")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Lurasidone for the treatment of bipolar depression: an evidence-based review
Authors Franklin R, Zorowitz S, Corse A, Widge A, Deckersbach T
Received 8 April 2015
Accepted for publication 12 June 2015
Published 19 August 2015 Volume 2015:11 Pages 2143—2152
DOI https://doi.org/10.2147/NDT.S50961
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Rachel Franklin,1 Sam Zorowitz,1 Andrew K Corse,1 Alik S Widge,2 Thilo Deckersbach1
1Division of Neurotherapeutics, Department of Psychiatry, Massachusetts General Hospital, Harvard Medical School, Charlestown, 2Picower Institute for Learning and Memory, Massachusetts Institute of Technology, Cambridge, MA, USA
Abstract: Bipolar disorder (BD) is a debilitating and difficult-to-treat psychiatric disease that presents a serious burden to patients’ lives as well as health care systems around the world. The essential diagnostic criterion for BD is episodes of mania or hypomania; however, the patients report that the majority of their time is spent in a depressive phase. Current treatment options for this component of BD have yet to achieve satisfactory remission rates. Lurasidone is a drug in the benzisothiazole class approved by the US Food and Drug Administration in June 2013 for the acute treatment of bipolar depression. Its pharmacological profile features high-affinity antagonism at D2, 5-HT2A, and 5-HT7 receptors; moderate-affinity antagonism at α2C-adrenergic receptors; low- to very low-affinity antagonism at α1A-adrenergic, α2A-adrenergic, H1, M1, and 5-HT2C receptors; and high-affinity partial agonism at 5-HT1A. Preliminary findings from two recent double-blinded clinical trials suggest that lurasidone is efficacious in treating bipolar I depression, with clinical effects manifesting as early as the first 2–3 weeks of treatment (as measured by the Montgomery–Åsberg Depression Rating Scale and Clinical Global Impressions Scale for use in bipolar illness). Its therapeutic benefit appears to be comparable to the current US Food and Drug Administration-indicated treatments: quetiapine and olanzapine–fluoxetine, according to a measure of effect size known as number needed to treat. These studies reported relatively limited extrapyramidal and metabolic side effects as a result of treatment with lurasidone, with the most common side effect being nausea. Safety data drawn from these studies, as well as a more extensive body of schizophrenia research, indicate that in comparison with other atypical antipsychotics, treatment with lurasidone is less likely to result in metabolic side effects such as weight gain or disturbances of serum glucose or lipid levels. Lurasidone holds clinical potential as a novel, efficacious pharmacological treatment for bipolar depression. However, current data on its use for the treatment of BD are limited, and more extensive research, both longer in duration as well as independently conducted, is needed.
Keywords: lurasidone, bipolar depression, bipolar disorder, atypical antipsychotic
Introduction
Bipolar disorder (BD) is a chronic and often severely disabling psychiatric condition. Collectively, forms of BD (type I, type II, or not otherwise specified) are estimated to affect approximately 4.4% of Americans or about 12.7 million people.1,2 Worldwide, BD was ranked 18th by the World Health Organization in worldwide causes of years lived with disability, surpassing all forms of cancer.3 The economic burden in terms of cost of health care for patients with BD is estimated to be four times greater than that for patients without mental disorders.4 Overall, the total economic burden of BD to the US economy is difficult to estimate, but one 24-year-old study approximated the figure to be nearly $45 billion,5 adjusted for inflation; in 2015, the estimate will be closer to $78 billion.6
The essential diagnostic criterion for BD is episodes of elevated/irritable mood, usually either mania (type I) or hypomania (type II).7 Despite this, BD patients have reported through surveys and clinical status ratings that they spend the majority of their time in a depressive phase8 and that these episodes are more disruptive to their functioning than mania.9 Bipolar depression is notably distinct from unipolar depression in that its phenomenological features more commonly include psychosis, depressive mixed state, anxiety, agitation, anergic depression, irritability, and anger attacks.10 Moreover, the increased risk of suicide in patients with BD (whose rates of suicide are one of the highest among patients with psychiatric illness)11,12 is observed predominantly during the depressive phase.11,12
Treatment of BD aims for remission of symptoms, both manic and depressive. Efficacious treatment, however, is not easy to achieve; the remission rate for bipolar mania and depression after acute treatment is reported to be approximately 40%–50% and 25%–60%, respectively.13,14 (This estimate is possibly misrepresentative of long-term remission, as a common feature of BD depression is insensitivity to acute treatment).10 Factors complicating treatment include tolerance to medications and significant likelihood of relapse,15 even with continual pharmacological maintenance.
Pharmacotherapy options for BD are typically grouped by the targeted symptoms. Lithium, first- and second-generation antipsychotics, valproate, and carbamazepine are usually prescribed for the treatment of acute mania, while quetiapine, olanzapine–fluoxetine, lamotrigine (maintenance), and antidepressants in conjunction with an antimanic agent (acute) are usually prescribed for the treatment of depressive symptoms.16,17 Currently, only quetiapine, olanzapine–fluoxetine combination, and lurasidone are approved by the Food and Drug Administration (FDA) to treat bipolar depression. Further treatment options are greatly needed, as bipolar depression, the most prevalent and fatal feature of BD, is often not well covered by these regimens and nonresponse to first-line options is perhaps as high as 40%.18 In a review of therapeutic options for treatment-resistant BD depression, Sienaert et al found that although promising, current research for this diagnosis is scarce.18 In this review, we aim to summarize recent available literature regarding the compound lurasidone and its role in the treatment of bipolar depression.
Pharmacology and pharmacokinetics
The biological basis of BD depression remains unknown: one current theory, developed by Fountoulakis et al19 postulates that unlike unipolar depression, norepinephrine reuptake and 5-HT1A agonism are heavily implicated as core deficits. Pharmacologically, lurasidone is appropriate to treat these features, though long-term clinical data in conjunction with neurobiological models remain to be established.
