Back to Journals » Journal of Inflammation Research » Volume 16
LPA2 Alleviates Septic Acute Lung Injury via Protective Endothelial Barrier Function Through Activation of PLC-PKC-FAK
Authors Bai R, Pei J ![]() , Pei S, Cong X, Chun J, Wang F, Chen X
, Pei S, Cong X, Chun J, Wang F, Chen X
Received 27 June 2023
Accepted for publication 27 September 2023
Published 8 November 2023 Volume 2023:16 Pages 5095—5109
DOI https://doi.org/10.2147/JIR.S419578
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Ruifeng Bai,1 Jianqiu Pei,1 Shengqiang Pei,1 Xiangfeng Cong,1 Jerold Chun,2 Fang Wang,1,3,4 Xi Chen1,3
1State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China; 2Neuroscience Drug Discovery, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA, USA; 3Diagnostic Laboratory Service, Fuwai Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China; 4Department of Clinical Laboratory, Fuwai Yunnan Cardiovascular Hospital, Kunming, People’s Republic of China
Correspondence: Xi Chen; Fang Wang, Email [email protected]; [email protected]
Background: Increased endothelial permeability of pulmonary vessels is a primary pathological characteristic of septic acute lung injury (ALI). Previously, elevated lysophosphatidic acid (LPA) levels and LPA2 (an LPA receptor) expression have been found in the peripheral blood and lungs of septic mice, respectively. However, the specific role of LPA2 in septic ALI remains unclear.
Methods: A lipopolysaccharide (LPS)-induced model of sepsis was established in wild-type (WT) and global LPA2 knockout (Lpar2−/−) mice. We examined mortality, lung injury, assessed endothelial permeability through Evans blue dye (EBD) assay in vivo, and transendothelial electrical resistance (TEER) of mouse lung microvascular endothelial cells (MLMECs) in vitro. Enzyme-linked immunosorbent assay (ELISA), histopathological, immunofluorescence, immunohistochemistry, and Western blot were employed to investigate the role of LPA2 in septic ALI.
Results: Lpar2 deficiency increased vascular endothelial permeability, impaired lung injury, and increased mortality. Histological examination revealed aggravated inflammation, edema, hemorrhage and alveolar septal thickening in the lungs of septic Lpar2−/− mice. In vitro, loss of Lpar2 resulted in increased permeability of MLMECs. Pharmacological activation of LPA2 by the agonist DBIBB led to significantly reduced inflammation, edema and hemorrhage, as well as increased expression of the vascular endothelial tight junction (TJ) protein zonula occludens-1 (ZO-1) and claudin-5, as well as the adheren junction (AJ) protein VE-cadherin. Moreover, DBIBB treatment was found to alleviate mortality by protecting against vascular endothelial permeability. Mechanistically, we demonstrated that vascular endothelial permeability was alleviated through LPA-LPA2 signaling via the PLC-PKC-FAK pathway.
Conclusion: These data provide a novel mechanism of endothelial barrier protection via PLC-PKC-FAK pathway and suggest that LPA2 may contribute to the therapeutic effects of septic ALI.
Keywords: septic ALI, LPA, vascular endothelial permeability, LPA2, lung microvascular endothelial cells
Introduction
Septic acute lung injury (ALI) is a potentially lethal medical condition, involves excessive accumulation of alveolar fluid in the pulmonary capillaries, refractory hypoxemia and respiratory embarrassment, resulting from disruption of the lung endothelial barrier integrity.1 Currently, ALI patients are treated with nitric oxide, mechanical ventilation, extracorporeal membrane oxygenation, and steroid therapy. However, significant advances in ALI recognition and management have been made, treatment options remain limited to enable substantial improvement in survival.2 Pulmonary vascular endothelial cells are vital constituents of the pulmonary capillary barrier and form a semi-permeable barrier at the interface between the blood in circulation and nearby tissues. The impairment of the endothelial barrier results in heightened vascular permeability, thus thought to be an important factor in septic ALI.3 When endothelial injury occurs, the release of inflammatory mediators disrupts the integrity of the vascular endothelial barrier which contribute to high mortality in septic ALI.4 Since increased pulmonary vascular permeability is the main pathophysiological characteristic of ALI, targeting endothelial barrier dysfunction is a potential approach to treat septic ALI.
Tight junctions (TJs) and adherens junction (AJs) are essential in facilitating cell-cell attachment and regulating endothelial permeability, therefore critical during ALI development.5,6 AJs common in capillary veins, consist of adhesion complexes comprised of α-, β-, and P120-catenin together with VE-cadherin. VE-cadherin has been shown to mediate the homeostasis of pulmonary fluids and integrity of endothelial junctions which expressed only in endothelial cells.7 TJs form a continuous intercellular network, which create a barrier that prevents pathogens from penetrating the epithelium.8 TJs consist of occludin and claudins which interact with intracellular zonulas (ZOs, including ZO-1, ZO-2, and ZO-3).9 ZO-1 is expressed on pulmonary vascular endothelial cells, interacts with almost all claudin transmembrane proteins, and regulates endothelial AJs and endothelial barrier formation. Claudin-5 is observed primarily in vascular endothelial cells.10
The extracellular lipid mediator lysophosphatidic acid (LPA) engages in signaling via six G-protein-coupled receptors (LPA1 - LPA6) to regulate multiple physiological function.11 Several reports have demonstrated that LPA is involved in stabilizing endothelial barrier function and reducing vascular endothelial permeability,12,13 as well as stimulating angiogenesis.14–16 We have previously shown that LPA2 promotes vascular endothelial homeostasis and heart regeneration following myocardial infarction, which shown to play a critical part in the regulation of vascular endothelial barrier function.17 Furthermore, LPA2 has been shown to have over 100 × affinity for LPA than other LPA receptors.18,19
LPA2 is a G-protein coupled receptor (GPCR) that acts through three different pathways: Gα12/13-Rho, Gαq /11-PLC-PKC and Gαi/o-PI3K or Ras-MAPK. Studies have shown that LPA promotes E-cadherin expression in HBEPCs through the PKC-FAK pathway.20 Thus, we found that LPA promotes VE-cadherin expression in MLMECs. However, the role of LPA2 in mediating endothelial permeability during septic ALI is unknown, and it is not clear whether the protective effect of LPA2 occurs during PLC-PKC-FAK mediated endothelial barrier dysfunction.
