Back to Journals » Journal of Inflammation Research » Volume 15
Lupus Nephritis: Current Perspectives and Moving Forward
Authors Lichtnekert J, Anders HJ, Lech M ![]()
Received 7 October 2022
Accepted for publication 22 November 2022
Published 2 December 2022 Volume 2022:15 Pages 6533—6552
DOI https://doi.org/10.2147/JIR.S363722
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Julia Lichtnekert, Hans-Joachim Anders, Maciej Lech
Nephrologisches Zentrum, Medizinische Klinik und Poliklinik IV, Klinikum der Universität München, LMU München, München, Germany
Correspondence: Maciej Lech, Medizinische Klinik und Poliklinik IV Klinikum der Universität München, Goethestr. 31, München, D-80336, Germany, Tel +49-89-218075842, Fax +49-89-218075860, Email [email protected]
Abstract: Lupus nephritis is a severe organ manifestation of systemic lupus erythematosus, and its pathogenesis involves complex etiology and mechanisms. Despite significant knowledge gains and extensive efforts put into understanding the development and relapsing disease activity, lupus nephritis remains a substantial cause of morbidity and mortality in lupus patients. Current therapies retain a significant unmet medical need regarding rates of complete response, preventing relapse of lupus nephritis, progression of chronic kidney disease to kidney failure, drug toxicity, and pill burden-related drug non-adherence. Connected to progression of chronic kidney disease are the associated risks for disabling or even lethal cardiovascular events, as well as chronic kidney disease-related secondary immunodeficiency and serious infections. In this regard, biomarkers are needed that can predict treatment response to specific drugs to enable personalized precision medicine. A series of clinical trials with innovative immunomodulatory drugs are ongoing and raise expectations for improvements in the management of lupus nephritis. Here, we review how new developments in pathogenesis connect with current and future perspectives for the management of lupus nephritis.
Keywords: systemic lupus erythematosus, glomerulonephritis, proteinuria, autoimmunity, steroids
Introduction
Systemic lupus erythematosus (SLE) is a complex autoimmune disease that despite recent advances in the understanding of pathogenesis remains challenging to treat. Its multifactorial origin, involving genetic, epigenetic, and environmental factors and the complex mechanisms underlying its development still need to be uncovered.1 SLE pathogenesis involves genetic variants that impair the elimination of dying cells and the persistence of nuclear antigens in extracellular compartments, where they trigger innate and adaptive immune responses. Other genetic predispositions affect the magnitude of these immune responses and checkpoints of immune tolerance that promote the priming and expansion of autoreactive T and B-lymphocytes.2,3 As a result, antinuclear and other autoantibodies circulate and form immune complexes, which further drive systemic inflammation and organ damage. In SLE, systemic inflammation causes general symptoms such as fever, fatigue, and myalgia, but immune complex deposits in the peripheral microvasculature can trigger inflammation and malfunction in multiple organs. The classification criteria released by the American College of Rheumatology and the European Alliance of Associations for Rheumatology (ACR/EULAR) in 2019 show much better sensitivity and overall specificity than previous versions.4 However, diagnosis remains difficult and relies on integrating information at many levels. Glucocorticoids and other drugs currently used for the management of SLE can have unfavorable side effects due to their non-selective mechanism of action that altogether implies significant unmet medical needs.5 In recent years, the understanding of the physiopathology of SLE and its frequent complication lupus nephritis (LN) has progressed, and biological treatments specifically targeting different cellular and inflammatory mechanisms have broadened the therapeutic arsenal. Suppression of SLE activity is the main therapeutic strategy to prevent onset of LN, prevent relapsing episodes of LN, and prevent the unfortunate consequences of chronic kidney disease (CKD), ie, progression to kidney failure, cardiovascular disease, and serious infections due to secondary immunodeficiency related to CKD (SIDKD).6 Here, we provide an update to the molecular pathophysiology of SLE/LN as a rationale for recently approved drugs and drugs in the pipeline of clinical trials as well as on the impact of reverse translation on lupus research.
Effect of Monogenic and Polygenic Defects on Autoimmunity
The different monogenic forms of SLE as well as experimental studies support the concept that many different diseases can lead to the formation of antinuclear antibodies and inflammatory organ damage currently referred to as a single disease SLE. However, SLE is rather a clinical syndrome representing a group of diverse molecular disorders.7 Nevertheless, the different monogenic disorders provide clues to shared molecular pathomechanisms of SLE. For example, altered type I interferon (IFN) signaling (interferonopathies) triggers SLE-like antinuclear antibodies and organ injuries. Other genetic alterations affecting immune cell activation, impaired clearance of dead cells, dysregulated immune cell expansion and antibody production also participate in the development of SLE and LN.8–13
Genetic Alterations Associated with SLE
Cell Death and Clearance of Cellular Debris
Autoimmunity is defined as a breakdown of immune tolerance to self-antigens. The retention of autoreactive lymphocyte clones represents one of the key elements of SLE’s development and persistence. Dysregulated cell death and clearance of cellular debris represent other important components in the pathogenesis of SLE.14 In SLE patients, apoptotic cells are not removed efficiently, and accumulation of apoptotic cellular compartments followed by immune responses can be observed.15 DNASE1, DNASE1L3, and TREX1 also known as DNAse3 are involved in the nucleic acid degradation pathway and determine susceptibility to SLE. Various SLE susceptibility genes regulating cell death and dead cell clearance including FcγRIIB, ITGAM, and ACP5 were linked to SLE.16–18 Moreover, polymorphisms in autophagy-related genes, such as ATG5, ATG7, IRGM, DRAM1, CDKA1B, APOL1, and MTMR3, were described.19–22 The most prominent appeared to be ATG5, which is crucial for the initiation of autophagosome formation.22–24
Nucleic Acids Recognition and Interferon Signaling
The pathways primarily involved in type I IFN regulation and the activation of antiviral immunity as well as receptors sensing nucleic acids contribute to the systemic activity of SLE.25,26 Most patients display an increased expression of type I IFN and their regulated genes (interferon signature). Since IFNs are present in low concentrations and characterized by quick consumption, often IFN-dependent genes are used to define the IFN signature.27–29 The known anti-viral pathways include the DNA-sensor “cyclic GMP, AMP synthase” (cGAS),30 the RNA sensors “retinoic acid-inducible gene I” (RIG-I),31 “melanoma differentiation-associated gene 5” (MDA5),32,33, and “RNA-activated protein kinase” (PKR),34,35 as well as Toll-like receptors (TLRs) 3, 7, 8, and 9 which are activated after receptor-mediated endocytosis of extracellular nucleic acids.3,36,37 IFN induction also elicits local effects in the kidney and further compromises glomerular function by enhancing the activation of endothelial cells, mesangial cells, podocyte apoptosis, and insufficient podocyte regeneration. Other pathways involved in the sensing and transfer of nucleic acid include DDX1, 21, 36, and 41, IFI16, SIDT2, and Aim2.38 Duplication or overexpression of TLR7 promotes autoimmunity,39–41 whereas the deletion of Tlr7 in lupus-prone MRL/lpr mice reduces autoantibody level and immune activation.42 In summary, human studies proved that genetic variants in TLR7, TLR regulatory molecules (UBE2L3), IFN signaling transcription factors (IRF5, IRF7/PHRF1, IRF8, and ETS1), cGAS, stimulator of IFN genes (STING) and downstream signaling of the IFN pathway, such as STAT4, IFIH1, and PRDM1, are associated with increased SLE susceptibility.12,13,43 A recent study by Brown et al described a novel missense TLR7Y264H gain of function mutation in a child with severe lupus that selectively increased the affinity of TLR7 for guanosine and cGMP and was sufficient to cause lupus when introduced into mice. The study proved that enhanced TLR7 signaling drives aberrant survival of B cell receptor (BCR)-activated B cells, accumulation of CD11c+ age-associated B cells and germinal center B cells, and highlights the crucial role of TLR7 in development of SLE.44
Inflammation-Related Pathways
Mutations in the complement system are often associated with both immunodeficiencies and autoimmunity.45 For instance, individuals with C1q deficiency develop early onset of lupus-like-phenotype.46 In addition, inflammatory signaling and changes in ubiquitination within NF-kB signaling pathways play a pivotal role in the development of SLE and often represent additional risk factors that accelerate the disease. For instance, mutations in tumor necrosis factor alpha-induced protein 3 or A20 (TNFAIP3) can trigger incorrect degradation and termination of pro-inflammatory responses.47 TNFAIP3 interacting protein 1 (TNIP1) has been described as another key repressor of inflammatory signaling and a potential factor in multiple autoimmune disorders. Mutations in TNIP1 lead to significant changes in NF-κB signaling and may initiate autoimmune processes.8,48 Polymorphisms in the gene encoding the A20-binding inhibitors of NF-κB (ABIN1) protein represent a predisposition of factors for autoimmune disease. For instance, ABIN1[D485N] knock-in mice show significant expansion of myeloid cells activated T- and B-lymphocytes, elevated serum Ig levels, lupus-like nephritis, and other obvious SLE-like phenotype features.49 Latest investigations suggest that another predominantly in lymphocytes expressed ubiquitin-carrier enzyme E2L3 (UBE2L3) is a risk gene that influences autoimmunity by modulating UBCH7 expression.50,51 Experimental studies in mice showed that the MCPIP1 protein negatively regulates c-Jun N-terminal kinase (JNK) and NF-κB activity by removing ubiquitin moieties from TRAF proteins, which determines autoantibodies in SLE.52,53 Furthermore, the interleukin-1 receptor (IL-1R) associated kinase (IRAK) family, which plays a crucial role in the protective response to pathogens and inflammatory processes, is involved in the susceptibility to SLE.54,55
Cellular Responses
In individuals with lupus, innate immune cells and autoreactive lymphocytes become overactive. Systemic inflammation affects the function and signaling of autoreactive B cells responsible for the production of SLE autoantibodies. For instance, genes involved in B cell signaling, differentiation, and proliferation such as BLK, BANK1, LYN, IRF8, ETS1, IKZF1, AFF1, RasGRP3, PRDM1, FcγRIIB, PRKCB, HLA-DR2, HLA-DR3, and NCF2 display genetic variants associated with the vulnerability to SLE.56,57 In addition, polymorphisms in genes encoding IL-10 and IL-21 enhance B and T lymphocytes’ survival and activity leading to antibody class switching and autoantibody production, respectively. Polymorphisms associated with SLE and T cell immunity include genes encoding ETS1, IKZF1, PRDM1, AFF1, TNFS4, PTPN22, HLA-DR2, HLA-DR3, CD44TYK2, and STAT4.58 Since T cell signaling combines both innate and adaptive immune responses, dysregulated T cells might represent an interesting target for future therapies. Innate immune responses also play a crucial role in the development of autoimmune disorders. As the first line of defense, the cells of the innate immune system infiltrate the injured tissues to orchestrate inflammation and the removal of self and foreign antigens. Furthermore, they directly initiate adaptive immune responses. For instance, proteins associated with development and phagocytic features of myeloid cells such as FcγRIIB, FcγIIIA/B, IL-10, and IRF8 are linked to SLE.59–61 Moreover, ITGAM and ICAM polymorphisms affect processes of migration and adhesion of neutrophils and monocytes in SLE patients.62,63 Previously mentioned mutations in DNase I are responsible for the increased NET formation and impaired degradation of NETs. These features enhance exposure of nucleic acids, activation of cells, and prolonged inflammation. Additional mechanisms such as production of NET-derived antimicrobial peptide LL37–DNA complexes trigger the development of B cells producing anti-LL37 antibodies.64 Thus, the overactivation of cellular activity and enhanced production of cytokines often determine long-term damage to important organs.
