Back to Journals » Lung Cancer: Targets and Therapy » Volume 12
Lung Cancer with MET exon 14 Skipping Mutation: Genetic Feature, Current Treatments, and Future Challenges
Authors Fujino T ![]() , Suda K
, Suda K ![]() , Mitsudomi T
, Mitsudomi T ![]()
Received 6 March 2021
Accepted for publication 10 April 2021
Published 20 May 2021 Volume 2021:12 Pages 35—50
DOI https://doi.org/10.2147/LCTT.S269307
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sai-Hong Ou
Toshio Fujino, Kenichi Suda, Tetsuya Mitsudomi
Division of Thoracic Surgery, Department of Surgery, Kindai University Faculty of Medicine, Osaka-Sayama, Japan
Correspondence: Tetsuya Mitsudomi
Kindai University Faculty of Medicine, Division of Thoracic Surgery, Department of Surgery, 377-2 Ohno-Higashi, Osaka-Sayama, 589-8511, Japan
Tel +81 72 366 0221
Fax +81 72 365 7161
Email [email protected]
Abstract: MET exon 14 skipping mutation (MET∆ex14) is present about 3% of non-small cell lung cancers (NSCLCs). NSCLC patients with MET∆ex14 are characterized by an average age of over 70 years at diagnosis, a smoking history and a higher frequency in pleomorphic carcinoma and adenosquamous cell carcinoma than in adenocarcinoma. It has also been reported that NSCLCs with MET∆ex14 often have codriver alterations such as EGFR amplification (6– 28%), FGFR1 alterations (5– 17%), KRAS alterations (∼ 8%), BRAF alterations (∼ 21%), or PIK3CA mutation/amplification (∼ 14%). In 2020, the approval of two MET-tyrosine kinase inhibitors (TKIs), capmatinib and tepotinib, for NSCLCs carrying MET∆ex14 dawned a new era for MET-targeted therapy. These drugs yielded progression-free survival of 5.4− 12.4 months in clinical trials; however, it has also been reported that one-third to half of patients show inherent resistance to MET-TKIs. In addition, the emergence of acquired resistance to MET-TKIs is inevitable. In this review, we summarize the clinical and molecular characteristics of NSCLCs with MET∆ex14, the efficacy and safety of capmatinib and tepotinib, the inherent and acquired resistance mechanisms to MET-TKIs, and new treatment strategies for NSCLCs with MET∆ex14 in the near future.
Keywords: non-small cell lung cancer, MET exon 14 skipping, capmatinib, tepotinib, resistance mechanisms, immune checkpoint inhibitors
Introduction
The MET proto-oncogene, located in the 7q31 locus of chromosome 7, encodes a receptor tyrosine kinase (RTK) for hepatocyte growth factor (HGF), also known as scatter factor. MET is essential for embryonic development, organogenesis and wound healing.1 The MET gene was originally discovered as a part of an oncogenic fusion with the TPR (translocated promoter region) gene in a chemically induced human osteosarcoma cell line in 1984.2 MET was named after the first three letters of the chemical mutagen “N-methyl-N’-nitro-N-nitrosoguanidine.” Subsequently, increased MET expression and/or MET gene copy number gain was reported to be correlated with a poor prognosis in several types of carcinoma,3–6 and thus, molecular targeted therapies against MET have been developed and tested in many clinical trials. However, the results of all these trials, which enrolled unselected populations or patients with MET protein overexpression, were disappointing.7–9 These failures are attributable mainly to the insufficient selection of patients with tumors that are truly driven by MET.
Several types of MET aberrations, such as MET gene amplification, point mutations, gene fusions, exon 14 skipping mutations, or protein overexpression, have been reported in many types of carcinoma.10–13 Among these MET aberrations, MET exon 14 skipping mutation (MET∆ex14) in non-small cell lung cancer (NSCLC) became the first target for which MET-targeted therapy was approved in 2020. In this review, we summarize the normal structure and function of MET, the activation mechanism of MET by exon 14 skipping, the clinicopathological characteristics of NSCLCs with MET∆ex14, the efficacies of currently available MET-TKIs (capmatinib and tepotinib) in this cohort, the inherent and acquired resistance mechanisms to MET-TKIs, and future directions to improve treatment outcomes of NSCLC patients with MET∆ex14.
Normal MET Structure and Function
In human cells, the MET protein is first synthesized as a 190 kDa single-chain precursor that is cleaved within the SEMA domain by the intracellular endoprotease furin during transport to form the mature MET protein. The mature MET protein consists of a 50 kDa α chain and a 145 kDa β chain connected through disulfide bonds (Figure 1).14 The extracellular domain contains the semaphorin (SEMA), plexin-semaphorin-integrin (PSI) and immunoglobulin-plexin-transcription (IPT) domains followed by a single-pass transmembrane segment. The intracellular domain contains juxtamembrane, tyrosine kinase, and multifunctional docking site domains.
 |
Figure 1 (A) Relationship between the MET protein and mRNA coding region and (B) the structure of normal MET. Mature MET consists of a 50 kDa alpha chain and a 145 kDa beta chain heterodimer through disulfide bonds. The extracellular domain of MET consists of the semaphorin (SEMA), plexin-semaphorin-integrin (PSI), and immunoglobulin-plexin-transcription (IPT) domains; the intracellular domain consists of juxtamembrane, tyrosine kinase and multifunctional docking site domains. |
HGF and its two shorter splicing isoforms (the N domain and kringle 1 and 2 (NK1 and NK2)) are the only known ligands for MET. NK1 acts as a partial agonist, while NK2 acts as an antagonist. The binding of HGF to the SEMA domain induces MET homodimerization, which causes the autophosphorylation of tyrosine residues at codons 1234 and 1235 (Y1234 and Y1235) in the activation loop of the tyrosine kinase domain. Subsequently, Y1349 and Y1356 in the carboxy-terminal tail are phosphorylated and serve as docking sites for several SRC (SRC proto-oncogene, non-receptor tyrosine kinase) homology (SH)2 domain-containing intracellular molecules, such as PI3K, GRB2 (growth factor receptor-bound protein 2), GAB1 (GrB2-associated binder 1), PLCγ (phospholipase C), SRC, STAT3 (signal transducer and activator of transcription 3), CRK (CT10 regulator of kinase), and SHP2 (Figure 1B). Recruitment of these molecules results in the activation of several downstream pathways, including the RAS/RAF/MEK/ERK and PI3K/AKT/mTOR pathways.15,16 The MET protein is often expressed on epithelial cells, while HGF is secreted by mesenchymal cells such as fibroblasts. HGF/MET signaling has important roles in cell motility, proliferation, embryogenesis, organogenesis, liver regeneration, and wound healing.15,17–19
Discovery of MET exon 14 Skipping and Its Activation Mechanisms
MET exon 14 skipping was originally discovered as an alternative splicing variant in cDNA isolated from normal mouse kidney, liver and brain tissues without any changes that disrupted the so-called splicing consensus sequence in 1994.20 More than 10 years later in 2005, MET∆ex14 was first reported in human NSCLC tissues as a result of a somatic mutation.21 In NSCLCs, MET∆ex14, deletion of the entire juxtamembrane amino acid (∆aa963-1009), is caused by several molecular aberrations, such as point mutations, insertions, deletions, or indels, that disrupt consensus sequences such as branch sites, polypyrimidine tracts, splice acceptors and splice donor sites for RNA splicing (Figure 2A).22 It has been reported that there are more than 500 different mutations at the genomic DNA level that cause MET exon 14 skipping from the analysis of 1387 patients carrying this MET mutation.23 Among these numerous aberrations, point mutations at the splice donor site are the most common. As expected, no phenotypic or therapeutic differences were recorded according to the difference in the molecular mechanisms.