Materials and Methods
Animals
Global LPA2 knockout (Lpar2−/−) mice were bred into a Balb/c background for at least ten generations. Wild-type (WT) and Lpar2 −/− mice (male, aged 8–10 weeks) were used and reared in the Fuwai Hospital Animal Experimental Center (Chinese Academy of Medical Sciences) in a specific pathogen free (SPF) animal room under a 12 h light-dark cycle at 22–26°C were given water libitum. The study was carried out in accordance with the “Regulation to the Care and Use of Experimental Animals” of the Beijing Council on Animal Care study (1996). The experiments were approved by Fuwai Hospital Animal Care and Use Committee.
Sepsis Model
The sepsis model was induced by intraperitoneal injection of lipopolysaccharide (LPS) (10mg/kg, Escherichia coli serotype 055: B5, L2880; Sigma-Aldrich, USA) in male WT and Lpar2−/− mice. An equivalent volume of phosphate-buffered saline (PBS) was intraperitoneally injected as the control group. Survival rate was recorded every 4 h after LPS injection for a 72 h period.
Lung Injury Score and Histological Examination
At 24 h after LPS treatment, lung tissue was isolated and fixed in 4% paraformaldehyde. Then embedded in paraffin and cut into 5 µm sections. After staining with hematoxylin eosin (HE), histopathological alterations were assessed using light microscopy. The lung injury score was determined following previously criteria.21
 |
|
Bronchoalveolar Lavage Fluid (BALF) Measurement
Mice were anesthetized with isoflurane after administration of LPS or PBS 24 h, and BALF was collected. Subsequently, BALF was centrifuged 15 min at 1500 g, and quantification of IL-6 (ab222503, Abcam, USA), CXCL15 (ab193720, Abcam, USA) and TNF-α (ab208348, Abcam) was performed with the corresponding ELISA kits within the supernatants. BCA protein assay Kit (cat. No. 23225; Thermo Fisher Scientific, USA) was used to assess the total protein level in the BALF.
Vascular Permeability Assessment
Vascular endothelial permeability was examined as previously described.22 Vascular leakage was assessed by injecting with 20 mg/kg Evans blue dye (EBD) through the tail vein 30 min prior to lungs collection. EBD was extracted from lung tissue samples by incubating with formamide at 55°C for 48 h. The EBD solution’s absorbance was determined at 620nm-740 nm by spectrophotometry.
Immunofluorescence Staining
Tissue sections were baked at 68°C for 45 min, followed by deparaffinization and dehydration. The samples were boiled in sodium citrate (pH 6.0, 10 mmol/L) for 2 min for antigen retrieval. Samples then permeabilized by incubating in Triton X-100 (0.1%) for 10 min. Next, the samples were blocked in normal goat serum for 1 h at room temperature.23 Sections were incubated with primary antibodies against VE-cadherin (1:200; ab205336, Abcam, USA), claudin-5 (1:200; ab15106, Abcam, USA), ZO-1 (1:200; ab216880, Abcam, USA) and CD31 (1:200, MA1-26196, Thermo Fisher, USA) overnight at 4°C, followed incubated with Alexa Fluor-488 (1:500; A-11001, Invitrogen, USA) or Alexa Fluor-594 (1:500; A-11012, Invitrogen, USA) secondary antibodies for 1 h at room temperature. Slides were counterstained with DAPI, coverslipped and fluorescent images captured using a laser-scanning confocal microscope (SP8, Leica).
Immunohistochemical Staining
Tissue slices were prepared as described previously.24 After dewaxing, antigen was extracted with citric acid buffer and samples were permeabilized with Triton X-100 (0.1%) for 10 min. The samples were then treated with 30% H2O2 to inhibit endogenous peroxidase activity. Subsequently, the samples were incubated with normal goat serum (10%) and subsequently with primary antibodies targeting VE-cadherin (1:200; ab282277, Abcam, USA), claudin-5 (1:200; ab15106, Abcam, USA) and ZO-1 (1:200; ab216880, Abcam, USA), at 4°C overnight. Samples were rewarmed at 37°C for 30 min, incubation with reaction enhancer solution was performed for 20 min at 37°C, followed by incubation with HRP-conjugated secondary antibodies for 30 min at 37°C. Diaminobenzidine (DAB) solution was added and incubated at room temperature and the nuclei were stained with hematoxylin. The stained sections were observed under a light microscope.
