Back to Journals » ImmunoTargets and Therapy » Volume 15
Lymphoproliferation in Inborn Errors of Immunity: Mechanisms, Manifestations and Clinical Management, with a Focus on ALPID
Authors Gualtiero G ![]() , Buso H, Coopmans EC, Milito C
, Buso H, Coopmans EC, Milito C ![]() , Firinu D, Dalm VA, Cinetto F
, Firinu D, Dalm VA, Cinetto F ![]()
Received 19 November 2025
Accepted for publication 27 February 2026
Published 7 March 2026 Volume 2026:15 582718
DOI https://doi.org/10.2147/ITT.S582718
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Giulia Gualtiero,1– 3,* Helena Buso,1,4,* Eva C Coopmans,2,3 Cinzia Milito,5,6 Davide Firinu,7 Virgil ASH Dalm,2,3 Francesco Cinetto1,4
1Department of Medicine (DIMED), University of Padua, Padua, Italy; 2Department of Internal Medicine, Division of Allergy and Clinical Immunology, Erasmus University Medical Center, Rotterdam, the Netherlands; 3Department of Immunology, Erasmus University Medical Center, Rotterdam, the Netherlands; 4Rare Diseases Referral Center, Internal Medicine 1, Ca’ Foncello Hospital, AULSS2 Marca Trevigiana, Treviso, Italy; 5Department of Molecular Medicine, “Sapienza” University of Rome, Rome, Italy; 6Centro di Riferimento per le Immunodeficienze Primitive, Azienda Ospedaliero Universitaria Policlinico Umberto I, Rome, Italy; 7Department of Medical Sciences and Public Health, University of Cagliari, Cagliari, Italy
*These authors contributed equally to this work
Correspondence: Cinzia Milito, Department of Molecular Medicine, “Sapienza” University of Rome, Viale Regina Elena, 291, Rome, 00185, Italy, Email [email protected]
Abstract: Lymphoproliferation represents a common and clinically relevant feature across many Inborn Errors of Immunity (IEIs), reflecting the underlying immune dysregulation that characterizes these disorders. In affected individuals, lymphoproliferation may manifest along a broad clinical spectrum, ranging from benign forms such as lymphadenopathy, splenomegaly, and hepatomegaly to malignant transformation, including lymphoma and leukemia. Multiple and heterogeneous pathogenetic mechanisms contribute to lymphoproliferation in IEIs; however, they can be conceptually grouped into two major categories: excessive lymphocyte activation and proliferation, and defective control of proliferation. The association of lymphoproliferation with autoimmune cytopenia classically defined the Autoimmune Lymphoproliferative Syndrome (ALPS), caused by an impairment in FAS-mediated apoptosis. However, a growing number of IEIs present with overlapping autoimmune lymphoproliferative phenotypes driven by diverse monogenic defects or, in many cases, without an identifiable genetic cause. This expanding spectrum has led to the concept of autoimmune lymphoproliferative immunodeficiencies (ALPID), encompassing a continuum of disorders characterized by lymphoproliferation, autoimmunity and diverse genetic backgrounds, requiring disease-specific therapeutic approaches. This narrative review provides an overview of the main IEIs associated with lymphoproliferation focusing on pathogenic pathways, diagnostic evaluation and classification, and current and emerging targeted therapeutic approaches.
Keywords: lymphoproliferation, IEIs, pathways, ALPS, ALPID
Introduction
Inborn Errors of Immunity (IEIs) represent a heterogeneous group of genetic disorders mainly caused by pathogenic germline variants affecting genes involved in immune function and regulation.1–3 IEIs specifically affect the function of one or various components of the human immune system and may present clinically with increased susceptibility to infections, autoinflammation, autoimmune disease, severe allergy and neoplasms.4,5 Although each IEI is individually rare, collectively their estimated prevalence reaches 1:1000 to 1:5000. To date, more than 500 different genes and up to 20 phenocopies have been detected to cause IEIs, and heterogeneity in resulting clinical phenotypes can be seen even for a single gene or variant. Thus, new challenges have emerged for identifying IEIs in patients with immune dysregulation and for understanding the link between a specific clinical phenotype and the underlying monogenic defect (Figure 1).6–9
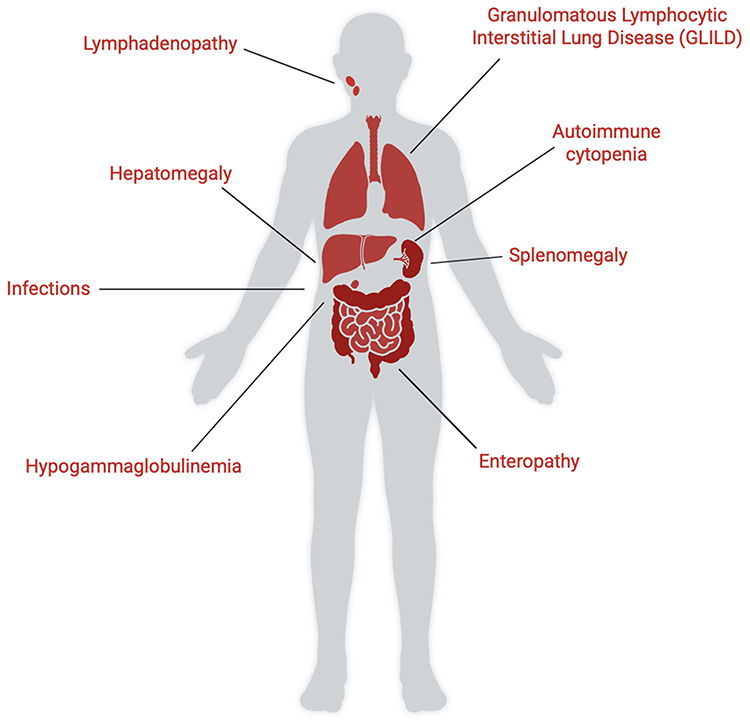 |
Figure 1 The clinical red flags of IEIs-associated lymphoproliferation. Anatomical mapping of key clinical manifestations which are indicative of lymphoproliferation in IEIs. Features include lymphadenopathy, hepatomegaly, infections, hypogammaglobulinemia, granulomatous lymphocytic interstitial lung disease (GLILD), autoimmune cytopenia, splenomegaly and enteropathy. The critical observation of these “red flags” is essential for early diagnosis and timely evaluation of IEIs. Created with BioRender.com. |
Lymphoproliferation in IEIs refers to persistent monoclonal, oligoclonal, or polyclonal proliferation of lymphoid cells.10,11 Patients with IEIs frequently experience both benign and malignant lymphoproliferation, which can present at disease onset or evolve over time, often in association with autoimmunity. This has recently led to the umbrella definition of autoimmune lymphoproliferative immunodeficiencies (ALPID).12
In the clinical setting, lymphoproliferation in patients with IEIs may present with lymphadenopathy, splenomegaly (defined as a palpable spleen or volume greater than 2 SDs at abdomen ultrasound compared with age-related normal values), hepatomegaly (defined as a palpable liver or volume greater than 2 SDs compared with age-related normal values), and/or proliferation of mucosa-associated lymphoid tissue (MALT). Benign lymphoproliferation is a prominent feature of immune dysregulation in many IEIs and may even be a dominant clinical feature in specific IEIs, a subgroup referred to as primary immune regulatory disorders (PRIDs).13,14 This condition frequently coexists with autoimmune manifestations, with a high prevalence of autoimmune cytopenias. In particular, autoimmune cytopenias comprise persistent or recurrent cytopenia marked by autoantibodies affecting at least one hematopoietic cell lineage or response to immunosuppression.15 Autoimmune lymphoproliferative syndrome (ALPS) caused by mutations in the genes responsible for the extrinsic apoptotic pathway is generally considered the paradigm of inherited disease presenting with ALPID.16–18 The ALPID study18 recently and prospectively investigated a cohort of pediatric patients with clearly defined symptoms of autoimmune lymphoproliferative disease initially lacking a genetical diagnosis. By applying strict clinical inclusion criteria this study improves the diagnostic classification of patients with ALPID to the diagnostic categories of ALPS, autosomal-dominant ALPID (AD-ALPID), other IEIs with lymphoproliferation, non-IEI diagnosis, unresolved ALPID (ALPID-U) and insufficient genetic workup.
Malignant lymphoproliferation is the most common neoplasm associated with IEIs. In patients with CVID, the most prevalent IEI, B cell non-Hodgkin lymphoma (NHL) is among the lymphoproliferative complications. The exact prevalence of lymphomas in CVID remains unclear; however, from patient registries it is possible to estimate that it may be up to 2.9%19–21 and from single-center studies up to 9.1%22–25 and that are associated with markedly reduced survival.21,22,26–28
Although the pathways of lymphoproliferation in IEIs are not entirely clear, both intrinsic (such as failed apoptotic signaling, hyperactivation of proliferation, or defective immune checkpoint control) and extrinsic factors (such as Epstein-Barr virus (EBV)) are known to contribute to the development of IEIs-associated lymphoproliferation.
This review highlights the pathobiology of the most frequent IEIs-associated lymphoproliferative disorders with a particular focus on updated genetic defects, related pathways and associated phenotypic characteristics, laboratory and immunological abnormalities, diagnostic strategies and therapeutic options.
Molecular Pathways Driving Lymphoproliferation in IEIs
The homeostasis of the lymphocyte compartment relies on a dynamic balance between signals which promote proliferation, differentiation and survival, and signals driving apoptosis and contraction of the immune response.29–38 The disruption of this tightly regulated balance, due to defective signaling or impaired apoptosis, represents a key mechanism underlying lymphoproliferation in IEIs.3,34,35,39–42 Several molecular pathways are critical in this pathological context, and genetic alterations related to them result in sustained lymphocyte accumulation, immune dysregulation, and increased risk of autoimmunity or malignancy (Figure 2).1,2,9,41,43
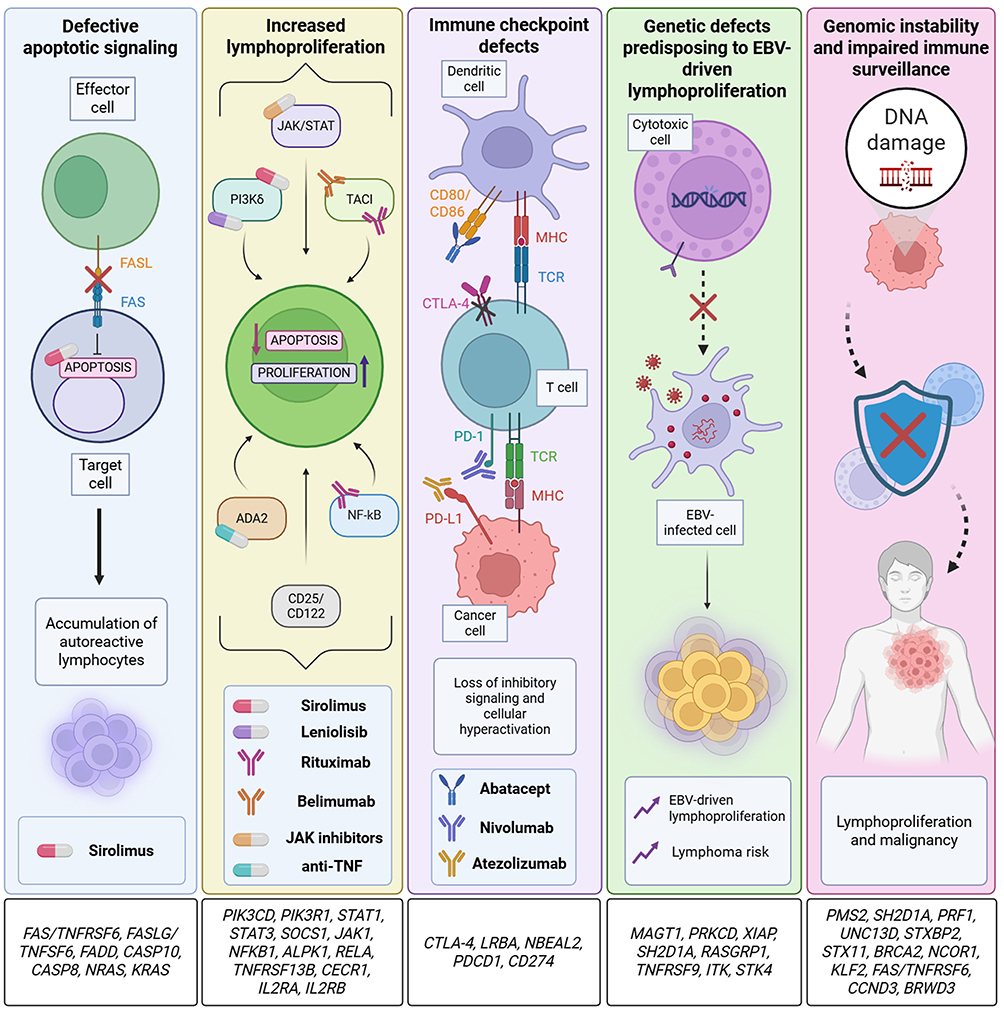 |
Figure 2 Molecular pathways driving lymphoproliferation in IEIs. Schematic overview of the key mechanisms underlying pathological lymphoproliferation in inborn errors of immunity. Each panel reports each single involved molecular pathway and the related biological implication in disease in bold text. From left to right: defective FAS-mediated apoptosis, dysregulated proliferative pathways (JAK/STAT, PI3Kδ, TACI, ADA2, NF-κB, CD25, CD122) cause aberrant lymphocyte proliferation, survival and activation. Other contributing factors include genetic defects leading to EBV driven proliferation and genomic instability affecting immune surveillance. Upward (↑), and diagonal (↗) arrows indicate an increase, downward (↓) arrows indicate a decrease. Crosses (x) denote inhibition or blockade of the corresponding pathway or process. All associated genes are listed above each panel. In the first three panels, from left to right, available treatment strategies are reported with representative symbols and related names in bold text. Created with BioRender.com. |
