Back to Journals » Journal of Inflammation Research » Volume 14
LRRK2-NFATc2 Pathway Associated with Neuroinflammation May Be a Potential Therapeutic Target for Parkinson’s Disease
Authors Wang Y, Zhang X, Chen F, Chen L, Wang J, Xie J
Received 12 January 2021
Accepted for publication 25 March 2021
Published 16 June 2021 Volume 2021:14 Pages 2583—2586
DOI https://doi.org/10.2147/JIR.S301531
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Youcui Wang,1– 3 Xiaoqin Zhang,1– 3 Fenghua Chen,1– 3 Leilei Chen,1– 3 Jun Wang,1– 3 Junxia Xie1– 3
1Institute of Brain Science and Disease, Qingdao University, Qingdao, People’s Republic of China; 2Shandong Provincial Key Laboratory of Pathogenesis and Prevention of Neurological Disorders, Qingdao University, Qingdao, People’s Republic of China; 3Shandong Provincial Collaborative Innovation Center for Neurodegenerative Disorders, Qingdao University, Qingdao, People’s Republic of China
Correspondence: Junxia Xie
Institute of Brain Science and Disease, Qingdao University, No. 308, Ningxia Road, Qingdao, 266071, People’s Republic of China
Email [email protected]
Abstract: Neuroinflammation plays an important role in the pathogenesis of Parkinson’s disease (PD). However, the molecular mechanisms involved in extracellular α‑synuclein-induced proinflammatory microglial responses through Toll-like receptor 2 (TLR2) are unclear. Leucine-rich repeat kinase 2 (LRRK2) is a serine/threonine kinase, and its mutations are closely related to autosomal dominant PD. Recently, Masliah et al characterized a novel-specific neuroinflammation cascade dependent on LRRK2-NFATc2 in microglia activated by neuron-released α-synuclein. LRRK2 selectively phosphorylated and induced nuclear translocation of NFATc2 to activate a neuroinflammation cascade. In this cascade, LRRK2 kinase was activated by neuron-released α-synuclein in microglia via TLR2. Further, NFATc2, as a kinase substrate for LRRK2, was directly phosphorylated, which accelerated nuclear translocation of NFATc2, where cytokine/chemokine gene expression including TNF-α and IL-6 is regulated by NFATc2 transcriptional activity, resulting in a neurotoxic inflammatory environment. Moreover, an abnormal increase of NFATc2 in nuclear was observed in the brains of patients and a mouse model of PD. Additionally, the administration of an LRRK2 inhibitor could ameliorate neuroinflammation, prevent neuronal loss, and improve motor function. Therefore, modulation of LRKK2-NFATc2 signaling cascade might be a potential therapeutic target for the treatment of PD.
Keywords: Parkinson’s disease, neuroinflammation, LRKK2, NFATc2, α-synuclein
Neuroinflammation plays an important role in the pathogenesis of Parkinson’s disease (PD),1 therefore, targeting crucial factors of different neuroinflammatory signaling pathways is a potential therapeutic strategy for PD. It is well known that PD is a chronic neurodegenerative disorder characterized by abnormal accumulation of α-synuclein in the degenerative dopaminergic neurons of substantia nigra, which can induce cell-autonomous neurotoxicity.2,3 Notably, a large number of studies have shown that α-synuclein can be secreted by neurons. This extracellular α‑synuclein can be directly transferred to neighboring neurons resulting in neurotoxic α‑synuclein deposition, and it can further lead to neuroinflammatory responses in microglia that can produce and release pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1beta (IL-1β), interleukin-6 (IL-6) and so on, which in turn aggravate neuronal degeneration and neuroinflammation.3 Previous studies have shown that extracellular α‑synuclein actives microglia through Toll-like receptor 2 (TLR2), which is a α-synuclein receptor expressed in neurons and microglia, leading to non-cell-autonomous neurotoxicity.4,5 The mechanism probably involves the production and release neurotoxic pro-inflammatory cytokines by activated microglia via nuclear factor B (NF-κB) and p38 mitogen-activated protein kinase (MAPK) downstream signaling pathways.4 Furthermore, increased expression of TLR2 was observed in the brains of patients and a mouse model of PD.6,7 However, the molecular mechanisms involved in extracellular α‑synuclein-induced neurotoxic proinflammatory microglial responses through TLR2 are unclear.