Isolation of Mouse Lung Microvascular Endothelial Cells (MLMECs)
MLMECs were isolated from 6–8 week mice by serial immunoselection using a purified rat anti-mouse CD31 antibody coupled with Dynabeads M-450 sheep anti-rat IgG as previously described.25 Briefly, lungs were removed from mice and washed with high-glucose DMEM containing 20% FBS. Next, the lung tissue was digested with collagenase I solution (2000 U/mL, 15 mL, C1639, Sigma, USA) in PBS for 45 min at 37°C, minced 15 times with a 20-gauge needle. The cell suspension was then passed through a 70 μm nylon filter then centrifuged for 8 min at 1300 g and 4°C. Primary MLMECs were plated in tissue culture flasks (T-25) precoated with human fibronectin (2.5 g/mL in aseptically filtered PBS, ECM001, Invitrogen, USA) and cultured at 37°C in a CO2 incubator.
MLMECs Treated with the Inhibitor
MLMECs were cultured on the luminal side of filters (0.4 μm pore size, Corning, USA) in gelatin-coated 12-well plates until confluent. They were then cultured in endothelial serum-free medium for 12 h and exposed to LPS (1μg/mL) for 24 h, then corresponding inhibitor and LPA (10μM) cultured for 20 h. Fluorescein isothiocyanate (FITC)-dextran fluorescein was used to confirm the permeability. FITC-dextran (1 mg/mL; 78331, Sigma, USA) was added to the upper compartments of the transwell cultures. A microplate reader was used to measure the values for absorbance of the lower chamber solution at 492 nm (excitation) and 520 nm (emission) wavelengths.
Western Blot Analysis
The left lungs of mice from each treatment group were stored in liquid nitrogen. After homogenization of tissue samples, protein extraction was conducted with an extraction reagent supplemented with protease inhibitors. The protein concentration was determined with a BCA Protein Assay Kit (23225, Thermo Fisher Scientific, USA). Proteins were separated with 10% SDS-PAGE, then transferred to PVDF membranes. The membranes were subsequently blocked in 5% BSA for 1 h, incubated overnight at 4℃ with primary rabbit polyclonal antibodies anti-VE-cadherin (1:1000; ab205336, Abcam, USA), anti-ZO-1 (1:1000; ab216880, Abcam, USA), anti-claudin-5 (1:1000; 35–2500, Thermo Fisher, USA), PKCδ (1:1000; ab182126, Abcam, USA), PKCζ (1:1000; ab108970, Abcam, USA), FAK (1:1000; ab40794, Abcam, USA), and p-FAK (1:1000; ab81298, Abcam, USA). The next day membranes were incubated with secondary antibodies, visualization was performed with SuperSignal West Pico Plus Substrate (34577, Thermo Fisher Scientific, USA). Protein band intensity was determined in ImageJ software.
Statistical Analysis
GraphPad Prism 8 and SPSS 21.0 software were used for statistical analysis. The data are expressed as mean ± SD. One-way ANOVA, together with Tukey’s test for multiple comparisons after the variance homogeneity test, was used to assess significant differences among groups. Differences were considered statistically significant when p < 0.05.
Results
Lpar2 Deficiency Decreases Survival and Aggravates LPS-Induced Septic ALI in Mice
The effects of LPA2 on LPS-induced sepsis were examined in WT and Lpar2−/− mice injected with LPS or PBS intraperitoneally. The survival rate was monitored every 4 h for 72 h. The survival rates of the WT and Lpar2−/− mice following LPS treatment were 70.8% and 41.7%, respectively (Figure 1A). Organs were removed from the LPS-induced septic mice 12 h later to examine the extent of organ damage. The lung tissue of LPS-Lpar2−/− mice displayed obvious edema and congestion, with hemorrhagic spots covering the surface of the lung tissue, an abundance of secretions and an outflow of pink fluid. In addition, lung injury was more severe in Lpar2−/− mice (Figure 1B). HE staining revealed that mice in the LPS-Lpar2−/− group experienced substantially exacerbated inflammatory cell infiltration, structural damage, alveolar interstitial edema, and alveolar wall thickening (Figure 1C-1). The lung injury score exceeded in the LPS-Lpar2−/− group than that observed in the LPS-WT group (Figure 1C-2). Furthermore, the BALF of LPS-Lpar2−/− mice exhibited a significant increase in neutrophil level (Figure 1D), as well as elevated level of IL-6 (Figure 1E-1), TNF-α (Figure 1E-2), and CXCL15 (Figure 1E-3). These results collectively indicate the crucial protective role of LPA2 in septic ALI.
