Back to Journals » Journal of Inflammation Research » Volume 14
LXA4 Inhibits Lipopolysaccharide-Induced Inflammatory Cell Accumulation by Resident Macrophages in Mice
Authors Mei HX, Ye Y, Xu HR, Xiang SY, Yang Q, Ma HY, Jin SW ![]() , Wang Q
, Wang Q ![]()
Received 10 January 2021
Accepted for publication 26 March 2021
Published 12 April 2021 Volume 2021:14 Pages 1375—1385
DOI https://doi.org/10.2147/JIR.S301292
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Hong-Xia Mei, Yang Ye, Hao-Ran Xu, Shu-Yang Xiang, Qian Yang, Hong-Yu Ma, Sheng-Wei Jin, Qian Wang
Department of Anesthesia and Critical Care, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, 325027, People’s Republic of China
Correspondence: Sheng-Wei Jin; Qian Wang
Department of Anesthesia and Critical Care, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, 109 Xueyuan Road, Wenzhou, Zhejiang Province, 325027, People’s Republic of China
Tel +86-577-88002806
Fax +86-577-88832693
Email [email protected]; [email protected]
Introduction: Alveolar macrophages that regulate the inflammatory response in lungs are the main target cell for the treatment of inflammatory pulmonary pathologies, such as acute respiratory distress syndrome (ARDS). Yolk sac derived alveolar resident macrophages play an important role in the pulmonary inflammatory response. With regards to anti-inflammatory actions, lipoxin A4 (LXA4) has been identified as an inflammatory “braking signal”.
Methods: In vivo, LXA4 (0.1 μg/mouse) was injected intraperitoneally after intratracheal (1 mg/kg) lipopolysaccharide (LPS) administration; flow cytometry was used to measure peripheral blood monocyte derived recruited macrophage and neutrophil numbers; resident alveolar macrophage was depleted by liposome clodronate; CXCL2, CCL2, MMP9 level was detected by RT-PCR and ELISA. In vitro, sorted resident macrophages (1× 106) were cultured with LPS (1 μg/mL) and LXA4 (100 nmol/mL) with or without BOC-2 (10 μM) for 24 h to gain a better understanding of the mechanisms of LXA4.
Results: LXA4 inhibited tumor necrosis factor-a (TNF-a) and interleukin-1β (IL-1β) production induced by LPS. LXA4 also mediated LPS-induced macrophage recruitment and showed that this was dependent on CCL2 secretion and release by resident macrophages. LXA4 protects lung tissue by inhibiting neutrophil recruitment, partly through the CXCL2/MMP-9 signaling pathway. CXCL2 and MMP-9 are mainly expressed by resident macrophages and neutrophils, respectively. Finally, LXA4’s beneficial effects were abrogated by BOC-2, an LXA4 receptor inhibitor.
Conclusion: These results suggest that LXA4 may be a promising therapy for preventing and treating ARDS.
Keywords: resident macrophage, recruited macrophage, neutrophil, lipoxin A4, lipopolysaccharide
Background
Alveolar macrophages are the front line of innate immune cells and play crucial roles in the response to pulmonary inflammation, such as that seen in acute respiratory distress syndrome (ARDS).1 There is a growing body of evidence suggesting that tissue resident macrophages, derived from the yolk sac, perform immune sentinel and homeostatic functions, especially in the pathogenesis of ARDS.2–4 Resident macrophages release chemokines to regulate inflammatory cells, and are key orchestrators of the response to inflammation.5
The chemokine C-C motif ligand 2 (CCL2) and C-X–C motif chemokine ligand 2 (CXCL2), which produced by multiple cell types,6,7 has been observed in response to a wide variety of inflammatory stimuli, including microbial infection and tissue damage.8–10 Matrix metalloproteinases (MMPs) play an important role in a variety of lung diseases.11–13 Previous study showed that MMP-9 expression was increased in the lung and bronchoalveolar fluid in ARDS.14 MMP-9 released by neutrophils promotes neutrophil transmembrane migration into the pulmonary alveoli in LPS-induced acute lung injury models.15,16
Lipoxins are arachidonic acid metabolites formed during inflammation and because of their anti-inflammatory actions, have been called inflammatory “braking signals”. One study showed that LXA4 limited leukocyte recruitment, induced neutrophils apoptosis, and enhanced efferocytosis.17 We have previously shown that in LPS-stimulated mice, LXA4 protects the lung,18 promotes alveolar fluid clearance,19 and accelerates lung epithelial repair after LPS administration.20 To date, no studies have examined LXA4’s effect on resident macrophages in LPS-induced lung injury.
We hypothesize that LXA4 attenuates LPS-induced lung injury via inhibiting resident macrophage-associated inflammatory cell recruitment. We also hypothesize that CCL2, CXCL2 and MMP-9, which released by resident macrophage and neutrophils, might be involved in regulating inflammatory cell recruitment. Finally, we have been suggested that the LXA4’s response is ALX (LXA4 receptor) dependent.
Methods
Materials
LXA4 was obtained from Cayman Chemical Company (Ann Arbor, MI), and LPS (Escherichia coli serotype 055:B5) from Sigma (St. Louis, MO). CCL2, CXCL2, MMP-9, TNF-α and IL-1β ELISA kits were obtained from R&D Systems (Minneapolis, MN). CCR2 inhibitor (CCR2i), CXCR2 inhibitor (CXCR2i), and MMP-9 inhibitor (MMP9i) were all from MedChem Express (Monmouth Junction, NJ). Anti-CXCL2, Anti-Goat IgG H&L (PE), anti-CCL2 PE-conjugated, anti-Ly6c APC-conjugated, anti-F4/80 PE-Cyanine7-conjugated, anti-Ly6G FITC-conjugated, anti-CD11b APC-conjugated and anti-CD11c PerCP-Cy5.5-conjugated antibodies were obtained from Invitrogen (Carlsbad, CA), Anti-CXCR2 PE-conjugated and anti-CCR2 PE-conjugated were from BioLegend (San Diego, CA) and the anti-MMP–9 antibody was purchased from Abcam (Cambridge, MA).