Lurasidone or (3aR,4S,7R,7aS)-2-{(1R,2R)-2-[4-(1,2-benzisothiazol-3-yl)piperazin-1-ylmethyl]cyclohexylmethyl}hexahydro-4,7-methano-2H-isoindole-1,3-dione hydrochloride20 (as it is known chemically) (Figure 1 for 3D chemical structure) is a chemical in the benzisothiazole class, structurally related to perospirone and ziprasidone, as well as the benzisoxazole derivative risperidone.21 In vitro assays have demonstrated that lurasidone is a full antagonist at D2 (Ki=1.68 nM)22 (Table 1) and 5-HT2A (Ki=2.03 nM) receptor subtypes, a property shared by other atypical antipsychotics such as risperidone, olanzapine, quetiapine, clozapine, and aripiprazole.22 In comparison to similar drugs, lurasidone has the highest binding affinity for the 5-HT7 receptor (Ki=0.5 nM).22 Other notable pharmacological properties include moderate-affinity α2C-adrenergic antagonism, partial agonism at the 5-HT1A receptor, and low affinity for the muscarinic (M1), histamine (H1) (both Ki values >1,000 nM), 5-HT2C (415 nM), α1A (47.9 nM), and α2A (40.7 nM) adrenergic receptors.23 Table 1 shows the pharmacological profile of lurasidone.
 | Figure 1 Three-dimensional structure of lurasidone, also known as (3aR,4S, 7R,7aS)-2-{(1R,2R)-2-[4-(1,2-benzisothiazol-3-yl)piperazin-1-ylmethyl]cyclohexylmethyl}hexahydro-4,7-methano-2H-isoindole-1,3-dione hydrochloride or Latuda. |
 | Table 1 Binding profile of the chemical lurasidone: endogenous neurotransmitter, characteristic activity type, and experimental Ki values associated with major receptors |
There is limited evidence that lurasidone may provide cognitive benefits due to several properties of its binding profile. Blocking activity at the 5-HT7 receptor (Ki=0.5 nM) may contribute to therapeutic benefits as suggested by a study of the drug’s effect on learning when given in conjunction with an N-methyl-D-aspartate blocker.22 Ishiyama et al found that rats were prevented from learning a passive-avoidance shock response when administered with N-methyl-D-aspartate receptor antagonist dizocilpine; this inhibition, however, was dose dependently reversed when the animals were given lurasidone, regardless of pre- or post-training administration. This effect suggests that lurasidone may contribute to restoration of the memory consolidation process and may therefore have benefits in reducing the cognitive impairments observed in bipolar depression.23
Additionally, the drug acts with moderate binding affinity as an antagonist at α2C-adrenergic receptor (Ki=10.8 nM), overexpression of which has been shown to increase impairment in the Morris water maze,24 further lending support to possible procognitive effects of lurasidone.
Lurasidone also interacts as a partial agonist at the 5-HT1A receptor (Ki=6.75 nM),22 activation of which has been shown to increase adult neuronal proliferation in the dentate gyrus of the hippocampus.25 This influence in neurogenesis may be of cognitive therapeutic benefit, as demonstrated by pilot clinical studies using antipsychotics with adjunct 5-HT1A agonists.26
Overall, based on neurobiological evidence, lurasidone may provide advantages in learning and memory via high-affinity 5-HT7 and moderate-affinity α2C-adrenergic antagonism, as well as partial agonism at the 5-HT1A receptor.22,23,27
The 5-HT7 receptor-blocking activity of lurasidone may also underlie antidepressant properties of the drug. Selective antagonists and experiments in 5-HT7 gene knockout animals have demonstrated anxiolytic-like and antidepressant-like effects in rodents, which demonstrated improvement on the Vogel drinking, elevated plus-maze, four-plate test, forced swimming, and tail suspension tests.28,29 It is far from proven that these tests translate well to human depression, unipolar or bipolar, but they may be useful screens. These mood effects may be mediated by a cortical and hippocampal dopamine efflux caused by activity at the 5-HT7 and 5-HT1A receptors.30
Similarly, lurasidone injected subcutaneously in adolescent rats modulates levels of brain-derived neurotrophic factor by preventing adult decreases in brain-derived neurotrophic factor expression normally seen in animals exposed to prenatal stress.31 This effect provides support for the neurotrophic hypothesis of depression32 and further implicates lurasidone as a potentially beneficial therapy for bipolar depression.
Clinically, one of the most promising features of lurasidone is its low affinity for muscarinic (M1), histamine (H1) (both Ki values >1,000 nM), 5-HT2C (415 nM), α1A- (47.9 nM), and α2A-(40.7 nM) adrenergic receptors.33 Well-known side effects of many antipsychotics, such as sedation, weight gain, and negative cognitive symptoms, have been only minimally observed in both animal and human trials of lurasidone (see “Safety and tolerability” section). This is thought to be due to the low levels of activity of lurasidone at H134 and 5-HT2C35 receptors.22 Decreased interaction with muscarinic and α-1 adrenergic receptors may prevent negative cognitive and cardiovascular side effects.36
Despite being a high-affinity D2 receptor antagonist, historically a harbinger of severe neurological side effects,33 in vivo studies of lurasidone to date have observed fewer central nervous system’s depressive effects, extrapyramidal symptoms, and anticholinergic side effects (such as dry mouth or amnesia)22 than other typical and even other atypical antipsychotics. This may be explained in part by the drug’s receptor saturation point. A study of lurasidone’s dopamine D2 receptor binding in healthy males using positron emission tomography demonstrated that doses less than 40 mg did not achieve adequate binding to reach antipsychotic effect;37 however, increasing the dose from 60 mg to 80 mg did not effectively change receptor occupancy (77%–84% and 73%–79%, respectively). This curve may explain, in part, why incidents of parkinsonism are infrequently seen, as there appears to be a dopamine receptor saturation point well below the threshold for extrapyramidal symptomology.