 |
Figure 1 Lpar2 deficiency increases the mortality, pulmonary damage and inflammation in LPS-induced mice. (A) Survival of WT and Lpar2−/− mice challenged with LPS (n = 24). (B) Representative images of the lung tissue in WT and Lpar2−/− mice. (C-1) Representative images of HE-stained lung tissue. Red arrows indicate inflammatory cell infiltration, black arrows indicate thickening of the alveolar walls. Scale bar, 100 μm. (C-2) The lung injury area was quantified by assessing the histological scores for each treatment group (n = 6). (D) Total neutrophil levels in the BALF (n = 6). (E1–3) CXCL15, TNF-α and IL-6 levels in the BALF (n = 6), *p < 0.05 and **p < 0.01 vs the LPS-WT group. |
Lpar2 Deficiency Augments Lung Vascular Endothelial Permeability
Endothelial barrier function has been associated with the pathogenesis of septic ALI. To explore whether LPA2 affects endothelial permeability during septic ALI, we measured endothelial permeability of WT mice and Lpar2−/− mice in vivo and in vitro, respectively. Endothelial permeability in vivo assessed using the wet-to-dry ratio (W/D), protein level in BALF and EBD extravasation in lung tissue. In vitro, an electric cell-substrate impedance sensing (ECIS) system was used to determine the transendothelial electrical resistance (TEER) in LPS-treated MLMECs.26 The W/D ratio was found to be significantly elevated in the lungs of LPS-Lpar2−/− mice (Figure 2C). Similarly, enhanced EBD leakage was observed in Lpar2−/− mice (Figure 2A and B). Consistent with our W/D ratio data, significantly higher level of total BALF protein were observed in LPS-Lpar2−/− mice (Figure 2D). In addition, increased permeability of LPS-Lpar2−/− than LPS-WT in MLMECs (Figure 2E). Finally, LPS exposure led to decreased TEER level in Lpar2−/− MLMECs (Figure 2F and G). Together, our findings suggest a protective role of LPA2 in mediating endothelial barrier integrity following LPS treatment.
 |
Figure 2 Lpar2 deficiency increases LPS-induced vascular endothelial permeability in mice lung tissue and MLMECs. (A) Representative images showing WT and Lpar2−/− mice lung tissue. (B) The vascular permeability of mouse lung tissue was quantified using the EBD assay (n = 6). (C) Quantification of mice pulmonary W/D ratio (n = 6). (D) Total BALF protein (n = 8). (E) The vascular permeability of MLMECs (n=3). (F) The permeability of MLMECs was assessed by measuring the TEER. (G) The statistical analysis of TEER in 24h, **p < 0.01 and ****p < 0.0001 vs LPS-WT group. |
Lpar2 Deficiency Decreases VE-Cadherin, ZO-1, and Claudin-5 Expression in Septic ALI Mice
To further investigate whether LPA2 regulates the expression of VE-cadherin, ZO-1, and claudin-5 and then affect the endothelial permeability, we detected the three proteins by Western blot, immunofluorescence and immunohistochemical. Our findings demonstrated a significant downregulation of VE-cadherin, ZO-1, and claudin-5 expression in the LPS-Lpar2−/− group with the results of Western blot (Figure 3B). Our immunofluorescence (Figure 3A1–A4) and immunohistochemical (Figure 3C1 and C2) data were consistent with Western blot results. The results indicated that LPA2 preserving vascular integrity and mitigating increased vascular permeability through the restoration of VE-cadherin, ZO-1, and claudin-5 expression in septic ALI mice.
 |
Figure 3 Lpar2 deficiency leads to a decrease in AJs and TJs in mice lung tissue. (A1–A3) Representative images showing immunofluorescence staining in mice lung tissue. White arrows indicate expression of VE-cadherin, ZO-1, and claudin-5 in vascular endothelium. Scale bar, 50 μm. (A4) Relative fluorescence intensity of the images in (A1–A3). (B) Representative Western blot and quantification of protein expression level (n = 6). (C-1) Representative images showing immunohistochemical staining in paraffin-embedded pulmonary tissue section. Scale bar, 100 μm. (C-2) The percentage of positively stained area, *p <0.05 and **p <0.01 vs LPS-WT group. |
LPA Has a Key Role in Promoting Expression of VE-Cadherin
Previous studies have shown that treatment of human bronchial epithelial cells with LPA promotes the cell-cell connection between E-cadherin/c-Met.20 Thus, we hypothesized that LPA may enhance the lung endothelial barrier function of septic mice. As shown in Figure 4A1 and A2, treatment with LPA increased the TEER of LPS-stimulated MLMECs. Western blot analysis revealed that expression of VE-cadherin markedly increased following LPA treatment of LPS-treated cells compared to the LPS alone treatment group (Figure 4B1 and B2). Taken together, our findings suggest that LPA reduces the permeability of MLMECs, thereby protecting endothelial barrier integrity.