Animal Studies
C57BL/6 mice (20–25 g) were obtained from the Slac Laboratory Animal (Shanghai, China) and the experimental protocols were approved by Wenzhou Medical University. Chinese guidelines for the welfare of the laboratory animals (GB/T 35823-2018), which was published by standardization administration of China, were followed in the present study.
Mice received LPS (atomization inhalation, 1 mg/kg) for 2 h, 6 h, 12 h, 24 h, or 48 h to elicit peak inflammation. Then, for the LPS and LPS+LXA4 group, mice received LPS with or without LXA4 (0.1 µg/mouse). Moreover, mice received the 2 mg/kg CXCR2i (CXCR2 inhibitor), 30 mg/kg CCR2i (CCR2 inhibitor), 10 mg/kg MMP9i (MMP9 inhibitor) or 10 μM BOC-2 (ALX inhibitor) intraperitoneal injection in the presence or absence of LXA4 after LPS administration. 24 h later, the bronchoalveolar lavage fluid (BALF) and lung tissue samples were collected.
Flow Cytometry
Anti-F4/80 PE-Cyanine7-conjugated and anti-Ly6c APC-conjugated antibodies, anti-Ly6G FITC-conjugated, anti-CD11b APC-conjugated and/or anti-CD11c PerCP-Cy5.5-conjugated antibodies were added to cell supernatant for 30 min then cells were incubated with fluorescence-activated cell sorting (FACS) solution for another 10 min, washed with PBS and analyzed by CytExpert 2.0 (Beckman Coulter). Resident macrophages were identified as F4/80+Ly6C−CD11b−CD11c+, recruited macrophages identified as F4/80+Ly6c+CD11b+CD11c−, and neutrophils identified as F4/80−Ly6c+Ly6g+.21,22
Resident Macrophage Depletion
50 μL of clodronate liposome (5 mg/mL) was used to deplete resident macrophages by intratracheal administration.23 PBS liposome was used as control. 72 h after depletion, mice were stimulated with 1 mg/kg LPS and/or 0.1 µg/mouse LXA4 for 24 h, after which the BALF was harvested.
Resident Macrophages Sorting
Resident macrophages were sorted using a 100 μm nozzle then cultured with LPS (1 μg/mL) in the presence or absence of LXA4 (100nM) for 24 h to measure CCL2 and CXCL2 mRNA expression.
Real-Time PCR
As previously described, total RNA was extracted from lung tissues with TRIzol reagent (Invitrogen, Carlsbad, CA), then reverse transcribed with the Super TaqMan OneStep RT-qPCR Kit (Life Technologies). qRT-PCR was performed on an Applied Biosystems 7500 real-time PCR system with the following cycling program: 40 cycles of incubation at 50 °C for 2 min and 95 °C for 10 min, followed by melting at 95°C for 15 s, then annealing at 60°C for 1 min. Target gene expression levels were normalized to the housekeeping gene, GAPDH, and the fold change was calculated using the 2−ΔΔCt method.
The primers sequences were as follows: CCL2: 5-CCACTCACCTGCTGCTACTCA-TTC-3, 5-CTGCTGCTGGTGATCCTCTTGTAG-3; CXCL2: 5′-CCACTCACCTGC-TGCTACTCATTC-3′ and reverse 5′-CTGCTGCTGGTGATCCTCTTGTAG-3′.
Western Blotting
After treatment, total protein from the BALF cells was extracted using RIPA lysis buffer and quantified using a BCA protein assay kit. Samples were separated by 10% sodium-dodecyl sulfonate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes. After incubation with primary (anti-MMP-9, 1:1000) and secondary antibodies, protein was detected using the UVP gel imaging system (Upland, CA).
ELISA
TNF-α, IL-1β, CCL2, CXCL2, and MMP-9 levels in BALF were measured by ELISA kits.
Statistical Analysis
Data are shown as the mean ± SEM. Data were assessed by one-way ANOVA, followed by Tukey’s test for post hoc comparison. A value of p<0.05 was considered significant. Statistical analyses were performed using SPSS 19.0 and Prism 6.0 software.
Results
LXA4 Inhibits LPS-Induced Accumulation of Inflammatory Cells
Resident macrophages were identified as F4/80+Ly6C−CD11b−CD11c+, recruited macrophages as F4/80+Ly6c+CD11b+CD11c−, and neutrophils as F4/80−Ly6c+Ly6g+ (Supplemental Figure 1). Intratracheal administration of 1 mg/kg LPS induced an increase in neutrophils in a time dependent manner, rising to a peak at 24 h, then declined to nearly normal at 48 h (Figure 1A and B). Treatment with LXA4 (intraperitoneal injection, ip) reduced both neutrophil levels and recruited macrophage accumulation (p<0.05) (Figure 1C and D), but not resident macrophage levels (p>0.05) (Figure 1E), at 24 h after LPS stimulation. LXA4 treatment also decreased TNF-α and IL-1β concentrations in BALF (p<0.05) (Figure 1F and G). LXA4 alone had no effect on neutrophil (C), recruited macrophage (D) and resident macrophage numbers (E), TNF-α (F) and IL-1β (G) level compared with CTR group (p>0.05), indicating that LXA4 reduced LPS-induced accumulation of inflammatory cells and production of cytokines to promote the resolution of inflammation.