Defective Apoptotic Signaling: FAS/FASL
The FAS/FASL pathway represents a critical extrinsic apoptotic pathway,44–49 essential for triggering activation-induced cell death (AICD),48,50 the process by which expanded T and B cell clones are eliminated after immune activation.50,51
This T cell-driven mechanism relies on cell-cell interactions, as apoptosis is initiated when FAS-expressing cells, primarily activated macrophages and T cells, bind to other cells expressing FASL on their surface, as in the case of activated T cells and NK cells.47,51–56 Following the binding of FAS by FASL, FAS undergoes trimerization. Consequently, the FAS-associated death domain adaptor protein (FADD) is recruited to the trimerized FAS cytoplasmic region, leading to the assembly of the death-inducing signaling complex (DISC) and the following activation of caspase-8 and −10, which initiate a cell-extrinsic apoptotic cascade, inducing activated caspase-8 release into the cytosol, thereby triggering apoptosis.51,57–64 This pathway is crucial not only for the termination of the immune response through the elimination of effector lymphocytes but also for the maintenance of peripheral self-tolerance by depleting potentially autoreactive clones.51,65,66
Conversely, alterations affecting FAS, FASL, or downstream caspases, impair this process, resulting in the development of ALPS, which is characterized by chronic lymphoproliferation, autoimmune cytopenias and increased risk of lymphoma.67
The most common genetic defects involve the FAS/TNFRSF6 gene (FAS, also reported as CD95/APO-1),45,48 where mutations can be germline or somatic.1,45,68,69 In particular, germline mutations (responsible for over 70% of cases) often act through dominant-negative effects or haploinsufficiency,44,45,68,70,71 whereas somatic mutations may confer a proliferative advantage to FAS-controlled T cells.45
Mutations in FASLG/TNFSF6 (FASL), FADD, CASP10 and, less frequently, in CASP81,12,68,72–75 also impair this apoptotic checkpoint and have been reported to cause ALPS or ALPS-like phenotypes.45,73 Notably, a significant proportion of patients (20–30%) meet the diagnostic criteria for ALPS in the absence of mutations in known causative genes (termed ALPS-U).76
Clinically, patients with ALPS typically present with signs of chronic lymphoproliferation, such as lymphadenopathy and/or splenomegaly, along with multi-lineage autoimmune cytopenias. Although ALPS most commonly manifests in early childhood, with a median age of onset around 3 years, late-onset cases in adulthood have also been documented, often in association with somatic mutations.77,78
The pathognomonic laboratory hallmark of ALPS is the expansion of autoreactive double-negative T cells (DNTs; CD3⁺ TCRαβ⁺ CD4− CD8−). While their precise role in disease pathogenesis remains incompletely understood, it is noteworthy that DNTs expansion has also been observed in other autoimmune disorders, such as systemic lupus erythematosus (SLE).79,80
Other relevant ALPS biomarkers are the elevation of vitamin B12, IL-10, sFASL and the impairment of FAS-mediated apoptosis.1,68,81
These biomarkers, in conjunction with the percentage of DNTs and the patient’s clinical features, are part of the latest ESID diagnostic criteria for ALPS (Table 1).82 Interestingly, a similar ALPS phenotype can also be observed in the case of RAS-associated autoimmune leukoproliferative disease (RALD), driven by mutations harbored in NRAS or KRAS genes.1,13,83
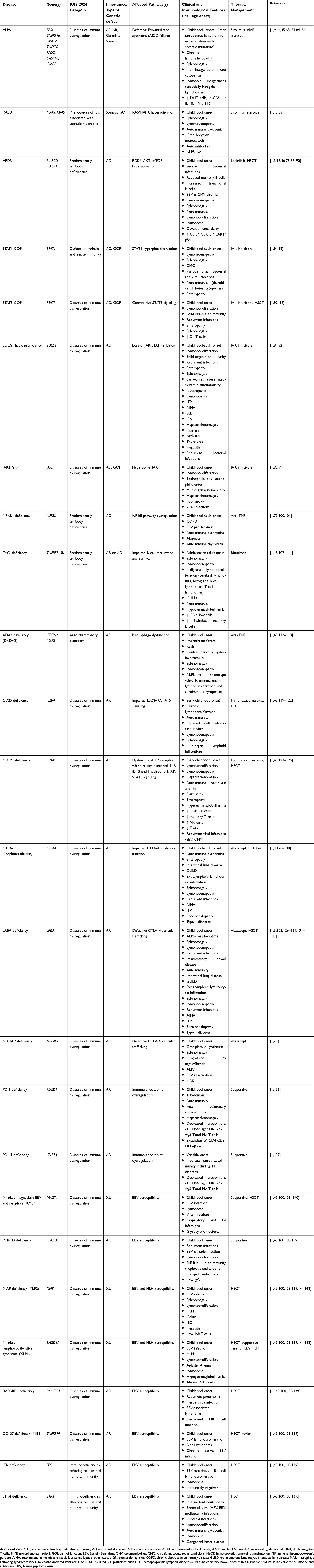 |
Table 1 Overview on Inborn Errors of Immunity Associated with Lymphoproliferation, Aligned with IUIS 2024 Classification |
Beyond the apoptotic function, it is important to note that FAS signals can also activate non-apoptotic pathways such as NF-kB, MAPK and PI3K/AKT/mTOR, which regulate immune responses and promote proliferation, survival and cell migration. Dysregulation of these pathways may further amplify immune activation and contribute to lymphoid malignancies.12,47
Indeed, patients with ALPS face a significantly elevated risk of developing lymphoma. Specifically, the relative risk for Hodgkin lymphoma is up to 40-fold higher than that of the general population, and the cumulative incidence of developing any type of lymphoma reaches approximately 15% by the third decade of life.143
Hyperactivated Signaling Pathways Driving Lymphoproliferation: PI3Kδ, JAK/STAT, NF-kB, TACI, ADA2, CD25/CD122
PI3Kδ
The phosphoinositide 3-kinase delta (PI3Kδ) pathway plays a key role in the regulation of lymphocyte activation, proliferation and survival.144–146 PI3Kδ is predominantly expressed in leukocytes and is activated as a downstream mediator of multiple receptors, including the T cell receptor (TCR), B cell receptor (BCR) and various cytokine receptors.144,145,147,148 Upon activation, PI3Kδ catalyzes the conversion of phosphatidylinositol-4,5-bisphosphate (PIP2) into phosphatidylinositol-3,4,5-trisphosphate (PIP3), a critical second messenger which triggers downstream signaling cascades, notably the AKT/mTOR pathway, thereby promoting metabolic fitness, cell growth and lymphocyte survival.149,150
Dysregulation of PI3Kδ signaling, due to genetic alterations which hyperactivate the pathway, underlies a group of IEIs collectively referred to as Activated PI3Kδ Syndrome (APDS).3,13,46,73 APDS results from either gain-of-function (GOF) mutations in PIK3CD (encoding the p110δ catalytic subunit; APDS1 phenotype) or functionally equivalent splice-site variants in PIK3R1 (encoding the p85ɑ regulatory subunit; APDS2 phenotype), both leading to constitutive PI3Kδ signalling.1,3,151,152
This persistent activation further drives the hyperactivation of downstream mTORC1, leading to excessive lymphocyte proliferation, impaired apoptosis and accumulation of terminally differentiated senescent effector T cells, including the expansion of CD57+CD8+ T cells. Biologically, in T and B cells of APDS patients these effects are reflected by the increased phosphorylation of AKT and S6 proteins (upstream and downstream regulators of the mTORC1 complex respectively), highlighting the crucial role of the PI3Kδ/AKT/mTOR/S6K axis in immune cell homeostasis.1,12,13,73,151,152 Clinically, this translates into immune dysregulation characterized by recurrent bacterial and viral infections, hypogammaglobulinemia, autoimmunity and a strong predisposition to lymphoproliferation, often manifesting as lymphadenopathy, severe hepatosplenomegaly, and an increased risk of B cell lymphomas.1
JAK/STAT
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway represents a central signaling axis in immunity and hematopoiesis, transducing signals from cytokine receptors into transcriptional programs which control lymphocyte proliferation, differentiation, and survival.91,153–155 After cytokine binding, receptor-associated JAKs become activated through transphosphorylation, subsequently phosphorylating STAT proteins, which dimerize and migrate into the nucleus to regulate gene expression.153,156 Given its central role in immune homeostasis, genetic dysregulation in the JAK/STAT signaling pathway can trigger a spectrum of IEIs characterized by immunodeficiency, autoimmunity and pathological lymphoproliferation.91,92,154
In particular, GOF mutations in STAT1 result in increased phosphorylation and delayed dephosphorylation of STAT1, disrupting the balance of cytokine signaling. This leads to immune dysregulation with lymphoproliferation, multi-organ autoimmunity and heightened susceptibility to chronic mucocutaneous candidiasis, as well as recurrent viral, bacterial and mycobacterial infections.91,92
In a similar way, GOF mutations in STAT3 represent a well-established cause of immune dysregulation syndromes, promoting aberrant transcriptional activity which drives enhanced lymphoproliferation, resulting in autoimmunity, enteropathy, and inflammation across multiple organ systems.1,92–95 Moreover, STAT3 GOF-associated lymphoproliferation leads to lymphadenopathy, splenomegaly, and elevated DNTs, as shown in cohort studies where about 73% of patients presented with lymphoproliferation.96–98
In addition, SOCS1 haploinsufficiency, which disrupts the negative regulation of JAK/STAT signaling, phenocopies STAT1 and STAT3 GOF mutations, with resulting STAT1 phosphorylation and comparable immune deregulatory manifestations.91,92 More rarely, GOF mutations in JAK1 have been reported as a cause of autosomal dominant immune dysregulation syndromes characterized by lymphoproliferation, eosinophilia, and multiorgan autoimmunity.99 Of note, these observations highlight how both the hyperactivation and defective regulation of the JAK/STAT signaling axis can converge into similar immune dysregulated phenotypes.