Genetic Alterations Associated with Lupus Nephritis
A series of studies describe gene polymorphisms in association with kidney disease in SLE.54,65–67 For instance, Chung et al described genetic variants in STAT4, ITGAM, K1AA1542, BANK1, and UBE2L3 that are associated with the presence of anti-dsDNA antibodies and may contribute to LN.68 Further, variants of BANK1 gene are linked to LN in SLE patients of European ancestry.39 In addition, there is an association of SNP rs7708392 and rs495881 in TNIP1 with LN in a large multi-racial cohort.54 Polymorphisms of APOL1 (apolipoprotein L1 gene) were shown to be significantly associated with the risk of developing end-stage kidney disease due to LN and other nephropathies in individuals of West African descent.21,69,70 STAT4 polymorphisms have been linked to LN in European cohorts and with severe renal insufficiency.71–73 In addition, there is a significant additive association between TNFSF4 alleles rs2205960 and rs10489265 and LN.74 Studies of multi-racial cohorts investigating pathways and risk of SNPs in LN showed variations in ZNF546, TRIM15, NFATC1, TRIMI0, and TTC34 to be significantly associated with LN.75 Another investigation conducted in 2021 identified a missense variant in IRF3 (rs7251), a key molecule in the type I IFN pathway, and linked it to LN.76 Each genetic defect mentioned above affects immune homeostasis. The identification of new genetic autoimmunity triggers and their validation in animal models is necessary to understand the molecular processes that are essential for the development of LN.
Targeting Lupus to Control Lupus Nephritis
With an increasing understanding of the pathogenesis of SLE and LN, therapeutics that target single immune elements or signaling pathways might replace conventional unspecific therapeutics such as glucocorticoids or unselective immunosuppressant drugs. SLE and LN patients display increased inflammatory responses and autoantibodies. Inhibition of cytokines or pathways with newly developed biological agents showed positive effects and might inspire future research to develop even more specific, targeted treatments (Figure 1).
 |
Figure 1 Diagram showing the immunopathogenic pathways leading to kidney damage in LN and presenting novel therapies targeting specific pathways. Abbreviations: DC, dendritic cell; pDC, plasmacytoid dendritic cell, APRIL, a proliferating-inducing ligand; BAFF, B cell activating factor; BAFFR, B cell activating factor receptor; BCMA, B cell maturation antigen; BTK, Bruton’s tyrosine kinase; CD, cluster of differentiation; IFN, interferon; IFNRI, interferon-type I receptor; JAK, Janus kinase; MHC, major histocompatibility complex; NETs, neutrophil extracellular traps; TACI, transmembrane activator and calcium modulator and cyclophilin ligand interactor; PRR, pattern recognition receptors, TLR, toll-like receptor; DAMPs, damage associated molecular patterns, PAMPs, pathogen associated molecular pattern; IL, interleukin; PAD, peptidylarginine deiminases. |
Targeting B-Lymphocytes
New therapies also target B cell activation. An efficient strategy would be to precisely remove autoreactive effector B cells and amplify the autoantigen-driven regulatory B cell population while maintaining correct immune responses. Inhibition of B-lymphocytes using anti-BAFF and APRIL and anti-CD19/FcγRIIb is part of such a strategy. B-lymphocytes need survival signals during their development. They are provided by various stimuli, and one of them is B-lymphocyte survival protein (BlyS), known as B Cell Activating Factor BAFF. This member of the TNF family acts through three receptors: Transmembrane Activator and Calcium Modulator and Cyclophilin Ligand Interactor (TACI), BAFFR, and B Cell Maturation Antigen (BCMA). A-Proliferation Inducing Ligand (APRIL) is another cytokine, similar to BAFF, which also binds the TACI receptor. Belimumab is a human IgG1 monoclonal antibody that binds BAFF and inhibits its activity. Two similarly designed 3-output phase studies, BLISS52 and BLISS76, showed a decreased significance of the lupus activity score. BLISS-52 randomized and treated 865, whereas BLISS-76 randomized and treated 819 SLE patients. The duration of therapy in the two studies was 52 weeks for BLISS-52 and 76 weeks for BLISS-76. Post hoc analyses demonstrated decreased proteinuria and a lower incidence of LN flares in patients who received belimumab.77 These findings initiated an additional trial, Belimumab International Study in LN (BLISS-LN), to assess the efficacy and safety of belimumab plus standard therapy (mycophenolate mofetil or cyclophosphamide and azathioprine) in patients with active but not severe LN. Patients who received belimumab plus standard therapy appeared to have a primary efficacy renal response than those who received standard therapy alone (BLISS-LN, NCT01639339). In summary, the belimumab trials demonstrated efficacy when added to either cyclophosphamide or MMF as the current standard of care in large parts of the world, opening the perspective for belimumab as an additional treatment for patients with LN.
Another approach involves the suppression of adaptive immunity by depleting B-lymphocytes, key antigen-presenting cells, and plasma cell precursors. Drugs used in this context include anti-CD20 monoclonal antibodies. For example, Rituximab, a chimeric antibody that depletes CD20+ B cells from the circulatory compartment, while depletion in lymphoid organs and tissues is limited. This may explain why the efficacy of Rituximab in SLE trials was limited. A Phase III clinical study to examine the efficacy and safety of rituximab in patients with active SLE Exploratory Phase II/III SLE Evaluation of Rituximab (EXPLORER)78 or SLE with active proliferative LN Assessment with Rituximab (LUNAR)79 did not show the advantage of rituximab over placebo. A frequently mentioned limitation of the EXPLORER studies and LUNAR is linked to their design. Patients included in these studies received high doses of glucocorticoids simultaneously, which limited the identification of a direct effect linked to rituximab. Interestingly, rituximab fails to deplete the B cells in lymphoid organs, however the remaining B cell population display reduced functional capacities.80 Another anti-CD20 monoclonal antibody with clinical potential in LN is obinutuzumab.81 The antibody displays efficient antibody-dependent cellular cytotoxicity due to its greater affinity for the FcγRIII on effector cells.82 Clinical studies and experimental models proved the advantage of obinutuzumab over rituximab during the treatment. Several clinical studies that use obinutuzumab in the SLE are under evaluation. This includes the Phase II trial NOBILITY (NCT02550652), which evaluated the safety and efficacy of obinutuzumab compared with a placebo in participants with LN. The clinical study evaluated the response to obinutuzumab in patients with LN treated with mycophenolate mofetil (MMF) and showed improved renal responses in patients with LN who received obinutuzumab plus standard therapies compared with standard therapies alone.81 The Phase 3 clinical trial REGENCY (NCT04221477) includes patients with proliferative LN class III or IV LN and evaluates further the effectiveness of obinutuzumab. The OBILUP trial (NCT04702256), initiated in 2021 investigates the efficacy of LN therapy with no added oral steroids, comparing oral corticosteroids plus MMF versus obinutuzumab and MMF. Another humanized anti-CD20 monoclonal antibody, ocrelizumab, was investigated in two clinical trials. First, the BEGIN placebo-controlled study (NCT00539838) evaluated the combination of ocrelizumab and standard therapy in SLE patients with moderate but not severe renal impairment, but the study was terminated prematurely as it did not show a benefit. Second, the BELONG study (NCT00626197) which assessed response to ocrelizumab in LN has also been terminated due to serious and opportunistic infections in the ocrelizumab group. Ofatumumab represents another alternative B-cell-depleting therapy using humanized anti-CD20 antibodies. According to case studies, patients with LN who were allergic to rituximab and received treatment with ofatumumab displayed a reduction in proteinuria.83 Ofatumumab has been approved for the treatment of chronic lymphocytic leukemia and multiple sclerosis therapy and could represent an alternative treatment for LN patients, but randomized controlled trials are needed to prove the efficacy of ofatumumab and to determine the optimal treatment regime.
Anti-CD19 chimeric antigen receptor (CAR) T cell therapy using autologous T cells from patients with SLE transduced with a lentiviral anti-CD19 CAR vector showed significant depletion of B cells, which was accompanied by improvement of clinical symptoms and levels of anti-double-stranded DNA antibodies. Importantly, drug-free remission was maintained during longer follow-up since reappearing B cells were naïve and showed non-class-switched B cell receptors.84,85 This study highlights the role of adaptive immune responses in the development of SLE and is an example of a tolerable and highly effective new therapeutic approach.