Lurasidone is primarily metabolized by CYP3A4, with the most common pathways being oxidative N-dealkylation, hydroxylation of the norbornane ring, and S-oxidation. The half-life, described in the product label as 18 hours, has been reported in some studies to be as long as 37 hours, given repeated oral doses at steady state.33 Several known pharmacologically active metabolites have been described such as ID-14283, ID-14326, and ID-11614 (25%, 3%, and <1% of parent exposure, respectively).33 In vitro studies demonstrated that both ID-14283 and ID-14236 showed affinity for D2 and 5-HT2A, as well as partial agonism at 5-HT1A and antagonism at 5-HT7. ID-14283 may contribute to the parent compound’s efficacy, but has a shorter half-life (7 hours).21
Therapeutic efficacy
Though there now exists a growing body of literature detailing the pharmacokinetic properties of lurasidone, a complementary body of literature documenting its efficacy for the treatment of bipolar I disorder is comparatively less due to the short time since initial approval.38 At the time of writing, only two controlled clinical trials have begun to investigate lurasidone as a treatment for bipolar I depression: as a monotherapy39 and as an adjunct treatment with lithium or valproate.40
The first of the two clinical trials was a randomized, double-blind, placebo-controlled, fixed-flexible dose study investigating the efficacy of lurasidone as a monotherapy treatment for bipolar I depression. Patients were randomly assigned to a 6-week treatment group of 20–60 mg/day of lurasidone (N=166; mean daily dose =31.8 mg), 80–120 mg/day of lurasidone (N=169; mean daily dose =82.0 mg), or a placebo (N=170). The primary outcome measure was the change between baseline and week 6 scores on the Montgomery–Åsberg Depression Rating Scale (MADRS); a secondary outcome measure was the change in the depression severity score on the Clinical Global Impressions Scale for use in bipolar illness (CGI-BP).
The investigators reported significantly greater decreases in MADRS scores in both the 20–60 mg/day group (−15.4) and in the 80–120 mg/day group (−15.4) as compared to the placebo group (−10.7) (Figure 2A). Moreover, a significantly greater reduction in core depressive symptoms (MADRS-6 subscale score) between baseline and week 6 was also observed for the lurasidone 20–60 mg group (−10.4) and the lurasidone 80–120 mg group (−10.4) as compared to the placebo group (−6.9). A similar pattern was found in the CGI-BP scores for the 20–60 mg/day group (−1.8) and the 80–120 mg/day group (−1.7) as compared to placebo (−1.1). Notably, these statistically significant decreases in MADRS and CGI-BP scores were observed in both dosage groups early in the course of treatment; differences between the groups in reported MADRS scores began at week 2, whereas differences in reported CGI-BP scores began at week 1 for the 80–120 mg group and week 2 for the 20–60 mg group. Caution is warranted, however, in interpreting these results as the authors included neither standard deviations nor confidence intervals in the differences in MADRS or CGI-BP scores. Nonetheless 53% and 51% of subjects met response criteria (defined as ≥50% reduction from baseline in MADRS total) after 6 weeks of treatment with lurasidone 20–60 mg and lurasidone 80–120 mg, respectively, as compared to only 30% in the placebo group. Finally, the lurasidone 20–60 mg and lurasidone 80–120 mg groups both showed proportionately higher remission rates (42%; 40%) than did the placebo group (25%).
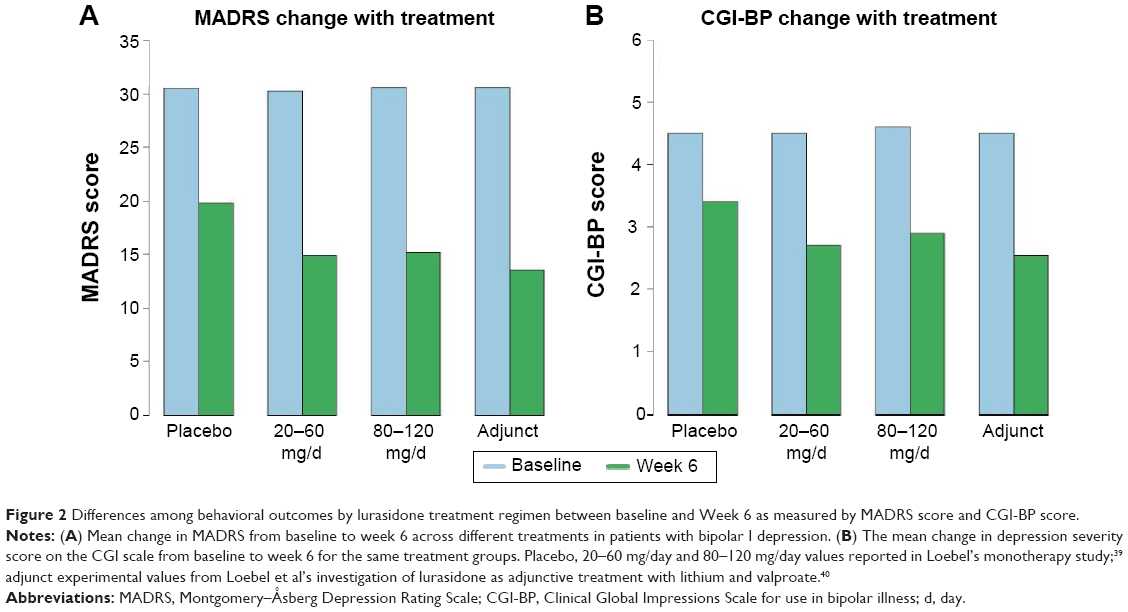 | Figure 2 Differences among behavioral outcomes by lurasidone treatment regimen between baseline and Week 6 as measured by MADRS score and CGI-BP score. |
The second of the two clinical trials was also double blinded, but investigated lurasidone as an adjunct to lithium or valproate in the treatment of bipolar I depression. Inclusion criteria required that patients should not have exhibited a response to a 28-day minimum trial of either lithium or valproate, prescribed at therapeutic levels, as determined by a health care professional. Qualifying patients were then randomly assigned to receive 6 weeks of lurasidone (N=183) or placebo (N=165) in addition to continuing their previously prescribed medication. Patients receiving lurasidone were initially administered a dosage of 60 mg; with each week after the first, health care professionals were allowed to adjust lurasidone doses by increments of 20 mg within a range of 20–120 mg/day (the mean daily dose was 66.3 mg). As in the monotherapy study, the primary and secondary outcomes of interest were changes in the MADRS and CGI-BP scores, respectively, between baseline and week 6 of the trial.
