOL.0000000000000485
45. Arends M, Biegstraaten M, Hughes DA, et al. Retrospective study of long-term outcomes of enzyme replacement therapy in Fabry disease: analysis of prognostic factors. PLoS One. 2017;12(8):e0182379. doi:10.1371/journal.pone.0182379
46. Huffaker MF, Liu AY, Enns GM, et al. Case series of sebelipase alfa hypersensitivity reactions and successful sebelipase alfa rapid desensitization. JIMD Rep. 2019;49(1):30–47. doi:10.1002/jmd2.12066
47. Wyatt K, Henley W, Anderson L, et al. The effectiveness and cost-effectiveness of enzyme and substrate replacement therapies: a longitudinal cohort study of people with lysosomal storage disorders. Health Technol Assess. 2012;16(39):1–543. doi:10.3310/hta16390
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.