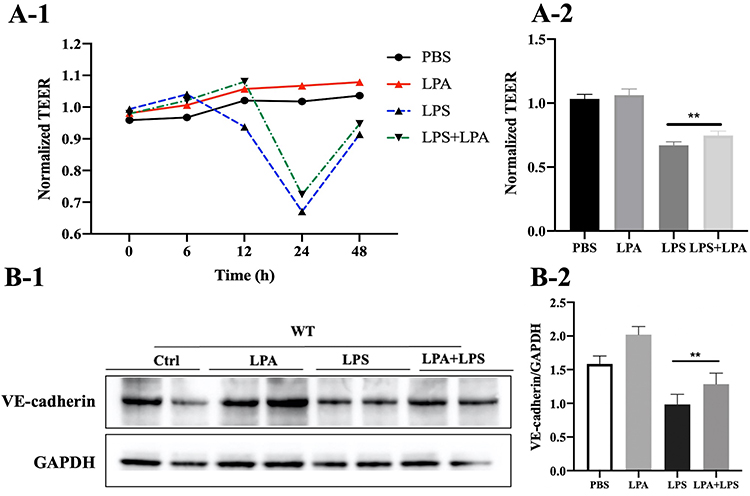 |
Figure 4 LPA facilitates expression of VE-cadherin in MLMECs. (A-1) The effects of LPA on the transmembrane resistance of MLMECs were examined by measuring the TEER. (A-2) The statistical analysis of TEER in 24h. (B-1) Western blot analysis to assess the effects of LPA treatment on expression of VE-cadherin. (B-2) Quantitative analysis of the Western blot data (n = 6), **p < 0.01 vs LPS group. |
LPA-LPA2 Promotes VE-Cadherin Expression Through the PLC-PKC-FAK Pathway
LPA2 is a G-protein coupled receptor (GPCR) that acts through three different pathways: Gα12/13-Rho, Gαq /11-PLC and Gαi/o-PI3K or Ras-MAPK (Figure 5A). We demonstrated that inhibition of PLC, PKC and FAK diminished the expression of VE-cadherin and increased permeability of MLMECs compared to MLMECs treated with LPS alone (Figure 5B). To further confirm that LPA promotes VE-cadherin expression in MLMECs through the PLC-PKC-FAK pathway, MLMECs were treated with PLC, PKC and FAK inhibitors and the expression of downstream signaling proteins was examined through Western blot. As shown in Figure 5C1 and C2, PKCδ, PKCζ, and pFAK/FAK expression levels were decreased following treatment with PLC inhibitors. Treatment with PKC inhibitors led to decreased VE-cadherin and pFAK/FAK expression levels (Figure 5D1 and D2), while VE-cadherin expression was reduced following treatment with FAK inhibitors (Figure 5E1 and E2). These results suggest that LPA-LPA2 may mediate the expression of VE-cadherin and endothelial cell permeability via the PLC-PKC-FAK pathway.
 |
Figure 5 LPA-LPA2 promotes VE-cadherin expression through the PLC-PKC-FAK signaling pathway in MLMECs. (A) Potential signaling pathways associated with LPA2. (B) MLMECs were treated with various pathway inhibitors (LPA2 inhibitor H2L518636, PLC inhibitor U73122, PI3K inhibitor LY294002, ROCK inhibitor Y27632, MAPK inhibitor SB203580, PKC inhibitor Go6983, FAK inhibitor PF-573228), and their effects on expression of VE-cadherin were analyzed by Western blot (n=6). (C-1) MLMECs were treated with the PLC inhibitor, and Western blot was performed to assess changes in the corresponding signaling pathway molecule. (C-2) Quantitative analysis of the Western blot data in (C-1) (n=6). (D-1) Expression levels of p-FAK, FAK and VE-cadherin were detected following treatment with PKCδ/PKCζ inhibitors. (D-2) Quantitative analysis of the Western blot data in D-1 (n=6). (E-1) Expression levels of VE-cadherin in MLMECs were assessed following treatment with the FAK pathway inhibitor. (E-2) Quantitative analysis of the Western blot data in E-1 (n=6), *p < 0.05, **p < 0.01, ***p < 0.001 vs LPA+LPS group. |
LPA2 Activation Reduces LPS-Induced Mice Lung Damage and Mortality
To determine whether activation of LPA2 alleviated the lungs damage and mortality in LPS-induced septic mice, we next administered DBIBB, a specific agonist of LPA2, followed by intraperitoneal injection of LPS to WT mice. We found that DBIBB treatment significantly reduced mortality with a survival rate of 79.2% in LPS + DBIBB mice compared to 58.3% in LPS mice (Figure 6A). Furthermore, DBIBB treatment resulted in alleviation of edema and congestion of the lung tissue, as well as a reduction in bleeding spots on the surface of the lung tissue and exudate secretions (Figure 6B). Histopathological analysis of the lung tissue revealed that DBIBB ameliorated alveolar interstitial edema, alveolar wall thickening and inflammatory cell infiltration (Figure 6C-1). Finally, the LPS + DBIBB group displayed a reduced lung injury score compared to the LPS group (Figure 6C-2). Together, these findings highlight the protective role of LPA2 in septic ALI.
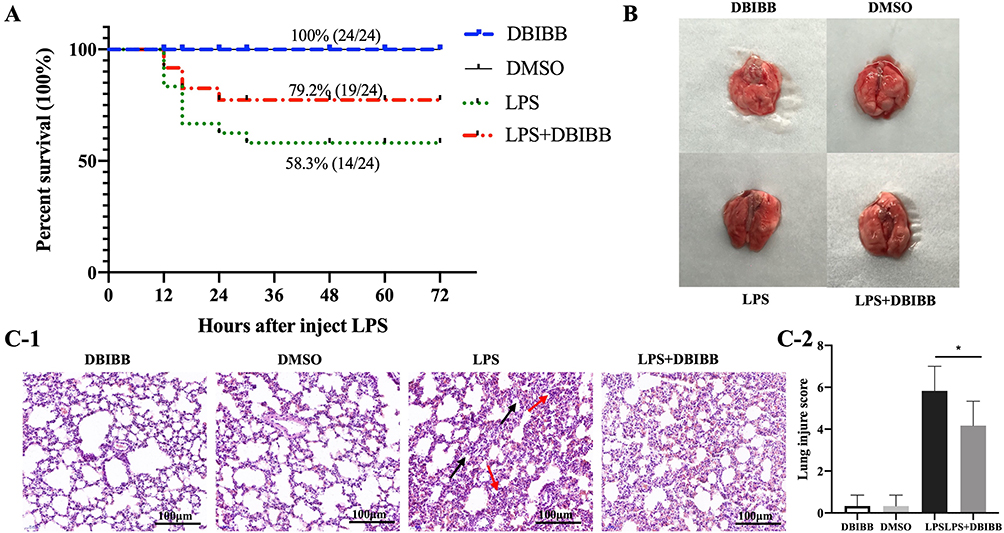 |
Figure 6 LPA2 reduces lung injury and mortality in LPS-treated mice. (A) Mice survival in all treatment groups (n = 24). (B) Representative images showing the mice lung tissue from each treatment group (n = 24). (C-1) Representative HE staining of the mice lung tissue for each treatment group. Red arrows indicate inflammatory cell infiltration, black arrows indicate thickening of the alveolar walls. Scale bar, 100 μm. (C-2) Quantification of the lung injury area was assessed by histological scores (n = 6), *p < 0.05 vs LPS group. |
Activation of LPA2 Decreases Vascular Endothelial Permeability in Septic ALI
Next, we examined whether activation of LPA2 alleviates endothelial barrier dysfunction in septic ALI using the W/D ratio and EBD. We found that DBIBB treatment substantially reversed the LPS-induced increased vascular permeability (Figure 7A). Similarly, the EBD concentration was significantly lower in the LPS + DBIBB group than the LPS group, suggesting that DBIBB treatment significantly weakened lung tissue permeability (Figure 7B). In addition, DBIBB treatment led to a significant reduction in W/D ratio values compared to LPS-treated mice (Figure 7C). Together, our findings suggested that treatment with an LPA2 agonist reduced endothelial barrier dysfunction in septic ALI mice.