Similar to the results of the monotherapy trial, significantly greater decreases in MADRS and CGI-BP scores were observed for the adjunct lurasidone group (−17.1; −1.96) than for the placebo group (−13.5; −1.51) (Figure 2B). These differences were first observed beginning at week 3 for the MADRS as well as at week 2 for the CGI-BP, and remained reliably different for the remainder of the trial. As mentioned earlier, caution is again warranted in interpreting these results as the authors included neither standard deviations nor confidence intervals in the differences in MADRS or CGI-BP scores. Finally, greater proportions of patients met response criteria (≥50% reduction from baseline in MADRS total) and remission (57%; 50%) than were observed in the placebo group (42%; 35%).
Although promising, these preliminary results regarding the therapeutic efficacy of lurasidone for the treatment of bipolar I depression should be considered with respect to the efficacy of preexisting treatments, specifically quetiapine and olanzapine–fluoxetine combination. Recently, Citrome et al41 compared the benefits of these three treatments using the number needed to treat (NNT) measure.42 As an indicator of effect size, NNT measures how many patients would need to be treated with one medication, on average, to observe one additional beneficial outcome of interest. Low NNTs are indicative of large effect sizes, with an example NNT of 2 indicating that on average, one of every two patients treated with a medication would receive the desired clinical benefit.
Citrome et al calculated the NNT for clinical response and remission for lurasidone based on the above two clinical trials, as well as the NNT for quetiapine (immediate and extended release) and olanzapine–fluoxetine combination based on published 8-week trials and product labeling (see article for details).43 Of the three reviewed quetiapine studies, two studies set dosing levels at 300 mg/day and 600 mg/day;43,44 the other prescribed quetiapine studies set dosing levels at 400 mg/day and 800 mg/day.45 In the two reviewed olanzapine–fluoxetine combination studies, the dosing levels were set at 6/25 mg/day, 6/50 mg/day, or 12/50 mg/day.46,47 Clinical response was defined as a ≥50% reduction from baseline on the MADRS, whereas remission was defined as a final MADRS score of ≤12. For clinical response, NNT values of 5 (95% CI 3–8) and 5 (95% CI 4–11) were found for patients prescribed monotherapy lurasidone at low (20–60 mg/day) or high (80–120 mg/day) doses; an NNT value of 7 (95%, 4–24) was found for adjunct lurasidone (20–120 mg/day). In comparison, NNT values of 6 and 5 were found for quetiapine and olanzapine–fluoxetine combination in clinical response, respectively. For clinical remission, an NNT value of 6 (95%, 4–14) was found for patients with a low dose (20–60 mg/day) of monotherapy lurasidone; an NNT value of 7 (95%, 4–21) was found for patients with a high dose (80–120 mg/day) of monotherapy lurasidone; and an NNT value of 7 (95%, 4–23) was found for patients with 20–120 mg/day adjunct dose of lurasidone. In comparison, NNT values of 6 and 4 were found for quetiapine and olanzapine–fluoxetine combination in clinical response, respectively.41,42 It is worth noting that 95% confidence intervals were similarly not reported for the NNT values of the comparative treatments. Perhaps related to this, Citrome et al offer a conservative conclusion regarding the therapeutic efficacy of lurasidone relative to its predecessors, stating only that it yielded comparable benefits for treatment.
In summary, preliminary findings from two recent double-blinded clinical trials suggest that lurasidone is efficacious in treating bipolar I depression, with clinical effects manifesting as early as the first 2–3 weeks of treatment (as measured by MADRS and CGI-BP). Its therapeutic benefit, however, appears to be comparable to other atypical antipsychotics, on average. This is in line with a recent meta-analysis of 212 clinical trials of antipsychotic drugs in schizophrenia, spanning more than 43,000 patients, that found lurasidone to be similar (if not weaker) than quetiapine and olanzapine in alleviating symptoms.48 Of course, it is still early in the investigation of lurasidone, with both clinical trials detailed above extended to 24-week trials and data from weeks 6 to 24 still unpublished. Future research will no doubt shed further light as to the true utility of lurasidone for bipolar I depression.
The tolerability and safety of lurasidone
If lurasidone is neither superior nor inferior to other atypicals, might it have some advantage on the basis of side effects? Evidence on the tolerability and safety of lurasidone comes from two clinical trials in patients with bipolar depression and many clinical trials in schizophrenia patients. Head-to-head randomized clinical trials need to be conducted in order to conclusively evidence the relative superior or inferior efficacy of lurasidone compared to other atypical antipsychotics. Although this article aims to review lurasidone in the context of bipolar depression, because there are only two published short-term studies of the drug in that patient population, it is important to also consider its safety and tolerability within schizophrenia patients.