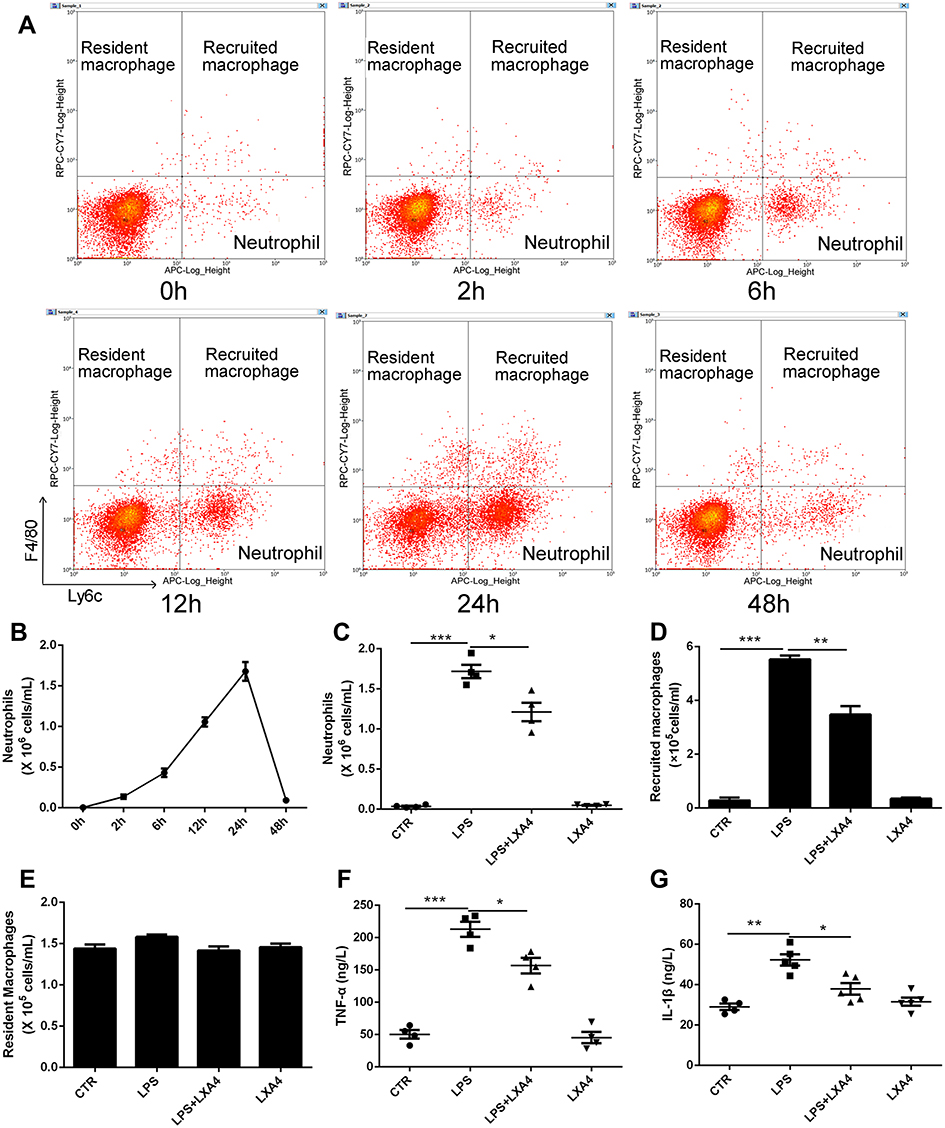 |
Figure 1 LXA4 reduces inflammatory cell infiltration to accelerate the resolution of LPS-induced pulmonary inflammation. Mice received 1 mg/kg LPS by intratracheal atomization then LXA4 (0.1 µg/mouse) by intraperitoneal injection. As assessed by flow cytometry, resident macrophages were F4/80+Ly6C−, recruited macrophages were F4/80+Ly6c+, and neutrophils were F4/80−Ly6c+ (A). LPS up-regulated neutrophil (B and C) and recruited macrophage (D) number, and increased TNF-α (F) and IL-1β (G) concentration in the BALF. LXA4 treatment markedly attenuated these effects. No changes in resident macrophage (E) numbers were seen in these groups. The data are presented as the mean ± SEM, n =6- 9. *p<0.05, **p<0.01,***p<0.001. |
LXA4 Inhibition of LPS-Induced Inflammatory Cell Accumulation is Associated with Resident Macrophages
Intratracheal administration of 50 μL clodronate liposome (5 mg/mL) resulted in a depletion of resident macrophages in a time dependent manner, reaching the lowest level on the third day (Figure 2A, Supplemental Figure 2). As shown in Figure 2B, clodronate liposome depleted the majority of resident macrophages by day 3 compared with PBS liposomes (p<0.05). In the PBS liposomes group, LXA4 treatment significantly decreased recruited macrophage and neutrophil numbers compared to the LPS group (p<0.05) (Figure 2C and D). However, after clodronate liposome intervention, LXA4 treatment had no effect on the number of recruited macrophages and neutrophils (p>0.05) (Figure 2C and D), suggesting that LXA4 inhibiting LPS-induced inflammatory cells accumulation was associated with resident macrophages.