 |
Figure 7 LPA2 protects against increased endothelial permeability in septic mice. (A) Representative images showing pulmonary tissue in LPS-induced WT and Lpar2−/− mice. (B) Vascular permeability was assessed in mice lung tissue using the EBD assay (n = 6). (C) Quantification of W/D ratio in mice lung tissue (n = 6), *p < 0.01 and **p < 0.05 vs LPS group. |
LPA2 Activation Promotes Expression of VE-Cadherin, ZO-1, and Claudin-5
VE-cadherin, ZO-1, and claudin-5 expression decreased after LPS treatment compared to PBS treatment. DBIBB treatment led to a notable increase expression of VE-cadherin, ZO-1, and claudin-5, as evidenced by the results obtained from immunofluorescence (Figure 8A1–A4), Western blot (Figure 8B) and immunohistochemical (Figure 8C1 and C2) assays. Thus, our results suggested that drug activation of LPA2 can protect against endothelial barrier dysfunction and decrease vascular endothelial permeability, thereby playing an important protective role in septic ALI.
 |
Figure 8 DBIBB treatment leads to an increase in AJs and TJs in septic mice. (A1–A3) Representative immunofluorescent staining images in lung sections. White arrows indicate expression of VE-cadherin, ZO-1, and claudin-5 in vascular endothelium. Scale bar, 50 μm. (A4) Relative fluorescence intensity of the images in (A1–A3). (B) Representative Western blot and corresponding densitometric quantification (n = 6). (C-1) Representative immunohistochemical images in paraffin-embedded lung sections. Scale bar, 100 μm. (C-2) The percentage of positively stained areas, *p <0.05 and **p <0.01 vs LPS group. |
Discussion
Herein, we provided evidence of a protective role of LPA2 in mitigating LPS-induced septic ALI by regulating the integrity of the endothelial barrier. We showed that Lpar2 deficiency increased vascular endothelial permeability of lungs in vivo, while activation of LPA2 with DBIBB in vitro promoted VE-cadherin expression through the PLC-PKC-FAK pathway. Together, our findings suggested that pharmacological activation of LPA2 with DBIBB could mitigate septic ALI.
The endothelial barrier function plays a critical role in modulating septic ALI, elevated endothelial permeability has been found in sepsis.27 Previous studies have reported a 6.2-fold increased of LPA2 in the carotid artery following ligation.28,29 Previously, we have shown that LPA2 is upregulated in myocardial infarction mice, and that the elevated LPA2 levels were mainly due to increased LPA2 expression on vascular endothelial cells.17 These findings are of particular interest because they highlight a potential underlying role of LPA2 in the pathogenesis of septic ALI. However, the precise mechanism of LPA2 in ALI etiopathogenesis during sepsis remains unclear.
Endothelial cells are critical in the maintenance of vascular homeostasis. An excessive increase in vascular permeability leads to vascular leakage, which causes an increase in blood cells and plasma proteins entering the interstitium, and ultimately organ failure and death. Here, we demonstrated that Lpar2 deficiency resulted in significantly higher mortality and impaired lung injury, as well as increased vascular endothelial permeability in the lungs of septic mice. Thus, our findings indicated that LPA2 had a significant effect on endothelial barrier function. Similarly, using primary MLMECs isolated from WT and Lpar2−/− mice, we demonstrated that loss of Lpar2 led to an increase in endothelial permeability in vitro measured by the fluorescence permeability assay and TEER. Together, our data suggested that Lpar2 deficiency may aggravate mortality and septic ALI through disruption of the endothelial barrier function, which leads to increased vascular endothelial permeability. Recent studies have revealed an important role of Lpar2 in allergic lung inflammation through negative regulation of dendritic cell activation, as well as evidence of a protective role for LPA2 in the lung.