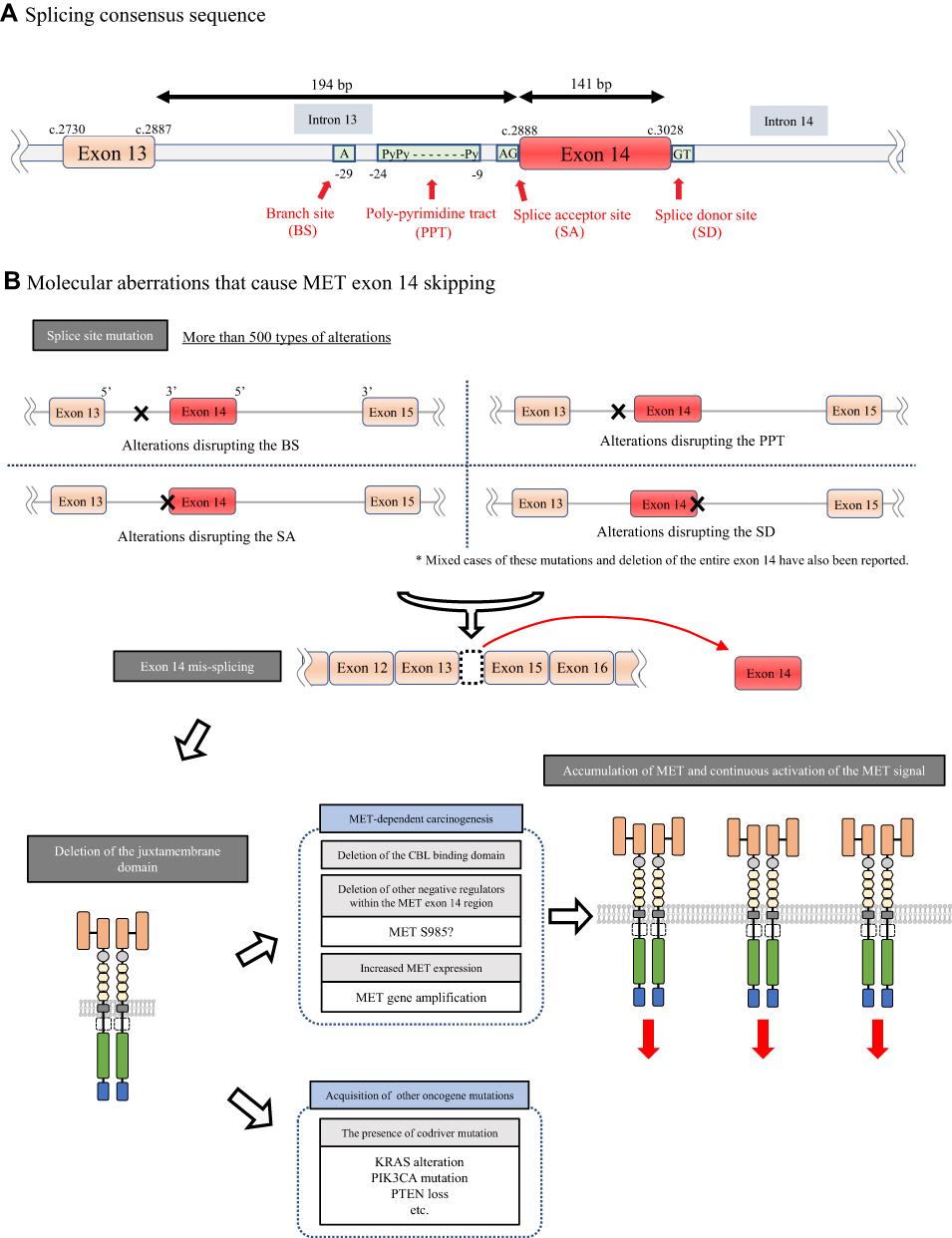 |
Figure 2 (A) Splicing consensus sequence consisting of a branch site, polypyrimidine tract, splice acceptor site and splice donor site. (B) Activation mechanism by MET exon 14 skipping. A large number of alterations, such as point mutations or insertions or deletions in the 3ʹ or 5ʹ splice site in MET exon 14, cause the mis-splicing of MET exon 14 by disrupting the splicing consensus, which results in an abnormal MET protein lacking a CBL-binding site. This causes the accumulation of shrinked MET receptors followed by increased MET signaling. |
The molecular mechanism by which MET∆ex14 elicits oncogenic activity in NSCLCs was clarified by Kong-Beltran et al in 2006.24 MET exon 14 contains Y1003, which forms a binding site for CBL, an E3 ubiquitin ligase, which was reported by Peschard et al.25 Therefore, when exon 14 is skipped, CBL-mediated MET protein degradation is impaired, leading to the accumulation of MET receptors and the aberrant activation of MET oncogenic signaling (Figure 2B).
However, later studies have suggested additional molecular mechanisms by which MET∆ex14 confers oncogenic activity. First, Lu et al showed that the half-life of the MET protein lacking the MET exon 14 region generated using the CRISPR/Cas9 system is extended only by 15% compared with the wild-type MET protein in airway epithelial cells.26 This result may suggest that accumulation of the MET protein is not the sole molecular mechanism of MET activation. The authors also observed that MET∆ex14 induced by editing the endogenous MET gene using the CRISPR/Cas9 system in Trp53flox mice was not oncogenic, whereas MET∆ex14 in Trp53flox mice induced by a lentivirus system that could express MET lacking the exon 14 region stably from cDNA successfully induced a cancer phenotype.26 This phenomenon suggests that the additive effect of increased MET expression, in addition to the skipping of exon 14, plays important roles in tumorigenesis driven by MET. In addition, other groups reported the role of the S985 residue, which is also located in exon 14 of MET. The phosphorylation of this amino acid residue by protein kinase C negatively regulates the kinase activity of MET (Figures 1B and 2B).27,28
Clinical Characteristics of NSCLC Patients with MET exon 14 Skipping
MET∆ex14 is present in up to 3% of NSCLCs, and this incidence is comparable to that of ALK fusions in NSCLCs.29–31 NSCLC patients with MET∆ex14 tend to be older and have a smoking history than patients with other driver mutations. In addition, they have a higher frequency in pleomorphic carcinoma and adenosquamous cell carcinoma than in adenocarcinoma.29,30,32 Among the histologic subtypes of lung adenocarcinoma, some studies have reported that MET∆ex14 is associated with the acinar or solid predominant subtype.30,33,34 The correlation between the frequency of MET∆ex14 and race, sex, stage, and histological grade has not yet been reported or is still controversial. Some studies reported the detection of MET∆ex14 in squamous cell carcinoma (~2%) and large cell carcinoma (0.8%).30,33,35,36
There are several methods to detect MET∆ex14 in NSCLCs. These include next-generation sequencing (NGS)-based panel tests with RNA-based or DNA-based technique. In Japan, an anchored multiplex PCR-based method (ArcherMET®) is approved to detect MET exon 14 skipping using RNA from tissue or ctDNA from liquid samples. Because there are more than 500 patterns of genomic DNA aberrations which cause MET exon 14 skipping as described above, as long as the quality of RNA is ensured, it has been reported that the sensitivity of RNA-based tests are better than DNA-based tests.37,38 Among DNA-based NGS panel tests, it is suggested that the sensitivity of hybrid capture based NGS (represented by Foundation One® that are approved in US and in Japan) is better than that of amplicon-based NGS.37
Clinical Efficacies of MET-TKIs in NSCLCs with MET exon 14 Skipping
In 2015, Paik et al first reported the clinical efficacy of crizotinib and cabozantinib as 3rd-line therapies in NSCLC patients with MET∆ex14.39 In this report, crizotinib showed antitumor activity in three of four patients, and cabozantinib showed stable disease in one patient, suggesting that tumors with MET∆ex14 depend on the MET pathway. Currently, there are many MET-TKIs under clinical development.40 In 2020 and 2021, two MET-TKIs, capmatinib and tepotinib, were approved in the USA and Japan for use as monotherapies in NSCLC patients carrying MET exon 14 skipping. Both MET-TKIs are potent and highly selective ATP competitors for MET in in vitro or in vivo models carrying MET∆ex14. Both drugs are classified as type Ib MET-TKIs and bind to an activated form of MET through interaction with the Y1230 residue in the activation loop of MET. It has also been reported that these type Ib MET-TKIs do not interact with the solvent front residue G1163 (homologous to G1202 and G2032 in the ALK and ROS1 genes, respectively).41–44
Capmatinib (Tabrecta®, INC280; Novartis)
The efficacy and safety of capmatinib were evaluated in the GEOMETRY mono-1 Phase II clinical trial (NCT02414139). A total of 97 NSCLC patients with MET∆ex14 were recruited, consisting of the untreated (n=69) and previously treated (n=28) cohorts (Table 1). In this study, MET∆ex14 was confirmed by qRT-PCR in a central laboratory using tumor tissues. Patients received a 400 mg dose of capmatinib twice daily. The objective response rates (ORRs) for the pretreated and treatment-naïve cohorts were 40.6% (95% CI, 28.9–53.1) and 67.9% (95% CI, 47.6–84.1), respectively. The median progression-free survival (PFS) times were 9.7 months (95% CI, 5.6–13.0) and 12.6 months (95% CI, 5.6-NE), respectively.45 Based on this result, health authorities in the USA and Japan approved the use of capmatinib for NSCLC patients with MET∆ex14 in May 2020 and June 2020, respectively.