 |
Figure 2 LXA4-mediated reduction in LPS-induced recruited macrophage and neutrophil infiltration depends on resident macrophages. Resident alveolar macrophages were depleted by intratracheal administration of 50 µL clodronate liposome (A). On day 3, when resident macrophages were most depleted (B), LXA4 (0.1 µg/mouse) was administered 10 min after LPS (1 mg/kg) stimulation and 24 hours later the numbers of recruited macrophage (C) and neutrophil (D) in the BAL fluid were measured by flow cytometry. The data are presented as the mean ± SEM, n =6- 9. *p<0.05, **p<0.01,***p<0.001. |
LXA4-Mediated Reduction of LPS-Induced Recruited Macrophage and Neutrophil Recruitment is Dependent on Resident Macrophage Secretion and Release of CCL2
As shown in Figure 3A, CCL2 was mainly expressed in resident macrophages. We saw that the LPS-mediated up-regulation of CCL2 mRNA in lung tissues (Figure 3B) and resident macrophage (Figure 3C), CCL2 protein in the BALF (Figure 3D), respectively, was reduced following LXA4 treatment. A significant decrease in macrophage recruitment was observed when mice were treated with LPS+LXA4+CCR2 inhibitor compared with LPS+LXA4 (p<0.05) (Figure 3E). LPS increased CCR2 expression in recruited macrophages, but LXA4 had no effect on CCR2 expression (Figure 3F). These data demonstrate that LXA4 inhibited CCL2 secretion and release, but not CCR2, on resident macrophages to reduce the accumulation of recruited macrophages in vivo.
 |
Figure 3 LXA4 down-regulated LPS-stimulated CCL2 secretion and release to reduce infiltration of recruited macrophages. Mice received 1 mg/kg LPS by intratracheal atomization then LXA4 (0.1 µg/mouse) by intraperitoneal injection. CCL2 was mainly expressed on resident macrophage (A). CCL2 mRNA (B) expression in lung tissue and CCL2 levels (D) in BALF were measured 24 h later. A CCR2 inhibitor was administered, and the number of recruited macrophage in the BAL fluid measured by flow cytometry (E). CCR2 expression by recruited macrophages was measured by flow cytometry (F). Resident macrophages were sorted and cultured with LPS (1 μg/mL) in the presence or absence of LXA4 (100 nM) for 24 h. CCL2 expression was measured by RT-PCR (C). The data are presented as the mean ± SEM, n =6- 9. *p<0.05, **p<0.01. |
LXA4-Mediated Reduction of LPS-Induced Neutrophil Recruitment is Dependent on CXCL2 Secretion and Release in Resident Macrophages
As shown in Figure 4A, CXCL2 was mainly expressed in resident macrophages. The LPS-mediated up-regulation of CXCL2 mRNA in lung tissues (Figure 4B) and resident macrophage (Figure 4C), CXCL2 protein levels in the BALF (Figure 4D), respectively, was reduced following LXA4 treatment (p<0.05). A significant decrease in neutrophil recruitment was seen in LPS+LXA4+CXCR2 inhibitor treated mice compared to LPS+LXA4 treatment (p<0.05) (Figure 4E). Moreover, while LPS increased CXCR2 expression, LXA4 had no effect on CCR2 expression (Figure 4F). These data show that LXA4 inhibited CXCL2 secretion and release, but not CCR2, in resident macrophages to reduce neutrophil accumulation in vivo.
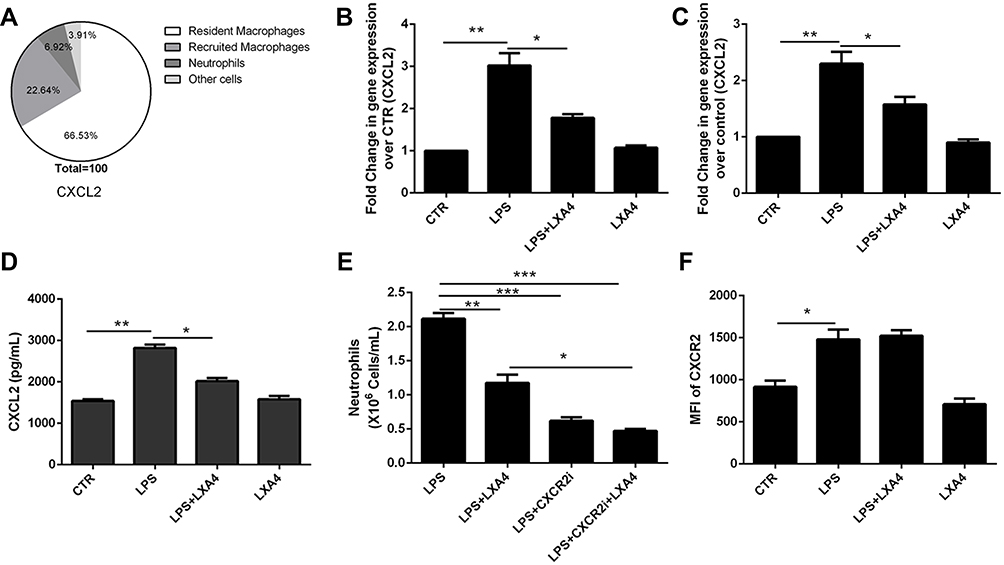 |
Figure 4 LXA4-mediated reduction of LPS-induced neutrophil recruitment is dependent on secretion and release of CXCL2 by resident macrophages. LXA4 (0.1 µg/mouse) was administrated 10 min after LPS (1 mg/kg) stimulation. Twenty-four hours later, CXCL2 was measured by flow cytometry (A). CXCL2 mRNA (B) level on lung tissues and CXCL2 protein level (D) on the BALF were measured by real-time PCR and Elisa. Mice received a CXCR2 inhibitor (2 mg/kg) in the presence or absence of LXA4 via intraperitoneal injection 10 min after LPS administration. The number of neutrophils in the BALF and the mean fluorescence intensity (MFI) (E) of CXCR2 (F) were assessed by flow cytometry. Resident macrophages were sorted and cultured with LPS (1 μg/mL) in the presence or absence of LXA4 (100 nM) for 24 h. CXCL2 expression was measured by RT-PCR (C). The data are presented as the mean ± SEM, n =6- 9. *p<0.05, **p<0.01,***p<0.001. |
LXA4-Mediated Reduction of LPS-Induced Neutrophil Transmigration is Associated with MMP-9 Expression
To examine whether MMP-9 was involved in the LXA4-mediated reduction of LPS-induced neutrophil transmigration, mice were pretreated with MMP-9 inhibitor (10 mg/kg) or CXCR2 inhibitor (2 mg/kg) 20 min prior to LPS (1 mg/kg) treatment with or without LXA4 (0.1 µg/mouse) for 24 h. As expected, MMP-9 expression in the lung homogenate was much lower in the LPS+LXA4 group compared to the LPS group (P< 0.05) (Figure 5A and B). MMP-9 concentration was lower in the LPS+LXA4+CXCR2i group compared to the LPS+LXA4 group, (P< 0.05), and no difference was seen between the LPS+LXA4+CXCR2i and LPS+CXCR2i groups (P> 0.05) (Figure 5B). As shown in Figure 5C, BALF neutrophil numbers were much lower in the LPS+LXA4+MMP9i group compared to the LPS+MMP9i group (P< 0.05). These data demonstrated that LXA4-mediated reduction of LPS-induced neutrophil transmigration is associated with MMP-9 levels. We also found that MMP-9 was mainly produced in F4/80− cells (almost 80%) (Figure 5D).