NF-κB
The NF-κB pathway represents a central signaling hub in immune homeostasis and its dysregulation contributes to a broad spectrum of IEIs.157 NF-κB activation is typically triggered by diverse stimuli, including antigen receptors, tumor necrosis factor (TNF) receptors, and pattern-recognition receptors (PRRs), leading to transcriptional programs that regulate lymphocyte survival, proliferation and inflammatory responses.157
Haploinsufficiency of NFKB1, encoding the p105 precursor of p50, is among the most frequent monogenic causes of CVID and is associated with EBV-driven lymphoproliferation, defective phosphorylation of p105, and reduced p50 levels.12,100
Conversely, gain-of-function (GOF) mutations in ALPK1 drive constitutive NF-κB activation, contributing to the pathogenesis of the autoinflammatory disorder ROSAH syndrome (retinal dystrophy, optic nerve edema, splenomegaly, anhidrosis and headache).1,158
Furthermore, dominant-negative mutations in RELA, which encodes the p65 subunit of NF-κB, have been identified in type I interferonopathies, thereby linking NF-κB dysfunction to aberrant interferon signaling, autoimmunity, and systemic autoinflammation.43,159
TACI
The TNFRSF13B gene, which encodes the TACI (Transmembrane Activator and CAML Interactor) protein, is widely recognized as being involved in inducing lymphoproliferation in certain IEIs, particularly in CVID.102,160–162 TACI is expressed on the surface of B cells and regulates their function, differentiation and survival.102,160 In addition, variants in TNFRSF13B have been associated with the development of granulomatous lymphocytic interstitial lung disease (GLILD), a peculiar complication of CVID which results from dysregulated B and T cell interactions at the pulmonary tissue level.103,104
Germline or somatic mutations of TNFRSF13B have been reported in numerous studies in B cell lymphomas. Specifically, mutations in TNFRSF13B are associated with lymphoproliferation and autoimmunity in CVID patients.102,105,106 In an analysis of 564 patients with antibody deficiency, 8.9% (50 patients) had at least one altered TNFRSF13B allele, with the majority (82%) showing heterozygous mutations.102 In this cohort, the prevalence of lymphoproliferation was significantly higher (p < 0.001) in the heterozygous C104R group compared to patients without a TNFRSF13B mutation.102
Common lymphoproliferative manifestations include splenomegaly, lymphadenopathy, and tonsillar hypertrophy, which often require surgical intervention.12,18,73 In one study, lymphoproliferation was found in 60% (30 out of 50) of CVID patients with TACI mutations. In more severe cases, malignant lymphoproliferation, including cerebral lymphomas, low-grade B cell lymphomas, and T cell lymphomas, has also been observed.1,46,107,108 It is important to note that, although TNFRSF13B mutations are strongly associated with hypogammaglobulinemia (p <0.001; relative risk 4.3), heterozygous variants can also be present in healthy individuals.102 This suggests that they may act more as “disease modifiers” rather than being “disease-causing” on their own,102,163 but they still predispose CVID patients to autoimmunity and lymphoproliferation.105,106 The mechanisms underlying this lymphoproliferation in cases of TACI deficiency appear to include the loss of pro-apoptotic factors and altered terminal differentiation.45,160
ADA2
Adenosine deaminase 2 (ADA2), encoded by the CECR1 gene, represents an extracellular enzyme responsible for purine metabolism and immune regulation.112,113 It plays a crucial role in macrophage differentiation and in modulating inflammatory responses through the adenosine signaling pathway.113,164 Biallelic mutations in the CECR1 gene are at the basis of ADA2 deficiency (DADA2), a complex systemic autoinflammatory disorder characterized by intermittent fevers, rash, central nervous system involvement and vasculitis which simulates polyarteritis nodosa (PAN). Of note, DADA2 frequently includes lymphoproliferation with splenomegaly and lymphadenopathy in its wide and variable phenotype.1,43 This condition has been specifically reported to cause an ALPS-like phenotype characterized by chronic non-malignant lymphoproliferation and autoimmune cytopenias.114
CD25 and CD122
A central, major role in immune regulation is embodied by the IL-2 cytokine, which interacts with different binding affinity types of IL-2 receptors (IL2Rs) expressed on the surface of T cells and NK cells. A single unit of the IL2R α chain forms the low-affinity IL2R (Kd ~ 10−8 M); the intermediate-affinity (Kd ~ 10−9 M) IL2R comes from the combination of β and ɣ chains, while the high-affinity conformation (Kd ~ 10−11 M) comprises all α, β and ɣ chains.42,165
CD25, encoded by the IL2RA gene, represents the ɑ chain of the IL-2 receptor complex and is a critical component of the IL-2/JAK/STAT5 signaling pathway.119,153,166 This pathway regulates T cell proliferation, differentiation and the maintenance of immune tolerance through regulatory T cells (Treg).167–169
In this context, CD25 deficiency, resulting from loss-of-function (LOF) mutations in the IL2RA gene, impair high-affinity IL-2 signaling, resulting in defective Treg development, function and uncontrolled lymphocyte proliferation. Since IL-2 signaling is crucial in sustaining FOXP3 expression and stability in the Treg cell subpopulation, its alteration further alters their suppressive action.119,120 The consequent immune dysregulation clinically manifests as chronic lymphoproliferation characterised by lymphadenopathy, splenomegaly and lymphoid infiltrations of various organs.1,43
In a similar way, LOF mutations in the IL2RB gene, encoding CD122, representing the β chain of the IL-2 receptor, disrupt IL-2 and IL-15 signaling, causing defective Treg and NK cellular functions coupled with a comparable clinical phenotype of immune dysregulation supported by chronic lymphoproliferation comparable to what observed in CD25 deficiency.42,123,124
Immune Checkpoint Defects: CTLA-4, PD-1, PD-L1
CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4) represents a central immune checkpoint receptor that maintains peripheral tolerance by restraining excessive T cell activation.45,170,171 Following T cell receptor (TCR) engagement, CTLA-4 is mobilized to the cell surface, where it competes with the co-stimulatory receptor CD28 for CD80/CD86 binding on antigen-presenting cells.170,172 By outcompeting CD28, CTLA-4 transduces inhibitory signals that limit proliferation, cytokine production, and effector responses, preventing immune hyperactivation and autoimmunity.173,174 Proper CTLA-4 expression, recycling and vesicular trafficking are therefore essential for immune homeostasis.175,176
Haploinsufficiency of CTLA-4 and LOF in LRBA (LPS-responsive beige-like anchor protein), which is required for CTLA-4 vesicular trafficking and prevention of lysosomal degradation, are among the most common monogenic causes of ALPS-like syndromes, accounting for nearly 50% of cases.3,126,127
These conditions clinically develop into severe lymphoproliferation typically manifesting as splenomegaly and lymphadenopathy. Autoimmune manifestations are also observed, sometimes mimicking Evans syndrome with autoimmune hemolytic anemia (AIHA) and immune thrombocytopenic purpura (ITP), as well as multi-organ autoimmunity and autoinflammation including enteropathy, encephalopathy, type 1 diabetes and GLILD. Moreover, both conditions often present with immunodeficiency, marked by hypogammaglobulinemia and inefficient class-switching of memory B cells leading to increased susceptibility to infections.103,131,132,177,178 Similarly, mutations in NBEAL2, encoding a protein involved in vesicular trafficking, have been shown to reduce CTLA-4 expression in activated conventional T cells, further underscoring the central role of this pathway in immune checkpoint regulation.73
The wide spectrum of immune dysregulation diseases in IEIs also includes other genes related to immune checkpoints, specifically PD-1 (Programmed cell Death-1) and PD-L1 (Programmed cell Death-Ligand 1). In particular, PD-1 deficiency (PDCD1), which follows an autosomal recessive inheritance pattern, is associated with severe IEIs phenotypes, including the expansion of CD4−CD8− double-negative (DN) ɑβ cells, tuberculosis and severe autoimmunity (such as type 1 diabetes and fatal pulmonary autoimmunity).1,136 On the other hand, PD-L1 (CD274) hereditary deficiency, shows to be clinically and immunologically less severe than PD-1 deficiency.1,137
Interestingly, the therapeutic blockade of immune checkpoints, as in the case of CTLA-4, PD-1 and PD-L1 in cancer immunotherapy, can phenocopy several features of the reported corresponding genetic checkpoint defects. This leads to the development of immune-related adverse events (irAEs) which mirror monogenic immune dysregulation syndromes, as in IEIs. This sheds light on the molecular mechanisms of immune homeostasis balance shared in both inherited and pharmacologically induced checkpoint failure.179–184
Genetic Defects Predisposing to EBV-Driven Lymphoproliferation
Susceptibility to Epstein-Barr Virus (EBV) infection and the resulting lymphoproliferation can arise from genetic defects which impair cytotoxic lymphocyte function.43,100,138,139 Genes such as MAGT1, PRKCD, XIAP, SH2D1A, RASGRP1, TNFRSF9, ITK and STK4 are critical for the proliferation, survival and cytotoxic activity of CD8+T cells, NK cells and NKT cells.1 Mutations in these genes compromise the ability of cytotoxic lymphocytes to recognize and eliminate EBV-infected cells, leading to uncontrolled viral expansion and chronic immune activation. Consequently, affected individuals often present with EBV-driven lymphoproliferation, which may progress to severe immune dysregulation or malignancy.1
Genomic Instability and Impaired Immune Surveillance in IEIs
Lymphoproliferative disorders in patients with IEIs can arise not only from dysregulated signaling pathways, but also from intrinsic genomic instability and impaired immune surveillance.12,39 Defects in DNA repair mechanisms and genome maintenance significantly increase the risk of malignancy.35,185 For instance, patients with Nijmegen Breakage Syndrome (NBS) or Ataxia-Telangiectasia (AT) exhibit chromosomal instability, combined B and T cell immunodeficiency, and hypersensitivity to ionizing radiation, predisposing to early-onset lymphoid neoplasms.186,187 Similarly, Bloom Syndrome (BLM) and homozygous mutations in PMS2, which impair mismatch repair, are associated with increased incidence of lymphomas, leukemias, and other tumors.188
Compromised immune surveillance further reinforces this risk. IEIs patients display a major susceptibility to malignancies, particularly lymphoid neoplasms, which account for over 60% of cancers observed in this population.35,107 Multiple mechanisms underlie this predisposition, including defective tumor immunosurveillance, uncontrolled lymphocyte proliferation and impaired apoptosis.1
Severe Combined Immunodeficiency (SCID) exemplifies the impact of lost T- and NK cell-mediated surveillance, which triggers unchecked EBV-driven B-cell expansion that can progress into lymphoma.35,43 More broadly, immune dysregulation in IEIs increases vulnerability to viral infections, including EBV, that can drive lymphoproliferation when cytotoxic responses are insufficient. Defective antiviral effector functions (impaired proliferation, co-stimulation and pathogen clearance) underlie EBV-associated lymphoproliferative syndromes.138
X-linked lymphoproliferative syndrome type 1 (XLP-1), caused by pathogenic variants in the SH2D1A gene, illustrates this concept, where viral infection triggers hemophagocytic lymphohistiocytosis (HLH) due to underlying molecular defects.189,190 In this biological context, HLH represents a peculiar hyperinflammatory syndrome arising from uncontrolled activation of cytotoxic T cells and macrophages. In IEIs, HLH is typically triggered in case of EBV infection due to defective cytotoxic lymphocyte function (as in XLP-1) or perforin pathway defects (PRF1, UNC13D, STXBP2, STX11).191–196 This setting results in cytokine storms and persistent immune activation leading to tissue damage, cytopenias, and progressive lymphoproliferation, as a bridge linking impaired immune surveillance and malignancy.197
Finally, recent studies reveal that somatic mutations are significantly enriched in IEIs-associated diffuse large B cell lymphomas (DLBCL) compared to sporadic DLBCL, involving genes such as BRCA2, NCOR1, KLF2, FAS, CCND3, and BRWD3.188 These findings indicate that both genomic instability and impaired immune surveillance converge to promote lymphoproliferation and malignancy in IEI.
The Diagnosis of IEIs in the Setting of Lymphoproliferation
Lymphoproliferation is a common feature of many conditions, including infections, hematological disorders, and rheumatological diseases. Since lymphoproliferation may itself represent the earliest sign of an underlying immune defect, a thorough evaluation aimed at identifying key warning signs of IEIs is essential. These include early age at onset, positive family history, consanguinity, failure to thrive, syndromic features, autoimmunity, autoinflammation and recurrent infections.18 A careful patient assessment is then required, ranging from comprehensive clinical evaluation to detailed lymphocyte subset analysis and genetic testing.
Recently, a step-by-step diagnostic workup has been proposed for pediatric patients with symptoms of autoimmunity and lymphoproliferation that should raise the suspicion for ALPID (Figure 3).73
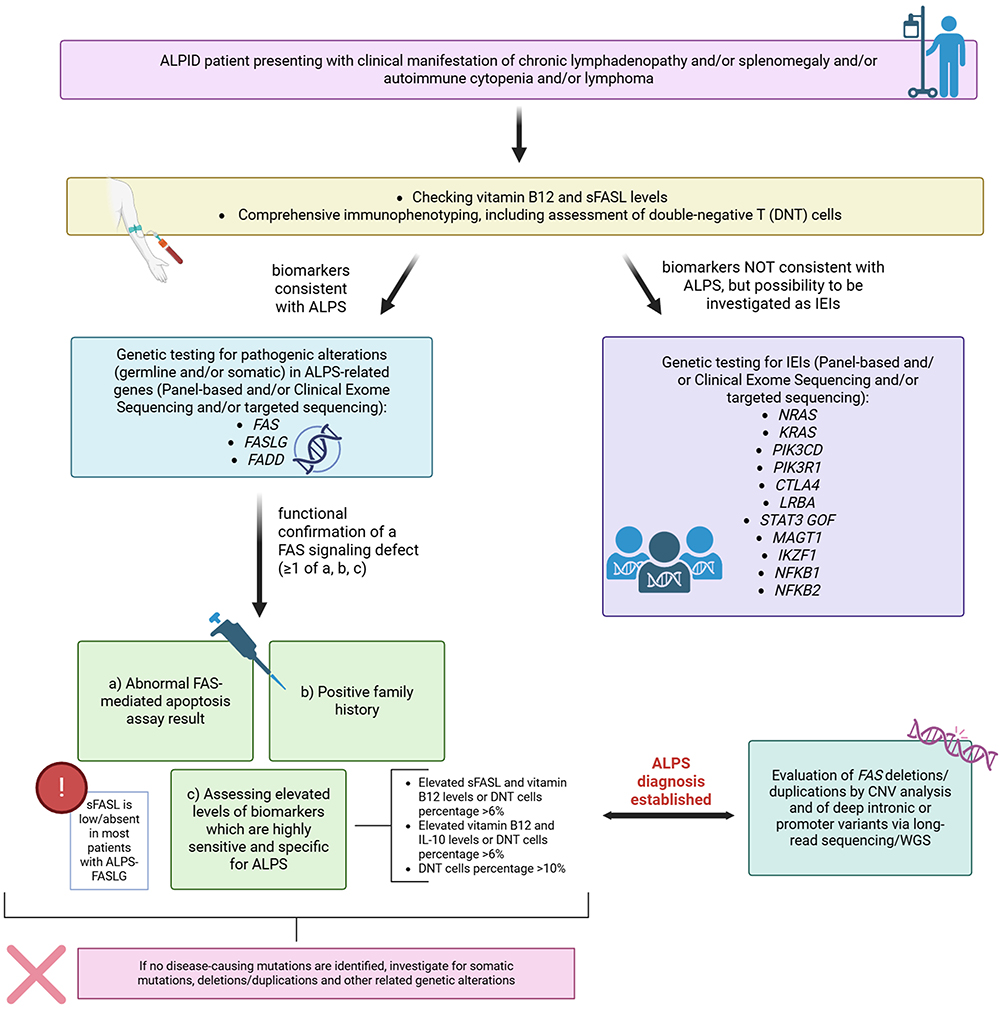 |
Figure 3 Proposed diagnostic workflow for the evaluation of genetically defined ALPID patients with suspected lymphoproliferation. Figure reporting a stepwise guided approach for assessing patients with chronic lymphadenopathy, splenomegaly, autoimmune cytopenias or lymphomas. The initial stages include the testing of vitamin B12 and sFASL levels, immunophenotyping with DNT cell assessment and biomarker evaluation. Based on the patient’s biomarker profile, targeted genetic testing for ALPS-related genes (FAS, FASLG, FADD) or broader IEIs paneling is performed. The further confirmation of the presence of a FAS signaling defect supports ALPS diagnosis, followed by CNV analysis or long-read sequencing/WGS when necessary. Adapted from Magerus et al.73 Created with BioRender.com. |
The proposed “warning symptoms” included: (1) lymphoproliferation and autoimmune cytopenia and/or lymphoma; (2) lymphoproliferation and at least one additional sign of an IEI (namely: increased susceptibility to infections, autoimmunity other than cytopenia, malignancy, syndromic manifestations, consanguinity, family history, and laboratory findings suggesting an IEI); (3) bilineage autoimmune cytopenia; (4) autoimmune cytopenia and/or lymphoma and at least one additional sign of an IEI. These criteria were proposed for pediatric patients, who have a higher likelihood of receiving a genetic diagnosis, while this likelihood appears to be much lower in adults with a similar clinical phenotype. Nonetheless, the warning signs remain generally applicable for defining an ALPID phenotype in adult patients as well.