LPA is involved in maintaining the function of the endothelial barrier through mediation of multiple pathological and physiological functions. Recently, elevated LPA level have been reported in patients with acute myocardial infarction, as well as acute coronary syndrome.30,31 Herein, we observed that septic ALI increased level of LPA, thus indicating that there is a conserved mechanism between the two species. Our studies have demonstrated that LPA exerts its protective effect on septic ALI by acting through LPA2 to regulate endothelial permeability. Sumitomo et al demonstrated that LPA protected the integrity of the endothelial cell barrier,32,33 consistent with our findings. Similarly, LPA has been shown to promote lung and corneal epithelial barrier integrity.20 In contrast, high concentrations of LPA have been found to increase vascular endothelial permeability after LPA exposure.34,35 These discrepancies may be due to differences between the studies in terms of diseases, organs and LPA concentration.
It remains unclear which LPA receptors are involved in mediating key events associated with vascular endothelial permeability in the lung. Although knockdown of LPA1 was found to mitigate pulmonary damage and fibrosis, treatment with an LPA1 antagonist unexpectedly exhibited an antifibrotic effect, resulting in increased lung endothelial barrier permeability.36 LPA has been shown to act through LPA6 to stimulate the generation of actin stress fibers, as well as enhance the capillary endothelial permeability of brain cells.37 Previously, we described a role for LPA2 in maintaining vascular endothelial homeostasis following cardiac ischemia.17 Thus, these studies indicate that different LPA receptors mediate various function of endothelial cells. Herein, we employed Lpar2−/− mice to determine the effects of LPA2 on endothelial permeability. Our results indicate that LPA stimulation facilitated the expression of VE-cadherin, ZO-1, and claudin-5 through LPA2.
Studies have indicated that LPA2 acts through Gα12/13-Rho, Gαq /11-PLC and Gαi/o-PI3K or Ras-MAPK pathways. We found that LPA2 acts mainly through the PLC-PKC pathway in septic ALI. On the other hand, LPA promotes E-cadherin expression in HBEPCs through the PKC-FAK pathway.20 In our study LPA-LPA2 promotes expression of VE-cadherin in MLMECs. Therefore, we hypothesized that LPA-LPA2 promoted the expression of VE-cadherin through PLC-PKC-FAK pathway, and our conjecture was confirmed by employing the PLC, PKC and FAK inhibitors separately. In summary, the protective effect of LPA2 occurs during PLC-PKC-FAK pathway mediated endothelial barrier dysfunction in septic ALI.
DBIBB, an agonist of LPA2, has been shown to mitigate gastrointestinal radiation syndrome by promoting survival of the intestinal crypt, enterocyte multiplication and decreasing apoptosis.19 In addition, DBIBB has been observed to participate in maintaining vascular endothelium homeostasis and heart regeneration following myocardial infarction.17 In the current study, DBIBB was found to reduce endothelial permeability in LPS-induced septic ALI mice. Thus, our findings suggest that LPA2 is a key mediator during septic ALI, consistent with the gastrointestinal radiation syndrome study.19 Interestingly, S1P1, which is highly homologous to the LPA receptor family, has been shown to maintain endothelial function. Furthermore, FTY720, agonist of S1P1, is considered to be a potential drug target for 4 FDA-approved medicines,38 Since no obvious adverse accidents have been found in small-scale human trials as a target.39,40 It is likely that targeting LPA2 may also be a potential clinical therapeutic strategy for the treatment of septic ALI.
The innovation of this study is the first time elucidate LPA2 regulates the expression of VE-cadherin, ZO-1 and claudin-5 to alleviate ALI and thus reduce the mortality of mice, which may find a new pathway for the regulation of septic ALI. This study has several limitations. Administration of LPS and cecum ligation and puncture (CLP) are the most commonly used in sepsis research. In our study, we identified the role for LPA2 in a LPS model of sepsis-induced ALI to assure a better consistency, which is considered a more highly controlled and standardized model. We only whereas its role in other sepsis models (such as CLP) requires further study. In addition, our study showed that LPA2 mediates lung injury through PLC-PKC-FAK dependent endothelial barrier pathway. However, multiple associated proteins such as ZO-1 and claudin-5, could lead to protect endothelial barrier.41 It is unclear LPA2 specifically through which pathway regulates ZO-1 and claudin-5. Further investigations are required to have a better understanding of the precise mechanisms in the future.
In summary, our data reveal for the first time a novel, selective and causal role of LPA2 in protecting against endothelial barrier disruption, lung impairment, and mortality in LPS-mediated sepsis model in vivo, and in mediating the endothelial barrier in LPS-induced MLMECs in vitro. The potential protective role of LPA2 results from activation of the PLC-PKC-FAK pathway, as well as restoration of the endothelial AJs and TJs proteins VE-cadherin, ZO-1 and claudin-5. In consistent, activation of LPA2 with DBIBB protects against endothelial barrier function and improve ALI in sepsis. These findings suggest that targeting LPA2 and its downstream signaling pathways may prove to be an innovative therapeutic strategy for the treatment of septic ALI.
Conclusion
In conclusion, the present study provides a novel mechanism of endothelial barrier protection and suggest that LPA2 may contribute to the clinical therapeutic effects of septic ALI. Mechanisms may be involved that LPA- LPA2 signaling alleviate vascular endothelial permeability through the PLC-PKC-FAK pathway (Figure 9).
 |
Figure 9 The protective role of LPA2 against septic ALI is mediated through the PLC-PKC-FAK signaling pathway. LPA acts through LPA2 to promote VE-cadherin expression through the PLC-PKC-FAK signaling pathway, thereby attenuating endothelial permeability, and alleviating sepsis-induced lung impairment. |
Ethical Statement
The Animal Care & Use Committee of Fuwai Hospital approved this study, which was conducted in accordance with the “Regulation to the Care and Use of Experimental Animals (1996)” guidelines of the Beijing Animal Care Council.