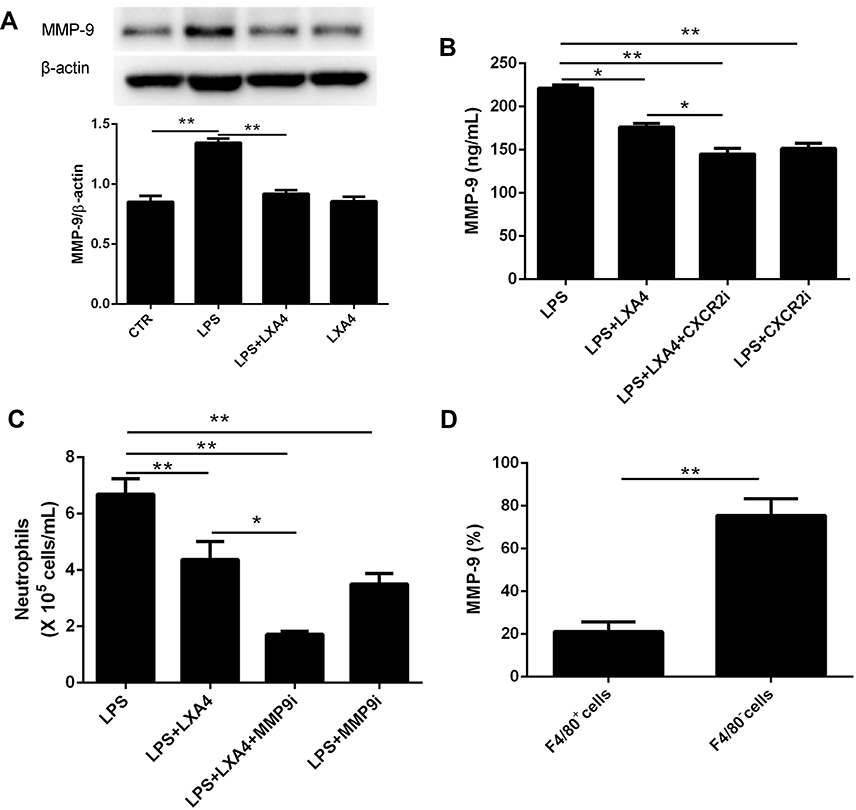 |
Figure 5 LXA4-mediated reduction in LPS-induced neutrophil transmigration was association with MMP-9 expression. Mice received MMP-9 (10 mg/kg) and CXCR2 inhibitors (2 mg/kg) 20 min prior to LPS (1 mg/kg) administration with or without LXA4 (0.1 µg/mouse) for 24 h. MMP-9 protein expression in the lung homogenates was measured by Western blot (A) and MMP-9 concentration in the BALF was measured by Elisa (B). Neutrophil numbers in the BALF (C) and the percentage of MMP-9 expression on the F4/80+ and F4/80− cells (D) was measured by flow cytometry. The data are presented as the mean ± SEM, n =6- 9. *p<0.05, **p<0.01. |
LXA4’s Protective Effect in LPS-Induced Lung Injury is Negated by BOC-2
To further investigate LXA4’s ALX-dependent actions in vivo, we co-administered LXA4 (0.1 µg/mouse) and BOC-2 (an ALX inhibitor) (10 μM) by intraperitoneal injection after LPS (1 mg/kg) administration. Twenty four hours later, BALF was collected. As shown in Figure 6A, LXA4’s protective effect against morphological changes in lung tissues was abolished by BOC-2. The ALI score in the LPS+LXA4 group was lower than that of the LPS+LXA4+BOC-2 group (P<0.05) (Figure 6B). A decrease was also seen in CCL2, CXCL2, MMP-9, TNF-α, IL-1β levels and neutrophil numbers in the LPS+LXA4 group compared to the LPS+LXA4+BOC-2 group (P<0.05) (Figure 6C,E,G–J), indicating that LXA4’s protective effect was abolished by BOC-2.