Although the B-cell antigen receptor (BCR) is responsible for maintaining the differentiation of mature B cells, co-receptor molecules play an important role in the regulation of B-cell activity. For instance, the co-receptor CD19 modulates B-cell fate at multiple stages of development. One of its important functions in mature B cells includes the regulatory pathway initiated by complement C3d-fixed antigens via complement receptor 2 (CR2/CD21) and BCR. Obexelimab/XmAb5871 (NCT02725515), a first-in-class monoclonal antibody that targets CD19 and FcγRIIB on B cells is inhibiting BCR complex aggregation without having a depleting effect on B cells. A Phase 2 study of obexelimab/XmAb5871 (NCT02725515) with 104 patients with moderate to severe, non-organ threatening SLE enrolled did not meet the primary endpoint (no loss of improvement/LOI) but achieved the secondary endpoints (time-to-LOI and safety).86 Due to the negative study results, obexelimab is not further investigated in a phase III trial in SLE. Thus, the depletion or inactivation of lymphocytes by anti-B–lymphocytes monoclonal antibodies are recognized as favorable in the management of patients with an LN, nevertheless controlled phase 3 studies that demonstrate a positive effect of these drugs are still pending.
Further worth mentioning studies involve molecules targeting the BAFF/TACI pathway, such as telitacicept and atacicept, are humanized recombinant soluble fusion proteins. Atacicept contains the extracellular ligand-binding domain of TACI, which is fused into the Fc portion of human IgG1, blocking both APRIL and BAFF. Telitacicept, also called RC18 is a novel recombinant dual-targeting drug TACI-Fc fusion protein binding to BAFF and APRIL. Both drugs seem to be promising, but the clinical trial investigating atacicept in patients with active LN (APRIL-LN; NCT00573157) has been terminated early due to severe infections associated with hypogammaglobulinemia. Telitacicept reached the primary endpoint of the clinical trial and showed a decrease in disease activity (NCT02885610).87 The study will continue in a phase III trial (NCT04082416). In 2021, Telitacicept got conditionally approved in China for the treatment of patients with active SLE. However, only data on pharmacokinetics have been published so far. Two further BAFF-targeting therapeutics were generated recently. Tabalumab (LY2127399), a monoclonal antibody that antagonizes BAFF in both membrane and soluble forms showed biological activity changes in anti-dsDNA, complement, B cells, and immunoglobulins. This finding indicates efficient inhibition of the BAFF pathway. However, key clinical efficacy endpoints have not been achieved in two phase III trials (Illuminate-1 and −2).88,89 Another interesting study focused on blisibimod (A-623), a fusion protein consisting of four BAFF binding domains. The data demonstrated that although the SRI-6 endpoint was not met, blisibimod led to successful steroid reduction, decreased proteinuria, and biomarker responses (NCT01395745).90 Although APRIL and BAFF have been considered very promising targets for LN, clinical trial evidence could not always convincingly demonstrate their sufficient efficacy. This could relate to the redundant relevance of the molecular target in SLE or problems in trial design, choice of endpoints, or populations studied.
Another potential candidate drug is Rozibafusp Alfa/AMG570. The ongoing phase 2 trial with 320 participants aims to determine if this first-in-class bispecific antibody–peptide conjugate inhibiting BAFF and the interaction of inducible ligand (ICOSL also known as B7-related protein-1 (B7RP-1)) could be a useful therapeutic agent in SLE (NCT04058028). Until now, multiple ascending doses of rozibafusp alfa have been safe and generally well tolerated. The pharmacokinetic and pharmacodynamic analysis demonstrated a nonlinear, target-mediated disposition consistent with cell surface target interaction (NCT03156023). Since the interaction between ICOS and its ligand plays an important role in T cell-dependent B cell activation, inhibition of this pathway has been targeted in various preclinical studies. Another monoclonal antibody that binds ICOSL is also a promising candidate for efficient SLE therapy: AMG557 showed safety and potential benefit in SLE patients with active arthritis but needs further evaluation (NCT01683695).91
The CD40–CD40L pathway in B cells was targeted with BI 655064, a humanized anti-CD40 monoclonal antibody that blocks the CD40 pathway and downregulates activated B cells. Additional effect of CD40 blocking therapy is an inhibition of myeloid cell activation including resident kidney cells, which may obstruct local inflammation and consequently, kidney injury. A double-blind, randomized, placebo-controlled trial evaluating the effect of BI 655064 administered as subcutaneous injections, on the renal response after 1 year of treatment, was initiated in patients with active LN. Thus, CD40 is an appealing therapeutic target in LN (NCT02770170).
Further new approaches for B-lymphocyte-directed therapy include fenebrutinib, iberdomide, and Mab5261. While fenebrutinib, a non-covalent and reversible BTK inhibitor, had an acceptable safety profile, the primary endpoint, SLE Responder Index SRI-4 defining disease activity, was not met despite evidence of strong pathway inhibition (NCT02908100).91 However, the drug showed treatment benefits in rheumatoid arthritis. The goal is to evaluate if a higher dose of the drug could be more efficient and lead to a greater reduction in IgM and IgG without risk of infection. Iberdomide (cc-220) is a cereblon ligand that stimulates proteasomal degradation of transcription factors Ikaros (IKZF1) and Aiolos (IKZF3). Iberdomide significantly reduced the activity of the type I IFN gene signature and B cell activation. Moreover, iberdomide triggers a boost of IL-2 production and consequently stimulates Tregs expansion, leading to a selective rebalancing of immune responses in SLE (NCT03161483).92 A 24-week phase 2 trial showed an SRI-4 response in patients with active SLE but did not meet secondary endpoints such as a significant change in the Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI)–Activity Score. There also might be an increased number of side effects and a potentially elevated risk of teratogenicity and thrombogenicity of iberdomide.93 Mab5261 aims to follow the strategy of antibody-mediated disruption of the CXCL13 signaling that is the major determinant for B cell recruitment. The drug interferes with the formation of ectopic lymphoid follicles in the target organs and inhibits autoimmune disease progression.94 This is an example of one of the many new strategies followed in pre-clinical development.
Targeting Plasma Cells
Activation of B cells triggers a massive clonal expansion and production of short-lived plasma cells in secondary lymphoid tissues.95 Simultaneously, the formation of both memory B cells and long-lived plasma cells (plasmablasts) takes place in germinal centers.96 During inflammatory processes, distinct plasma cell subpopulations can be found in human tissues.97–99 Some plasma cell subpopulations express CD20 and can be depleted by anti-CD20 monoclonal antibodies. However, mature human plasma cells do not express CD20 and significantly contribute to antibody production.100,101 Thus, long-lived plasma cells also produce autoantibodies,102 and cannot be targeted by conventional immunosuppressive drugs such as steroids, antimetabolites, and cyclophosphamide or by B cell depletion.103 Novel monoclonal antibodies directly target plasma cells. One example is daratumumab that targets CD38, which is known to be expressed particularly by plasma cells. CD38 appeared to be already an important target in the treatment of multiple myeloma since this antigen is also over-expressed in malignant plasma cells. Two clinical trials are now ongoing to test daratumumab in refractory SLE patients (DARALUP/NCT04810754) and patients with active LN (NCT04868838). The second most important drug is bortezomib, a proteasome inhibitor that is also used in oncology for the treatment of multiple myeloma. Recently, a case study showed the use of daratumumab in two patients with life-threatening lupus. The drug-induced substantial clinical response and triggered significant depletion of long-lived plasma cells and reduction of IFN type I activity.104 Controlled clinical studies are necessary to prove the efficacy and safety of the drug. Another study showed that bortezomib might be an effective option for refractory LN. In the study, 12 female patients diagnosed with class IV or IV/V LN with acute or rapidly progressive kidney injury (n = 11) and/or severe nephrotic syndrome (n = 1) were treated with bortezomib.105 All patients with acute or rapidly progressive kidney injury showed a significant reduction in disease activity, manifested by a significant decrease in anti-ds DNA antibody titers, serum creatinine, and proteinuria. A clinical trial including patients with myasthenia gravis, rheumatoid arthritis, or SLE was, however, terminated due to recruitment difficulties (TAVAB, NCT02102594).
Targeting the Type I Interferon Pathway
The above-mentioned IFN signature characterizes the majority of patients affected by SLE. This results in an increased secretion of type I IFNs and consequently a high expression of genes regulated by IFNs. The mechanism that is responsible for this SLE phenomenon is associated with the presence of endogenous IFN inducers, hyper-activation of IFN-producing cell types, a variety of genetic predispositions promoting IFN production, and dysregulated negative feedback mechanisms. The continuous stimulation of immune response including the IFN pathway leads to a break of tolerance and the development of autoimmune response. Therefore, efficient therapy for patients with SLE must include a strategy aiming at downregulation of the IFN system. Anifrolumab, a monoclonal antibody-binding subunit 1 of the type I IFN receptor (IFNR1), showed benefit in two randomized controlled studies, TULIP1 and TULIP2. The patients included in the study suffered from moderate to severe lupus, without severe active LN. Further study TULIP-LN showed that administration of anifrolumab together with oral steroids and MMF had no effect on proteinuria levels at 52 weeks, the primary response criterion and end-point of the TULIP-LN. Since proteinuria is a relevant predictor for long-term outcomes in patients with LN, it is substantial to remark that other therapies are more potent in inducing proteinuria response. For instance, voclosporin has direct effects on podocytes and therefore reduces glomerular damage by affecting proteinuria more strongly than other immunosuppressive drugs.106 Recent findings and perspectives from TULIP trials are characterized in detail in work issued in 2022.107,108
Other therapies that neutralize circulating IFN-α, such as sifalimumab and rontalizumab, are no longer in clinical development. The efficacy and safety of sifalimumab were assessed in a phase randomised, double-blind, placebo-controlled study (NCT01283139) of 431 patients with moderate to severe active SLE. At first, the drug was selected as a promising treatment for SLE patients since the improvement was consistent across various clinical endpoints, including global and organ-specific measures of disease activity.109 However, further evaluation was terminated due to the internal competition in the phase III study of anifrolumab. Another humanized IgG1 anti-IFN α monoclonal antibody rontalizumab was evaluated in patients with moderate-to-severe SLE. Except for disease activity also in this study, the efficacy was additionally examined by IFN-regulated gene expression (interferon signature metric, ISM). The primary and secondary endpoints of this trial were not met in all patients or patients with high ISM scores. Nevertheless, rontalizumab therapy was associated with improvements in disease activity, reduced flares, and decreased steroid use in patients with SLE with low ISM scores (NCT00962832).110 In summary, the above-mentioned molecules that bind to IFN-α can significantly decrease the INF-α signature and decrease skin lesions in biopsies. Unfortunately, further clinical phase did not reveal clinical efficacy beyond that of placebo. Nevertheless, this initiated similar approaches (such as MEDI 545) that are currently being tested in a scleroderma trial (NCT00930683), and a phase III trial in SLE is planned. Another example is IFN-α–Kinoid (IFN-K; Neovacs) which is evaluated for the safety and clinical impact on SLE in mild to moderate disease manifestation (NCT01058343). Moreover, Novo Nordisk develops two anti-IFN agents. Another INFa targeting drug is completing the Phase I trial of 40 patients (NCT00960362). Finally, AMG 811 represents an anti–IFN-γ monoclonal antibody that is under evaluation in an LN trial (NCT01164917).