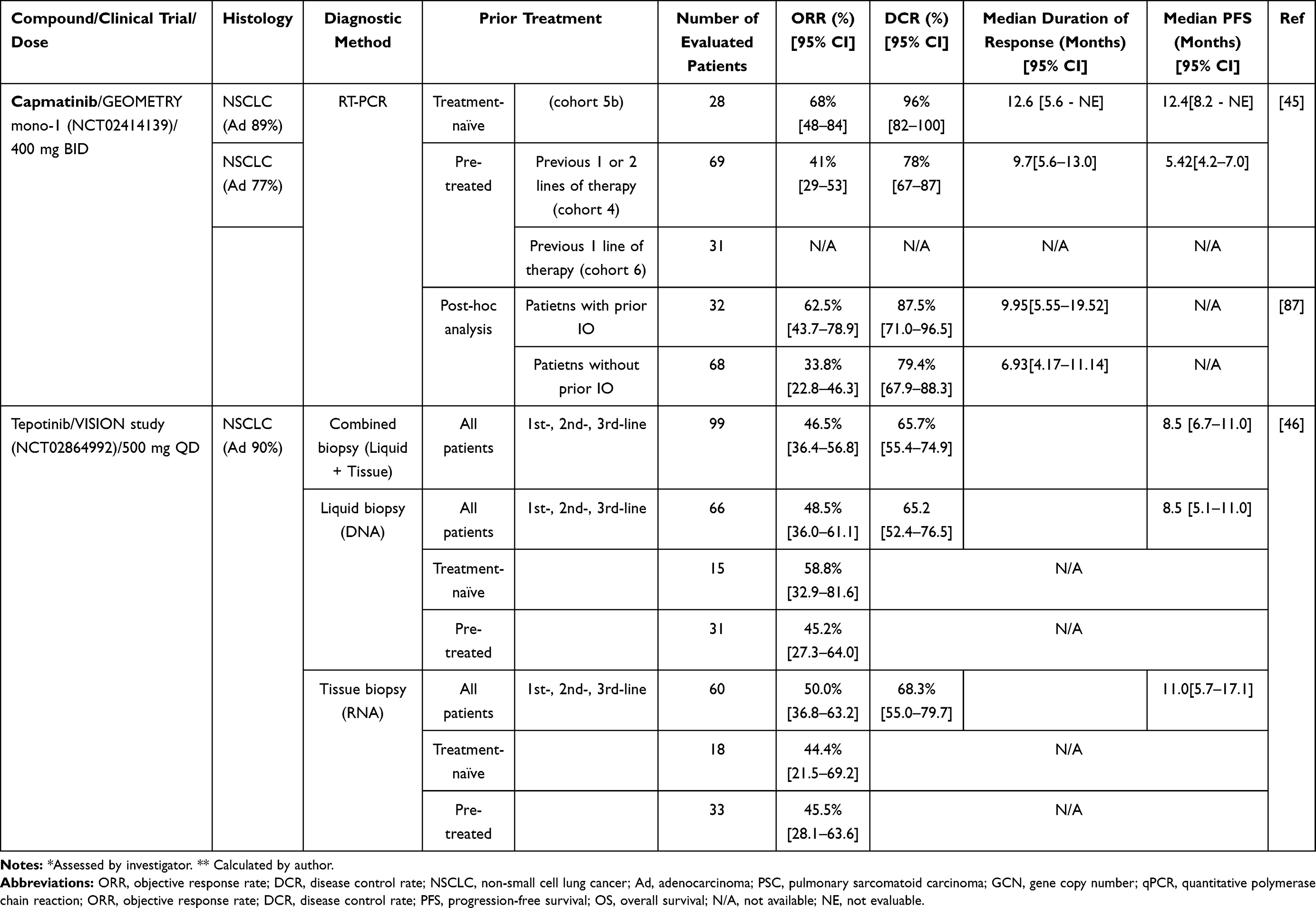 |
Table 1 Clinical Efficacy of Capmatinib and Tepotinib |
Tepotinib (TEPMETKO®, EMD1214063, MSC2156119J; Merck [Darmstadt, Germany])
The efficacy and safety of tepotinib were evaluated in a phase II trial (VISION trial, NCT02864992). A total of 152 NSCLC patients with MET∆ex14 were recruited regardless of previous treatment (Table 1). The patients were divided into two cohorts: those diagnosed by liquid biopsy (DNA-based assay) and those diagnosed by tissue biopsy (RNA-based assay). These patients received a 500 mg dose of tepotinib once daily.
In the total cohort, the ORR and median PFS were 46% (95% CI, 36–57) and 11.1 months (95% CI, 7.2-NE), respectively. The ORRs of patients diagnosed by liquid biopsy and tissue biopsy were 48% (95% CI, 36–61) and 50% (95% CI, 37–63), respectively.46 In addition, the median PFS times were 8.5 months (95% CI, 6.7–11.0) and 11.0 months (95% CI, 5.7–17.1), respectively. The patients who received tepotinib in the first-line setting (n=43) showed an ORR of 44.2% (95% CI, 29.1–60.1), and those who received tepotinib in the second-line or later setting (n=56) showed an ORR of 48.2% (95% CI, 34.7–62.0).46
Based on this favorable result, in March 2020, tepotinib became the first approved MET-TKI for NSCLCs with MET∆ex14 in Japan. In February 2021, the USA FDA also approved its use. In addition, the European Medicines Agency is now validating the approval of tepotinib for the treatment of advanced NSCLCs carrying MET∆ex14.
Toxicities of Type Ib MET-TKIs
Common toxicities that will lead to the dose reduction or discontinuation of capmatinib and tepotinib are peripheral edema and increased serum creatinine.45,46 Because peripheral edema has also been observed in clinical trials of antibody drugs targeting HGF or MET,47,48 it is considered an on-target side effect of MET-HGF axis inhibition. Growth factors, including HGF, increase vascular endothelial barrier function, and inhibition of this barrier function is speculated to be a potential molecular mechanism.49
The increase in serum creatinine is suspected to be due to the inhibitory effects of organic cation transporters (OCTs) and multidrug and toxin extrusion protein transporters (MATEs) in the human kidney by capmatinib and tepotinib. OCTs and MATEs are known as the major transporters for cation drugs (such as capmatinib and tepotinib) from the blood and into the urine.50 Because 10–20% of eliminated creatinine is due to creatinine secretion via these transporters in the renal tubules,51 it is hypothesized that capmatinib or tepotinib will antagonize serum creatinine. Therefore, it is believed that the increases in serum creatinine levels are due to the inhibition of creatinine transport by capmatinib or tepotinib and are not due to true renal function failure.
Inherent and Acquired Resistance Mechanisms to MET-TKIs
With the approval of capmatinib and tepotinib, these drugs will be used for the treatment of NSCLC patients carrying MET∆ex14 in clinical practice. However, the results of clinical trials have shown that approximately one-third to one-half of patients show initial resistance to capmatinib or tepotinib.45,46 In addition, even in patients who show an initial clinical response to capmatinib or tepotinib, the emergence of acquired resistance is almost inevitable.52 In this section, we summarize inherent and acquired resistance mechanisms to MET-TKIs and potential therapeutic strategies to overcome resistance.
Potential Factors Related to Inherent Resistance to MET-TKIs
As described above, the ORRs of two approved MET-TKIs in NSCLC patients carrying MET∆ex14 in each clinical trial were 44–68%, even in treatment-naïve patients.45,46 These numbers are much lower than those in epidermal growth factor receptor (EGFR)-mutated NSCLC patients treated with EGFR-TKIs or NSCLC patients with ALK fusions treated with ALK-TKIs.53–56 Therefore, some clinical trials of MET-TKIs for NSCLCs with MET∆ex14 have explored factors that are associated with the efficacy of MET-TKIs by performing molecularly defined subgroup analyses based on (1) the location of the splicing site mutation at the genomic DNA level, (2) the coexistence of MET amplification, and (3) the presence of codriver gene alterations. Among them, it has been reported that the mutation site at the genomic DNA is not associated with the efficacy of MET-TKIs.46,57
The frequency of coexisting MET amplification is reportedly approximately 4–40% among NSCLCs with MET∆ex14 (Table 2). 29,30,35,45,46,57–59 Although EGFR amplification or ALK amplification has been reported as an acquired resistance mechanism to EGFR-TKIs or ALK-TKIs, respectively, some studies have reported that the ORRs of MET-TKIs are better in MET∆ex14-positive NSCLC patients with coexisting MET amplification than in patients without MET amplification. This result may indicate that the coexistence of MET amplification suggests that tumors depend solely on MET signaling.60,61 On the other hand, Guo et al reported that some patients with MET∆ex14 had no detectable MET protein expression on MET immunohistochemistry (IHC)/mass spectrometry.61 The authors found that these tumors, without a detectable MET protein, had a high frequency of codriver alterations in the RAS/RAF/MAPK or PI3K/AKT pathway, suggesting that these tumors are refractory to MET-targeted therapies.