Lurasidone was granted FDA approval for the treatment of schizophrenia in October 2010 and then in June 2013 for bipolar depression.49 In reviewing relevant literature from 2009 to present, lurasidone has some evidence for a safety advantage over other recently approved second-generation atypical antipsychotics, including paliperidone, iloperidone, and asenapine.41,50,51 De Hert et al showed that treatment with lurasidone in BD and schizophrenia is less likely to result in hypercholesterolemia, hyperglycemia, hyperlipidemia, and other metabolic side effects such as weight gain.41,50,51 Short-term studies of lurasidone in schizophrenia patients suggest that it is tolerable, safe, and has a low discontinuation rate.21 In schizophrenia trials, the most common adverse events included nausea, somnolence, akathisia, sedation, and parkinsonism.52 In an open-label 6-month study in which 198 schizophrenic patients who were stable but symptomatic were switched to lurasidone, a total of 66.2% of patients had at least one adverse event, most of which were only mild or moderate.53 The study concluded that the drug was generally well tolerated and the investigators did not identify any new safety concerns. A handful of 1-year studies suggest that the long-term use of lurasidone is well tolerated in patients with schizophrenia, having similar discontinuation rates to patients receiving treatment with quetiapine XR (7% vs 5%).54,55 In this population, lurasidone appears to cause weight gain, but only modestly.21 Short-term clinical trials reported a mean increase in body weight of 26 kg in subjects treated with placebo and an increase in body weight of 75 kg in subjects treated with lurasidone. Only 5.6% of subjects in the lurasidone group showed a weight gain of 7% or more, as compared with 4% of subjects in the placebo group.56 Caccia et al found that the weight gain was not dose related.21 In Citrome’s 6-month open-label study, 17 subjects reported weight loss (>7% of their body weight) from the study baseline, whereas only 14 patients reported weight gain of the same proportion.53 Although there is not much evidence regarding weight gain in long-term use of lurasidone in schizophrenia patients, lurasidone showed a more favorable effect on body weight compared with quetiapine and risperidone in two long-term trials.50,51
Evidence does not suggest that lurasidone induces clinically significant changes in glucose levels or other metabolic parameters.57,58 In a pooled data analysis from short-term studies on schizophrenia, Cucchiaro et al showed that the mean increases in fasting glucose levels were 1.4 mg/dL in subjects receiving lurasidone and 0.6 mg/dL in subjects receiving placebo, which is not a statistically significant difference.59 A long-term (12-month) study comparing lurasidone with quetiapine XR showed that there was no negative influence on glucose levels when taking lurasidone for an extended period.54
In short-term studies of lurasidone, there were no adverse events related to hyperlipidemia and there were no increases in low-density lipoprotein cholesterol, level of total cholesterol, or triglycerides.59 In fact, one double-blind comparison study found that a decrease in triglycerides (−2.6 mg/dL) was associated with lurasidone, compared to an increase (+22.4 mg/dL) associated with ziprasidone.60 In a long-term comparison study of risperidone and lurasidone, the median changes in low-density lipoprotein cholesterol levels and triglycerides in the lurasidone group were −2.0 mg/dL and −3.5 mg/dL, respectively.55
Lurasidone is absorbed quickly, reaching peak serum concentration in 1–3 hours.38 Maximum concentration increases three-fold when administered with food, indicating that the site of absorption is likely the stomach.61 In a study aiming to elucidate the target amount of food needed to maximize serum concentration, the mean Cmax was 52.9 ng/mL in the fasted state, 161 ng/mL for the 350 kcal/high-fat meal, 135 ng/mL for the 500 kcal/high-fat meal, and 131 ng/mL for the 800–1,000 kcal/high-fat meal.62 Therefore, lurasidone is advised to be taken with about 350 kcal, although fat content per se does not appear to have an influence. Once absorbed, 99.8% of it is bound to albumin and α-1-glycoprotein.33
Significant increases in prolactin levels were associated with lurasidone in several short-term studies in schizophrenia.21 Pooled data from five short-term studies indicated that mean changes in prolactin levels were −0.6 ng/mL in the placebo group and +1.1 ng/mL in the lurasidone group.59 However, the prolactin changes due to lurasidone may be lower than those due to other similar drugs. One study found that mean changes in prolactin levels were greater in patients receiving haloperidol (+8.5 ng/mL) and olanzapine (+3.7 ng/mL) than lurasidone (+1.1 ng/mL).63 A long-term study that compared risperidone with lurasidone showed median changes in prolactin levels in the 0 ng/mL lurasidone group in male subjects and +0.95 ng/mL lurasidone group among female subjects. These levels were lower than those of the risperidone group, which were +7.50 ng/mL in males and +26.40 ng/mL in females.55