After warning signs recognition and clinical evaluation, the subsequent biomarker and genetic assessment should aim to confirm the suspicion of ALPID, ruling out non-IEI diagnosis, and to further define it as ALPS or autosomal-dominant ALPID (AD-ALPID), or other IEIs with lymphoproliferation. Among ALPID, ALPS is unique since it is associated with characteristic biomarker profile and immunophenotype. The initial evaluation should thus include the measurement of vitamin B12 (proposed cut-off > 1500 ng/L) and sFASL levels (>200 pg/mL), CD3+TCRαβ+CD4−CD8−double negative (DN) T cells (>6%) and IL-10 (>20 pg/mL). These alterations reflect impaired FAS signaling, being therefore recapitulated in all genetic constellations of ALPS with the only exception of the absence of sFASL in case of FASLG mutations.44
If biomarker profile and immunophenotype are consistent with ALPS the next step is to perform genetic testing for IEIs (panel based and/or clinical exome sequencing and/or targeted sequencing) and for germline variants in ALPS-causative genes (FAS, FASLG or FADD). If no germline variants are found while biomarker profile and immunophenotype are consistent with ALPS, genetic testing for somatic FAS mutations by deep next-generation sequencing (NGS), droplet PCR or using sorted DNT cells is recommended. If no somatic variants are found, genetic testing for FAS deletion or duplication using CNV analysis and for deep intronic or promoter variants using long-read sequencing/whole genome sequencing (WGS) is recommended.
Characteristic lymph node pathologic findings in ALPS include paracortical expansion due to infiltration by polyclonal TCRαβ+-DNT cells accompanied by follicular hyperplasia and polyclonal plasmacytosis.198 The marked TCRαβ+-DNT cell infiltration may result in disruption of lymph nodes architecture and possibly extend to bone marrow and spleen.76 In contrast to lymph node pathologic findings, bone marrow biopsies show a more heterogeneous picture. Although lymphocytosis is a predominant feature, patterns of lymphoid cell infiltration may vary, with formation of aggregates comprising T cells or B cells alone or a mixture of T and B cells. Of note DNT cells may only be detected by immunohistochemistry in the minority of cases.199 If biomarker profile and immunophenotype are not consistent with ALPS but there is a clinical decision to rule out IEIs, the next step is to perform a broader genetic testing for IEIs (panel based and/or clinical exome sequencing and/or targeted sequencing) in order to identify AD-ALPIDs. If no disease-causing mutations is identified, further investigation for somatic mutations, deletions/duplications, etc., is recommended to establish the diagnosis.
With the exception of ALPS, to date there are no well-recognized biomarkers for ALPID disease.73
APDS patients frequently present increased CD57+CD8+ T cell levels together with elevated transitional B cells and IgM levels,73 while in STAT3 GOF disease the increase of CD57+CD8+ T cells levels is more frequently accompanied by increased numbers of DNT cells, and elevated CD21low B cells and reduced Treg cell levels are more common in CTLA-4 deficiency.73 However, the specificity of these markers is too low to guide the diagnostic work-up. Thus, in patients with non-ALPS ALPID, large-scale molecular genetic efforts, including trio whole exome sequencing or whole exome sequencing as preferred methods, are needed to establish a diagnosis (Figure 3).
Despite a thorough laboratory and genetic workup, a number of patients may still not reach a definite classification, and the classification of ALPID-U (“unresolved despite genetic analysis”) has been suggested by Hagele et al.18
Notably, and particularly in adult patients, the ALPID-U category might also include CVID patients with lymphoproliferative phenotype and without a specific genetic finding. Thus, at this point of the process, CVID diagnostic criteria should also be applied for classification purposes in patients with hypogammaglobulinemia. For ALPID-U patients who do not fulfill the CVID criteria, particularly if pediatric, an immunological follow-up should be recommended.
Treatment Strategies
The treatment of IEIs with lymphoproliferation requires a personalized approach that carefully balances the need to control immune dysregulation with the increased risk of infections associated with both the underlying disease and the use of immunosuppressants. In this context, clinical management should integrate infection-preventive measures alongside immunosuppressive regimens. These include immunoglobulin replacement therapy (IgRT), antibiotic prophylaxis and vaccination.200–206
Focusing on immunosuppression, recent advances in the characterization of pathogenic immune pathways have driven a transition from broad-spectrum immunosuppressive agents, such as corticosteroids and DMARDs, to more selective, pathway-specific targeted therapies.
The use of sirolimus (rapamycin), an inhibitor of the mammalian target of rapamycin (mTOR), has revolutionized the management of ALPS and is now considered a key steroid-sparing agent for patients requiring chronic therapy, owing to a better efficacy profile in comparison with mycophenolate mofetil. Indeed, by inhibiting mTOR, sirolimus reduces non-malignant lymphoproliferation, the inappropriate survival of autoreactive B cells and autoimmune cytopenia, effects that are mirrored by a reduction in DNT cells and ALPS biomarkers. Periodic monitoring of 24-hour trough levels of rapamycin is essential to individualize dosing, with the target nadir trough concentration typically maintained between 7.5 and 15 ng/mL.84–86,207
Given that APDS is similarly driven by hyperactivation of mTOR signaling downstream of PI3Kδ, sirolimus has also demonstrated good efficacy in controlling lymphoproliferation in this disorder. However, non-lymphoproliferative complications such as cytopenias and enteropathy are generally less responsive to mTOR inhibition.87
Recent advances have emerged in the treatment of PI3K-related disorders with the discovery of leniolisib, a selective PI3Kδ inhibitor. Indeed, in a randomized, placebo-controlled Phase III trial, leniolisib was well tolerated and showed efficacy in reducing lymphadenopathy and restoring physiological B cell distribution by increasing the proportion of naïve B cells.88 Although it was a secondary outcome, cytopenias also showed a trend toward improvement. These results were consistent in both adolescents and adults.89 Furthermore, an interim analysis of the open-label extension study demonstrated sustained clinical benefits, with durable outcomes maintained for up to five years of follow-up.90
Moving to IEIs related to the dysregulation of JAK/STAT signaling pathway, a recent ESID/EBMT-IEWP retrospective study showed that the treatment with JAK inhibitors, resulted in a partial or complete remission of symptoms in 87% of STAT1 GOF and in 90% of STAT3 GOF patients. Notably, approximately 50% of STAT3 GOF patients presented with lymphadenopathy and/or splenomegaly, with clinical improvement observed in over 80% of these cases.92
Currently, no targeted therapies are available for the treatment of lymphoproliferation in patients with NF‑κB pathway dysregulation. This is likely because lymphoproliferation in these disorders may result from either increased or decreased NF‑κB activity, leading to highly variable and context-dependent mechanisms that hinder the identification of effective therapeutic strategies. Theoretically, anti-TNF drugs could represent a potential therapeutic strategy in cases of NF-κB hyperactivation by inhibiting the TNF-mediated NF-κB activation; however, its role remains to be explored.101
Given the role of B lymphocytes as key drivers of immune dysregulation in patients with CVID, rituximab is emerging as a potentially safe and effective therapeutic option to manage immune dysregulation manifestations, such as GLILD, including those with NF‑κB pathway defects.101
This strategy is further supported in other CVID patients, also with TACI mutations, where it has been shown that BAFF-driven B cell hyperplasia is correlated with the progression of ILD.109
In line with this, recent retrospective studies suggest that rituximab, alone or in combination therapy, may be effective for managing GLILD in CVID patients, even in those without a definitive genetic diagnosis. However, data on the durability of this response remain limited.110,111,131
Belimumab, an anti-BAFF monoclonal antibody, represents a promising therapeutic alternative due to the mentioned role of BAFF; however, its potential has yet to be explored.
In patients with DADA2, anti-TNF therapy has proven highly effective in treating inflammatory manifestations driven by cytokine overproduction, mediated through interferon and NF-κB signaling pathways. However, its effectiveness in managing hematological features appears limited.115–117
In the future, restoring wild-type ADA2 production through gene therapy and infusing recombinant ADA2 via enzyme replacement therapy are promising treatment options.118
Indeed, a recent preclinical study demonstrates that lentiviral ADA2 gene therapy restores ADA2 enzyme activity and cellular function in patient-derived hematopoietic stem cells, showing efficient, multilineage engraftment and a favorable safety profile in humanized NBSGW mice.208
Another important targeted therapeutic approach is the use of abatacept, a fusion protein that mimics endogenous CTLA-4 function by inhibiting CD28-mediated T cell co-stimulation. In patients with CTLA-4 or LRBA insufficiency, abatacept has demonstrated superior efficacy compared to traditional immunosuppressive treatment strategies in managing disease manifestations over time, especially when treatment is initiated early, compared to traditional immunosuppressants. Of note, also patients with GLILD experienced improvement of clinical symptoms, lung function and radiologic findings.127–129
Indeed, from an immunological perspective, treatment with abatacept has been shown to increase thymic output and promote the expansion of naïve T and B cells. Additionally, it reduces memory T cell subsets, decreases CD4+ T cell cytokine production, and lowers the levels of autoreactive B cells. Recent multi-modal transcriptomic and proteomic analyses have also demonstrated that abatacept reverses the abnormal upregulation of inflammatory mediators, including CHI3L1, CXCL13, and CSF1, in these patients.209
Ongoing studies are further characterizing the effects of CTLA-4–Ig therapy in CTLA-4 insufficiency, and the ABACHAI trial is expected to clarify its efficacy and durability of response.210
The use of targeted therapies for patients with lymphoproliferation related to IEIs without a specific genetic diagnosis can be challenging. One possible approach is to analyse the overall phenotype, focusing on the connection between unique manifestations and the underlying immunological background, such as lymphocyte subsets. This can provide insights into potential alterations in a specific immune pathway. Indeed, in CVID patients with autoimmunity, reduced expression of CTLA-4 and LRBA, has been observed, even without identifiable genetic mutations, that can be targeted by abatacept.211
Another example is the effectiveness of sirolimus in treating refractory autoimmune cytopenias in patients without a confirmed genetic defect in ALPS related genes.212
Despite the specificity and efficacy of these targeted therapies, none of the aforementioned agents offer a definitive cure, and long-term follow-up data remain limited. Therefore, Hematopoietic Stem Cell Transplantation (HSCT) should be considered as a potential curative option in a subset of patients, based on a thorough risk–benefit evaluation.121,122,125,130,133–135,140,213
Given the considerable procedure-related morbidity, HSCT should be reserved for patients in whom adequate disease control cannot be achieved with conventional immunosuppressive therapy. Conversely, early transplantation, performed prior to the onset of irreversible organ damage, is essential to maximize the likelihood of a favorable long-term outcome.214,215
While HSCT is generally not recommended for patients with ALPS, and only a few cases have been reported in the literature,216,217 more evidence is available for APDS, in which HSCT should be considered for patients presenting with severe clinical manifestations that show no or inadequate response to conventional therapies.218,219 Specifically, Dimitrova et al retrospectively analyzed 57 patients with APDS 1/2 treated with HSCT, reporting a two-year overall survival rate of 86%.220
In this context, the potential role of sirolimus or leniolisib as a bridge therapy to transplantation has yet to be elucidated. Indeed, the use of targeted therapies as a pre-HSCT bridge to mitigate immune dysregulation has shown promising results in other settings. For example, in IEIs affecting the JAK/STAT signaling pathway, conditions in which HSCT has been associated with a high incidence of graft rejection and poor survival, with a reported overall survival of only 40% in patients with STAT1 GOF,221 pre-transplant use of JAK inhibitors has been shown to improve outcomes, with survival rates reaching up to 90%.92
Moreover, in a recent multicenter retrospective study pre-treatment with JAK inhibitors was associated with a better event free survival.222
To date, only a few cases of CVID patients with associated lymphoproliferation, such as GLILD, treated with HSCT have been reported in the literature, generally showing poor outcomes.131
This is likely due to the high burden of comorbidities and organ damage in those selected for HSCT, particularly lung involvement (e.g., GLILD and bronchiectasis) and hepatopathy. Supporting this, a recent multicenter IEWP study focusing on adolescent and adult patients with IEIs who underwent HSCT found that overall survival and event-free survival were lower in patients with primary antibody deficiencies. Specifically, overall survival in this group was 59%, compared to 68% for combined immunodeficiencies and 78% for phagocyte disorders.223
Moving to CTLA-4 or LRBA insufficiency, HSCT has been reported to be a potential curative treatment that prevents disease progression and mortality.133,224
In the largest published cohort to date, including 40 patients with CTLA-4 insufficiency, the 3-year overall survival was 76.7%, and the disease-free survival was 74.4%. As observed in other IEIs, uncontrolled disease activity prior to transplantation, objectively assessed using the Immune Dysregulation Disease Activity (IDDA) score, was associated with poorer overall and disease-related survival.224
Notably, the IDDA score takes into account the presence of autoimmune cytopenia, lymphoproliferation, and hepatosplenomegaly.225 Unexpectedly, pre-transplant use of abatacept was not directly associated with improved survival outcomes. This apparent lack of effect may be due to a small sample size, the limitations of CTLA-4–Ig in reversing existing organ damage and patient selection.224
Finally, in patients with XLP1 and XLP2, disorders in which target therapies are not available, HSCT represents the only curative option, and should be undertaken in all patients with HLH.141,142
Conclusions
Lymphoproliferation represents a “transversal” feature across IEIs, arising from distinct, sometimes undefined, genetic and functional backgrounds that can result in overlapping clinical features. This requires a deep integration of genetic, immunologic and clinical evaluations, since these apparently similar phenotypes often require different therapeutic strategies, ideally tailored to the underlying molecular and immunological mechanisms rather than to the clinical presentation alone. Recognizing the clinical phenotypes and deciphering the functional immunological background of disease, particularly in genetically undefined cases, is crucial to enable more precise and individualized therapeutic approaches.