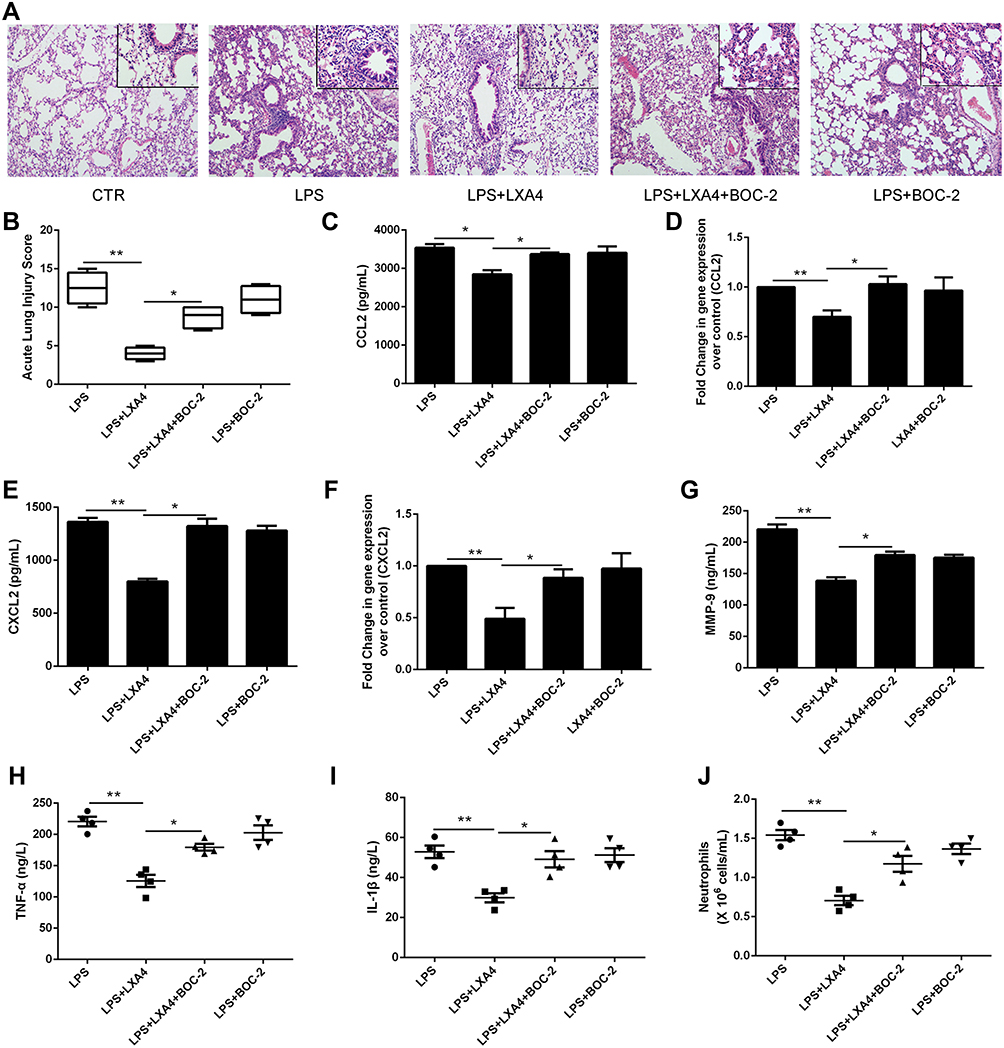 |
Figure 6 LXA4’s protective effect on LPS-induced lung injury was abrogated by BOC-2. Mice were intraperitoneally injected with BOC-2 (50 μg/kg) 2 h before LPS (1 mg/kg) administration with or without of LXA4 (0.1 µg/mouse). The beneficial effects of LXA4 were abrogated by BOC-2, as assessed by histological analysis (A), acute lung injury score (B), CCL2 (C), CXCL2 (E), MMP-9 (G), TNF-α (H) and IL-1β (I) protein expression, and the number of neutrophils (J) in the BALF. Resident macrophages were sorted and pretreated with BOC-2 (10 μM) 30 min before cultured with LPS (1 μg/mL) in the presence or absence of LXA4 (100 nM) for 24 h. CCL2 (D) and CXCL2 (F) mRNA level were measured by RT-PCR. The data are presented as the mean ± SEM, n =6- 9. *p<0.05, **p<0.01. |
To further investigate LXA4’s ALX-dependent actions in vitro, resident macrophages were sorted and pretreated with BOC-2 (10 μM) 30 min before cultured with LPS (1 μg/mL) in the presence or absence of LXA4 (100 nM) for 24 h. As shown in Figure 6D and F, LXA4 induced CCL2 and CXCL2 down-regulation in the primary resident macrophage was abrogated by BOC-2, suggesting that the effect of LXA4 might be mediated via an ALX‐dependent pathway.
Discussion
In this study, we showed that inflammation peaked at 24h after LPS administration, and that LXA4 inhibited the pro-inflammatory cytokines TNF-α and IL-1β, and reduced the recruited macrophage and neutrophil accumulation induced by the expression and release of CCL2 and CXCL2 by resident macrophages. We also showed that MMP-9 levels in neutrophils are associated with neutrophil transmembrane migration and that LXA4 inhibition of inflammatory cell infiltration was receptor dependent.
Acute lung injury is a devastating disease, characterized by pulmonary edema and infiltration of excessive inflammatory cells and factors.24,25 Pulmonary infiltration is a hallmark of LPS-induced lung injury, and it is widely accepted that resolution of alveolar infiltration is an essential step in patient survival. Timely resolution of the uncontrolled inflammation is therefore a key to repair lung injury. Previous studies have shown that neutrophils and resident and recruited macrophages are involved in acute lung injury and that resident macrophages play critical roles in this process.26,27 In this study we found that neutrophils were significantly recruited to the BALF from 2 h after LPS-induced lung injury, peaked at 24 h, then returned to their original level at 48 h. This suggests that inflammation peaked at 24 h after LPS administration. LXA4 treatment reduced numbers of neutrophils and recruited macrophages 24 h after LPS stimulation, but resident macrophage numbers were unchanged, consistent with a previous study which showed that resident alveolar macrophage levels remained constant for 14 days in endotoxin-induced lung injury.28 LXA4 treatment also inhibited TNF-α and IL-1β levels, but LXA4 alone has no effect on levels of inflammatory cells and factors, suggesting that resident macrophages play a crucial role in the resolution of inflammation.