Janus Kinase Inhibitors
Cytoplasmic Janus protein tyrosine kinases (JAKs) are components of diverse signaling cascades that participate in various signal transduction pathways such as cellular survival, proliferation, inflammation, and differentiation. Recent studies indicate that not only STAT signaling but also phosphorylation of cytokine and growth factor receptors enabling the interaction of cytokine/IFN receptors with their ligands might be a mechanism of action for JAK kinases. Importantly, loss of JAK kinase function seems to lead to immunodeficiency. Therefore, small-molecule therapies such as JAK inhibitors may represent an efficient way for the treatment of diverse immune and inflammatory diseases. Their potential in treating SLE was used by the application of drugs such as tofacitinib and baricitinib. These molecules are commonly used to treat arthritis rheumatoid and minimize cell signaling from various pro-inflammatory mediators including IFNs. A randomized clinical trial from 2018 showed a benefit of baricitinib regarding skin and joint manifestation of SLE.111 A Phase 1 study to test safety and tolerability of tofacitinib in SLE (NCT02535689) enrolled 30 patients and found tofacitinib to be safe in SLE and to improve cardiometabolic and immunologic parameters.112 Another trial testing tofacitinib in moderate and severe skin involvement in young adults with lupus is currently recruiting patients (NCT03288324). Also, tofacitinib has been shown to modulate the formation of NETs and reduce vascular damage in LN MRL/lpr-mice (MRL/lpr background). It improved disease activity, including nephritis, skin inflammation, and autoantibody production.113 Recently, a small Phase II, randomized, double-blind study (NCT03285711) assessed the Janus kinase 1 (JAK1) inhibitor filgotinib and the spleen tyrosine kinase inhibitor lanraplenib in patients with lupus membranous nephropathy (LMN).114 Due to the small number of patients (n=9), only limited conclusions can be drawn. Nevertheless, the study showed a therapeutic benefit with filgotinib treatment measured in percent change in 24-h urine protein from baseline to week 16.
Targeting Complement
In recent years, the therapy of SLE and LN has been improved by the introduction of highly specific biological drugs targeting, among others, B cells and plasma cells as well as IFN signaling pathways. One of the main features of SLE is autoantibodies produced by self-reactive B cells and consequently, immune complexes deposited in the tissues and activating the complement system and inflammatory response.115,116 An efficient complement system is crucial to performing safe and highly controlled clearance of immune complexes, apoptotic cells, and cellular debris.117 These processes protect against exposure to self-antigens and reduce autoantibody load. Many components of complement function as opsonins and ensure the safe removal of cellular material. For instance, the CR1 receptor expressed on erythrocytes (E-CR1) seems to be involved in LN pathology. Complement-opsonized ICs bound to E-CR1 cannot deposit in the organs and trigger inflammation.118 E-CR1 consumption in patients at flare correlated with fewer signs of renal involvement, suggesting that E-CR1 protective function in SLE nephritis.119 Novel therapeutics considered for the treatment of LN aim to modulate complement function. Inhibition of selected components might be beneficial for LN patients. A human monoclonal antibody Narsoplimab/OMS721 that targets the lectin pathway and mannan-binding lectin serine protease 2 (MASP-2), is one of the new drugs that target patients with glomerular diseases including active LN (NCT02682407). Another example is Ravulizumab, which targets the epitope of C5 and displays extended antibody recycling through enhanced affinity to the neonatal Fc receptor. The initial investigations proved the efficacy of terminal complement inhibition in participants with LN or IgA nephropathy (NCT04564339).
Targeting Cytokines and T Cells
T cells represent an interesting target of immunosuppressive treatments since activation of T cells may instruct (auto)-immune responses by orchestrating B cells and other T cells whose differentiation pathways they have to initiate. Importantly, humoral autoimmunity depends on autoreactive Th cells since autoantibody-producing B-lymphocytes undergo T-lymphocyte-dependent affinity maturation. Furthermore, autoreactive T cells play a direct role in local tissue damage by producing various inflammatory mediators. Therefore, not only regulatory T cells but also in vivo application of monoclonal antibodies targeting other T cells subpopulations might be beneficial for SLE patients since there is an imbalance between effector T cells and regulatory T cells that represent an essential cell population for immune tolerance and are altered in SLE. These cells are strongly dependent on IL2 signaling that maintains their expansion and survival. To target them, the administration of a low-dose IL-2 therapy for SLE was conducted. The primary endpoint was the SLE Responder Index-4 (SRI-4) at week 12, and secondary endpoints were other clinical responses, such as safety and dynamics of immune cell subsets.120 Low-dose IL-2 treatment appeared to be effective and well-tolerated. It resulted in 53.85% complete remission in patients with LN, compared with 16.67% (2/12) in the placebo group (p=0.036). Besides the expansion of regulatory T cells, low-dose IL-2 may also sustain cellular immunity with enhanced natural killer cells (NCT02465580 and NCT02932137). IL2 treatment could represent an alternative for patients with severe SLE who are resistant to standard treatments. Unlike Tregs, IL17-producing Th17 lymphocytes are hyperactive and over-proportionally abundant in SLE. These cells are significantly involved in the maintenance of inflammation and tissue damage.121 Anti-IL17, secukinumab is currently undergoing a phase 3 study (SELUNE, NCT04181762) as an additional treatment in LN. Another T cell cytokine-specific antibody, Ustekinumab, targets the p40 subunit common to the cytokines IL23 and IL12 and affects the expansion of Th1 and Th17 lymphocytes. Although current treatment involves autoimmune diseases, the phase 3 study in patients with SLE could not show the expected results and it was terminated (LOTUS, NCT02349061).
Previously mentioned CD40 is a receptor expressed in particular on the surface of B-lymphocytes. The interaction with its ligand CD40L, expressed mainly by T-lymphocytes, but also by dendritic cells is necessary to allow stable activation and antibody production. Since CD40-CD40L (CD154) interactions maintain immune cells’ cross-talk targeting this signaling pathway could provide substantial benefits to LN patients. For instance, dapirolizumab which targets CD40L and inhibits its interaction with CD40 was recently enrolled in various SLE studies. Two phase 2 studies without a primary focus on renal function were designed, the first (NCT02804763) is a double-blind placebo-controlled study that evaluates the efficacy and safety of dapirolizumab in patients with moderate to severe SLE.122 Another randomized clinical trial examines the response to dapirolizumab as treatment support in patients with moderate to severe SLE, excluding those with severe renal impairment (NCT04294667).
The mechanism of action of another T-cell-designed fusion protein abatacept targets human cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4). This selective modulator with inhibitory activity on T lymphocytes has already been approved for the treatment of rheumatoid arthritis, however, failed to achieve satisfactory results in SLE clinical trial. According to experts’ opinion despite disappointing results and based on its mechanism of action, abatacept seems to have a role in LN, and new trials lacking design pitfalls could be conducted in the future.