Acknowledgments
The authors acknowledge ERN RITA Network: Ca’ Foncello Hospital, Erasmus University Medical Center and Azienda Ospedaliero Universitaria Policlinico Umberto I represent ERN RITA reference centers for IEIs.
Giulia Gualtiero, Helena Buso and Francesco Cinetto gratefully acknowledge the support of FoRiBiCa ETS. Francesco Cinetto and Virgil A.S.H. Dalm equally contributed to this work as last authors.
Disclosure
Francesco Cinetto reports personal fees from GSK, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Poli MC, Aksentijevich I, Bousfiha AA, et al. Human inborn errors of immunity: 2024 update on the classification from the international union of immunological societies expert committee. J Hum Immun. 2025;1(1):e20250003. doi:10.70962/jhi.20250003
2. Notarangelo LD, Bacchetta R, Casanova JL, Su HC. Human inborn errors of immunity: an expanding universe. Sci Immunol. 2020;5(49):eabb1662. doi:10.1126/sciimmunol.abb1662
3. Delmonte OM, Castagnoli R, Calzoni E, Notarangelo LD. Inborn errors of immunity with immune dysregulation: from bench to bedside. Front Pediatr. 2019;7. doi:10.3389/fped.2019.00353
4. Wang JJF, Dhir A, Hildebrand KJ, et al. Inborn errors of immunity in adulthood. Allergy Asthma Clin Immunol off. 2024;20(1):6. doi:10.1186/s13223-023-00862-8
5. Akalu YT, Bogunovic D. Inborn errors of immunity: an expanding universe of disease and genetic architecture. Nat Rev Genet. 2024;25(3):184–30. doi:10.1038/s41576-023-00656-z
6. Karabiber E, Baris S. Delineating the clinical and immunologic characteristics: a comparative study of inborn errors of immunity in adult versus pediatric diagnosed. Int Arch Allergy Immunol. 2024;185(11):1123–1135. doi:10.1159/000540538
7. Slade CA, Bosco JJ, Binh Giang T, et al. Delayed diagnosis and complications of predominantly antibody deficiencies in a cohort of australian adults. Front Immunol. 2018;9:694. doi:10.3389/fimmu.2018.00694
8. IJspeert H, Edwards ESJ, O’Hehir RE, Dalm VASH, van Zelm MC. Update on inborn errors of immunity. J Allergy Clin Immunol. 2025;155(3):740–751. doi:10.1016/j.jaci.2024.12.1075
9. Bucciol G, Delafontaine S, Meyts I, Poli C. Inborn errors of immunity: a field without frontiers. Immunol Rev. 2024;322(1):15–27. doi:10.1111/imr.13297
10. van Krieken JH, van Krieken JH. Lymphoproliferative disease associated with immune deficiency in children. Am J Clin Pathol. 2004;122(Suppl):S122–127. doi:10.1309/MEVXGV99DTTRGAE4
11. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the world health organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. doi:10.1182/blood-2016-01-643569
12. Toskov V, Ehl S. Autoimmune lymphoproliferative immunodeficiencies (ALPID) in childhood: breakdown of immune homeostasis and immune dysregulation. Mol Cell Pediatr. 2023;10(1):11. doi:10.1186/s40348-023-00167-1
13. López-Nevado M, González-Granado LI, Ruiz-García R, et al. Primary immune regulatory disorders with an autoimmune lymphoproliferative syndrome-like phenotype: immunologic evaluation, early diagnosis and management. Front Immunol. 2021;12:671755. doi:10.3389/fimmu.2021.671755
14. Chan AY, Torgerson TR. Primary immune regulatory disorders: a growing universe of immune dysregulation. Curr Opin Allergy Clin Immunol. 2020;20(6):582–590. doi:10.1097/ACI.0000000000000689
15. Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood. 2009;113(26):6511–6521. doi:10.1182/blood-2009-01-129155
16. Rieux-Laucat F, Le Deist F, Hivroz C, et al. Mutations in fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268(5215):1347–1349. doi:10.1126/science.7539157
17. Fisher GH, Rosenberg FJ, Straus SE, et al. Dominant interfering fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81(6):935–946. doi:10.1016/0092-8674(95)90013-6
18. Hägele P, Staus P, Scheible R, et al. Diagnostic evaluation of paediatric autoimmune lymphoproliferative immunodeficiencies (ALPID): a prospective cohort study. Lancet Haematol. 2024;11(2):e114–e126. doi:10.1016/S2352-3026(23)00362-9
19. Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–286. doi:10.1182/blood-2007-11-124545
20. Gathmann B, Mahlaoui N, Gérard L, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014;134(1):116–126. doi:10.1016/j.jaci.2013.12.1077
21. Mayor PC, Eng KH, Singel KL, et al. Cancer in primary immunodeficiency diseases: cancer incidence in the United States immune deficiency network registry. J Allergy Clin Immunol. 2018;141(3):1028–1035. doi:10.1016/j.jaci.2017.05.024
22. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119(7):1650–1657. doi:10.1182/blood-2011-09-377945
23. Kralickova P, Milota T, Litzman J, et al. CVID-associated tumors: czech nationwide study focused on epidemiology, immunology, and genetic background in a cohort of patients with CVID. Front Immunol. 2018;9:3135. doi:10.3389/fimmu.2018.03135
24. Pulvirenti F, Pecoraro A, Cinetto F, et al. Gastric cancer is the leading cause of death in italian adult patients with common variable immunodeficiency. Front Immunol. 2018;9:2546. doi:10.3389/fimmu.2018.02546
25. Kiaee F, Azizi G, Rafiemanesh H, et al. Malignancy in common variable immunodeficiency: a systematic review and meta-analysis. Expert Rev Clin Immunol. 2019;15(10):1105–1113. doi:10.1080/1744666X.2019.1658523
26. Jolles S. The variable in common variable immunodeficiency: a disease of complex phenotypes. J Allergy Clin Immunol Pract. 2013;1(6):545–556;quiz557. doi:10.1016/j.jaip.2013.09.015
27. Riaz IB, Faridi W, Patnaik MM, Abraham RS. A systematic review on predisposition to lymphoid (B and T cell) neoplasias in patients with primary immunodeficiencies and immune dysregulatory disorders (Inborn Errors of Immunity). Front Immunol. 2019;10:777. doi:10.3389/fimmu.2019.00777
28. Mellemkjaer L, Hammarstrom L, Andersen V, et al. Cancer risk among patients with IgA deficiency or common variable immunodeficiency and their relatives: a combined Danish and Swedish study. Clin Exp Immunol. 2002;130(3):495–500. doi:10.1046/j.1365-2249.2002.02004.x
29. Xu G, Shi Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res. 2007;17(9):759–771. doi:10.1038/cr.2007.52
30. Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272(5258):60–66. doi:10.1126/science.272.5258.60
31. Scheiermann C, Frenette PS, Hidalgo A. Regulation of leucocyte homeostasis in the circulation. Cardiovasc Res. 2015;107(3):340–351. doi:10.1093/cvr/cvv099
32. Yanes RE, Gustafson CE, Weyand CM, Goronzy JJ. Lymphocyte generation and population homeostasis throughout life. Semin Hematol. 2017;54(1):33–38. doi:10.1053/j.seminhematol.2016.10.003
33. Grossman Z, Paul WE. Dynamic tuning of lymphocytes: physiological basis, mechanisms, and function. Annu Rev Immunol. 2015;33:677–713. doi:10.1146/annurev-immunol-032712-100027
34. Makkoukdji N, Satnarine T, De Almeida AX, et al. Inborn Errors of Immunity in Apoptosis. Front Biosci-Landmark. 2025;30(5):27231. doi:10.31083/FBL27231
35. Sharma S, Pilania RK, Anjani G, et al. Lymphoproliferation in inborn errors of immunity: the eye does not see what the mind does not Know. Front Immunol. 2022;13:856601. doi:10.3389/fimmu.2022.856601
36. Heinzel S, Marchingo JM, Horton MB, Hodgkin PD. The regulation of lymphocyte activation and proliferation. Curr Opin Immunol. 2018;51:32–38. doi:10.1016/j.coi.2018.01.002
37. Cantrell D. Signaling in lymphocyte activation. Cold Spring Harb Perspect Biol. 2015;7(6):a018788. doi:10.1101/cshperspect.a018788
38. Janeway CA, Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994;76(2):275–285. doi:10.1016/0092-8674(94)90335-2
39. Costagliola G, De Marco E, Consonni F, et al. Inborn errors of immunity presenting with lymphoproliferation: lessons from a case series. Ann Hematol. 2025;104(6):3117–3127. doi:10.1007/s00277-025-06456-1
40. Costagliola G, De Marco E, Massei F, et al. The etiologic landscape of lymphoproliferation in childhood: proposal for a diagnostic approach exploring from infections to inborn errors of immunity and metabolic diseases. Ther Clin Risk Manag. 2024;20:261–274. doi:10.2147/TCRM.S462996
41. Sogkas G, Atschekzei F, Adriawan IR, Dubrowinskaja N, Witte T, Schmidt RE. Cellular and molecular mechanisms breaking immune tolerance in inborn errors of immunity. Cell Mol Immunol. 2021;18(5):1122–1140. doi:10.1038/s41423-020-00626-z
42. Ren A, Yin W, Miller H, et al. Novel discoveries in immune dysregulation in inborn errors of immunity. Front Immunol. 2021;12:725587. doi:10.3389/fimmu.2021.725587
43. Bousfiha AA, Jeddane L, Moundir A, et al. The 2024 update of IUIS phenotypic classification of human inborn errors of immunity. J Hum Immun. 2025;1(1):e20250002. doi:10.70962/jhi.20250002
44. Maccari ME, Schneider P, Smulski CR, et al. Revisiting autoimmune lymphoproliferative syndrome caused by Fas ligand mutations. J Allergy Clin Immunol. 2023;151(5):1391–1401.e7. doi:10.1016/j.jaci.2022.11.028
45. Consonni F, Gambineri E, ALPS FC. FAS, and beyond: from inborn errors of immunity to acquired immunodeficiencies. Ann Hematol. 2022;101(3):469–484. doi:10.1007/s00277-022-04761-7
46. Rao VK, Pittaluga S, Uzel G. Beyond FAScinating: advances in diagnosis and management of autoimmune lymphoproliferative syndrome and activated PI3 kinase δ syndrome. Hematology. 2024;2024(1):126–136. doi:10.1182/hematology.2024000537
47. Hu L, Lu J, Fan H, et al. FAS mediates apoptosis, inflammation, and treatment of pathogen infection. Front Cell Infect Microbiol. 2025:15. doi:10.3389/fcimb.2025.1561102
48. Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30(2):180–192. doi:10.1016/j.immuni.2009.01.001
49. Matson DR, Yang DT. Autoimmune Lymphoproliferative Syndrome: an Overview. Arch Pathol Lab Med. 2020;144(2):245–251. doi:10.5858/arpa.2018-0190-RS
50. Waring P, Müllbacher A. Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunol Cell Biol. 1999;77(4):312–317. doi:10.1046/j.1440-1711.1999.00837.x
51. Arakaki R, Yamada A, Kudo Y, Hayashi Y, Ishimaru N. Mechanism of activation-induced cell death of T cells and regulation of FasL expression. Crit Rev Immunol. 2014;34(4):301–314. doi:10.1615/critrevimmunol.2014009988
52. Nagata S, Golstein P. The fas death factor. Science. 1995;267(5203):1449–1456. doi:10.1126/science.7533326
53. Park DR, Thomsen AR, Frevert CW, et al. Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J Immunol (Baltimore, MD). 2003;170(12):6209–6216. doi:10.4049/jimmunol.170.12.6209
54. Itoh N, Yonehara S, Ishii A, et al. The polypeptide encoded by the cDNA for human cell surface antigen fas can mediate apoptosis. Cell. 1991;66(2):233–243. doi:10.1016/0092-8674(91)90614-5
55. Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75(6):1169–1178. doi:10.1016/0092-8674(93)90326-l
56. Malyshkina A, Littwitz-Salomon E, Sutter K, et al. Fas ligand-mediated cytotoxicity of CD4+ T cells during chronic retrovirus infection. Sci Rep. 2017;7(1):7785. doi:10.1038/s41598-017-08578-7
57. Tourneur L, Chiocchia G. FADD: a regulator of life and death. Trends Immunol. 2010;31(7):260–269. doi:10.1016/j.it.2010.05.005
58. Micheau O, Solary E, Hammann A, Dimanche-Boitrel MT. Fas ligand-independent, FADD-mediated activation of the fas death pathway by anticancer drugs. J Biol Chem. 1999;274(12):7987–7992. doi:10.1074/jbc.274.12.7987
59. Wang L, Yang JK, Kabaleeswaran V, et al. The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat Struct Mol Biol. 2010;17(11):1324–1329. doi:10.1038/nsmb.1920
60. Suzuki E, Takashina T, Nakayama M. Engineered FADD induces apoptosis via an artificial Death-Inducing Signaling Complex (DISC). Int J Biomed Sci IJBS. 2009;5(3):237–245.