To date, there have been two peer-reviewed articles that investigated lurasidone in the treatment of bipolar I depression. Both of these were 6-week, randomized, double-blind, and placebo-controlled trials conducted by Loebel et al as reviewed earlier. In the monotherapy study, dropout rates due to adverse events were similar across all three groups, ranging from 5.9% to 6.6%. There were no statistically significant differences in the effect of treatment on metabolic parameters reported, including weight, lipid, prolactin or cholesterol levels, waist circumference, glycemic control, or electrocardiogram readings.39 The percent of patients with a 7% or greater increase in weight from baseline was 0.7% in both the 80–120 mg and placebo groups and 4.2% in the 20–60 mg group. In general, the high-dose group experienced more adverse effects. Nausea was the most common (N=29, 17.4%), followed by akathisia (N=18, 10.8%), headache (N=15, 9.0%), extrapyramidal events (N=15, 9.0%), and sedation (N=12, 7.2%).39 Rates of serious adverse events were low across groups, and there were no deaths, suicidal behaviors, or suicides during the course of the study.39 In the second clinical trial, discontinuation rates were lower in the lurasidone + lithium/valproate group (6%) compared to the placebo + lithium/valproate group (7.9%).40 The lurasidone + lithium/valproate group experienced more adverse effects than did the placebo + lithium/valproate group. In the group receiving lurasidone, the most common adverse event was nausea (N=32, 17.5%), followed by extrapyramidal events (N=28, 15.3%), somnolence (N=16, 8.7%), tremor (N=15, 8.2%), and akathisia (N=14, 7.7%).40 The percentage of subjects receiving lurasidone + lithium/valproate who experienced at least one adverse event was 63.9%, while the percentage of placebo + lithium/valproate subjects experiencing at least one adverse event was 57.7%. In this study, treatment with lurasidone was not associated with significant increases in weight, lipids, glucose, or prolactin levels.40
Dose adjustments are recommended for patients with either liver or kidney problems. Most of the deactivation of the drug is hepatic, and it should be taken with caution when in conjunction with strong CYP3A4 inhibitors (such as ketoconazole) or inducers (such as rifampin).38 Smoking is not expected to have an effect on the pharmacokinetics of lurasidone because it is not a substrate for CYP1A2.37 For patients with hepatic impairment, maximum doses should be 40 mg/day or 80 mg/day, depending on the Child–Pugh score. For patients with renal impairment, a maximum dose of 80 mg/day should be observed.38 Safety and tolerability have not been established in pediatric or geriatric populations. However, according to industry data, no dose adjustment was needed in 65–85-year old schizophrenia patients.64
The adverse effects experienced by patients receiving lurasidone for bipolar depression in the two clinical trials were similar to those experienced by schizophrenia patients receiving lurasidone. These two bipolar depression studies suggest that lurasidone could be safely used both as a monotherapy and as an adjunctive therapy.27 However, there is no evidence for long-term safety of lurasidone in bipolar patients, and additional clinical trials need to be conducted to establish sustained safety and tolerability within the bipolar patient population. There is reason for optimism; ziprasidone, a chemically similar agent, was suggested to have reduced metabolic side effects in early data, and these impressions have since been verified in much larger scale studies. A 2004 comparison study of risperidone and ziprasidone by Addington et al showed that ziprasidone had a significantly smaller effect on weight gain. In total, 16.0% (N=20) of risperidone patients experienced significant weight gain (≥7%), while only 8.2% of ziprasidone patients did.65 A 2014 meta-analysis by Oh et al of 73 randomized clinical trials corroborated these early results, showing that treatment with ziprasidone, compared to six other antipsychotics, resulted in a substantially smaller chance of significant weight gain (odds ratios ranging from only 0.02 to 0.44).66
Conclusion
Lurasidone is a member of the second-generation atypical antipsychotics. Its recent approval by the FDA for the treatment of bipolar depression has led to limited but potentially promising potential therapeutic efficacy for this disabling, prevalent, treatment-resistant disease. Lurasidone interacts as an antagonist with high affinity to D2 and 5-HT2A receptors, as do other members of its pharmaceutical class. Lurasidone, however, somewhat uniquely binds with high affinity to the 5-HT7 receptor, as well as acts as a partial agonist at the 5-HT1A receptor. These neurobiological properties may lend procognitive, antidepressant, or anxiolytic effects to lurasidone. In addition, lurasidone interacts negligibly with receptors implicated in sedation, weight gain, and cardiovascular symptoms associated with other atypical antipsychotics.
Limited data are available concerning lurasidone’s clinical efficacy. Two 6-week double-blinded and randomized trials published promising decreases in MADRS and GCI-BP scores after treatment with lurasidone as a monotherapy as well as an adjunctive therapy with lithium or valproate. Moreover, these studies reported relatively limited extrapyramidal and metabolic side effects as a result of treatment with lurasidone, with the most common side effect being nausea. The caveat is that both studies were funded and overseen by lurasidone’s manufacturer and have not yet been independently replicated. The average weight change throughout the course of both studies was statistically insignificant. These advantages over other drugs within the same class, if further validated, could someday make lurasidone a preferable agent for patients with bipolar depression.
The safety of lurasidone for the treatment of bipolar depression is not strongly established, as there are few clinical data available. Safety data drawn from schizophrenia research indicate that, in comparison with other atypical antipsychotics, treatment with lurasidone is less likely to result in metabolic side effects such as weight gain or disturbances of serum glucose or lipid levels. Lurasidone is best absorbed with approximately 350 kcal of food and is hepatically metabolized by CYP3A4; thus, it should not be taken in conjunction with strong CYP3A4 inhibitors or inducers. Overall, the rates of adverse effects have been low; however, the data with bipolar depression patients remain limited.
In conclusion, lurasidone holds clinical potential as a novel, efficacious pharmacological treatment for bipolar depression. However, current data on its use in the bipolar I population are limited; more extensive research, both longer in duration as well as independently conducted, is needed.