mTOR/Rapamycin/Sirolimus
Rapamycin inhibits antigen-induced T-cell proliferation but also has a role in B cells and macrophages.123 SLE patients have an altered T-cell profile associated with the activation of the mammalian target of rapamycin (mTOR). Also, the activated mTOR pathway may be a marker of disease activity and prognosis. In a retrospective study, rapamycin (mTOR) activation in renal tissue of 187 LN patients was elevated compared to patients with diabetic nephropathy of minimal change disease. MTORC1 was activated in the glomerular compartment including mesangial cells, endothelial cells, and podocytes and was associated with altered biomarkers of LN like serum albumin, complement C3, proteinuria, crescent formation, interstitial inflammation, and fibrosis.124 In lupus-prone New Zealand Black/White F1 (NZB/W F1) mice rapamycin was beneficial to LN development as it markedly decreased proteinuria and serum anti-double-stranded DNA antibody levels and improved renal function.125 Rapamycin in addition to MMF reduced fibrosis in LN in NZB/W F1 mice.126 A single-arm, open-label, phase 1/2 trial of sirolimus in 31 active SLE patients without LN confirmed a reduction in disease activity (SLEDAI and BILAG scores) and ameliorated a pro-inflammatory T cell profile during 12 months of sirolimus treatment.127
Calcineurin Inhibitors
Calcineurin inhibitors (CNI), including tacrolimus, cyclosporin A (CsA) and voclosporin act on T cells by inhibiting the calcineurin activity and NFAT (nuclear factor of activated T cells) dephosphorylation in cytosolic immunophilins.128 However, the main therapeutic effect of CNIs we believe is regulating the podocyte skeleton by inhibiting the dephosphorylation and degradation of synaptopodin and therefore improving the podocyte foot process barrier function and proteinuria in the patient.106 However, CNIs are associated with side effects like hypertension and nephrotoxicity. Recently, a new CNI voclosporin, an analog of CsA has been studied in LN patients to be beneficial in renal outcome. Voclosporin is showing more stable pharmacokinetic and pharmacodynamic characteristics compared to tacrolimus including enhanced action against calcineurin and improved metabolic profile.129 In the phase III study (AURORA 1) voclosporin was combined with MMF standard of care and with a consecutive faster tapering and low-dose corticosteroid regimen for 52 weeks. This resulted in a superior and faster renal response defined by change of proteinuria (urine protein creatinine ratio of 0.5 mg/mg or less) and stable renal function (eGFR ≥60 mL/min/1.73 m2 or no decrease from baseline in eGFR of >20%) compared to MMF and corticosteroids alone. The safety profile was similar in both groups.130 In the ongoing phase 3 study AURORA 2 (NCT03597464) additional 24 months are investigated to assess long-term safety and tolerability of voclosporin continuing under the same conditions as at the end of the AURORA 1 study. In the interim analysis reported by the company patients in the voclosporin-treated group, sustained meaningful reductions in proteinuria, and kidney function remained stable without any unexpected side effects compared to the control group. In summary, this drug may be beneficial adding to standard care with a benign pharmacologic profile that eliminates the need for therapeutic drug monitoring. Moreover, the advantage of voclosporin includes an improved lipid, glucose, and metabolic profile. In addition, voclosporin does not interact with MMF. The rapid reduction in proteinuria in the voclosporin group is an outcome. However, it will be important to assess the long-term outcome in terms of the efficacy and safety of voclosporin. Voclosporin was approved by the FDA in January 2021 and received a positive statement by the EMA in July 2022 and is indicated in combination with MMF for the treatment of adult patients with active class III, IV including mixed class III/V and IV/V LN. There are few randomized trials on other CNI such as tacrolimus in patients with LN. 40 patients with class V+IV LN were treated with a multitarget therapy including tacrolimus, MMF and corticosteroids and compared to cyclophosphamide for 6 and 9 months. That study showed a higher rate of complete remission with multitarget therapy at both 6 and 9 months than with cyclophosphamide.131 A comparable study assessed the efficacy and safety of a multitarget therapy consisting of tacrolimus, MMF, and steroid compared with intravenous cyclophosphamide and steroid as induction therapy for LN for 24 weeks with a higher complete remission rate in the multitarget group (45.9%) than in the cyclophosphamide group.132 In the follow-up trial, patients continued to receive multitarget therapy (tacrolimus, 2–3 mg/d; MMF, 0.50–0.75 g/d; prednisone, 10 mg/d), and patients who had received intravenous cyclophosphamide induction treatment received azathioprine (2 mg/kg per day) plus prednisone (10 mg/d) for another 18 months which showed a lower renal relapse rate and fewer adverse events, suggesting that multitarget therapy is an effective and safe maintenance treatment for patients with LN.133 A trial with 81 patients with LN compared the efficacy and safety of tacrolimus versus intravenous cyclophosphamide as induction therapy for 6 months. The tacrolimus group revealed higher cumulative probabilities of complete remission than the cyclophosphamide group, showing that induction therapy tacrolimus was non-inferior to cyclophosphamide in the background of corticosteroids. As expected, proteinuria was significantly decreased in tacrolimus- versus cyclophosphamide-treated patients after the first month of treatment.134 A similar trial with 60 patients compared MMF and tacrolimus compared with intravenous cyclophosphamide in induction treatment for active LN with similar complete remission rates in the three groups.135
In a trial with 150 patients, tacrolimus was also demonstrated to be non-inferior to MMF, when combined with prednisolone, for induction therapy of active LN after 6 months of treatment.136 Long-term data of the 10-year outcome confirmed non-inferiority of tacrolimus to MMF as induction therapy of LN with maintenance therapy with azathioprine for 5 years in almost 80% of the patients.137 Recently, a phase 3 study with 314 LN patients supports oral tacrolimus to be non-inferior to cyclophosphamide in induction therapy for LN, showing a more favorable safety profile than cyclophosphamide.138 In LN patients who failed to achieve complete response after standard induction therapy, thus having a refractory condition, might profit from multitarget therapy, especially if proteinuria persists. This treatment can help patients achieve a renal response and reduce the use of steroids.139,140 Very few randomized controlled trials demonstrated beneficial outcomes for patients with LN with treatment with CsA. In one trial, 42 patients with lupus membranous nephropathy were treated either with alternate-day prednisone alone or in combination with low-dose CsA for 1 year or with cyclophosphamide for 6 doses. The cumulative probability of remission was 27% with prednisone, 60% with IV CYC, and 83% with CsA after 1 year. However, the relapse rate of nephrotic syndrome was significantly higher after completion of CsA than after cyclophosphamide.141 CsA was as effective as cyclophosphamide in the trial of sequential induction and maintenance treatment in patients with proliferative LN and preserved renal function.142 In the Cyclofa-Lune study, 40 patients with proliferative LN and preserved renal function were randomized to cyclophosphamide or CsA for induction and maintenance treatment in patients. The study showed that CsA was non-inferior to cyclophosphamide at 9 and 18 months.142 In general, reported side effects of CNI are mostly a slightly increased blood pressure and a reversible decrease in glomerular filtration rate. Discontinuation often results in a relapse of proteinuria. Regular drug level monitoring is necessary for both CsA and Tacrolimus to avoid (nephro)toxicity.
The characterized therapies for LN include T- and B-lymphocyte signaling, their proliferation and activation, as well as inflammatory responses (Table 1). Many of biological agents mentioned here are still in phase II and III clinical stages and must still be evaluated regarding efficacy and tolerability. Importantly, many trials mentioned here included moderate or severe LN patients. It is still not sufficiently tested (eg, in preclinical studies) if some of the agents could be combined to achieve better effects or if better classification of patients could improve the results.
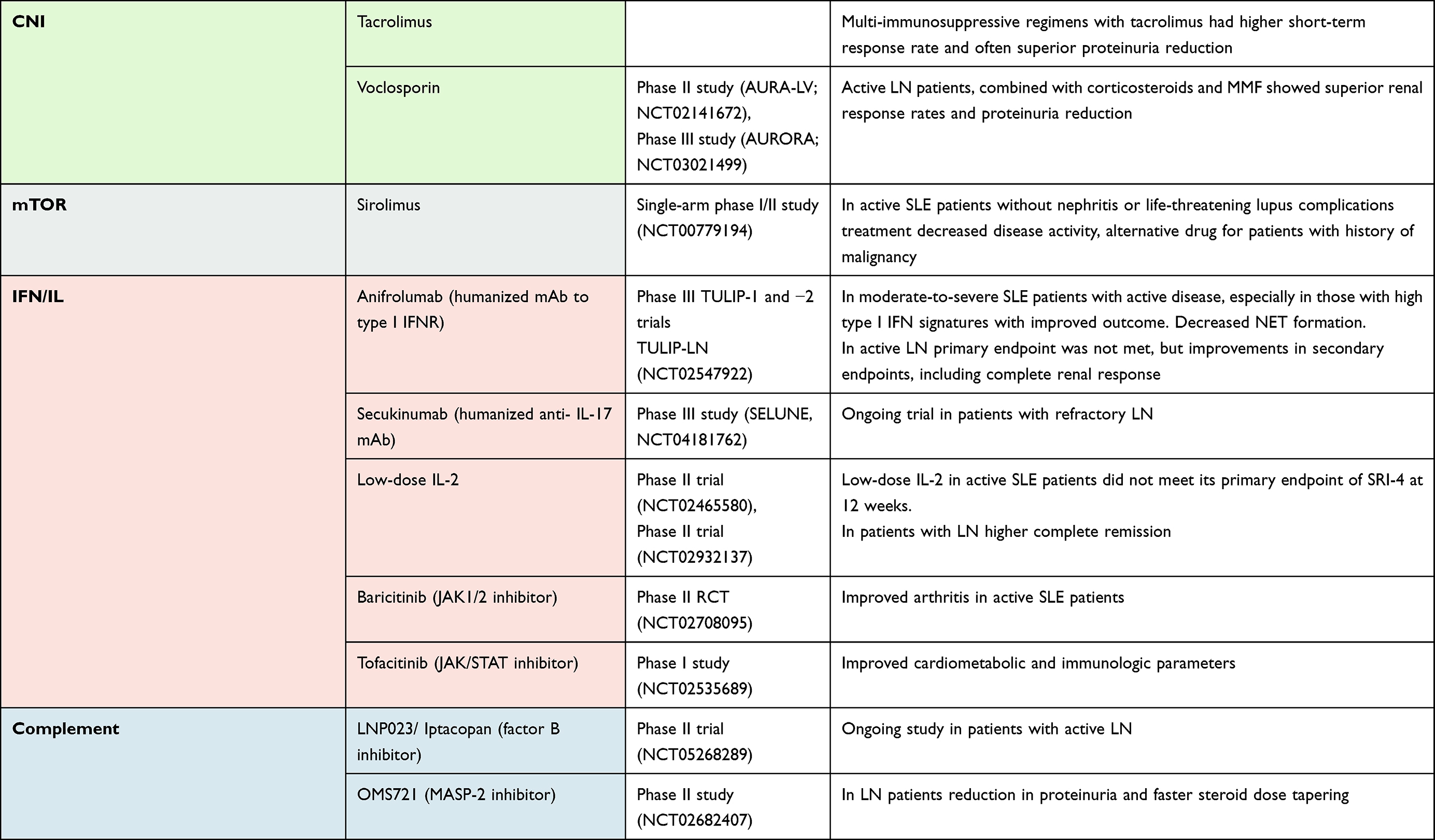 |
Table 1 Relevant Clinical Trials in Patients with Lupus Nephritis |
Conclusion
In recent years, clinical trials evaluating the value of biological drugs in the treatment of SLE and LN have multiplied. Studies in progress offer encouraging examples of targets and perspectives for new therapies. Moreover, novel targeted treatments are precise and often display fewer side effects than standard immunosuppressants. However, the difficulty of obtaining positive results beyond phase 2 within studies might be associated with heterogeneity in patients with SLE as well as a study design. A further understanding of SLE pathology remains crucial to identify new targets and at the same time clinical and molecular phenotypes, which will favor the administration of individualized biological therapies. Upcoming years will surely deliver some exciting new findings regarding the treatment of SLE and LN. For instance, ALXN2050 in proliferative LN and Immunoglobulin A Nephropathy (IgAN), NCT05097989 and EQ001 (Itolizumab) in SLE with or without active proliferative nephritis (EQUALISE), NCT04128579 are currently recruiting patients for the phase 2 and 1, respectively. A further exciting approach is a randomized, phase III multicenter trial investigating induction therapy for LN with no added oral steroids: a trial comparing oral corticosteroids plus MMF versus obinutuzumab and MMF, NCT04702256. Zanubrutinib, also known as BGB-3111/Brukinsa, which blocks BCR signaling by forming a covalent bond with a cysteine residue in the BTK active site, is also being investigated in LN. Participants with active proliferative LN are currently recruited in a phase 2 study to evaluate the safety and efficacy of Zanubrutinib (NCT04643470). Further upcoming trials include the study of efficacy and safety of LNP023/Iptacopan in participants with active LN Class III–IV, ± V, (NCT05268289), the evaluation of VIB4920 for active LN (VIBRANT, NCT05201469), and Ravulizumab trial that investigate the termination of complement pathway by binding to C5 with high affinity in proliferative LN or Immunoglobulin A Nephropathy (IgAN) (SANCTUARY, NCT04564339).