 | 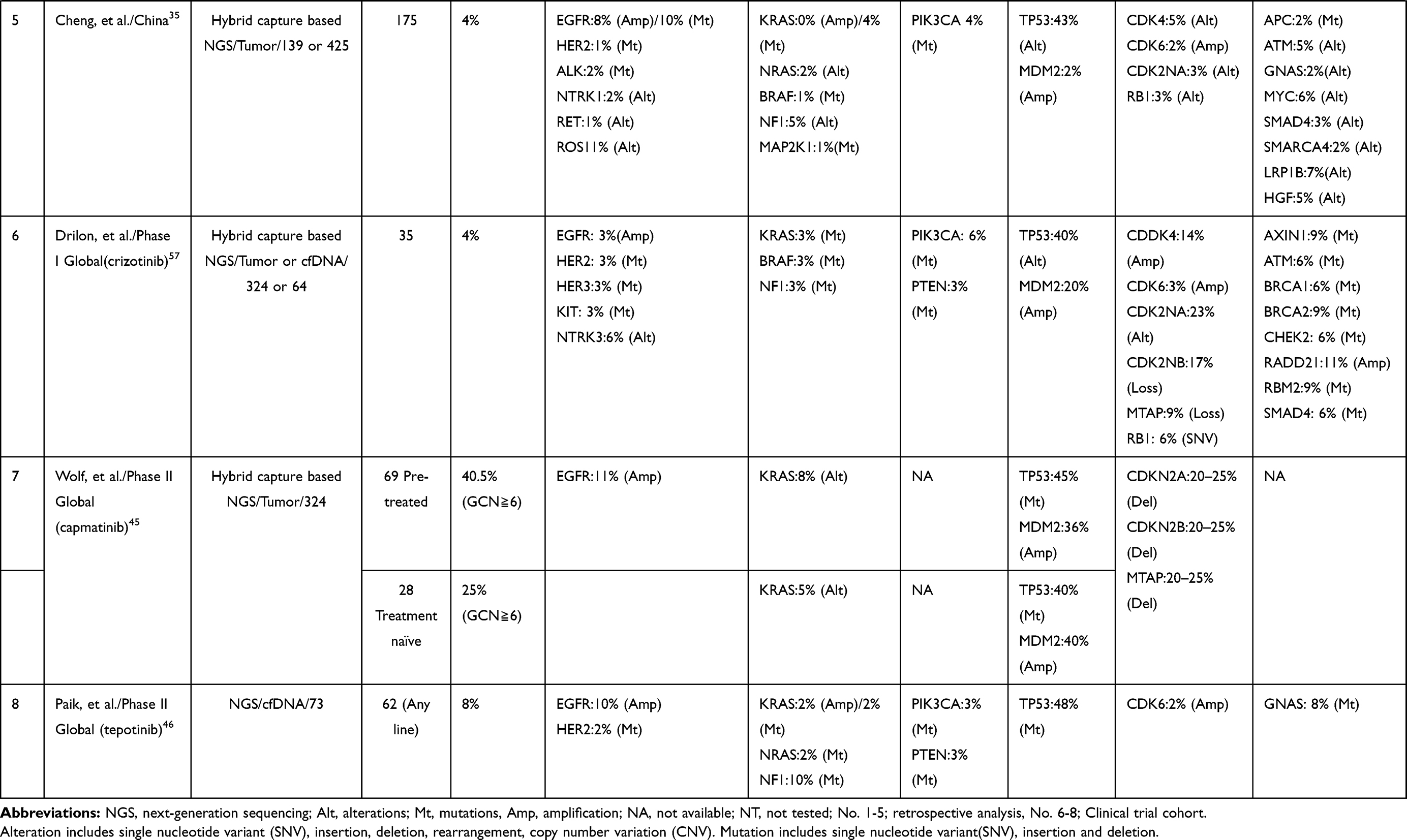 |  |
Table 2 Exploration of Biomarker for Efficacy of MET-TKI |
Another possible reason for the low sensitivity of NSCLCs with MET∆ex14 to MET-TKIs is (3) the presence of codriver gene alterations. As summarized in Table 2, tumors with MET∆ex14 often harbor codriver mutations/amplifications. Potential codrivers include alterations of other RTKs, such as EGFR amplification (6.4–28.5%) or FGFR1 alteration (4.8–16.6%); aberrant activation of the RAS-RAF-MAPK pathway, such as KRAS alteration (~8%) or BRAF alteration (~21.4%); and activation of the PI3K-AKT pathway, such as PIK3CA mutation/amplification (~14.2%).58,59 As a preclinical model, the NCI-H596 lung cancer cell line harbors MET∆ex14; however, this cell line is resistant to MET inhibition. The coexistence of PIK3CA mutation is the mechanism of resistance to MET inhibition, and it was reported that NCI-H596 cells were effectively killed by the combination of a PI3K inhibitor and a MET-TKI.62,63 Indeed, a retrospective analysis reported that the coexistence of these mutations resulted in primary resistance to MET-TKIs or a short response duration in NSCLC patients with MET∆ex14.58,59,61,64
Mutations of the TP53 gene (27–50%) and the amplification of MDM2 (2–46%), which is an E3 ubiquitin ligase for TP53, are frequently identified in NSCLCs with MET∆ex14. In addition, they are reportedly mutually exclusive.13 Although the coexistence of TP53 mutation is associated with reduced efficacy in EGFR-mutated NSCLCs treated with EGFR-TKIs,65 we could not find evidence that showed the impact of TP53 or MDM2 alterations on the efficacy of MET-TKIs in NSCLC patients with MET∆ex14. Taken together, these results indicate that the coexistence of gene aberrations in the p53 pathway may be involved in the process of oncogenic transformation in NSCLC with MET∆ex14.
Acquired Resistance Mechanisms to MET-TKIs
As with other TKI therapies, such as EGFR-TKIs or ALK-TKIs, for NSCLCs with a driver mutation, acquired resistance to MET-TKIs is also inevitable Some studies have reported acquired resistance mechanisms to MET-TKIs in patient specimens obtained after MET-TKI treatment failure. In addition, we reported potential acquired resistance mechanisms that were identified by in vitro experiments. Acquired resistance mechanisms to MET-TKIs can be classified into secondary mutations of MET (on-target resistance mechanisms) and activation of bypass signaling (off-target resistance mechanisms). Recondo et al analyzed resistance mechanisms to MET-TKIs (mainly crizotinib) in 20 patients with MET∆ex14 and reported that the on-target and off-target mechanisms accounted for 35% and 45%, respectively.52 However, it is not clear whether more specific MET-TKIs, capmatinib and tepotinib show similar frequencies of on-target and off-target resistance mechanisms because crizotinib is a multitarget TKI.
As acquired resistance mechanisms to capmatinib or tepotinib, we found through an in vitro analysis using Ba/F3 models that MET secondary mutations involving D1228 or Y1230 in the activation loop are common as on-target resistance mechanisms.43 In a clinical case report, it was reported that a variety of amino acid substitutions at codons D1228 and Y1230 occurred in a single patient.66 As an acquired resistance mechanism to crizotinib, the MET solvent front mutation G1163R has been reported.67 However, it has been shown that capmatinib and tepotinib do not interact with the MET G1163 residue; therefore, secondary mutations involving G1163 will not occur as a resistance mechanism to capmatinib or tepotinib.43 Our in vitro study also showed that the potential on-target resistance mechanisms to capmatinib or tepotinib, D1228 or Y1230 secondary mutations, can be overcome by so-called type II MET-TKIs43 such as merestinib, cabozantinib and glesatinib, which bind to the inactive state of MET.68 However, in clinical cases, type II MET-TKIs, cabozantinib and glesatinib showed antitumor activity against Y1230X-mediated resistance but not against D1228X-mediated resistance.52,69–71
As off-target resistance mechanisms to MET-TKIs, genetic alterations that cause activation of the RAS/RAF/MAPK pathway (such as KRAS amplification or KRAS mutations) and/or the PI3K/AKT pathway (such as PIK3CA mutation) have been reported.52,58,64,72 Preclinical studies have shown that combination therapy with trametinib, a MEK inhibitor, or GDC0941, a PI3K inhibitor, can overcome these acquired resistance mechanisms to MET-TKIs.46,59,64,72 A few studies reported that EGFR, HER3, and MDM2 amplification was detected after acquired resistance to MET-TKIs; however, it is not clear whether amplification of these genes is truly associated with acquired resistance to MET-TKIs.58,66
Future Treatment Strategies for NSCLCs with MET exon 14 Skipping
At present, after capmatinib or tepotinib treatment failure, NSCLCs with MET∆ex14 are treated following the recommendations for NSCLCs with no detectable driver mutation or an unknown mutational status. As a future treatment strategy, MET antibody drugs are now being evaluated in clinical trials. In addition, some recent studies have reported the superior efficacy of immune checkpoint inhibitors (ICIs) in NSCLC patients with MET∆ex14. In this section, we summarize the efficacies of these treatments in NSCLC patients with MET∆ex14.