Leucine-rich repeat kinase 2 (LRRK2) is a serine/threonine kinase,8 and its mutations are closely related to autosomal dominant PD.9,10 Mounting evidence indicates that LRRK2 is highly expressed in immune cells and is frequently associated with neuroinflammation in PD.11,14 It has been reported that the production of pro-inflammatory cytokines TNF-α and IL-1β decreased in primary microglia with LRRK2 knockdown or kinase inhibition.12,13 Therefore, an understanding of the interplay between LRRK2-mediated neuroinflammatory responses and PD is essential to clearly define the upstream and downstream factors of this pathway. Although knockdown of LRRK2 can reduce the transcriptional activity of NF-κB in activated microglia,14 it might raise the question whether there are other LRRK2-mediated neuroinflammatory signaling pathways in activated microglia via extracellular α‑synuclein.
Recently, Masliah et al characterized a novel neuroinflammation cascade dependent on LRRK2-nuclear factor of activated T cells, cytoplasmic 2 (LRRK2-NFATc2) in microglia activated by neuron-released α-synuclein15 (Figure 1). In this study, the level of LRRK2 phosphorylation and activity increased in mouse primary microglia with extracellular α-synuclein treatment. However, the increase disappeared when the microglia were pretreated with TLR2 function blocking antibody T2.5, suggesting that extracellular α-synuclein activated LRRK2 by TLR2 in microglial cultures. Furthermore, Lrrk2 knockout significantly suppressed α-synuclein-mediated microglial neurotoxicity by decreasing of TNF-α and IL-6 expression. It has been reported that TLR2 is a pattern recognition receptor that plays a key role in the innate immune response to micro-organisms.16,17 Single-nucleotide polymorphisms of Lrrk2 or its adjacent genes are susceptible to leprosy infection in humans.18 Although evidence suggests that TLR2 and LRRK2 are involved in the innate immune response to micro-organisms, the underlying interaction between them is not completely understood. It has been shown that marked phosphorylation of LRRK2 at Ser910 and Ser935 is induced by the canonical IκB kinase family during TLR signaling.19 While Masliah et al did not provide additional information regarding the interactions between TLR2 and LRRK2, further studies are needed to elucidate this mechanism.15
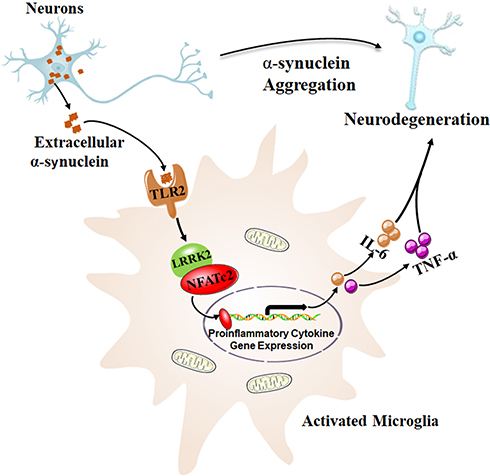 |
Figure 1 Neuron-released α-synuclein activated microglia through TLR2, and subsequently activated LRRK2 kinase. NFATc2, as a kinase substrate of LRRK2, was directly phosphorylated, which accelerated nuclear translocation of NFATc2 where cytokine/chemokine gene expression such as IL-6 and TNF-α, were upregulated by NFATc2 transcriptional activity. This resulted in a neurotoxic inflammatory environment, which in return aggravated the neuronal degeneration in a mouse model of PD. Data from Kim et al.15 |
To address the mechanisms underlying the role of LRRK2 in microglial activation via extracellular α-synuclein, Masliah et al analyzed transcriptome data (GSE26532) obtained from primary rat microglia exposed to extracellular α-synuclein. Forty-three up-regulated genes, which involved in TLR signaling and other immune signaling pathways, were selected and a signaling network was reconstructed to describe their interactions using functional gene enrichment analysis and information on protein–protein interactions.15 According to the analysis results, Masliah et al hypothesized that LRRK2 and NFATc2 selectively modulate cytokine and chemokine expression in extracellular α-synuclein-mediated microglia via TLR2 signaling pathway.15 It has been shown that NFATc2, as an immune transcription factor, can translocate to the nucleus, and NFAT responsive elements are located in the promoter region of multiple immune-related genes such as TNF-α, IL-6 and so on.20,21 Besides, calcineurin/NFAT signaling cascade is frequently associated with the neuroinflammation in mouse models of Alzheimer’s disease (AD)22 and PD.23 Additionally, phosphorylation of NFATc1 is closely related to the transcription of genes associated with neuroinflammation in AD mice.24 Notably, the NFATc2 isoform was the most highly expressed in murine microglia cultures, and the ability of the microglia to secrete cytokines was attenuated by deletion of NFATc2 in mouse models of AD.25 The above findings are strong evidence to support the hypothesis that LRRK2 and NFATc2 selectively modulate pro-inflammatory cytokine expression in extracellular α-synuclein-mediated microglia via TLR2 signaling pathway.