Acknowledgment
HJA received support from the Deutsche Forschungsgemeinschaft (AN372/30-1) and the Volkswagen Foundation (Nr. 97-744).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
HJA received honoraries from GSK, Novartis, Janssen, Otsuka, Kezar, and AstraZeneca. He also reports grants and/or personal fees from Bayer, Boehringer Ingelheim, Previpharma, and Lilly. The authors report no other conflicts of interest in this work.
References
1. Anders H-J, Saxena R, Zhao M-H, et al. Lupus nephritis. Nat Rev Dis Primers. 2020;6:7. doi:10.1038/s41572-019-0141-9
2. Anders H-J, Fogo AB. Immunopathology of lupus nephritis. Semin Immunopathol. 2014;36:443–459. doi:10.1007/s00281-013-0413-5
3. Lorenz G, Lech M, Anders H-J. Toll-like receptor activation in the pathogenesis of lupus nephritis. Clin Immunol. 2017;185:86–94. doi:10.1016/j.clim.2016.07.015
4. Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 2019;71(9):1400–1412. doi:10.1002/art.40930
5. Fanouriakis A, Tziolos N, Bertsias G, Boumpas DT. Update on the diagnosis and management of systemic lupus erythematosus. Ann Rheum Dis. 2021;80(1):14–25. doi:10.1136/annrheumdis-2020-218272
6. Steiger S, Rossaint J, Zarbock A, Anders H-J. Secondary Immunodeficiency Related to Kidney Disease (SIDKD)-definition, unmet need, and mechanisms. J Am Soc Nephrol. 2022;33:259–278.
7. Demirkaya E, Sahin S, Romano M, Zhou Q, Aksentijevich I. New horizons in the genetic etiology of systemic lupus erythematosus and lupus-like disease: monogenic lupus and beyond. J Clin Med. 2020;9:E712.
8. Guerra SG, Vyse TJ, Cunninghame Graham DS. The genetics of lupus: a functional perspective. Arthritis Res Ther. 2012;14:211. doi:10.1186/ar3844
9. Tiffin N, Adeyemo A, Okpechi I. A diverse array of genetic factors contribute to the pathogenesis of systemic lupus erythematosus. Orphanet J Rare Dis. 2013;8:2. doi:10.1186/1750-1172-8-2
10. Deng Y, Tsao BP. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat Rev Rheumatol. 2010;6:683–692. doi:10.1038/nrrheum.2010.176
11. Kelley JM, Edberg JC, Kimberly RP. Pathways: strategies for susceptibility genes in SLE. Autoimmun Rev. 2010;9:473–476. doi:10.1016/j.autrev.2010.02.003
12. Rullo OJ, Tsao BP. Recent insights into the genetic basis of systemic lupus erythematosus. Ann Rheum Dis. 2013;72(Suppl 2):ii56–61. doi:10.1136/annrheumdis-2012-202351
13. Costa-Reis P, Sullivan KE. Genetics and epigenetics of systemic lupus erythematosus. Curr Rheumatol Rep. 2013;15:369. doi:10.1007/s11926-013-0369-4
14. Yang F, He Y, Zhai Z, Sun E. Programmed cell death pathways in the pathogenesis of systemic lupus erythematosus. J Immunol Res. 2019;3638562:2019.
15. Mistry P, Kaplan MJ. Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin Immunol. 2017;185:59–73. doi:10.1016/j.clim.2016.08.010
16. Hepburn AL, Mason JC, Wang S, et al. Both Fcγ and complement receptors mediate transfer of immune complexes from erythrocytes to human macrophages under physiological flow conditions in vitro. Clin Exp Immunol. 2006;146(1):133–145. doi:10.1111/j.1365-2249.2006.03174.x
17. MacPherson M, Lek HS, Prescott A, Fagerholm SC. A systemic lupus erythematosus-associated R77H substitution in the CD11b chain of the Mac-1 integrin compromises leukocyte adhesion and phagocytosis. J Biol Chem. 2011;286:17303–17310. doi:10.1074/jbc.M110.182998
18. Nath SK, Han S, Kim-Howard X, et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet. 2008;40:152–154. doi:10.1038/ng.71
19. Yang W, Tang H, Zhang Y, et al. Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in Asians. Am J Hum Genet. 2013;92:41–51. doi:10.1016/j.ajhg.2012.11.018
20. Harley JB, Alarcón-Riquelme ME, Criswell LA; International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN). Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–210. doi:10.1038/ng.81
21. Freedman BI, Langefeld CD, Andringa KK, et al. End-stage renal disease in African Americans With Lupus Nephritis Is Associated With APOL1. Arthritis Rheumatol. 2014;66:390–396. doi:10.1002/art.38220
22. Zhou X, Lu X-L, Lv J-C, et al. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis. 2011;70:1330–1337. doi:10.1136/ard.2010.140111
23. Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–1036. doi:10.1038/nature03029
24. Gateva V, Sandling JK, Hom G, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41(11):1228–1233. doi:10.1038/ng.468
25. Allam R, Anders H-J. The role of innate immunity in autoimmune tissue injury. Curr Opin Rheumatol. 2008;20:538–544. doi:10.1097/BOR.0b013e3283025ed4
26. Lech M, Anders H-J. The pathogenesis of lupus nephritis. J Am Soc Nephrol. 2013;24:1357–1366. doi:10.1681/ASN.2013010026
27. Rodero MP, Decalf J, Bondet V, et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J Exp Med. 2017;214(5):1547–1555. doi:10.1084/jem.20161451
28. Nehar-Belaid D, Hong S, Marches R, et al. Mapping systemic lupus erythematosus heterogeneity at the single-cell level. Nat Immunol. 2020;21(9):1094–1106. doi:10.1038/s41590-020-0743-0
29. Banchereau R, Hong S, Cantarel B, et al. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell. 2016;165(3):551–565. doi:10.1016/j.cell.2016.03.008
30. Gao D, Li T, Li X-D, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A. 2015;112(42):E5699–E5705. doi:10.1073/pnas.1516465112
31. Yoneyama M, Kikuchi M, Matsumoto K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175(5):2851–2858. doi:10.4049/jimmunol.175.5.2851
32. Loo Y-M, Gale M. Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi:10.1016/j.immuni.2011.05.003
33. Keating SE, Baran M, Bowie AG. Cytosolic DNA sensors regulating type I interferon induction. Trends Immunol. 2011;32:574–581. doi:10.1016/j.it.2011.08.004
34. Radetskyy R, Daher A, Gatignol A. ADAR1 and PKR, interferon stimulated genes with clashing effects on HIV-1 replication. Cytokine Growth Factor Rev. 2018;40:48–58. doi:10.1016/j.cytogfr.2018.03.007
35. Wang TT, Li ZG, Wang Q. The RNA-specific adenosine deaminase ADAR1 Inhibits Human Protein Kinase R Activation. Viral Immunol. 2018;31:537–538. doi:10.1089/vim.2018.0056
36. Patole PS, Zecher D, Pawar RD, et al. G-rich DNA suppresses systemic lupus. J Am Soc Nephrol. 2005;16(11):3273–3280. doi:10.1681/ASN.2005060658
37. Anders HJ. A Toll for lupus. Lupus. 2005;14:417–422. doi:10.1191/0961203305lu2102rr
38. Vance RE. Cytosolic DNA sensing: the field narrows. Immunity. 2016;45:227–228. doi:10.1016/j.immuni.2016.08.006
39. Bolin K, Imgenberg-Kreuz J, Leonard D, et al. Variants in BANK1 are associated with lupus nephritis of European ancestry. Genes Immun. 2021;22(3):194–202. doi:10.1038/s41435-021-00142-8
40. Pisitkun P, Deane JA, Difilippantonio MJ, et al. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–1672. doi:10.1126/science.1124978
41. Christensen SR, Shupe J, Nickerson K, et al. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25(3):417–428. doi:10.1016/j.immuni.2006.07.013
42. Deane JA, Pisitkun P, Barrett RS, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27(5):801–810. doi:10.1016/j.immuni.2007.09.009
43. Rönnblom L. The type I interferon system in the etiopathogenesis of autoimmune diseases. Ups J Med Sci. 2011;116:227–237. doi:10.3109/03009734.2011.624649
44. Brown GJ, Cañete PF, Wang H, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. 2022;605(7909):349–356. doi:10.1038/s41586-022-04642-z
45. Chen M, Daha MR, Kallenberg CGM. The complement system in systemic autoimmune disease. J Autoimmun. 2010;34:J276–J286. doi:10.1016/j.jaut.2009.11.014
46. Stegert M, Bock M, Trendelenburg M. Clinical presentation of human C1q deficiency: how much of a lupus? Mol Immunol. 2015;67:3–11. doi:10.1016/j.molimm.2015.03.007
47. Musone SL, Taylor KE, Lu TT, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet. 2008;40:1062–1064. doi:10.1038/ng.202
48. Kawasaki A, Ito S, Furukawa H, et al. Association of TNFAIP3 interacting protein 1, TNIP1 with systemic lupus erythematosus in a Japanese population: a case-control association study. Arthritis Res Ther. 2010;12(5):R174. doi:10.1186/ar3134
49. Zhou J, Wu R, High AA, et al. A20-binding inhibitor of NF-κB (ABIN1) controls Toll-like receptor-mediated CCAAT/enhancer-binding protein β activation and protects from inflammatory disease. Proc Natl Acad Sci U S A. 2011;108(44):E998–E1006. doi:10.1073/pnas.1106232108
50. Moynihan TP, Cole CG, Dunham I, et al. Fine-mapping, genomic organization, and transcript analysis of the human ubiquitin-conjugating enzyme gene UBE2L3. Genomics. 1998;51(1):124–127. doi:10.1006/geno.1998.5257
51. Kim T, Bae S-C, Kang C. Synergistic activation of NF-κB by TNFAIP3 (A20) reduction and UBE2L3 (UBCH7) augment that synergistically elevate lupus risk. Arthritis Res Ther. 2020;22:93. doi:10.1186/s13075-020-02181-4
52. Liang J, Saad Y, Lei T, et al. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-kappaB signaling. J Exp Med. 2010;207:2959–2973. doi:10.1084/jem.20092641
53. Dobosz E, Lorenz G, Ribeiro A, et al. Murine myeloid cell MCPIP1 suppresses autoimmunity by regulating B-cell expansion and differentiation. Dis Model Mech. 2021;14(3):dmm047589. doi:10.1242/dmm.047589
54. Caster DJ, Korte EA, Nanda SK, et al. ABIN1 dysfunction as a genetic basis for lupus nephritis. J Am Soc Nephrol. 2013;24(11):1743–1754. doi:10.1681/ASN.2013020148
55. Lech M, Kantner C, Kulkarni OP, et al. Interleukin-1 receptor-associated kinase-M suppresses systemic lupus erythematosus. Ann Rheum Dis. 2011;70(12):2207–2217. doi:10.1136/ard.2011.155515
56. Szelinski F, Lino AC, Dörner T. B cells in systemic lupus erythematosus. Curr Opin Rheumatol. 2022;34:125–132. doi:10.1097/BOR.0000000000000865
57. Harley ITW, Allison K, Scofield RH. Polygenic autoimmune disease risk alleles impacting B cell tolerance act in concert across shared molecular networks in mouse and in humans. Front Immunol. 2022;13:953439.