MET Antibodies
Antibodies targeting MET are designed to bind to the SEMA domain of MET, which is important in HGF binding to MET. These MET antibodies are also expected to promote receptor internalization and degradation, resulting in inhibition of the MET signaling pathway, and to enhance complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC). MET antibodies have been clinically developed for some time.7,73 In a Phase III trial (NCT01456325) that compared the efficacy of onartuzumab (a MET monoclonal antibody) plus erlotinib with placebo plus erlotinib in advanced NSCLC patients exhibiting MET expression (>50% by IHC), improved clinical outcomes were not observed. This disappointing result could be attributed to insufficient patient selection. At present, it is believed that MET overexpression itself does not necessarily indicate a truly MET-driven state from the two findings that MET overexpression coexists with various oncogenic mutations74 and that MET overexpression is independent of MET∆ex14.74,75 On the other hand, these findings may show antitumor activity for NSCLC patients with MET∆ex14, which results in accumulation of the MET protein. Selecting patients with both MET∆ex14 and Met overexpression may be a useful strategy to test anti-MET antibodies.
One MET antibody being developed for use in NSCLC patients with MET∆ex14 is Sym015 (Symphogen, Copenhagen, Denmark) (Table 3). This drug consists of two recombinant humanized IgG1 monoclonal antibodies targeting different epitopes of MET. MET internalization/degradation and the stimulation of CDC/ADCC have been observed in in vitro and in vivo experiments after treatment with Sym015.76 A phase I/II trial (NCT02648724) of Sym015 in NSCLC patients with MET∆ex14 who progressed on a MET-TKI is currently ongoing.
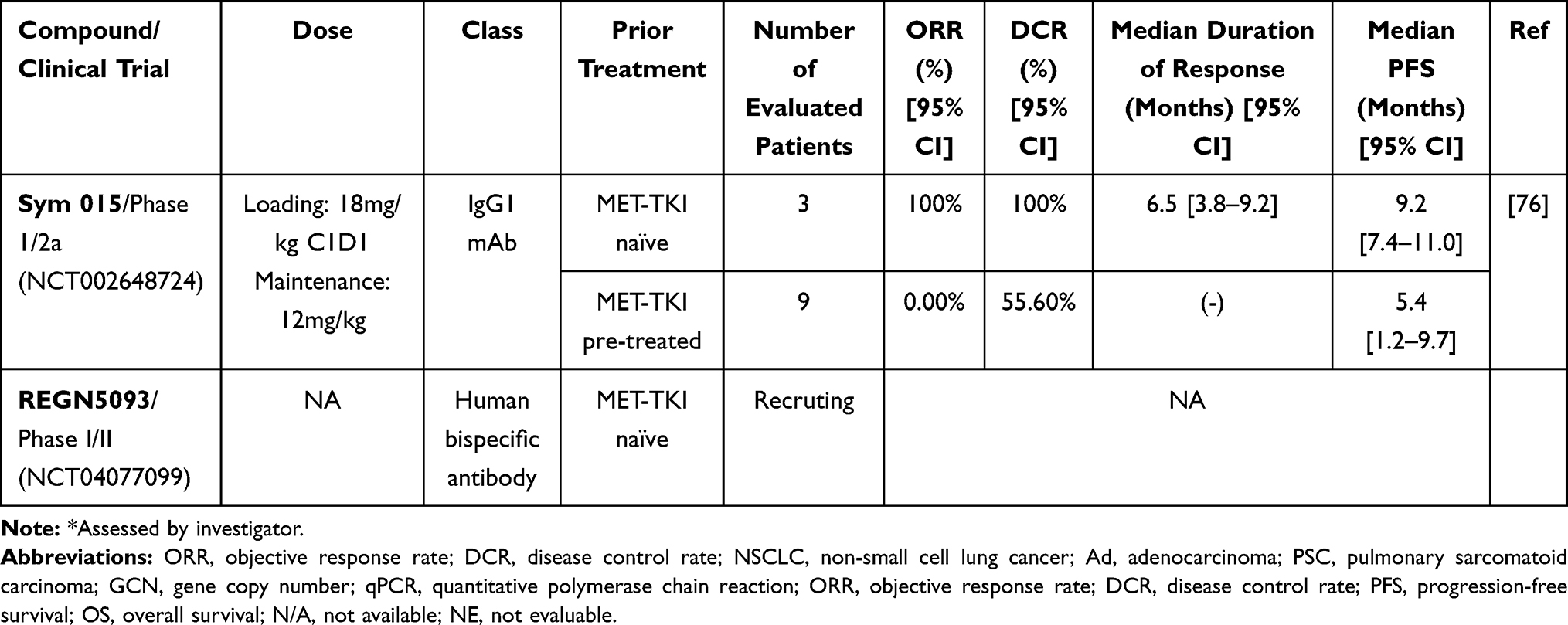 |
Table 3 MET Antibody Under Clinical Development for NSCLC Patients Carrying MET Exon 14 Skipping Mutation |
Immunotherapies
Immunotherapy with/without chemotherapy has become a standard front-line therapy for NSCLCs without detectable driver mutations. On the other hand, it has been reported that the efficacy of immunotherapies is low in NSCLC patients with EGFR mutations or ALK fusions.77,78 However, as described above, the clinicopathological characteristics of NSCLC patients with MET∆ex14, such as smoking status or histology, are different from those of NSCLC patients with EGFR mutations or ALK fusions. Therefore, it is not surprising that some retrospective studies showed that the expression of PD-L1, a potential biomarker used to predict the efficacy of ICIs, is high in NSCLCs with MET∆ex14 (43–91% if 1% ≧ PD-L1 is used as a cutoff) (Table 4).23,79–85 This high PD-L1 expression may be due not only to so-called adaptive immune resistance but also to activated MET signaling; a preclinical study showed that MET activation induced the expression of several immune checkpoints, including PD-L1, through a JAK2-independent pathway.86 Therefore, the efficacy of ICIs in NSCLC patients with MET∆ex14 has received a great deal of attention. Some retrospective studies have reported the efficacy of ICI monotherapy in NSCLC patients with MET∆ex14. However, the efficacy of ICI monotherapy is controversial because any reported results were obtained from retrospective, small cohort analyses, and the efficacy varied depending on the report.79–81 On the other hand, the efficacy of ICI + chemotherapy in NSCLCs with MET∆ex14 is currently unknown, and further analysis is warranted.
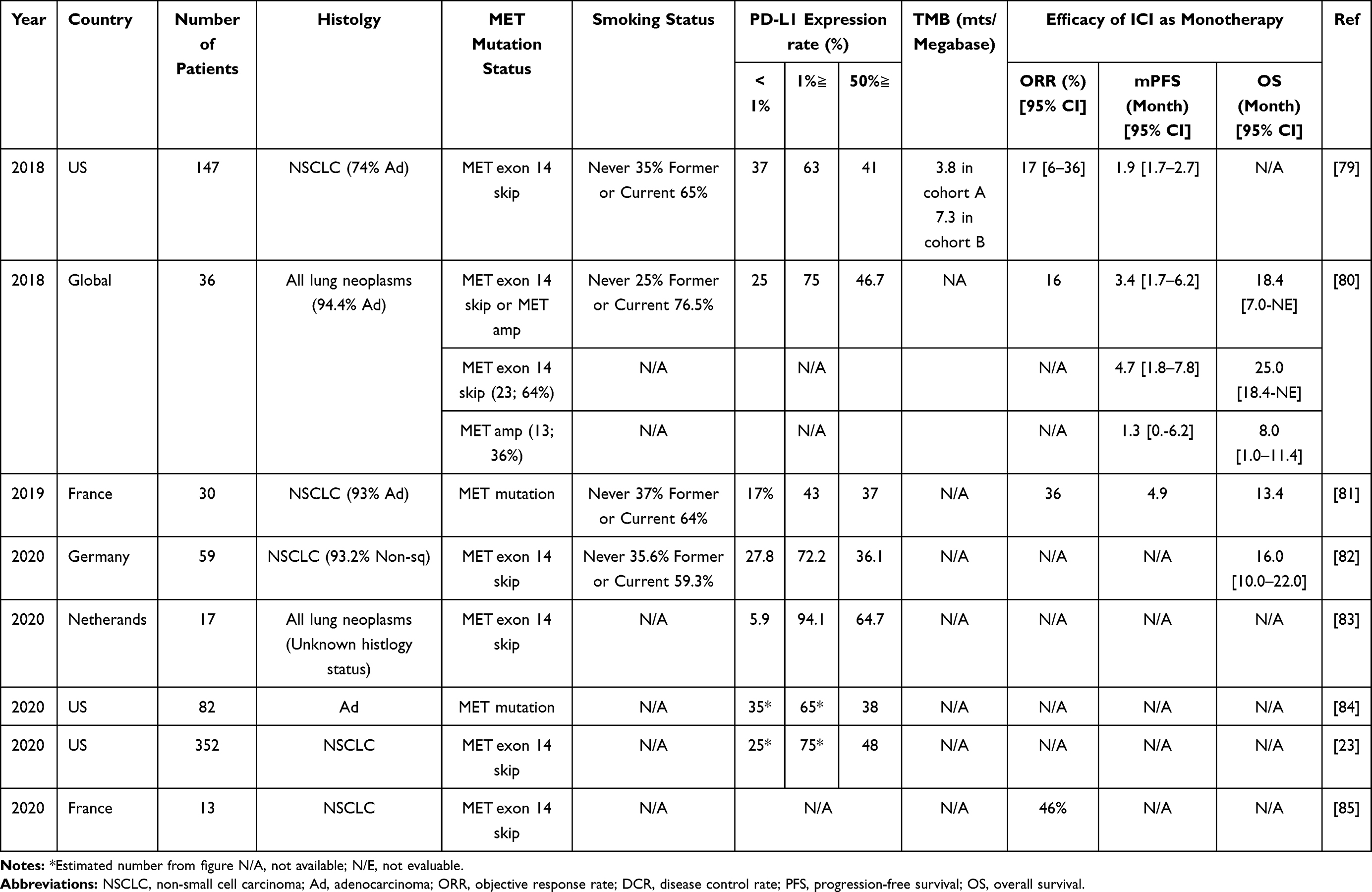 |
Table 4 Summary of Studies Which Analyzed PL-L1 Status in NSCLC with MET Aberrations |
Combination or sequential treatments involving ICIs and MET-TKIs could be promising strategies in the treatment of NSCLC patients with MET∆ex14. In the post hoc analysis that compared the type of prior therapy before capmatinib in the GEOMETRY phase II trial, there was a large difference in the response rate to capmatinib: 32% in the chemotherapy-pretreated group and 64% in the ICI-pretreated group (Table 1).87 One possible reason for this difference is that the residual effect of ICIs used in the previous treatment was boosted by the use of MET-TKIs because MET signaling reportedly affects the immune system.88 For example, Glodde e2t al showed the synergistic efficacy of MET inhibition and ICIs regardless of the tumor’s MET status in vivo.89 The authors reported that MET-expressing neutrophils are mobilized from the bone marrow to tumors in response to ICIs, these neutrophils confer immunosuppressive properties in tumors, and MET inhibition impairs reactive neutrophil recruitment to tumors. It has also been reported that the combination of ICIs plus MET-TKIs is tolerable in NSCLC patients.90 In addition, in January 2021, the FDA approved the combination of nivolumab and cabozantinib, a type II MET-TKI, as a first-line treatment for patients with advanced renal cell carcinoma based on the results of a phase III trial.91
Conclusion