Disclosure
The authors report no conflicts of interest in this work.
References
Merikangas KR, Akiskal HS, Angst J, et al. Lifetime and 12-month prevalence of bipolar spectrum disorder in the national comorbidity survey replication. Arch Gen Psychiatry. 2007;64(5):543–552. | ||
2007 World Population Data Sheet. Available from: http://www.prb.org/Publications/Datasheets/2007/2007WorldPopulationDataSheet.aspx. Accessed March 23, 2015. | ||
Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet. 2013;380(9859):2163–2196. | ||
Altamura AC, Serati M, Albano A, Paoli RA, Glick ID, Dell’Osso B. An epidemiologic and clinical overview of medical and psychopathological comorbidities in major psychoses. Eur Arch Psychiatry Clin Neurosci. 2011;261(7):489–508. | ||
Kleinman LS, Lowin A, Flood E, Gandhi G, Edgell E, Revicki DA. Costs of bipolar disorder. Pharmacoeconomics. 2003;21(9):601–622. | ||
Inflation Calculator: Bureau of Labor Statistics. Available from: http://www.bls.gov/data/inflation_calculator.htm. Accessed March 26, 2015. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM). 5th ed. Arlington, VA: American Psychiatric Publishing; 2013. | ||
Judd LL, Akiskal HS, Schettler PJ, et al. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry. 2002;59(6):530–537. | ||
Calabrese JR, Hirschfeld R, Frye MA, Reed ML. Impact of depressive symptoms compared with manic symptoms in bipolar disorder: results of a US community-based sample. J Clin Psychiatry. 2004;65(11):1499–1504. | ||
El-Mallakh RS, Ghaemi SN. Bipolar Depression: A Comprehensive Guide. American Psychiatric Publishing; 2007. Available from: http://books.google.com/books?id=fFKgiZU2KscC | ||
Tondo L, Isacsson G, Baldessarini RJ. Suicidal behaviour in bipolar disorder. CNS Drugs. 2003;17(7):491–511. | ||
Malhi GS, Adams D, Berk M. Medicating mood with maintenance in mind: bipolar depression pharmacotherapy. Bipolar Disord. 2009;11(s2):55–76. | ||
Beyer JL. An evidence-based medicine strategy for achieving remission in bipolar disorder. J Clin Psychiatry. 2007;69:31–37. | ||
Woo YS, Wang HR, Bahk W-M. Lurasidone as a potential therapy for bipolar disorder. Neuropsychiatr Dis Treat. 2013;9:1521. | ||
Gitlin MJ, Swendsen J, Heller TL, Hammen C. Relapse and impairment in bipolar disorder. Am J Psychiatry. 1995;152(11):1635–1640. | ||
Fountoulakis KN, Kasper S, Andreassen O, et al. Efficacy of pharmacotherapy in bipolar disorder: a report by the WPA section on pharmacopsychiatry. Eur Arch Psychiatry Clin Neurosci. 2012;262(1):1–48. | ||
Bauer M, Ritter P, Grunze H, Pfennig A. Treatment options for acute depression in bipolar disorder. Bipolar Disord. 2012;14(s2):37–50. | ||
Sienaert P, Lambrichts L, Dols A, De Fruyt J. Evidence-based treatment strategies for treatment-resistant bipolar depression: a systematic review. Bipolar Disord. 2013;15(1):61–69. | ||
Fountoulakis KN, Gazouli M, Kelsoe J, Akiskal H. The pharmacodynamic properties of lurasidone and their role in its antidepressant efficacy in bipolar disorder. Eur Neuropsychopharmacol. 2014;25(3):335–342. | ||
Lurasidone HCl | C28H37ClN4O2S – PubChem. Available from: http://pubchem.ncbi.nlm.nih.gov/compound/11237860. Accessed March 23, 2015. | ||
Caccia S, Pasina L, Nobili A. Critical appraisal of lurasidone in the management of schizophrenia. Neuropsychiatr Dis Treat. 2012;8:155. | ||
Ishibashi T, Horisawa T, Tokuda K, et al. Pharmacological profile of lurasidone, a novel antipsychotic agent with potent 5-hydroxytryptamine 7 (5-HT7) and 5-HT1A receptor activity. J Pharmacol Exp Ther. 2010;334(1):171–181. | ||
Ishiyama T, Tokuda K, Ishibashi T, Ito A, Toma S, Ohno Y. Lurasidone (SM-13496), a novel atypical antipsychotic drug, reverses MK-801-induced impairment of learning and memory in the rat passive-avoidance test. Eur J Pharmacol. 2007;572(2):160–170. | ||
Björklund M, Sirviö J, Riekkinen M, Sallinen J, Scheinin M, Riekkinen P. Overexpression of alpha2C-adrenoceptors impairs water maze navigation. Neuroscience. 1999;95(2):481–487. | ||
Banasr M, Hery M, Printemps R, Daszuta A. Serotonin-induced increases in adult cell proliferation and neurogenesis are mediated through different and common 5-HT receptor subtypes in the dentate gyrus and the subventricular zone. Neuropsychopharmacology. 2004;29(3):450–460. | ||
Schreiber R, Newman-Tancredi A. Improving cognition in schizophrenia with antipsychotics that elicit neurogenesis through 5-HT 1A receptor activation. Neurobiol Learn Mem. 2014;110:72–80. | ||
Citrome L. Lurasidone in schizophrenia: new information about dosage and place in therapy. Adv Ther. 2012;29(10):815–825. | ||
Wesolowska A, Nikiforuk A, Stachowicz K, Tatarczyńska E. Effect of the selective 5-HT 7 receptor antagonist SB 269970 in animal models of anxiety and depression. Neuropharmacology. 2006;51(3):578–586. | ||
Guscott M, Bristow LJ, Hadingham K, et al. Genetic knockout and pharmacological blockade studies of the 5-HT 7 receptor suggest therapeutic potential in depression. Neuropharmacology. 2005;48(4):492–502. | ||
Huang M, Horiguchi M, Felix AR, Meltzer HY. 5-HT1A and 5-HT7 receptors contribute to lurasidone-induced dopamine efflux. Neuroreport. 2012;23(7):436–440. | ||
Luoni A, Berry A, Calabrese F, et al. Delayed BDNF alterations in the prefrontal cortex of rats exposed to prenatal stress: preventive effect of lurasidone treatment during adolescence. Eur Neuropsychopharmacol. 2014;24(6):986–995. | ||
Castrén E, Rantamäki T. The role of BDNF and its receptors in depression and antidepressant drug action: reactivation of developmental plasticity. Dev Neurobiol. 2010;70(5):289–297. | ||
Meyer JM, Loebel AD, Schweizer E. Lurasidone: a new drug in development for schizophrenia. Expert Opin Investig Drugs. 2009;18(11):1715–1726. | ||
Kroeze WK, Hufeisen SJ, Popadak BA, et al. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology. 2003;28(3):519–526. | ||
Reynolds GP, Hill MJ, Kirk SL. The 5-HT2C receptor and antipsychoticinduced weight gain–mechanisms and genetics. J Psychopharmacol. 2006;20(4 suppl):15–18. | ||