58. Suárez-Fueyo A, Bradley SJ, Tsokos GC. T cells in Systemic Lupus Erythematosus. Curr Opin Immunol. 2016;43:32–38. doi:10.1016/j.coi.2016.09.001
59. Karimifar M, Akbari K, ArefNezhad R, et al. Impacts of FcγRIIB and FcγRIIIA gene polymorphisms on systemic lupus erythematosus disease activity index. BMC Res Notes. 2021;14(1):455. doi:10.1186/s13104-021-05868-2
60. Rzeszotarska E, Sowinska A, Stypinska B, et al. IL-1β, IL-10 and TNF-α polymorphisms may affect systemic lupus erythematosus risk and phenotype. Clin Exp Rheumatol. 2022;40:1708–1717.
61. Chrabot BS, Kariuki SN, Zervou MI, et al. Genetic variation near IRF8 is associated with serologic and cytokine profiles in systemic lupus erythematosus and multiple sclerosis. Genes Immun. 2013;14(8):471–478. doi:10.1038/gene.2013.42
62. Kim K, Brown EE, Choi CB, et al. Variation in the ICAM1-ICAM4-ICAM5 locus is associated with systemic lupus erythematosus susceptibility in multiple ancestries. Ann Rheum Dis. 2012;71:1809–1814.
63. Roberts AL, Thomas ER, Bhosle S, et al. Resequencing the susceptibility gene, ITGAM, identifies two functionally deleterious rare variants in systemic lupus erythematosus cases. Arthritis Res Ther. 2014;16(3):R114. doi:10.1186/ar4566
64. Moreno-Angarita A, Aragón CC, Tobón GJ. Cathelicidin LL-37: a new important molecule in the pathophysiology of systemic lupus erythematosus. J Transl Autoimmun. 2020;3:100029.
65. Borchers AT, Leibushor N, Naguwa SM, et al. Lupus nephritis: a critical review. Autoimmun Rev. 2012;12:174–194.
66. Schwartzman-Morris J, Putterman C. Gender differences in the pathogenesis and outcome of lupus and of lupus nephritis. Clin Dev Immunol. 2012;604892:2012.
67. Maroz N, Segal MS. Lupus nephritis and end-stage kidney disease. Am J Med Sci. 2013;346:319–323.
68. Chung SA, Taylor KE., Graham RR, et al. Differential genetic associations for systemic lupus erythematosus based on anti-dsDNA autoantibody production. PLoS Genet. 2011;7:e1001323.
69. Freedman BI, Kopp JB, Langefeld CD, et al. The Apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J Am Soc Nephrol. 2010;21:1422–1426. doi:10.1681/ASN.2010070730
70. Lin CP, Adrianto I, Lessard CJ, et al. Role of MYH9 and APOL1 in African and non-African populations with lupus nephritis. Genes Immun. 2012;13(3):232–238. doi:10.1038/gene.2011.82
71. Alonso-Perez E, Suarez-Gestal M, Calaza M, et al. Further evidence of subphenotype association with systemic lupus erythematosus susceptibility loci: a European cases only study. PLoS One. 2012;7(9):e45356. doi:10.1371/journal.pone.0045356
72. Taylor KE, Remmers EF, Lee AT, et al. Specificity of the STAT4 genetic association for severe disease manifestations of systemic lupus erythematosus. PLoS Genet. 2008;4:e1000084.
73. Bolin K, Sandling JK, Zickert A, et al. Association of STAT4 polymorphism with severe renal insufficiency in lupus nephritis. PLoS One. 2013;8(12):e84450. doi:10.1371/journal.pone.0084450
74. Zhou X, Cheng F, Qi Y, Zhao M, Zhang H. A replication study from Chinese supports association between lupus-risk allele in TNFSF4 and renal disorder. Biomed Res Int. 2013;597921:2013.
75. Lanata CM, Nititham J, Taylor KE, et al. Genetic contributions to lupus nephritis in a multi-ethnic cohort of systemic lupus erythematosus patients. PLoS One. 2018;13(6):e0199003. doi:10.1371/journal.pone.0199003
76. Zhang F, Wang Y-F, Zhang Y, et al. Independent Replication on Genome-Wide Association Study Signals Identifies IRF3 as a Novel Locus for Systemic Lupus Erythematosus. Front Genet. 2020;11:600. doi:10.3389/fgene.2020.00600
77. Dooley MA, Houssiau F, Aranow C, et al. Effect of belimumab treatment on renal outcomes: results from the phase 3 belimumab clinical trials in patients with SLE. Lupus. 2013;22(1):63–72. doi:10.1177/0961203312465781
78. Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62:222–233.
79. Rovin BH, Furie R, Latinis K, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012;64(4):1215–1226. doi:10.1002/art.34359
80. Kamburova EG, Koenen HJPM, Borgman KJE, et al. A single dose of rituximab does not deplete B cells in secondary lymphoid organs but alters phenotype and function. Am J Transplant. 2013;13:1503–1511. doi:10.1111/ajt.12220
81. Furie RA, Aroca G, Cascino MD, et al. B-cell depletion with obinutuzumab for the treatment of proliferative lupus nephritis: a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2022;81(1):100–107. doi:10.1136/annrheumdis-2021-220920
82. Mössner E, Brünker P, Moser S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. 2010;115:4393–4402. doi:10.1182/blood-2009-06-225979
83. Haarhaus ML, Svenungsson E, Gunnarsson I. Ofatumumab treatment in lupus nephritis patients. Clin Kidney J. 2016;9:552–555. doi:10.1093/ckj/sfw022
84. Mackensen A, Müller F, Mougiakakos D, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. 2022. doi:10.1038/s41591-022-02017-5
85. Mougiakakos D, Krönke G, Völkl S, et al. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N Engl J Med. 2021;385(6):567–569. doi:10.1056/NEJMc2107725
86. Merrill JT, Wallace DJ, Wax S, et al. Efficacy and safety of atacicept in patients with systemic lupus erythematosus: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled, parallel-arm, Phase IIb study. Arthritis Rheumatol. 2018;70(2):266–276. doi:10.1002/art.40360
87. Wu D, Li J, Xu D, et al. A human recombinant fusion protein targeting B lymphocyte stimulator (BlyS) and a proliferation-inducing ligand (April), telitacicept (RC18), in systemic lupus erythematosus (SLE): results of a phase 2b study [abstract no. L18]. Arthritis Rheumatol. 2019;71(Suppl10):5262–5264.
88. Merrill JT, Van Vollenhoven RF, Buyon JP, et al. Efficacy and safety of subcutaneous tabalumab, a monoclonal antibody to B-cell activating factor, in patients with systemic lupus erythematosus: results from ILLUMINATE-2, a 52-week, Phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2016;75:332–340.
89. Isenberg DA, Petri M, Kalunian K, et al. Efficacy and safety of subcutaneous tabalumab in patients with systemic lupus erythematosus: results from ILLUMINATE-1, a 52-week, phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2016;75(2):323–331. doi:10.1136/annrheumdis-2015-207653
90. Merrill JT, Shanahan WR, Scheinberg M, et al. Phase III trial results with blisibimod, a selective inhibitor of B-cell activating factor, in subjects with systemic lupus erythematosus (SLE): results from a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2018;77(6):883–889. doi:10.1136/annrheumdis-2018-213032
91. Cheng LE, Amoura Z, Cheah B, et al. Brief report: a randomized, double-blind, parallel-group, placebo-controlled, multiple-dose study to evaluate AMG 557 in patients with systemic lupus erythematosus and active lupus arthritis. Arthritis Rheumatol. 2018;70:1071–1076.
92. Lipsky PE, Vollenhoven RV, Dörner T, et al. Biological impact of iberdomide in patients with active systemic lupus erythematosus. Ann Rheum Dis Annrheumdis. 2022;81(8):1136–1142. doi:10.1136/annrheumdis-2022-222212
93. Merrill JT, Werth VP, Furie R, et al. Phase 2 trial of iberdomide in systemic lupus erythematosus. N Engl J Med. 2022;386:1034–1045.
94. Klimatcheva E, Pandina T, Reilly C, et al. CXCL13 antibody for the treatment of autoimmune disorders. BMC Immunol. 2015;16:6.