61. Davis AR, Lotocki G, Marcillo AE, Dietrich WD, FasL KRW. Fas, and death-inducing signaling complex (DISC) proteins are recruited to membrane rafts after spinal cord injury. J Neurotrauma. 2007;24(5):823–834. doi:10.1089/neu.2006.0227
62. Lee EW, Kim JH, Ahn YH, et al. Ubiquitination and degradation of the FADD adaptor protein regulate death receptor-mediated apoptosis and necroptosis. Nat Commun. 2012;3:978. doi:10.1038/ncomms1981
63. Lavrik IN, Krammer PH. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 2012;19(1):36–41. doi:10.1038/cdd.2011.155
64. Sahoo G, Samal D, Khandayataray P, Murthy MK. A review on caspases: key regulators of biological activities and apoptosis. Mol Neurobiol. 2023;60(10):5805–5837. doi:10.1007/s12035-023-03433-5
65. Ju ST, Matsui K, Ozdemirli M. Molecular and cellular mechanisms regulating T and B cell apoptosis through Fas/FasL interaction. Int Rev Immunol. 1999;18(5–6):485–513. doi:10.3109/08830189909088495
66. Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in fas antigen that mediates apoptosis. Nature. 1992;356(6367):314–317. doi:10.1038/356314a0
67. Sneller MC, Wang J, Dale JK, et al. Clinical, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood. 1997;89(4):1341–1348.
68. Magerus A, Bercher-Brayer C, Rieux-Laucat F. The genetic landscape of the FAS pathway deficiencies. Biomed J. 2021;44(4):388–399. doi:10.1016/j.bj.2021.06.005
69. Holzelova E, Vonarbourg C, Stolzenberg MC, et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med. 2004;351(14):1409–1418. doi:10.1056/NEJMoa040036
70. Ben-Mustapha I, Agrebi N, Barbouche MR. Novel insights into FAS defects underlying autoimmune lymphoproliferative syndrome revealed by studies in consanguineous patients. J Leukoc Biol. 2018;103(3):501–508. doi:10.1002/JLB.5MR0817-332R
71. Rodríguez-Bayona B, Lucena-Soto JM, Croché-Santander B, Olbrich P, González-Escribano MF, Neth O. Autoimmune lymphoproliferative syndrome (Alps) due to a novel dominant negative germline mutation in the FAS gene. Immunol Res. 2024;72(1):162–166. doi:10.1007/s12026-023-09411-2
72. Rieux-Laucat F, Magérus-Chatinet A, Neven B. The autoimmune lymphoproliferative syndrome with defective FAS or FAS-ligand functions. J Clin Immunol. 2018;38(5):558–568. doi:10.1007/s10875-018-0523-x
73. Magerus A, Rensing-Ehl A, Rao VK, Teachey D, Rieux-Laucat F, Ehl S. Autoimmune-lymphoproliferative immunodeficiencies (ALPID). J Allergy Clin Immunol. 2024;153(1):67–76. doi:10.1016/j.jaci.2023.11.004
74. Consonni F, Moreno S, Vinuales Colell B, et al. Study of the potential role of CASPASE-10 mutations in the development of autoimmune lymphoproliferative syndrome. Cell Death Dis. 2024;15(5):315. doi:10.1038/s41419-024-06679-6
75. Pellé O, Moreno S, Lorenz MR, et al. Combined germline and somatic human FADD mutations cause autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol. 2024;153(1):203–215. doi:10.1016/j.jaci.2023.09.028
76. Oliveira JB, Bleesing JJ, Dianzani U, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (Alps): report from the 2009 NIH International Workshop. Blood. 2010;116(14):e35–e40. doi:10.1182/blood-2010-04-280347
77. Lambotte O, Neven B, Galicier L, et al. Diagnosis of autoimmune lymphoproliferative syndrome caused by FAS deficiency in adults. Haematologica. 2013;98(3):389–392. doi:10.3324/haematol.2012.067488
78. Deutsch M, Tsopanou E, Dourakis SP. The autoimmune lymphoproliferative syndrome (Canale-Smith) in adulthood. Clin Rheumatol. 2004;23(1):43–44. doi:10.1007/s10067-003-0830-2
79. Poddighe D, Maulenkul T, Dossybayeva K, Zhubanova G, Mukusheva Z, Akhmaltdinova L. Double-negative T cells in pediatric rheumatic diseases. Clin Exp Pediatr. 2024;67(12):632–640. doi:10.3345/cep.2023.01760
80. Poddighe D, Dossybayeva K, Kozhakhmetov S, Rozenson R, Assylbekova M. Double-negative T (DNT) cells in patients with systemic lupus erythematosus. Biomedicines. 2024;12(1):166. doi:10.3390/biomedicines12010166
81. Caminha I, Fleisher TA, Hornung RL, et al. Using biomarkers to predict the presence of FAS mutations in patients with features of the autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol. 2010;125(4):946–949.e6. doi:10.1016/j.jaci.2009.12.983
82. Seidel MG, Kindle G, Gathmann B, et al. The European Society for Immunodeficiencies (ESID) registry working definitions for the clinical diagnosis of inborn errors of immunity. J Allergy Clin Immunol Pract. 2019;7(6):1763–1770. doi:10.1016/j.jaip.2019.02.004
83. Xie W, Fan G, Wiszniewska J, Press RD, Yang F. Histopathological and molecular findings in a patient with Ras-associated autoimmune leukoproliferative disorder. Hum Pathol Rep. 2022;29:300670. doi:10.1016/j.hpr.2022.300670
84. Klemann C, Esquivel M, Magerus-Chatinet A, et al. Evolution of disease activity and biomarkers on and off rapamycin in 28 patients with autoimmune lymphoproliferative syndrome. Haematologica. 2017;102(2):e52–e56. doi:10.3324/haematol.2016.153411
85. Bride KL, Vincent T, Smith-Whitley K, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016;127(1):17–28. doi:10.1182/blood-2015-07-657981
86. Völkl S, Rensing-Ehl A, Allgäuer A, et al. Hyperactive mTOR pathway promotes lymphoproliferation and abnormal differentiation in autoimmune lymphoproliferative syndrome. Blood. 2016;128(2):227–238. doi:10.1182/blood-2015-11-685024
87. Maccari ME, Abolhassani H, Aghamohammadi A, et al. Disease evolution and response to rapamycin in activated phosphoinositide 3-kinase δ syndrome: the european society for immunodeficiencies-activated phosphoinositide 3-kinase δ syndrome registry. Front Immunol. 2018:9. doi:10.3389/fimmu.2018.00543
88. Rao VK, Webster S, Šedivá A, et al. A randomized, placebo-controlled Phase 3 trial of the PI3Kδ inhibitor leniolisib for activated PI3Kδ syndrome. Blood. 2023;141(9):971–983. doi:10.1182/blood.2022018546
89. Rao VK, Šedivá A, Dalm VASH, et al. A randomised, placebo-controlled, phase III trial of leniolisib in activated phosphoinositide 3-kinase delta (PI3Kδ) syndrome (APDS): adolescent and adult subgroup analysis. Clin Immunol Orlando Fla. 2025;270:110400. doi:10.1016/j.clim.2024.110400
90. Rao VK, Kulm E, Šedivá A, et al. Interim analysis: open-label extension study of leniolisib for patients with APDS. J Allergy Clin Immunol. 2024;153(1):265–274.e9. doi:10.1016/j.jaci.2023.09.032
91. Chaimowitz NS, Smith MR, Forbes Satter LR. JAK/STAT defects and immune dysregulation, and guiding therapeutic choices. Immunol Rev. 2024;322(1):311–328. doi:10.1111/imr.13312
92. Fischer M, Olbrich P, Hadjadj J, et al. JAK inhibitor treatment for inborn errors of JAK/STAT signaling: an ESID/EBMT-IEWP retrospective study. J Allergy Clin Immunol. 2024;153(1):275–286.e18. doi:10.1016/j.jaci.2023.10.018
93. Flanagan SE, Haapaniemi E, Russell MA, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet. 2014;46(8):812–814. doi:10.1038/ng.3040
94. Vogel TP, Leiding JW, Cooper MA, Forbes Satter LR. STAT3 gain-of-function syndrome. Front Pediatr. 2023;10. doi:10.3389/fped.2022.770077
95. Todaro F, Tamassia N, Pinelli M, et al. Multisystem autoimmune disease caused by increased STAT3 phosphorylation and dysregulated gene expression. Haematologica. 2019;104(7):e322–e325. doi:10.3324/haematol.2018.202374
96. Leiding JW, Vogel TP, Santarlas VGJ, et al. Monogenic early-onset lymphoproliferation and autoimmunity: natural history of STAT3 gain-of-function syndrome. J Allergy Clin Immunol. 2023;151(4):1081–1095. doi:10.1016/j.jaci.2022.09.002
97. Jägle S, Heeg M, Grün S, et al. Distinct molecular response patterns of activating STAT3 mutations associate with penetrance of lymphoproliferation and autoimmunity. Clin Immunol Orlando Fla. 2020;210:108316. doi:10.1016/j.clim.2019.108316
98. Erdős M, Tsumura M, Kállai J, et al. Novel STAT‐3 gain‐of‐function variant with hypogammaglobulinemia and recurrent infection phenotype. Clin Exp Immunol. 2021;205(3):354–362. doi:10.1111/cei.13625
99. Bel KLD, Ragotte RJ, Saferali A, et al. JAK1 gain-of-function causes an autosomal dominant immune dysregulatory and hypereosinophilic syndrome. J Allergy Clin Immunol. 2017;139(6):2016–2020.e5. doi:10.1016/j.jaci.2016.12.957
100. Boztug H, Hirschmugl T, Holter W, et al. NF-κB1 haploinsufficiency causing immunodeficiency and EBV-driven lymphoproliferation. J Clin Immunol. 2016;36:533–540. doi:10.1007/s10875-016-0306-1
101. Lorenzini T, Fliegauf M, Klammer N, et al. Characterization of the clinical and immunologic phenotype and management of 157 individuals with 56 distinct heterozygous NFKB1 mutations. J Allergy Clin Immunol. 2020;146(4):901–911. doi:10.1016/j.jaci.2019.11.051
102. Salzer U, Bacchelli C, Buckridge S, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. 2009;113(9):1967–1976. doi:10.1182/blood-2008-02-141937
103. Cinetto F, Scarpa R, Carrabba M, et al. Granulomatous Lymphocytic Interstitial Lung Disease (GLILD) in Common Variable Immunodeficiency (CVID): a multicenter retrospective study of patients from Italian PID referral centers. Front Immunol. 2021;12:627423. doi:10.3389/fimmu.2021.627423
104. Perlman DM, Sudheendra MT, Racilla E, Allen TL, Joshi A, Bhargava M. Granulomatous-lymphocytic interstitial lung disease mimicking sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2021;38(3):e2021025. doi:10.36141/svdld.v38i3.11114
105. Seshasayee D, Valdez P, Yan M, Dixit VM, Tumas D, Grewal IS. Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity. 2003;18(2):279–288. doi:10.1016/s1074-7613(03)00025-6
106. Castigli E, Wilson S, Garibyan L, et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet. 2007;39(4):430–431. doi:10.1038/ng0407-430
107. Wehr C, Houet L, Unger S, et al. Altered spectrum of lymphoid neoplasms in a single-center cohort of common variable immunodeficiency with immune dysregulation. J Clin Immunol. 2021;41(6):1250–1265. doi:10.1007/s10875-021-01016-4
108. Bonilla FA, Barlan I, Chapel H, et al. International Consensus Document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2016;4(1):38–59. doi:10.1016/j.jaip.2015.07.025
109. Maglione PJ, Gyimesi G, Cols M, et al. BAFF-driven B cell hyperplasia underlies lung disease in common variable immunodeficiency. JCI Insight. 2019;4(5). doi:10.1172/jci.insight.122728
110. Tessarin G, Baronio M, Gazzurelli L, et al. Rituximab monotherapy is effective as first-line treatment for Granulomatous Lymphocytic Interstitial Lung Disease (GLILD) in CVID patients. J Clin Immunol. 2023;43(8):2091–2103. doi:10.1007/s10875-023-01587-4
111. Moratti M, Schifino G, Baccelli F, et al. Granulomatous lymphocytic interstitial lung disease in common variable immune deficiency: an in-depth clinical, immunological, functional and radiological exploration with a focus on its management, challenged by chronic CMV infection. Front Immunol. 2025;16. doi:10.3389/fimmu.2025.1589052
112. Zavialov AV, Engström A. Human ADA2 belongs to a new family of growth factors with adenosine deaminase activity. Biochem J. 2005;391(Pt 1):51–57. doi:10.1042/BJ20050683
113. Sauer AV, Brigida I, Carriglio N, Aiuti A. Autoimmune dysregulation and purine metabolism in adenosine deaminase deficiency. Front Immunol. 2012;3:265. doi:10.3389/fimmu.2012.00265
114. Barzaghi F, Minniti F, Mauro M, et al. Alps-like phenotype caused by ADA2 deficiency rescued by allogeneic hematopoietic stem cell transplantation. Front Immunol. 2018;9:2767. doi:10.3389/fimmu.2018.02767