Terry AV, Mahadik SP. Time-dependent cognitive deficits associated with first and second generation antipsychotics: cholinergic dysregulation as a potential mechanism. J Pharmacol Exp Ther. 2007;320(3):961–968. | ||
Uchida H, Takeuchi H, Graff-Guerrero A, Suzuki T, Watanabe K, Mamo DC. Dopamine D2 receptor occupancy and clinical effects: a systematic review and pooled analysis. J Clin Psychopharmacol. 2011;31(4):497–502. | ||
Sunovion Pharmaceuticals Inc. Latuda® (lurasidone HCI tablets) [prescribing information]. 2013. | ||
Loebel A, Cucchiaro J, Silva R, et al. Lurasidone monotherapy in the treatment of bipolar I depression: a randomized, double-blind, placebo-controlled study. Am J Psychiatry. 2014;171(2):160–168. | ||
Loebel A, Cucchiaro J, Silva R, et al. Lurasidone as adjunctive therapy with lithium or valproate for the treatment of bipolar I depression: a randomized, double-blind, placebo-controlled study. Am J Psychiatry. 2014;171(2):169–177. | ||
Citrome L, Ketter TA, Cucchiaro J, Loebel A. Clinical assessment of lurasidone benefit and risk in the treatment of bipolar I depression using number needed to treat, number needed to harm, and likelihood to be helped or harmed. J Affect Disord. 2014;155:20–27. | ||
Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412–419. | ||
Thase ME, Macfadden W, Weisler RH, et al; BOLDER II Study Group. Efficacy of quetiapine monotherapy in bipolar I and II depression: a double-blind, placebo-controlled study (the BOLDER II study). J Clin Psychopharmacol. 2006;26(6):600–609. | ||
Calabrese JR, Keck PE Jr, Macfadden W. A randomized, double-blind, placebo-controlled trial of quetiapine in the treatment of bipolar I or II depression. Am J Psychiatry. 2005;162(7):1351–1360. | ||
AstraZeneca. Seroquel (quetiapine fumarate); 2011. Available from: http://www1.astrazeneca-us.com/pi/Seroquel.pdf. Accessed May 25, 2015. | ||
Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry. 2003;60(11):1079–1088. | ||
Eli L. Symbyax (symbyax (olanzapine and fluoxetine hydrochloride); 2013. Available from: http://pi.lilly.com/us/symbyax-pi.pdf. Accessed May 25, 2015. | ||
Leucht S, Cipriani A, Spineli L, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;382(9896):951–962. | ||
Sunovion Pharmaceuticals Inc. The US Food and Drug Administration approved Latuda® (lurasidone HCl) for the treatment of adult patients with depressive episodes associated with bipolar I disorder (bipolar depression), both as monotherapy and as an adjunct to lithium or valproate; 2013. Available from: http://www.sunovion.com/news/latuda-press-room.html. Accessed March 21, 2015. | ||
Citrome L. Lurasidone for schizophrenia: a review of the efficacy and safety profile for this newly approved second-generation antipsychotic. Int J Clin Pract. 2011;65(2):189–210. | ||
De Hert M, Yu W, Detraux J, Sweers K, van Winkel R, Correll CU. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone and paliperidone in the treatment of schizophrenia and bipolar disorder. CNS Drugs. 2012;26(9):733–759. | ||
Citrome L. Iloperidone, asenapine, and lurasidone: a brief overview of 3 new second-generation antipsychotics. Postgrad Med. 2011;123(2):153–162. | ||
Citrome L, Weiden PJ, McEvoy JP, et al. Effectiveness of lurasidone in schizophrenia or schizoaffective patients switched from other antipsychotics: a 6-month, open-label, extension study. CNS Spectr. 2014;19(04):330–339. | ||
Loebel A, Cucchiaro J, Xu J, Sarma K, Pikalov A, Kane JM. Effectiveness of lurasidone vs quetiapine XR for relapse prevention in schizophrenia: a 12-month, double-blind, noninferiority study. Schizophr Res. 2013;147(1):95–102. | ||
Citrome L, Cucchiaro J, Sarma K, et al. Long-term safety and tolerability of lurasidone in schizophrenia: a 12-month, double-blind, active-controlled study. Int Clin Psychopharmacol. 2012;27(3):165–176. | ||
Samalin L, Garnier M, Llorca P-M. Clinical potential of lurasidone in the management of schizophrenia. Ther Clin Risk Manag. 2011;7:239. | ||
McIntyre RS, Cha DS, Alsuwaidan M, McIntosh D, Powell AM, Jerrell JM. A review of published evidence reporting on the efficacy and pharmacology of lurasidone. Expert Opin Pharmacother. 2012;13(11):1653–1659. | ||
Kantrowitz JT, Citrome L. Lurasidone for Schizophrenia: What’s different? Expert Review of Neurotherapeutics. 2012;12(3):265–273. | ||
Cucchiaro C, Josephine Pikalov A, Ogasa M. Safety of lurasidone: pooled analysis of five placebo-controlled trials in patients with schizophrenia. Int J Neuropsychopharmacol. 2010;13(suppl 1):217. | ||
Potkin SG, Ogasa M, Cucchiaro J, Loebel A. Double-blind comparison of the safety and efficacy of lurasidone and ziprasidone in clinically stable outpatients with schizophrenia or schizoaffective disorder. Schizophr Res. 2011;132(2):101–107. | ||
Findlay LJ, El-Mallakh P, El-Mallakh RS. Management of bipolar I depression: clinical utility of lurasidone. Ther Clin Risk Manag. 2015;11:75. | ||
Preskorn S, Ereshefsky L, Chiu Y-Y, Poola N, Loebel A. Effect of food on the pharmacokinetics of lurasidone: results of two randomized, open-label, crossover studies. Hum Psychopharmacol. 2013;28(5):495–505. | ||
Ehret MJ, Sopko MA Jr, Lemieux T. Focus on lurasidone: a new atypical antipsychotic for the treatment of schizophrenia. Formulary. 2010;45:313–317. | ||
Rado J, Janicak PG. Pharmacological and clinical profile of recently approved second-generation antipsychotics. Drugs Aging. 2012;29(10):783–791. | ||
Addington DE, Pantelis C, Dineen M, Benattia I, Romano SJ. Efficacy and tolerability of ziprasidone versus risperidone in patients with acute exacerbation of schizophrenia or schizoaffective disorder: an 8-week, double-blind, multicenter trial. J Clin Psychiatry. 2004;65(12):1624–1633. | ||
Oh GH, Yu J-C, Choi K-S, Joo E-J, Jeong SH. Simultaneous comparison of efficacy and tolerability of second-generation antipsychotics in schizophrenia: mixed-treatment comparison analysis based on head-to-head trial data. Psychiatry Investig. 2015;12(1):46–54. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.