The approval of two MET-TKIs, capmatinib and tepotinib, for NSCLCs with MET∆ex14 marked a new revolution of MET-targeted therapy. However, as summarized in this review, NSCLCs with this mutation often have codriver mutations and are highly heterogeneous; therefore, it is understandable that some patients show inherent resistance to MET-TKIs. In addition, some studies reported on-target and off-target mechanisms of acquired resistance to MET-TKIs. In addition to immunotherapy, novel treatments, including novel MET-TKIs, MET antibodies, and novel combination therapies, are now being evaluated in clinical trials.
Disclosure
Dr. Fujino has received research funding from Apollomics and lecture fee from Novartis during the study. Dr. Suda has received honorarium from Boehringer Ingelheim, has been on the advisory board of AstraZeneca, and has received research funding from Boehringer Ingelheim and Rain Therapeutics during the study. Dr. Mitsudomi has received lecture fees from AstraZeneca, Boehringer Ingelheim, Chugai, and Pfizer, Bristol-Myers Squibb, Dohme, Eli Lilly and Merck Sharp and research funding from Astra Zeneca, Boehringer Ingelheim, Chugai, Daiichi Sankyo, Ono Pharmaceutical and Taiho during the study; he has been on the advisory board of Novartis and has a patent KU22015PCT pending. The authors report no other conflicts of interest in this work.
References
1. Comoglio PM, Trusolino L, Boccaccio C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat Rev Cancer. 2018;18(6):341–358. doi:10.1038/s41568-018-0002-y
2. Cooper CS, Park M, Blair DG, et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311(5981):29–33. doi:10.1038/311029a0
3. Okuda K, Sasaki H, Yukiue H, et al. Met gene copy number predicts the prognosis for completely resected non-small cell lung cancer. Cancer Sci. 2008;99(11):2280–2285. doi:10.1111/j.1349-7006.2008.00916.x
4. Park S, Choi YL, Sung CO, et al. High MET copy number and MET overexpression: poor outcome in non-small cell lung cancer patients. Histol Histopathol. 2012;27:197–207. doi:10.14670/HH-27.197
5. Liu Y, Yu XF, Zou J, et al. Prognostic value of c-Met in colorectal cancer: a meta-analysis. World J Gastroenterol. 2015;21:3706–3710. doi:10.3748/wjg.v21.i12.3706
6. Kim JH, Kim BJ, Kim HS. Clinicopathological impacts of high c-Met expression in head and neck squamous cell carcinoma: a meta-analysis and review. Oncotarget. 2017;8.
7. Spigel DR, Edelman MJ, O’Byrne K, et al. Results From the Phase III Randomized Trial of Onartuzumab Plus Erlotinib Versus Erlotinib in Previously Treated Stage IIIB or IV Non–Small-Cell Lung Cancer: mETLung. J Clin Oncol. 2016;35:412–420. doi:10.1200/JCO.2016.69.2160
8. Scagliotti G, von Pawel J, Novello S, et al. Phase III Multinational, Randomized, Double-Blind, Placebo-Controlled Study of Tivantinib (ARQ 197) Plus Erlotinib Versus Erlotinib Alone in Previously Treated Patients With Locally Advanced or Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J Clin Oncol. 2015;33:2667–2674. doi:10.1200/JCO.2014.60.7317
9. Schoffski P, Gordon M, Smith DC, et al. Phase II randomised discontinuation trial of cabozantinib in patients with advanced solid tumours. Eur J Cancer. 2017;86:296–304. doi:10.1016/j.ejca.2017.09.011
10. Consortium APG. AACR Project GENIE: powering Precision Medicine through an International Consortium. Cancer Discov. 2017;7:818–831.
11. Schmidt L, Duh FM, Chen F, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68–73. doi:10.1038/ng0597-68
12. Di Renzo MF, Olivero M, Martone T, et al. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene. 2000;19(12):1547–1555. doi:10.1038/sj.onc.1203455
13. Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5(8):850–859. doi:10.1158/2159-8290.CD-15-0285
14. Giordano S, Di Renzo MF, Narsimhan RP, et al. Biosynthesis of the protein encoded by the c-met proto-oncogene. Oncogene. 1989;4(11):1383–1388.
15. Sipeki S, Bander E, Buday L, et al. Phosphatidylinositol 3-kinase Contributes to Erk1/Erk2 MAP Kinase Activation Associated with Hepatocyte Growth Factor-induced Cell Scattering. Cell Signal. 1999;11(12):885–890. doi:10.1016/S0898-6568(99)00060-1
16. Ponzetto C, Bardelli A, Zhen Z, et al. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell. 1994;77(2):261–271. doi:10.1016/0092-8674(94)90318-2
17. Birchmeier C, Birchmeier W, Gherardi E, et al. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4(12):915. doi:10.1038/nrm1261
18. Eder JP, Vande Woude GF, Boerner SA, et al. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res. 2009;15(7):2207–2214. doi:10.1158/1078-0432.CCR-08-1306
19. Zhang YW, Wang LM, Jove R, et al. Requirement of Stat3 signaling for HGF/SF-Met mediated tumorigenesis. Oncogene. 2002;21(2):217–226. doi:10.1038/sj.onc.1205004
20. Lee CC, Yamada KM. Identification of a novel type of alternative splicing of a tyrosine kinase receptor. Juxtamembrane deletion of the c-met protein kinase C serine phosphorylation regulatory site. J Biol Chem. 1994;269:19457–19461. doi:10.1016/S0021-9258(17)32190-7
21. Ma PC, Jagadeeswaran R, Jagadeesh S, et al. Functional Expression and Mutations of c-Met and Its Therapeutic Inhibition with SU11274 and Small Interfering RNA in Non–Small Cell Lung Cancer. Cancer Res. 2005;65(4):1479. doi:10.1158/0008-5472.CAN-04-2650
22. Onozato R, Kosaka T, Kuwano H, et al. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol. 2009;4(1):5–11. doi:10.1097/JTO.0b013e3181913e0e
23. Mark M, Awad JKL. Characterization of 1387 NSCLCs with MET exon 14 (METex14) skipping alterations (SA) and potential acquired resistance (AR) mechanisms. 2020 ASCO Virtual Scientific Program. 2020.