We then investigated the mechanism by which LXA4 inhibits inflammatory cell recruitment. Resident macrophages play crucial role in the pathogenesis of lung injury. When resident macrophages were depleted by clodronate liposomes, we found that neutrophil and recruited macrophage levels were also reduced, suggesting that inflammatory cell infiltration was associated with resident macrophages. In addition, LXA4 had no effect on inflammatory cell numbers after resident macrophage depletion, indicating that its ability to reduce infiltration of inflammatory cells requires resident macrophages.
Chemokines play a central role in inflammatory cell trafficking. CCL2 can regulate the migration and infiltration of monocytes and CXCL2 is able to control neutrophil recruitment during tissue inflammation.29,30 Therefore, we investigated the chemoattractant role of CCL2 and CXCL2 in LPS-induced lung injury in vivo and in vitro. We showed that CCL2 and CXCL2 expression was rapidly induced in LPS-stimulated lung tissues, BALF and primary resident macrophage, but LXA4 treatment reduced their levels. This was consistent with previous studies showing that CCL2 was significantly elevated in BALF recovered from LPS-challenged volunteers and patients with ARDS,31 and that LXA4 decreased the production of MIP-2/CXCL2 in mice.32 We also saw that the inhibitory effects of LXA4 on recruited macrophages and neutrophils were abolished by CCR2 and CXCR2 inhibitors, respectively, and that LXA4 had no effect on CCR2 and CXCR2 expression in recruited macrophages and neutrophils. We also found that CCL2 and CXCL2 were mainly secreted by resident macrophages. These results, taken together, indicate that LXA4 reduced recruited macrophage and neutrophil accumulation via secretion and release CCL2 and CXCL2, respectively, by resident macrophages.
MMP-9 was recently found to play an important role in ALI/ARDS, by regulating neutrophil transmigration into the alveolar space.16 We found that LXA4 inhibited MMP-9 expression and reduced neutrophil accumulation directly, which was consistent with previous study that LXA4 inhibits MMP-9 mRNA and protein levels.33 MMP-9 expression is known to be impacted by the activation of TNF-α. Previously, Ganguly et al reported that TNF-α, and IL-1β expressions were significantly associated with MMP-9 transcriptions during gastric damage.34 We also found that LXA4 inhibited TNF-α level, which in turn reduced the up-regulation of MMP-9. In addition, MMP-9 is mainly produced by neutrophils and its levels can be reduced with a CXCR2 inhibitor. This shows that CXCL2/MMP-9 is involved in LPS-induced neutrophil infiltration, and LXA4 reducing neutrophil recruitment partly through CXCL2/MMP-9 signaling pathway to protect lung tissues.
ALX was the first receptor being identified as a target of LXA4. In this study, we found that LXA4’s protective effect was abrogated by BOC-2, an ALX inhibitor. The LXA4-mediated reduction of CCL2, CXCL2, MMP-9, TNF-α, and IL-1β expression and neutrophil numbers was also abolished by BOC-2, indicating that LXA4’s response is ALX dependent.
There are some shortages to this study. First, there is no genetically-modified animal models used in the study, the mechanism conclusions were from chemical inhibitors. Second, the resident macrophages were sorted by flow cytometry for in vitro experiments, which does not adequately represent the in vivo alveolar environment.
Conclusion
Our study demonstrates that LXA4 exerts protective effects on LPS-induced lung injury by inhibiting the accumulation of recruited macrophages and neutrophils induced by the secretion and release of CCL2 and CCL2, respectively, by resident macrophages. We also showed that MMP-9, which is mainly expressed in neutrophils, was associated with neutrophil migration, and that LXA4 effects are ALX dependent. These results suggest that LXA4 may be a promising therapeutic for preventing and treating ARDS by directly targeting resident macrophage actions.
Acknowledgments
This work was sponsored by grants from the National Natural Science Foundation of China (no. 81870065) and by the Natural Science Foundation of Zhejiang Province (LY18H010005), Provincial Medical and health science and technology project (2021455293) and Research Fund for Lin He’s Academician Workstation of New Medicine and Clinical Translation (19331102).
Disclosure
The authors declare no conflicts of interest.
References
1. Jackson MV, Morrison TJ, Doherty DF, et al. Mitochondrial transfer via tunneling nanotubes is an important mechanism by which mesenchymal stem cells enhance macrophage phagocytosis in the in vitro and in vivo models of ARDS. Stem Cells. 2016;34:L2210–L2223.
2. Duan M, Li WC, Vlahos R, Maxwell MJ, Anderson GP, Hibbs ML. Distinct macrophage subpopulations characterize acute infection and chronic inflammatory lung disease. J Immunol. 2012;189:L946–L955.
3. Aggarwa L, King LS, D’Alessio FR. Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol. 2014;306:L709–L725.
4. Davies LC. Taylor PR:Tissue-resident macrophages: then and now. Immunology. 2015;1444:L541–8.
5. Huang X, Xiu H, Zhang S, Zhang G. The role of macrophages in the pathogenesis of ALI/ARDS. Mediators Inflamm. 2018;13:12649138.
6. Abe Y, Yamamoto N, Nakamura K, et al. IL-13 attenuates early local CXCL2-dependent neutrophil recruitment for Candida albicans clearance during a severe murine systemic infection. Immunobiology. 2019;224:L15–L29.
7. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29:L313–L326.
8. Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:L421–L452.