95. Hiepe F, Alexander T, Voll RE. Plasmazellen [Plasma cells]. Z Rheumatol. 2015;74:20–25. German. doi:10.1007/s00393-014-1438-4
96. Odendahl M, Mei H, Hoyer BF, et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood. 2005;105(4):1614–1621. doi:10.1182/blood-2004-07-2507
97. Muehlinghaus G, Cigliano L, Huehn S, et al. Regulation of CXCR3 and CXCR4 expression during terminal differentiation of memory B cells into plasma cells. Blood. 2005;105(10):3965–3971. doi:10.1182/blood-2004-08-2992
98. Arce S, Luger E, Muehlinghaus G, et al. CD38 low IgG-secreting cells are precursors of various CD38 high-expressing plasma cell populations. J Leukoc Biol. 2004;75(6):1022–1028. doi:10.1189/jlb.0603279
99. Rodríguez-Bayona B, Ramos-Amaya A, Bernal J, Campos-Caro A, Brieva JA. Cutting edge: IL-21 derived from human follicular helper T cells acts as a survival factor for secondary lymphoid organ, but not for bone marrow, plasma cells. J Immunol. 2012;188:1578–1581. doi:10.4049/jimmunol.1102786
100. Kehrl JH, Riva A, Wilson GL, Thévenin C. Molecular mechanisms regulating CD19, CD20 and CD22 gene expression. Immunol Today. 1994;15:432–436. doi:10.1016/0167-5699(94)90273-9
101. Leandro MJ. B-cell subpopulations in humans and their differential susceptibility to depletion with anti-CD20 monoclonal antibodies. Arthritis Res Ther. 2013;15(Suppl 1):S3. doi:10.1186/ar3908
102. Hoyer BF, Moser K, Hauser AE, et al. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med. 2004;199(11):1577–1584. doi:10.1084/jem.20040168
103. Neubert K, Meister S, Moser K, et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med. 2008;14(7):748–755. doi:10.1038/nm1763
104. Ostendorf L, Burns M, Durek P, et al. Targeting CD38 with Daratumumab in Refractory Systemic Lupus Erythematosus. N Engl J Med. 2020;383(12):1149–1155. doi:10.1056/NEJMoa2023325
105. Alexander T, Sarfert R, Klotsche J, et al. The proteasome inhibitor bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann Rheum Dis. 2015;74(7):1474–1478. doi:10.1136/annrheumdis-2014-206016
106. Faul C, Donnelly M, Merscher-Gomez S, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14(9):931–938. doi:10.1038/nm.1857
107. Jayne D, Rovin B, Mysler EF, et al. Phase II randomised trial of type I interferon inhibitor anifrolumab in patients with active lupus nephritis. Ann Rheum Dis. 2022;81(4):496–506. doi:10.1136/annrheumdis-2021-221478
108. Steiger S, Anders H-J. Interferon blockade in lupus: effects on antiviral immunity. Nat Rev Nephrol. 2022;18:415–416. doi:10.1038/s41581-022-00581-0
109. Khamashta M, Merrill JT, Werth VP, et al. Sifalimumab, an anti-interferon-α monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2016;75(11):1909–1916. doi:10.1136/annrheumdis-2015-208562
110. Kalunian KC, Merrill JT, Maciuca R, et al. A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-α) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis. 2016;75(1):196–202. doi:10.1136/annrheumdis-2014-206090
111. Wallace DJ, Furie RA, Tanaka Y, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2018;392(10143):222–231. doi:10.1016/S0140-6736(18)31363-1
112. Hasni SA, Gupta S, Davis M, et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat Commun. 2021;12(1):3391. doi:10.1038/s41467-021-23361-z
113. Furumoto Y, Smith CK, Blanco L, et al. Tofacitinib Ameliorates Murine Lupus and Its Associated Vascular Dysfunction. Arthritis Rheumatol. 2017;69(1):148–160. doi:10.1002/art.39818
114. Baker M, Chaichian Y, Genovese M, et al. Phase II, randomised, double-blind, multicentre study evaluating the safety and efficacy of filgotinib and lanraplenib in patients with lupus membranous nephropathy. RMD Open. 2020;6(3):e001490. doi:10.1136/rmdopen-2020-001490
115. Bao L, Cunningham PN, Quigg RJ. Complement in Lupus Nephritis: new Perspectives. Kidney Dis. 2015;1:91–99. doi:10.1159/000431278
116. Li NL, Birmingham DJ, Rovin BH. Expanding the Role of Complement Therapies: the Case for Lupus Nephritis. J Clin Med. 2021;10:626. doi:10.3390/jcm10040626
117. Martin M, Blom AM. Complement in removal of the dead - balancing inflammation. Immunol Rev. 2016;274:218–232. doi:10.1111/imr.12462
118. Birmingham DJ, Hebert LA. CR1 and CR1-like: the primate immune adherence receptors. Immunol Rev. 2001;180:100–111. doi:10.1034/j.1600-065X.2001.1800109.x
119. Birmingham DJ, Gavit KF, McCarty SM, et al. Consumption of erythrocyte CR1 (CD35) is associated with protection against systemic lupus erythematosus renal flare. Clin Exp Immunol. 2006;143(2):274–280. doi:10.1111/j.1365-2249.2005.02983.x
120. He J, Zhang R, Shao M, et al. Efficacy and safety of low-dose IL-2 in the treatment of systemic lupus erythematosus: a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2020;79(1):141–149. doi:10.1136/annrheumdis-2019-215396
121. Koga T, Ichinose K, Kawakami A, Tsokos GC. The role of IL-17 in systemic lupus erythematosus and its potential as a therapeutic target. Expert Rev Clin Immunol. 2019;15:629–637. doi:10.1080/1744666X.2019.1593141
122. Morel T, Cano S, Bartlett SJ, et al. The FATIGUE-PRO: a new patient-reported outcome instrument to quantify fatigue in patients affected by systemic lupus erythematosus. Rheumatology. 2022;61(8):3329–3340. doi:10.1093/rheumatology/keab920
123. Oaks Z, Winans T, Huang N, Banki K, Perl A. Activation of the mechanistic target of rapamycin in SLE: explosion of evidence in the last five years. Curr Rheumatol Rep. 2016;18:73. doi:10.1007/s11926-016-0622-8
124. Mao Z, Tan Y, Tao J, et al. Renal mTORC1 activation is associated with disease activity and prognosis in lupus nephritis. Rheumatology. 2022;61(9):3830–3840. doi:10.1093/rheumatology/keac037
125. Lui SL, Yung S, Tsang R, et al. Rapamycin prevents the development of nephritis in lupus-prone NZB/W F 1 mice. Lupus. 2008;17(4):305–313. doi:10.1177/0961203307088289
126. Zhang C, Chan CY, Cheung K, et al. Effect of mycophenolate and rapamycin on renal fibrosis in lupus nephritis. Clin Sci. 2019;133(15):1721–1744. doi:10.1042/CS20190536
127. Lai Z-W, Kelly R, Winans T, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet. 2018;391(10126):1186–1196. doi:10.1016/S0140-6736(18)30485-9
128. Dutta D, Barr VA, Akpan I, et al. Recruitment of calcineurin to the TCR positively regulates T cell activation. Nat Immunol. 2017;18(2):196–204. doi:10.1038/ni.3640
129. Sin FE, Isenberg D. An evaluation of voclosporin for the treatment of lupus nephritis. Expert Opin Pharmacother. 2018;19:1613–1621. doi:10.1080/14656566.2018.1516751
130. Rovin BH, Teng YKO, Ginzler EM, et al. Efficacy and safety of voclosporin versus placebo for lupus nephritis (Aurora 1): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2021;397(10289):2070–2080. doi:10.1016/S0140-6736(21)00578-X
131. Bao H, Liu Z-H, Xie H-L, et al. Successful treatment of class V+IV lupus nephritis with multitarget therapy. J Am Soc Nephrol. 2008;19(10):2001–2010. doi:10.1681/ASN.2007121272
132. Liu Z, Zhang H, Liu Z, et al. Multitarget therapy for induction treatment of lupus nephritis: a randomized trial. Ann Intern Med. 2015;162(1):18–26. doi:10.7326/M14-1030
133. Zhang H, Liu Z, Zhou M, et al. Multitarget therapy for maintenance treatment of lupus nephritis. J Am Soc Nephrol. 2017;28(12):3671–3678. doi:10.1681/ASN.2017030263
134. Chen W, Tang X, Liu Q, et al. Short-term outcomes of induction therapy with tacrolimus versus cyclophosphamide for active lupus nephritis: a multicenter randomized clinical trial. Am J Kidney Dis. 2011;57(2):235–244. doi:10.1053/j.ajkd.2010.08.036
135. Li X, Ren H, Zhang Q, et al. Mycophenolate mofetil or tacrolimus compared with intravenous cyclophosphamide in the induction treatment for active lupus nephritis. Nephrol Dial Transplant. 2012;27(4):1467–1472. doi:10.1093/ndt/gfr484
136. Mok CC, Ying KY, Yim CW, et al. Tacrolimus versus mycophenolate mofetil for induction therapy of lupus nephritis: a randomised controlled trial and long-term follow-up. Ann Rheum Dis. 2016;75(1):30–36. doi:10.1136/annrheumdis-2014-206456
137. Mok CC, Ho LY, Ying SKY, et al. Long-term outcome of a randomised controlled trial comparing tacrolimus with mycophenolate mofetil as induction therapy for active lupus nephritis. Ann Rheum Dis. 2020;79(8):1070–1076. doi:10.1136/annrheumdis-2020-217178
138. Zheng Z, Zhang H, Peng X, et al. Effect of tacrolimus vs intravenous cyclophosphamide on complete or partial response in patients with lupus nephritis: a randomized clinical trial. JAMA Netw Open. 2022;5(3):e224492. doi:10.1001/jamanetworkopen.2022.4492
139. Choi C-B, Won S, Bae S-C. Outcomes of multitarget therapy using mycophenolate mofetil and tacrolimus for refractory or relapsing lupus nephritis. Lupus. 2018;27:1007–1011. doi:10.1177/0961203318758505
140. Yap DYH, Li PH, Tang C, et al. Long-term results of triple immunosuppression with tacrolimus added to mycophenolate and corticosteroids in the treatment of lupus nephritis. Kidney Int Rep. 2022;7(3):516–525. doi:10.1016/j.ekir.2021.12.005
141. Austin HA, Illei GG, Braun MJ, Balow JE. Randomized, controlled trial of prednisone, cyclophosphamide, and cyclosporine in lupus membranous nephropathy. J Am Soc Nephrol. 2009;20:901–911. doi:10.1681/ASN.2008060665
142. Zavada J, Pešickova SS, Ryšava R, et al. Cyclosporine A or intravenous cyclophosphamide for lupus nephritis: the Cyclofa-Lune study. Lupus. 2010;19(11):1281–1289. doi:10.1177/0961203310371155
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.