24. Kong-Beltran M, Seshagiri S, Zha J, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006;66(1):283–289. doi:10.1158/0008-5472.CAN-05-2749
25. Peschard P, Fournier TM, Lamorte L, et al. Mutation of the c-Cbl TKB Domain Binding Site on the Met Receptor Tyrosine Kinase Converts It into a Transforming Protein. Mol Cell. 2001;8(5):995–1004. doi:10.1016/S1097-2765(01)00378-1
26. Lu X, Peled N, Greer J, et al. MET exon 14 Mutation Encodes an Actionable Therapeutic Target in Lung Adenocarcinoma. Cancer Res. 2017;77(16):4498–4505. doi:10.1158/0008-5472.CAN-16-1944
27. Gandino L, Longati P, Medico E, et al. Phosphorylation of serine 985 negatively regulates the hepatocyte growth factor receptor kinase. J Biol Chem. 1994;269(3):1815–1820. doi:10.1016/S0021-9258(17)42099-0
28. Hashigasako A, Machide M, Nakamura T, et al. Bi-directional regulation of Ser-985 phosphorylation of c-met via protein kinase C and protein phosphatase 2A involves c-Met activation and cellular responsiveness to hepatocyte growth factor. J Biol Chem. 2004;279(25):26445–26452. doi:10.1074/jbc.M314254200
29. Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J Clin Oncol. 2016;34:721–730. doi:10.1200/JCO.2015.63.4600
30. Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 Patients with Lung Cancer Harboring MET Exon 14 Skipping Alterations. J Thorac Oncol. 2016;11(9):1493–1502. doi:10.1016/j.jtho.2016.06.004
31. Jordan EJ, Kim HR, Arcila ME, et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov. 2017;7(6):596–609. doi:10.1158/2159-8290.CD-16-1337
32. Tong JH, Yeung SF, Chan AW, et al. MET Amplification and Exon 14 Splice Site Mutation Define Unique Molecular Subgroups of Non-Small Cell Lung Carcinoma with Poor Prognosis. Clin Cancer Res. 2016;22:3048–3056. doi:10.1158/1078-0432.CCR-15-2061
33. Gow CH, Hsieh MS, Wu SG, et al. A comprehensive analysis of clinical outcomes in lung cancer patients harboring a MET exon 14 skipping mutation compared to other driver mutations in an East Asian population. Lung Cancer. 2017;103:82–89. doi:10.1016/j.lungcan.2016.12.001
34. Lee GD, Lee SE, Oh DY, et al. MET Exon 14 Skipping Mutations in Lung Adenocarcinoma: clinicopathologic Implications and Prognostic Values. J Thorac Oncol. 2017;12(8):1233–1246. doi:10.1016/j.jtho.2017.04.031
35. Cheng T, Gu Z, Song D, et al. Genomic and clinical characteristics of MET exon14 alterations in a large cohort of Chinese cancer patients revealed distinct features and a novel resistance mechanism for crizotinib. J Cancer. 2021;12(3):644–651. doi:10.7150/jca.49391
36. Liu SY, Gou LY, Li AN, et al. The Unique Characteristics of MET Exon 14 Mutation in Chinese Patients with NSCLC. J Thorac Oncol. 2016;11:1503–1510. doi:10.1016/j.jtho.2016.05.016
37. Davies KD, Lomboy A, Lawrence CA, et al. DNA-Based versus RNA-Based Detection of MET Exon 14 Skipping Events in Lung Cancer. J Thorac Oncol. 2019;14:737–741. doi:10.1016/j.jtho.2018.12.020
38. Kim EK, Kim KA, Lee CY, et al. Molecular Diagnostic Assays and Clinicopathologic Implications of MET Exon 14 Skipping Mutation in Non-small-cell Lung Cancer. Clin Lung Cancer. 2019;20:e123–e132. doi:10.1016/j.cllc.2018.10.004
39. Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5:842–849. doi:10.1158/2159-8290.CD-14-1467
40. Fujino T, Suda K, Mitsudomi T. Emerging MET tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. Expert Opin Emerg Drugs. 2020;25(3):229–249. doi:10.1080/14728214.2020.1791821
41. Liu X, Wang Q, Yang G, et al. A novel kinase inhibitor, INCB28060, blocks c-MET-dependent signaling, neoplastic activities, and cross-talk with EGFR and HER-3. Clin Cancer Res. 2011;17:7127–7138. doi:10.1158/1078-0432.CCR-11-1157
42. Baltschukat S, Schacher Engstler B, Huang A, et al. Capmatinib (INC280) Is Active Against Models of Non-Small Cell Lung Cancer and Other Cancer Types with Defined Mechanisms of MET Activation. Clin Cancer Res. 2019;25(10):3164–3175. doi:10.1158/1078-0432.CCR-18-2814
43. Fujino T, Kobayashi Y, Suda K, et al. Sensitivity and Resistance of MET Exon 14 Mutations in Lung Cancer to Eight MET Tyrosine Kinase Inhibitors In Vitro. J Thoracic Oncol. 2019;14(10):1753–1765. doi:10.1016/j.jtho.2019.06.023
44. Bladt F, Faden B, Friese-Hamim M, et al. EMD 1214063 and EMD 1204831 constitute a new class of potent and highly selective c-Met inhibitors. Clin Cancer Res. 2013;19(11):2941–2951. doi:10.1158/1078-0432.CCR-12-3247
45. Wolf J, Seto T, Han J-Y, et al. Capmatinib in MET Exon 14–Mutated or MET-Amplified Non–Small-Cell Lung Cancer. N Eng J Med. 2020;383:944–957. doi:10.1056/NEJMoa2002787
46. Paik PK, Felip E, Veillon R, et al. Tepotinib in Non–Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N Engl J Med. 2020;383(10):931–943. doi:10.1056/NEJMoa2004407
47. Spigel DR, Ervin TJ, Ramlau RA, et al. Randomized Phase II Trial of Onartuzumab in Combination With Erlotinib in Patients With Advanced Non–Small-Cell Lung Cancer. J Clin Oncol. 2013;31(32):4105–4114. doi:10.1200/JCO.2012.47.4189
48. Patnaik A, Weiss GJ, Papadopoulos KP, et al. Phase I ficlatuzumab monotherapy or with erlotinib for refractory advanced solid tumours and multiple myeloma. Br J Cancer. 2014;111(2):272–280. doi:10.1038/bjc.2014.290
49. Woo PC, Lau SK, Wong SS, et al. Two cases of continuous ambulatory peritoneal dialysis-associated peritonitis due to Plesiomonas shigelloides. J Clin Microbiol. 2004;42(2):933–935. doi:10.1128/JCM.42.2.933-935.2004
50. Motohashi H, Inui K. Organic cation transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney. AAPS J. 2013;15(2):581–588. doi:10.1208/s12248-013-9465-7
51. Chu X, Bleasby K, Chan GH, et al. The Complexities of Interpreting Reversible Elevated Serum Creatinine Levels in Drug Development: does a Correlation with Inhibition of Renal Transporters Exist? Drug Metab Dispos. 2016;44(9):1498–1509. doi:10.1124/dmd.115.067694
52. Recondo G, Bahcall M, Spurr LF, et al. Molecular Mechanisms of Acquired Resistance to MET Tyrosine Kinase Inhibitors in Patients with MET Exon 14–Mutant NSCLC. Clin Cancer Res. 2020;26(11):2615. doi:10.1158/1078-0432.CCR-19-3608
53. Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or Chemotherapy for Non–Small-Cell Lung Cancer with Mutated EGFR. N Eng J Med. 2010;362(25):2380–2388. doi:10.1056/NEJMoa0909530
54. Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised Phase 3 trial. Lancet Oncol. 2010;11(2):121–128. doi:10.1016/S1470-2045(09)70364-X
55. Yang JC-H, Wu Y-L, Schuler M, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015;16:141–151. doi:10.1016/S1470-2045(14)71173-8
56. Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med. 2018;378:113–125. doi:10.1056/NEJMoa1713137
57. Drilon A, Clark JW, Weiss J, et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat Med. 2020;26(1):47–51. doi:10.1038/s41591-019-0716-8
58. Rotow JK, Gui P, Wu W, et al. Co-occurring alterations in the RAS-MAPK pathway limit response to MET inhibitor treatment in MET exon 14 skipping mutation-positive lung cancer. Clin Cancer Res. 2019;26(2):439–449. doi:10.1158/1078-0432.CCR-19-1667
59. Jamme P, Fernandes M, Copin M-C, et al. Alterations in the PI3K pathway drive resistance to MET inhibitors in NSCLC harboring MET exon 14 skipping mutations. J Thoracic Oncol. 2020;15(5):741–751. doi:10.1016/j.jtho.2020.01.027
60. Lu S, Fang J, Cao L, et al. Abstract CT031: preliminary efficacy and safety results of savolitinib treating patients with pulmonary sarcomatoid carcinoma (PSC) and other types of non-small cell lung cancer (NSCLC) harboring MET exon 14 skipping mutations. Cancer Res. 2019;79:CT031.
61. Guo R, Offin M, Brannon AR, et al. MET exon 14-altered lung cancers and MET inhibitor resistance. Clin Cancer Res. 2020.