115. Ayan G, Basaran O, Firlatan B, et al. Long-term follow-up of anti-TNF treatment in adult and pediatric DADA2 patients: insights from real-world data. Int J Rheum Dis. 2024;27(11):e15377. doi:10.1111/1756-185X.15377
116. Kumar S, Chugh A, Kumar SC, et al. Clinical spectrum of and outcomes for Indian children with deficiency of adenosine deaminase 2 (DADA2): a multicentric study. Rheumatol Oxf Engl. 2025;64(4):2179–2184. doi:10.1093/rheumatology/keae489
117. Deuitch NT, Yang D, Lee PY, et al. TNF inhibition in vasculitis management in adenosine deaminase 2 deficiency (DADA2). J Allergy Clin Immunol. 2022;149(5):1812–1816.e6. doi:10.1016/j.jaci.2021.10.030
118. Lee PY, Aksentijevich I, Zhou Q. Mechanisms of vascular inflammation in deficiency of adenosine deaminase 2 (DADA2). Semin Immunopathol. 2022;44(3):269–280. doi:10.1007/s00281-022-00918-8
119. Goudy K, Aydin D, Barzaghi F, et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol Orlando Fla. 2013;146(3):248–261. doi:10.1016/j.clim.2013.01.004
120. Consonni F, Favre C, Gambineri E. IL-2 signaling axis defects: how many faces? Front Pediatr. 2021;9:669298. doi:10.3389/fped.2021.669298
121. Vignoli M, Ciullini Mannurita S, Fioravanti A, et al. CD25 deficiency: a new conformational mutation prevents the receptor expression on cell surface. Clin Immunol. 2019;201:15–19. doi:10.1016/j.clim.2019.02.003
122. Alzubedy M, Osman AS, Alajlan H, Alazami AM, Al-Mousa H. Expanding the clinical spectrum of interleukin-2 receptor alpha chain deficiency: two novel cases with long-term hematopoietic stem cell transplantation outcome and literature review. Front Immunol. 2026;17:1716101. doi:10.3389/fimmu.2026.1716101
123. Fernandez IZ, Baxter RM, Garcia-Perez JE, et al. Correction: a novel human IL2RB mutation results in T and NK cell–driven immune dysregulation. J Exp Med. 2019;216(6):1465. doi:10.1084/jem.2018201505102019c
124. Zhang Z, Gothe F, Pennamen P, et al. Human interleukin-2 receptor β mutations associated with defects in immunity and peripheral tolerance. J Exp Med. 2019;216(6):1311–1327. doi:10.1084/jem.20182304
125. AlQahtani F, AlGhamdi M, AlZahrani M, AlAzami AM, Al-Buhairi S, Al-Mousa H. Hematopoietic Stem Cell Transplantation Corrects IL-2Rβ Deficiency. J Clin Immunol. 2025;45(1):68. doi:10.1007/s10875-025-01860-8
126. Flinn AM, Gennery AR. Primary immune regulatory disorders: undiagnosed needles in the haystack? Orphanet J Rare Dis. 2022;17(1):99. doi:10.1186/s13023-022-02249-1
127. Lo B, Zhang K, Lu W, et al. Autoimmune Disease. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. 2015;349(6246):436–440. doi:10.1126/science.aaa1663
128. Taghizade N, Babayeva R, Kara A, et al. Therapeutic modalities and clinical outcomes in a large cohort with LRBA deficiency and CTLA4 insufficiency. J Allergy Clin Immunol. 2023;152(6):1634–1645. doi:10.1016/j.jaci.2023.08.004
129. Kiykim A, Ogulur I, Dursun E, et al. Abatacept as a long-term targeted therapy for LRBA deficiency. J Allergy Clin Immunol Pract. 2019;7(8):2790–2800.e15. doi:10.1016/j.jaip.2019.06.011
130. Drabko K, Zarychta J, Kowalczyk A, Cienkusz M. Case report: pediatric patient with severe clinical course of CTLA-4 insufficiency treated with HSCT. Front Immunol. 2024;15:1484467. doi:10.3389/fimmu.2024.1484467
131. Lamers OAC, Smits BM, Leavis HL, et al. Treatment strategies for GLILD in common variable immunodeficiency: a systematic review. Front Immunol. 2021;12:606099. doi:10.3389/fimmu.2021.606099
132. Hurst JR, Verma N, Lowe D, et al. British lung foundation/united kingdom primary immunodeficiency network consensus statement on the definition, diagnosis, and management of granulomatous-lymphocytic interstitial lung disease in common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2017;5(4):938–945. doi:10.1016/j.jaip.2017.01.021
133. Tesch VK, Abolhassani H, Shadur B, et al. Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. J Allergy Clin Immunol. 2020;145(5):1452–1463. doi:10.1016/j.jaci.2019.12.896
134. Shadur B, NasserEddin A, Zaidman I, et al. Successful haematopoietic stem cell transplantation for LRBA deficiency with fludarabine, treosulfan, and thiotepa-based conditioning. J Clin Immunol. 2024;45(1):3. doi:10.1007/s10875-024-01770-1
135. Seidel MG, Hirschmugl T, Gamez-Diaz L, et al. Long-term remission after allogeneic hematopoietic stem cell transplantation in LPS-responsive beige-like anchor (LRBA) deficiency. J Allergy Clin Immunol. 2015;135(5):1384–1390.e1–8. doi:10.1016/j.jaci.2014.10.048
136. Ogishi M, Yang R, Aytekin C, et al. Inherited PD-1 deficiency underlies tuberculosis and autoimmunity in a child. Nat Med. 2021;27(9):1646–1654. doi:10.1038/s41591-021-01388-5
137. Johnson MB, Ogishi M, Domingo-Vila C, et al. Human inherited PD-L1 deficiency is clinically and immunologically less severe than PD-1 deficiency. J Exp Med. 2024;221(6):e20231704. doi:10.1084/jem.20231704
138. Barman P, Basu S, Goyal T, et al. Epstein-Barr virus-driven lymphoproliferation in inborn errors of immunity: a diagnostic and therapeutic challenge. Expert Rev Clin Immunol. 2024;20(11):1331–1346. doi:10.1080/1744666X.2024.2386427
139. Rivalta B, Amodio D, Milito C, et al. Case report: EBV chronic infection and lymphoproliferation in four APDS patients: the challenge of proper characterization, therapy, and follow-up. Front Pediatr. 2021;9:703853. doi:10.3389/fped.2021.703853
140. de Groot PF, Kwakernaak AJ, van Leeuwen EMM, et al. Case report: XMEN disease: a patient with recurrent Hodgkin lymphoma and immune thrombocytopenia. Front Med. 2023;10:1264329. doi:10.3389/fmed.2023.1264329
141. Booth C, Gilmour KC, Veys P, et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood. 2011;117(1):53–62. doi:10.1182/blood-2010-06-284935
142. Yang L, Booth C, Speckmann C, et al. Phenotype, genotype, treatment, and survival outcomes in patients with X-linked inhibitor of apoptosis deficiency. J Allergy Clin Immunol. 2022;150(2):456–466. doi:10.1016/j.jaci.2021.10.037
143. Price S, Shaw PA, Seitz A, et al. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood. 2014;123(13):1989–1999. doi:10.1182/blood-2013-10-535393
144. Fung-Leung WP. Phosphoinositide 3-kinase delta (PI3Kδ) in leukocyte signaling and function. Cell Signal. 2011;23(4):603–608. doi:10.1016/j.cellsig.2010.10.002
145. Preite S, Gomez-Rodriguez J, Cannons JL, Schwartzberg PL. T and B-cell signaling in activated PI3K delta syndrome: from immunodeficiency to autoimmunity. Immunol Rev. 2019;291(1):154–173. doi:10.1111/imr.12790
146. Balakrishnan K, Peluso M, Fu M, et al. The phosphoinositide-3-kinase (PI3K)-delta and gamma inhibitor, IPI-145 (Duvelisib), overcomes signals from the PI3K/AKT/S6 pathway and promotes apoptosis in CLL. Leukemia. 2015;29(9):1811–1822. doi:10.1038/leu.2015.105
147. Hemmati S, Sinclair T, Tong M, et al. PI3 kinase alpha and delta promote hematopoietic stem cell activation. JCI Insight. 2019;5(13):e125832,125832. doi:10.1172/jci.insight.125832
148. So L, Fruman DA. PI3K signaling in B and T lymphocytes: new developments and therapeutic advances. Biochem J. 2012;442(3):465–481. doi:10.1042/BJ20112092
149. Berglund LJ. Modulating the PI3K signalling pathway in activated PI3K delta syndrome: a clinical perspective. J Clin Immunol. 2024;44(1):34. doi:10.1007/s10875-023-01626-0
150. Nunes-Santos CJ, Uzel G, Rosenzweig SD. PI3K pathway defects leading to immunodeficiency and immune dysregulation. J Allergy Clin Immunol. 2019;143(5):1676–1687. doi:10.1016/j.jaci.2019.03.017
151. Lucas CL, Zhang Y, Venida A, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med. 2014;211(13):2537–2547. doi:10.1084/jem.20141759
152. Deau MC, Heurtier L, Frange P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest. 2014;124(9):3923–3928. doi:10.1172/JCI75746
153. Samra S, Bergerson JRE, Freeman AF, Turvey SE. JAK-STAT signaling pathway, immunodeficiency, inflammation, immune dysregulation, and inborn errors of immunity. J Allergy Clin Immunol. 2025;155(2):357–367. doi:10.1016/j.jaci.2024.09.020
154. O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013;368(2):161–170. doi:10.1056/NEJMra1202117
155. Philips RL, Wang Y, Cheon H, et al. The JAK-STAT pathway at 30: much learned, much more to do. Cell. 2022;185(21):3857–3876. doi:10.1016/j.cell.2022.09.023
156. Glassman CR, Tsutsumi N, Saxton RA, Lupardus PJ, Jude KM, Garcia KC. Structure of a Janus kinase cytokine receptor complex reveals the basis for dimeric activation. Science. 2022;376(6589):163–169. doi:10.1126/science.abn8933
157. Barnabei L, Laplantine E, Mbongo W, Rieux-Laucat F, Weil R. NF-κB: at the borders of autoimmunity and inflammation. Front Immunol. 2021;12. doi:10.3389/fimmu.2021.716469
158. Kozycki CT, Kodati S, Huryn L, et al. Gain-of-function mutations in ALPK1 cause an NF-κB-mediated autoinflammatory disease: functional assessment, clinical phenotyping and disease course of patients with ROSAH syndrome. Ann Rheum Dis. 2022;81(10):1453–1464. doi:10.1136/annrheumdis-2022-222629
159. Moriya K, Nakano T, Honda Y, et al. Human RELA dominant-negative mutations underlie type I interferonopathy with autoinflammation and autoimmunity. J Exp Med. 2023;220(9):e20212276. doi:10.1084/jem.20212276
160. Salzer U, Grimbacher B. TACI deficiency — a complex system out of balance. Curr Opin Immunol. 2021;71:81–88. doi:10.1016/j.coi.2021.06.004
161. Cheng J, Dávila Saldaña BJ, Chandrakasan S, Keller M. Pediatric lymphoproliferative disorders associated with inborn errors of immunity. Clin Immunol. 2024;266:110332. doi:10.1016/j.clim.2024.110332
162. Poodt AEJ, Driessen GJA, de Klein A, van Dongen JJM, van der Burg M, de Vries E. TACI mutations and disease susceptibility in patients with common variable immunodeficiency. Clin Exp Immunol. 2009;156(1):35–39. doi:10.1111/j.1365-2249.2008.03863.x
163. Salzer U, Chapel HM, Webster ADB, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005;37(8):820–828. doi:10.1038/ng1600
164. Signa S, Bertoni A, Penco F, et al. Adenosine deaminase 2 deficiency (DADA2): a crosstalk between innate and adaptive immunity. Front Immunol. 2022;13:935957. doi:10.3389/fimmu.2022.935957
165. Spolski R, Li P, Leonard WJ. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol. 2018;18(10):648–659. doi:10.1038/s41577-018-0046-y
166. Linka RM, Risse SL, Bienemann K, et al. Loss-of-function mutations within the IL-2 inducible kinase ITK in patients with EBV-associated lymphoproliferative diseases. Leukemia. 2012;26(5):963–971. doi:10.1038/leu.2011.371
167. Semenzato G, Pizzolo G, Zambello R. The interleukin-2/interleukin-2 receptor system: structural, immunological, and clinical features. Int J Clin Lab Res. 1992;22(1):133–142. doi:10.1007/BF02591413
168. Hinks A, Ke X, Barton A, et al. Association of the IL2RA/CD25 gene with juvenile idiopathic arthritis. Arthritis Rheum. 2009;60(1):251–257. doi:10.1002/art.24187
169. Kanai T, Jenks J, Nadeau KC. The STAT5b Pathway Defect and Autoimmunity. Front Immunol. 2012;3. doi:10.3389/fimmu.2012.00234
170. Chikuma S. CTLA-4, an Essential Immune-Checkpoint for T-Cell Activation. Curr Top Microbiol Immunol. 2017;410:99–126. doi:10.1007/82_2017_61
171. Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–182. doi:10.1111/j.1600-065X.2008.00662.x