Interestingly, the direct interaction between NFATc2 and LRRK2 was first identified by automated image analysis in this study. Specifically, LRRK2 selectively phosphorylated and induced the nuclear translocation of NFATc2 to activate a neuroinflammation cascade. In this cascade, LRRK2 kinase was activated by neuron-released α-synuclein in microglia via TLR2. Further, NFATc2, as a kinase substrate for LRRK2, was directly phosphorylated, which accelerated the nuclear translocation of NFATc2 where the cytokine/chemokine gene expression including IL-6 and TNF-α is regulated by NFATc2 transcriptional activity, resulting in a neurotoxic inflammatory environment15 (Figure 1). Moreover, an abnormal increase of NFATc2 in the nucleus was observed in the brains of patients and mouse models of PD.15 Additionally, the administration of an LRRK2 inhibitor (HG-10-102-01) could ameliorate neuroinflammation, prevent neuronal loss, and improve motor function in a mouse model of PD.15 Although LRRK2 inhibitor clinical trials provide opportunities to refine our understanding of LRRK2 in human immune function, it is unknown whether LRRK2 inhibition increases the risk of opportunistic infections.11 In addition, it is unclear whether LRRK2 inhibitors have disease modifier role in PD patients with LRRK2 mutations.11 Although it is difficult to develop specific antibodies targeting NFATc2 because of multiple phosphorylation residues, modulation of the LRKK2-NFATc2 signaling cascade might be a potential therapeutic target for the treatment of PD.
Altogether, this study is the first to report a novel LRKK2-NFATc2 signaling cascade that plays an important role in neuroinflammation in PD, and provides new clues for the clinical treatment of PD.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31900745, 31771124, 31800893), Shandong Provincial Natural Science Foundation (ZR2017BC031), and Special Fund for Youth of Applied Foundational Research Program of Qingdao (No.18-2-2-44-jch). The authors also thank Qian Jiao (Department of Physiology, Shandong Provincial Key Laboratory of Pathogenesis and Prevention of Neurological Disorders and State Key Disciplines: Physiology, School of Basic Medicine, Qingdao University) for her skilful technical assistance in the figure design.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gelders G, Baekelandt V, Van der Perren A. Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J Immunol Res. 2018;2018:4784268. doi:10.1155/2018/4784268
2. Przedborski S. The two-century journey of Parkinson disease research. Nat Rev Neurosci. 2017;18(4):251–259. doi:10.1038/nrn.2017.25
3. Lee HJ, Bae EJ, Lee SJ. Extracellular α–synuclein – a novel and crucial factor in Lewy body diseases. Nat Rev Neurol. 2014;10:92–98. doi:10.1038/nrneurol.2013.275
4. Kim C, Ho DH, Suk JE, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562. doi:10.1038/ncomms2534
5. Kim C, Lee HJ, Masliah E, Lee SJ. Non-cell-autonomous neurotoxicity of α-synuclein through microglial toll-like receptor 2. Exp Neurobiol. 2016;25:113–119. doi:10.5607/en.2016.25.3.113
6. Dzamko N, Gysbers A, Perera G, et al. Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. 2017;133(2):303–319. doi:10.1007/s00401-016-1648-8
7. Drouin-Ouellet J, St-Amour I, Saint-Pierre M, et al. Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson’s disease. Int J Neuropsychopharmacol. 2014;18(6):18. doi:10.1093/ijnp/pyu103
8. Cookson MR. LRRK2 pathways leading to neurodegeneration. Curr Neurol Neurosci Rep. 2015;15(7):42. doi:10.1007/s11910-015-0564-y
9. Singleton AB, Gasser T. The discovery of LRRK2 mutations as a cause of Parkinson’s disease. Mov Disord. 2020;35:551–554. doi:10.1002/mds.27999