62. Tanizaki J, Okamoto I, Okamoto K, et al. MET tyrosine kinase inhibitor crizotinib (PF-02341066) shows differential antitumor effects in non-small cell lung cancer according to MET alterations. J Thorac Oncol. 2011;6(10):1624–1631. doi:10.1097/JTO.0b013e31822591e9
63. Liu X, Jia Y, Stoopler MB, et al. Next-Generation Sequencing of Pulmonary Sarcomatoid Carcinoma Reveals High Frequency of Actionable MET Gene Mutations. J Clin Oncol. 2016;34:794–802. doi:10.1200/JCO.2015.62.0674
64. Suzawa K, Offin M, Lu D, et al. Activation of KRAS Mediates Resistance to Targeted Therapy in MET Exon 14-mutant Non-small Cell Lung Cancer. Clin Cancer Res. 2019;25:1248–1260. doi:10.1158/1078-0432.CCR-18-1640
65. Canale M, Petracci E, Delmonte A, et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin Cancer Res. 2017;23:2195–2202. doi:10.1158/1078-0432.CCR-16-0966
66. Recondo G, Bahcall M, Spurr LF, et al. Molecular mechanisms of acquired resistance to MET tyrosine kinase inhibitors in patients with MET exon 14 mutant NSCLC. Clin Cancer Res. 2020;26(11):2615–2625.
67. Zhang Y, Yin J, Peng F. Acquired resistance to crizotinib in advanced lung adenocarcinoma with MET exon 14 skipping. Lung Cancer. 2017;113:69–71. doi:10.1016/j.lungcan.2017.09.006
68. Fujino T, Kobayashi Y, Suda K, et al. Sensitivity and Resistance of MET Exon 14 Mutations in Lung Cancer to Eight MET Tyrosine Kinase Inhibitors In Vitro. J Thorac Oncol. 2019;14(10):1753–1765.
69. Kang J, Chen HJ, Wang Z, et al. Osimertinib and Cabozantinib Combinatorial Therapy in an EGFR-Mutant Lung Adenocarcinoma Patient with Multiple MET Secondary-Site Mutations after Resistance to Crizotinib. J Thorac Oncol. 2018;13:e49–e53. doi:10.1016/j.jtho.2017.10.028
70. Piper-Vallillo AJ, Halbert BT, Rangachari D, et al. Acquired resistance to osimertinib plus savolitinib is mediated by MET-D1228 and Y1230 mutations in EGFR mutated MET amplified lung cancer. JTO Clin Res Rep. 2020;1(4):100071. doi:10.1016/j.jtocrr.2020.100071
71. Engstrom L, Aranda R, Lee M, et al. Glesatinib Exhibits Antitumor Activity in Lung Cancer Models and Patients Harboring MET Exon 14 Mutations and Overcomes Mutation-mediated Resistance to Type I MET Inhibitors in Nonclinical Models. Clin Cancer Res. 2017;23(21):6661–6672. doi:10.1158/1078-0432.CCR-17-1192
72. Bahcall M, Awad MM, Sholl LM, et al. Amplification of Wild-type KRAS Imparts Resistance to Crizotinib in MET Exon 14 Mutant Non–Small Cell Lung Cancer. Clin Cancer Res. 2018;24(23):5963–5976. doi:10.1158/1078-0432.CCR-18-0876
73. Scagliotti G, Moro-Sibilot D, Kollmeier J, et al. A Randomized-Controlled Phase 2 Study of the MET Antibody Emibetuzumab in Combination with Erlotinib as First-Line Treatment for EGFR Mutation-Positive NSCLC Patients. J Thorac Oncol. 2020;15:80–90. doi:10.1016/j.jtho.2019.10.003
74. Baldacci S, Figeac M, Antoine M, et al. High MET overexpression does not predict the presence of MET exon 14 splice mutations in NSCLC: results from the IFCT Predict.amm study. J Thorac Oncol. 2019;14(10):S851. doi:10.1016/j.jtho.2019.08.1838
75. Guo R, Berry LD, Aisner DL, et al. MET IHC Is a Poor Screen for MET Amplification or MET Exon 14 Mutations in Lung Adenocarcinomas: data from a Tri-Institutional Cohort of the Lung Cancer Mutation Consortium. J Thorac Oncol. 2019;14(9):1666–1671. doi:10.1016/j.jtho.2019.06.009
76. Camidge DR, Janku F, Martinez-Bueno A, et al. Safety and preliminary clinical activity of the MET antibody mixture, Sym015 in advanced non-small cell lung cancer (NSCLC) patients with MET amplification/exon 14 deletion (METAmp/Ex14∆). J Clin Oncol. 2020;38:9510. doi:10.1200/JCO.2020.38.15_suppl.9510
77. Gainor JF, Shaw AT, Sequist LV, et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: a Retrospective Analysis. Clin Cancer Res. 2016;22:4585–4593. doi:10.1158/1078-0432.CCR-15-3101
78. Lee CK, Man J, Lord S, et al. Clinical and Molecular Characteristics Associated With Survival Among Patients Treated With Checkpoint Inhibitors for Advanced Non-Small Cell Lung Carcinoma: a Systematic Review and Meta-analysis. JAMA Oncol. 2018;4:210–216. doi:10.1001/jamaoncol.2017.4427
79. Sabari JK, Leonardi GC, Shu CA, et al. PD-L1 Expression, Tumor Mutational Burden, and Response to Immunotherapy in Patients with MET exon 14 Altered Lung Cancers. Ann Oncol. 2018;29(10):2085–2091. doi:10.1093/annonc/mdy334
80. Mazieres J, Drilon A, Lusque A, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol. 2019;30(8):1321–1328. doi:10.1093/annonc/mdz167
81. Guisier F, Dubos-Arvis C, Vinas F, et al. Efficacy and safety of anti-PD-1 immunotherapy in patients with advanced Non Small Cell Lung Cancer with BRAF, HER2 or MET mutation or RET-translocation. GFPC 01-2018. J Thorac Oncol. 2020;15(4):628–636. doi:10.1016/j.jtho.2019.12.129
82. Kron A, Scheffler M, Heydt C, et al. Genetic Heterogeneity of MET-aberrant Non-Small Cell Lung Cancer and its Impact on the Outcome of Immunotherapy. J Thoracic Oncol. 2020;16(4):572–582. doi:10.1016/j.jtho.2020.11.017
83. Pruis MA, Geurts-Giele WRR, von der TJH, et al. Highly accurate DNA-based detection and treatment results of MET exon 14 skipping mutations in lung cancer. Lung Cancer. 2020;140:46–54. doi:10.1016/j.lungcan.2019.11.010
84. Schoenfeld AJ, Rizvi H, Bandlamudi C, et al. Clinical and molecular correlates of PD-L1 expression in patients with lung adenocarcinomas. Ann Oncol. 2020;31(5):599–608. doi:10.1016/j.annonc.2020.01.065
85. Mayenga M, Assie JB, Monnet I, et al. Durable responses to immunotherapy of non-small cell lung cancers harboring MET exon-14-skipping mutation: a series of 6 cases. Lung Cancer. 2020;150:21–25. doi:10.1016/j.lungcan.2020.09.008
86. Saigi M, Alburquerque-Bejar JJ, Mc Leer-Florin A, et al. MET-Oncogenic and JAK2-Inactivating Alterations Are Independent Factors That Affect Regulation of PD-L1 Expression in Lung Cancer. Clin Cancer Res. 2018;24:4579–4587. doi:10.1158/1078-0432.CCR-18-0267
87. Vansteenkiste JF, Smit EF, Groen HJM, et al. 1285P Capmatinib in patients with METex14-mutated advanced non-small cell lung cancer who received prior immunotherapy: the phase II GEOMETRY mono-1 study. Ann Oncol. 2020;2:31.
88. Ilangumaran S, Villalobos-Hernandez A, Bobbala D, et al. The hepatocyte growth factor (HGF)-MET receptor tyrosine kinase signaling pathway: diverse roles in modulating immune cell functions. Cytokine. 2016;82:125–139. doi:10.1016/j.cyto.2015.12.013
89. Glodde N, Bald T, van den Boorn-konijnenberg D, et al. Reactive Neutrophil Responses Dependent on the Receptor Tyrosine Kinase c-MET Limit Cancer Immunotherapy. Immunity. 2017;47(4):789–802. doi:10.1016/j.immuni.2017.09.012
90. Felip E. P76.03 - Efficacy and Safety of Capmatinib Plus Nivolumab in Pretreated Patients with EGFR Wild-Type Non–Small Cell Lung Cancer.
91. Choueiri TK, Powles T, Burotto M, et al. 696O_PR Nivolumab + cabozantinib vs sunitinib in first-line treatment for advanced renal cell carcinoma: first results from the randomized phase III CheckMate 9ER trial. Ann Oncol. 2020;1:31.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.