9. Carson WF, Salter-Green SE, Scola MM, Joshi A, Gallagher KA, Kunkel SL. Enhancement of macrophage inflammatory responses by CCL2 is correlated with increased miR-9 expression and downregulation of the ERK1/2 phosphatase Dusp6. Cell Immunol. 2017;314:L63–L72.
10. Martinu T, Gowdy KM, Nugent JL, et al. Role of C-C motif ligand 2 and C-C motif receptor 2 in murine pulmonary graft-versus-host disease after lipopolysaccharide inhalations. Am J Respir Cell Mol Biol. 2014;51:L510–L521.
11. Soccal PM, Gasche Y, Pache JC, et al. Matrix metalloproteinases correlate with alveolar-capillary permeability alteration in lung ischemia-reperfusion injury. Transplantation. 2000;70:L998–L1005.
12. Perez-Ramos J, Segura-Valdez ML, Vanda B, Selman M, Pardo A. Matrix metalloproteinases 2, 9, and 13, and tissue inhibitors of metalloproteinases 1 and 2 in experimental lung silicosis. Am J Respir Crit Care Med. 1999;160:L1274–L1282.
13. Fligiel SE, Standiford T, Fligiel HM, et al. Matrix metalloproteinases and matrix metalloproteinase inhibitors in acute lung injury. Hum Pathol. 2006;37:L422–L430.
14. Hsu AT, Barrett CD, DeBusk GM, et al. Kinetics and role of plasma matrix metalloproteinase-9 expression in acute lung injury and the acute respiratory distress syndrome. Shock. 2015;44:L128–L136.
15. Huang CH, Yang ML, Tsai CH, Li YC, Lin YJ, Kuan YH. Ginkgo biloba leaves extract (EGB 761) attenuates lipopolysaccharide-induced acute lung injury via inhibition of oxidative stress and NF-kB-dependent matrix metalloproteinase-9 pathway. Phytomedicine. 2013;20:L303–L309.
16. Delclaux C, Delacourt C, Ortho D, et al. Role of gelatinase B and elastase in human polymorphonuclear neutrophil migration across basement membrane. Am J Respir Cell Mol Biol. 1996;14:L288–L295.
17. Barnig C, Frossard N, Levy BD. Towards targeting resolution pathways of airway inflammation in asthma. Pharmacol Ther. 2018;186:L98–L113.
18. Yang CY, Lian QQ, Yang L, et al. Contribution of CFTR to alveolar fluid clearance by lipoxin A4 via PI3K/Akt pathway in LPS-induced acute lung injury. Mediators Inflamm. 2013;862628.
19. Wang Q, Lian QQ, Li R, et al. Lipoxin A(4) activates alveolar epithelial sodium channel,Na,K-ATPase, and increases alveolar fluid clearance. Am J Respir Cell Mol Biol. 2013;48:L610–L618.
20. Zheng S, D’Souza VK, Bartis D, et al. Lipoxin A4 promotes lung epithelial repair whilst inhibiting fibroblast proliferation. ERJ Open Res. 2016;2:00079–2015.
21. Fukuda S, Nagano M, Yamashita T, et al. Functional endothelial progenitor cells selectively recruit neurovascular protective monocyte-derived F4/80(+)/Ly6c(+) macrophages in a mouse model of retinal degeneration. Stem Cells. 2013;31:L2149–L2161.
22. Ueno M, Maeno T, Nishimura S, et al. Alendronate inhalation ameliorates elastase-induced pulmonary emphysema in mice by induction of apoptosis of alveolar macrophage. Nat Commun. 2015;6:6332.
23. Venosa A, Katzen J, Tomer Y, et al. Epithelial expression of an interstitial lung disease-associated mutation in surfactant Protein-C modulates recruitment and activation of key myeloid cell populations in mice. J Immunol. 2019;2029:L2760–2771.
24. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:L1334–L1349.
25. Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet. 2007;369:1553–1564.
26. Lomas-Neira J, Chung CS, Perl M, Gregory S, Biffl W, Ayala A. Role of alveolar macrophage and migrating neutrophils in hemorrhage-induced priming for ALI subsequent to septic challenge. Am J Physiol Lung Cell Mol Physiol. 2006;290:L51–L58.
27. Johnston LK, Rims CR, Gill SE, McGuire JK, Manicone AM. Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am J Respir Cell Mol Biol. 2012;47:L417–L426.
28. Janssen WJ, Barthel L, Muldrow A, et al. Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am J Respir Crit Care Med. 2011;184:L547–L560.
29. Lee YG, Jeong JJ, Nyenhuis S, et al. Recruited alveolar macrophages, in response to airway epithelial-derived monocyte chemoattractant protein 1/CCl2, regulate airway inflammation and remodeling in allergic asthma. Am J Respir Cell Mol Biol. 2015;52:L772–L784.
30. KDe F, Dudeck A, Hasenberg M, et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. 2013;121:L4930–L4937.
31. Williams AE, José RJ, Mercer PF, et al. Evidence for chemokine synergy during neutrophil migration in ARDS. Thorax. 2017;72:L66–L73.
32. Fang X, Abbott J, Cheng L, et al. Human mesenchymal Stem (Stromal) cells promote the resolution of acute lung injury in part through Lipoxin A4. J Immunol. 2015(195):L875–L881.
33. Zong L, Li J, Chen X, et al. Lipoxin A4 attenuates cell invasion by inhibiting ROS/ERK/MMP pathway in pancreatic cancer. Oxid Med Cell Longev. 2016;6815727.
34. Ganguly K, Swarnakar S. Induction of matrix metalloproteinase-9 and −3 in nonsteroidal anti-inflammatory drug-induced acute gastric ulcers in mice: regulation by melatonin. J Pineal Res. 2009;471:1.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.