10. Li H, Su L-Y, Yang L, et al. A cynomolgus monkey with naturally occurring Parkinson’s disease. Nat Sci Rev. 2020.
11. Alessi DR, Sammler E. LRRK2 kinase in Parkinson’s disease. Science. 2018;360(6384):36–37. doi:10.1126/science.aar5683
12. Moehle MS, Webber PJ, Tse T, et al. LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci. 2012;32(5):1602–1611. doi:10.1523/JNEUROSCI.5601-11.2012
13. Russo I, Berti G, Plotegher N, et al. Leucine-rich repeat kinase 2 positively regulates inflammation and down-regulates NF-κB p50 signaling in cultured microglia cells. J Neuroinflammation. 2015;12(1):230. doi:10.1186/s12974-015-0449-7
14. Russo I, Bubacco L, Greggio E. LRRK2 and neuroinflammation: partners in crime in Parkinson’s disease? J Neuroinflammation. 2014;11(1):52. doi:10.1186/1742-2094-11-52
15. Kim C, Beilina A, Smith N, et al. LRRK2 mediates microglial neurotoxicity via NFATc2 in rodent models of synucleinopathies. Sci Transl Med. 2020;12:12. doi:10.1126/scitranslmed.aay0399
16. Dunne A, O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE. 2003;2003(171):re3. doi:10.1126/stke.2003.171.re3
17. West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-like receptors. Annu Rev Cell Dev Biol. 2006;22(1):409–437. doi:10.1146/annurev.cellbio.21.122303.115827
18. Dzamko NL. LRRK2 and the immune system. Adv Neurobiol. 2017;14:123–143.
19. Dzamko N, Inesta-Vaquera F, Zhang J, et al. The IkappaB kinase family phosphorylates the Parkinson’s disease kinase LRRK2 at Ser935 and Ser910 during Toll-like receptor signaling. PLoS One. 2012;7(6):e39132. doi:10.1371/journal.pone.0039132
20. Fric J, Zelante T, Wong AY, Mertes A, Yu HB, Ricciardi-Castagnoli P. NFAT control of innate immunity. Blood. 2012;120:1380–1389. doi:10.1182/blood-2012-02-404475
21. Müller MR, Rao A. NFAT immunity and cancer: a transcription factor comes of age. Nat Rev Immunol. 2010;10:645–656. doi:10.1038/nri2818
22. Sompol P, Norris CM. Ca(2+), astrocyte activation and calcineurin/NFAT signaling in age-related neurodegenerative diseases. Front Aging Neurosci. 2018;10:199. doi:10.3389/fnagi.2018.00199
23. Manocha GD, Floden AM, Puig KL, Nagamoto-Combs K, Scherzer CR, Combs CK. Defining the contribution of neuroinflammation to Parkinson’s disease in humanized immune system mice. Mol Neurodegener. 2017;12(1):17. doi:10.1186/s13024-017-0158-z
24. Vasilopoulou F, Griñán-Ferré C, Rodríguez-Arévalo S, et al. I(2) imidazoline receptor modulation protects aged SAMP8 mice against cognitive decline by suppressing the calcineurin pathway. Geroscience. 2020. doi:10.1007/s11357-020-00281-2
25. Manocha GD, Ghatak A, Puig KL, Kraner SD, Norris CM, Combs CK. NFATc2 modulates microglial activation in the AβPP/PS1 mouse model of alzheimer’s disease. J Alzheimers Dis. 2017;58(3):775–787. doi:10.3233/JAD-151203
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.