172. Brunner-Weinzierl MC, Rudd CE. CTLA-4 and PD-1 control of T-cell motility and migration: implications for tumor immunotherapy. Front Immunol. 2018;9:2737. doi:10.3389/fimmu.2018.02737
173. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009;229(1):12–26. doi:10.1111/j.1600-065X.2009.00770.x
174. Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. 2001;1(3):220–228. doi:10.1038/35105024
175. McCoy KD, Le Gros G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol Cell Biol. 1999;77(1):1–10. doi:10.1046/j.1440-1711.1999.00795.x
176. Thompson CB, Allison JP. The emerging role of CTLA-4 as an immune attenuator. Immunity. 1997;7(4):445–450. doi:10.1016/S1074-7613(00)80366-0
177. Bhattar K, Agrawal P, Valencia Uribe J, Elizabeth Salazar P. Systemic granulomatous disease in CVID with LRBA deficiency: long-term management strategies. Am J Respir Crit Care Med. 2025;211(Abstracts):A5515–A5515. doi:10.1164/ajrccm.2025.211.Abstracts.A5515
178. Jamee M, Hosseinzadeh S, Sharifinejad N, et al. Comprehensive comparison between 222 CTLA-4 haploinsufficiency and 212 LRBA deficiency patients: a systematic review. Clin Exp Immunol. 2021;205(1):28–43. doi:10.1111/cei.13600
179. Ramos-Casals M, Brahmer JR, Callahan MK, et al. Immune-related adverse events of checkpoint inhibitors. Nat Rev Dis Primer. 2020;6(1):38. doi:10.1038/s41572-020-0160-6
180. Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer Oxf Engl. 2016;54:139–148. doi:10.1016/j.ejca.2015.11.016
181. Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378(2):158–168. doi:10.1056/NEJMra1703481
182. Tetzlaff MT, Nelson KC, Diab A, et al. Granulomatous/sarcoid-like lesions associated with checkpoint inhibitors: a marker of therapy response in a subset of melanoma patients. J Immunother Cancer. 2018;6(1):14. doi:10.1186/s40425-018-0323-0
183. Haanen J, Obeid M, Spain L, et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2022;33(12):1217–1238. doi:10.1016/j.annonc.2022.10.001
184. Gkiozos I, Kopitopoulou A, Kalkanis A, Vamvakaris IN, Judson MA, Syrigos KN. Sarcoidosis-like reactions induced by checkpoint inhibitors. J Thorac Oncol. 2018;13(8):1076–1082. doi:10.1016/j.jtho.2018.04.031
185. Tiri A, Masetti R, Conti F, et al. Inborn errors of immunity and cancer. Biology. 2021;10(4):313. doi:10.3390/biology10040313
186. Kondratenko I, Paschenko O, Polyakov A, Bologov A. Nijmegen breakage syndrome. Adv Exp Med Biol. 2007;601:61–67. doi:10.1007/978-0-387-72005-0_6
187. Shiloh Y. Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu Rev Genet. 1997;31:635–662. doi:10.1146/annurev.genet.31.1.635
188. Ye X, Maglione PJ, Wehr C, et al. Genomic characterization of lymphomas in patients with inborn errors of immunity. Blood Adv. 2022;6(18):5403–5414. doi:10.1182/bloodadvances.2021006654
189. Panchal N, Booth C, Cannons JL, Schwartzberg PL. X-linked lymphoproliferative disease type 1: a clinical and molecular perspective. Front Immunol. 2018;9:666. doi:10.3389/fimmu.2018.00666
190. Yang X, Wada T, Imadome KI, et al. Characterization of Epstein-Barr virus (EBV)-infected cells in EBV-associated hemophagocytic lymphohistiocytosis in two patients with X-linked lymphoproliferative syndrome type 1 and type 2. Herpesviridae. 2012;3(1):1. doi:10.1186/2042-4280-3-1
191. Caldirola MS, Raccio AG, Giovanni DD, Gaillard MI, Preciado MV. Pediatric inborn errors of immunity causing hemophagocytic lymphohistiocytosis: case report and review of the literature. J Leukoc Biol. 2022;112(4):607–615. doi:10.1002/JLB.5MR0622-037R
192. Planas R, Felber M, Vavassori S, Pachlopnik Schmid J. The hyperinflammatory spectrum: from defects in cytotoxicity to cytokine control. Front Immunol. 2023;14:1163316. doi:10.3389/fimmu.2023.1163316
193. Boyarchuk O, Volokha A, Yarema N, et al. Fatal HLH in patients with X-linked lymphoproliferative disease 1 due to a novel variant in SH2D1A: case report. Front Immunol. 2025;16:1602107. doi:10.3389/fimmu.2025.1602107
194. Cox MF, Mackenzie S, Low R, et al. Diagnosis and investigation of suspected haemophagocytic lymphohistiocytosis in adults: 2023 Hyperinflammation and HLH Across Speciality Collaboration (HiHASC) consensus guideline. Lancet Rheumatol. 2024;6(1):e51–e62. doi:10.1016/S2665-9913(23)00273-4
195. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503–1516. doi:10.1016/S0140-6736(13)61048-X
196. Cetica V, Sieni E, Pende D, et al. Genetic predisposition to hemophagocytic lymphohistiocytosis: report on 500 patients from the Italian registry. J Allergy Clin Immunol. 2016;137(1):188–196.e4. doi:10.1016/j.jaci.2015.06.048
197. Janka GE, Lehmberg K. Hemophagocytic lymphohistiocytosis: pathogenesis and treatment. Hematol Am Soc Hematol Educ Program. 2013;2013:605–611. doi:10.1182/asheducation-2013.1.605
198. Lim MS, Straus SE, Dale JK, et al. Pathological findings in human autoimmune lymphoproliferative syndrome. Am J Pathol. 1998;153(5):1541–1550. doi:10.1016/S0002-9440(10)65742-2
199. Xie Y, Pittaluga S, Price S, et al. Bone marrow findings in autoimmune lymphoproliferative syndrome with germline FAS mutation. Haematologica. 2017;102(2):364–372. doi:10.3324/haematol.2015.138081
200. Jolles S, Rojavin MA, Lawo JP, et al. Long-Term efficacy and safety of hizentra® in patients with primary Immunodeficiency in Japan, Europe, and the United States: a review of 7 phase 3 trials. J Clin Immunol. 2018;38(8):864–875. doi:10.1007/s10875-018-0560-5
201. Orange JS, Grossman WJ, Navickis RJ, Wilkes MM. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: a meta-analysis of clinical studies. Clin Immunol Orlando Fla. 2010;137(1):21–30. doi:10.1016/j.clim.2010.06.012
202. Buso H, Firinu D, Gambier RF, et al. Lung function trajectories in common variable immunodeficiencies: an observational retrospective multicenter study. J Allergy Clin Immunol. 2025;155(3):1027–1035. doi:10.1016/j.jaci.2024.10.037
203. Milito C, Pulvirenti F, Cinetto F, et al. Double-blind, placebo-controlled, randomized trial on low-dose azithromycin prophylaxis in patients with primary antibody deficiencies. J Allergy Clin Immunol. 2019;144(2):584–593.e7. doi:10.1016/j.jaci.2019.01.051
204. Eibl MM, Wolf HM. Vaccination in patients with primary immune deficiency, secondary immune deficiency and autoimmunity with immune regulatory abnormalities. Immunotherapy. 2015;7(12):1273–1292. doi:10.2217/IMT.15.74
205. Shearer WT, Fleisher TA, Buckley RH, et al; Medical Advisory Committee of the Immune Deficiency Foundation. Recommendations for live viral and bacterial vaccines in immunodeficient patients and their close contacts. J Allergy Clin Immunol. 2014;133(4):961–966. doi:10.1016/j.jaci.2013.11.043
206. van Assen S, Agmon-Levin N, Elkayam O, et al. EULAR recommendations for vaccination in adult patients with autoimmune inflammatory rheumatic diseases. Ann Rheum Dis. 2011;70(3):414–422. doi:10.1136/ard.2010.137216
207. Teachey DT, Greiner R, Seif A, et al. Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br J Haematol. 2009;145(1):101–106. doi:10.1111/j.1365-2141.2009.07595.x
208. Hong Y, Burleigh A, Liao A, et al. Preclinical evaluation of lentiviral gene therapy for adenosine deaminase 2 deficiency (DADA2): engraftment efficiency and biodistribution in humanised NBSGW mice. Gene Ther. 2025:1–8. doi:10.1038/s41434-025-00547-4
209. Catak MC, Surucu N, Bayram Catak F, et al. Abatacept restores dysregulated transcriptomic and proteomic profile in disorders of CTLA-4 insufficiency. J Allergy Clin Immunol. 2025:S0091–6749(25)00946–7. doi:10.1016/j.jaci.2025.08.027
210. Krausz M, Uhlmann A, Rump IC, et al. The ABACHAI clinical trial protocol: safety and efficacy of Abatacept (s.c.) in patients with CTLA-4 insufficiency or LRBA deficiency: a non controlled Phase 2 clinical trial. Contemp Clin Trials Commun. 2022;30:101008. doi:10.1016/j.conctc.2022.101008
211. Salami F, Fekrvand S, Yazdani R, et al. Evaluation of expression of LRBA and CTLA-4 proteins in common variable immunodeficiency patients. Immunol Invest. 2022;51(2):381–394. doi:10.1080/08820139.2020.1833029
212. Miano M, Scalzone M, Perri K, et al. Mycophenolate mofetil and Sirolimus as second or further line treatment in children with chronic refractory primitive or secondary autoimmune cytopenias: a single centre experience. Br J Haematol. 2015;171(2):247–253. doi:10.1111/bjh.13533
213. Lankester AC, Albert MH, Booth C, et al. EBMT/ESID inborn errors working party guidelines for hematopoietic stem cell transplantation for inborn errors of immunity. Bone Marrow Transplant. 2021;56(9):2052–2062. doi:10.1038/s41409-021-01378-8
214. Laberko A, Shelikhova L, Vashura A, et al. Risk factors for hematopoietic stem cell transplantation in inborn errors of immunity. Transplant Cell Ther. 2025:S2666–6367(25)01518–0. doi:10.1016/j.jtct.2025.10.015
215. Burns SO, Morris EC. How I use allogeneic HSCT for adults with inborn errors of immunity. Blood. 2021;138(18):1666–1676. doi:10.1182/blood.2020008187
216. Sleight BJ, Prasad VS, DeLaat C, et al. Correction of autoimmune lymphoproliferative syndrome by bone marrow transplantation. Bone Marrow Transplant. 1998;22(4):375–380. doi:10.1038/sj.bmt.1701306
217. Benkerrou M, Le Deist F, de Villartay JP, et al. Correction of Fas (CD95) deficiency by haploidentical bone marrow transplantation. Eur J Immunol. 1997;27(8):2043–2047. doi:10.1002/eji.1830270831
218. Nademi Z, Slatter MA, Dvorak CC, et al. Hematopoietic stem cell transplant in patients with activated PI3K delta syndrome. J Allergy Clin Immunol. 2017;139(3):1046–1049. doi:10.1016/j.jaci.2016.09.040
219. Jamee M, Moniri S, Zaki-Dizaji M, et al. Clinical, immunological, and genetic features in patients with activated pi3kδ syndrome (APDS): a systematic review. Clin Rev Allergy Immunol. 2020;59(3):323–333. doi:10.1007/s12016-019-08738-9
220. Dimitrova D, Nademi Z, Maccari ME, et al. International retrospective study of allogeneic hematopoietic cell transplantation for activated PI3K-delta syndrome. J Allergy Clin Immunol. 2022;149(1):410–421.e7. doi:10.1016/j.jaci.2021.04.036
221. Leiding JW, Okada S, Hagin D, et al. Hematopoietic stem cell transplantation in patients with gain-of-function signal transducer and activator of transcription 1 mutations. J Allergy Clin Immunol. 2018;141(2):704–717.e5. doi:10.1016/j.jaci.2017.03.049
222. Buddingh EP, Slatter M, Aldave Becerra JC, et al. Improved outcome of HSCT in STAT1 gain-of-function disease following JAK inhibition bridging. J Hum Immun. 2025;1(3):e20250027. doi:10.70962/jhi.20250027
223. Albert MH, Sirait T, Eikema DJ, et al. Hematopoietic stem cell transplantation for adolescents and adults with inborn errors of immunity: an EBMT IEWP study. Blood. 2022;140(14):1635–1649. doi:10.1182/blood.2022015506
224. Tsilifis C, Speckmann C, Lum SH, et al. Hematopoietic stem cell transplantation for CTLA-4 insufficiency across Europe: a European Society for Blood and Marrow Transplantation Inborn Errors Working Party study. J Allergy Clin Immunol. 2024;154(6):1534–1544. doi:10.1016/j.jaci.2024.08.020
225. Seidel MG, Tesch VK, Yang L, et al. Correction to: the Immune Deficiency and Dysregulation Activity (IDDA2.1 ‘Kaleidoscope’) Score and other clinical measures in inborn errors of immunity. J Clin Immunol. 2022;42(3):499. doi:10.1007/s10875-022-01220-w
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.