Acknowledgments
The current work received funding through grants obtained from the Natural Science Foundation of Beijing (7232077), the Natural Science Foundation of China (81770304, 82172334) and the CAMS Innovation Fund for Medical Sciences (CIFMS) (2017- I2M-3-003).
Disclosure
The authors declare no conflicts of interest associated with this work.
References
1. Cheng Z, Abrams ST, Toh J, et al. The critical roles and mechanisms of immune cell death in sepsis. Front Immunol. 2020;11:1918. doi:10.3389/fimmu.2020.01918
2. Fukatsu M, Ohkawara H, Wang X, et al. The suppressive effects of Mer inhibition on inflammatory responses in the pathogenesis of LPS-induced ALI/ARDS. Sci Signal. 2022;15(724):eabd2533. doi:10.1126/scisignal.abd2533
3. Dolmatova EV, Wang K, Mandavilli R, Griendling KK. The effects of sepsis on endothelium and clinical implications. Cardiovasc Res. 2021;117(1):60–73. doi:10.1093/cvr/cvaa070
4. Li Z, Wang Y, Feng Q, et al. Inhibition of the C3a receptor attenuates sepsis-induced acute lung injury by suppressing pyroptosis of the pulmonary vascular endothelial cells. Free Radic Biol Med. 2022;184:208–217. doi:10.1016/j.freeradbiomed.2022.02.032
5. Yang L, Liu S, Han S, et al. The HDL from septic-ARDS patients with composition changes exacerbates pulmonary endothelial dysfunction and acute lung injury induced by cecal ligation and puncture (CLP) in mice. Respir Res. 2020;21(1):293. doi:10.1186/s12931-020-01553-3
6. Abraham S, Yeo M, Montero-Balaguer M, et al. VE-Cadherin-mediated cell-cell interaction suppresses sprouting via signaling to MLC2 phosphorylation. Curr Biol. 2009;19(8):668–674. doi:10.1016/j.cub.2009.02.057
7. Chen D, Shen M, Wang H, et al. Sirt3 maintains microvascular endothelial adherens junction integrity to alleviate sepsis-induced lung inflammation by modulating the interaction of VE-cadherin and β-catenin. Oxid Med Cell Longev. 2021;2021:8978795. doi:10.1155/2021/8978795
8. Li Y, Chen H, Shu R, et al. Hydrogen treatment prevents lipopolysaccharide-induced pulmonary endothelial cell dysfunction through RhoA inhibition. Biochem Biophys Res Commun. 2020;522(2):499–505. doi:10.1016/j.bbrc.2019.11.101
9. Liu Y, Mu S, Li X, Liang Y, Wang L, Ma X. Unfractionated heparin alleviates sepsis-induced acute lung injury by protecting tight junctions. J Surg Res. 2019;238:175–185. doi:10.1016/j.jss.2019.01.020
10. Koval M. Claudin heterogeneity and control of lung tight junctions. Annu Rev Physiol. 2013;75(1):551–567. doi:10.1146/annurev-physiol-030212-183809
11. Wang F, Liu S, Pei J, et al. LPA (3)-mediated lysophosphatidic acid signaling promotes postnatal heart regeneration in mice. Theranostics. 2020;10(24):10892–10907. eCollection 2020. doi:10.7150/thno.47913
12. Yin F, Watsky MA. LPA and S1P increase corneal epithelial and endothelial cell transcellular resistance. Invest Ophthalmol Vis Sci. 2005;46(6):1927–1933. doi:10.1167/iovs.04-1256
13. Minnear F, Patil S, Bell D, Gainor JP, Morton CA. Platelet lipid(s) bound to albumin increases endothelial electrical resistance: mimicked by LPA. Am J Physiol Lung Cell Mol Physiol. 2001;281(6):L1337–L1344. doi:10.1152/ajplung.2001.281.6.L1337
14. Meeteren LA, Ruurs P, Stortelers C, et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol Cell Biol. 2006;26(13):5015–5022. doi:10.1128/MCB.02419-05
15. Tanaka M, Okudaira S, Kishi S, et al. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J Biol Chem. 2006;281(35):25822–25830. doi:10.1074/jbc.M605142200
16. Teo ST, Teo S, Yung YC, Herr DR, Chun J. Lysophosphatidic acid in vascular development and disease. IUBMB Life. 2009;61(8):791–799. doi:10.1002/iub.220
17. Pei J, Cai L, Wang F, et al. LPA2 contributes to vascular endothelium homeostasis and cardiac remodeling after myocardial infarction. Circ Res. 2022;131(5):388–403. doi:10.1161/CIRCRESAHA.122.321036
18. Kihara Y, Maceyka M, Spiegel S, et al. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br J Pharmacol. 2014;171(15):3575–3594. doi:10.1111/bph.12678
19. Patil R, Szabó E, Fells JI, et al. Combined mitigation of the gastrointestinal and hematopoietic acute radiation syndromes by an LPA2 receptor-specific nonlipid agonist. Chem Biol. 2015;22(2):206–216. doi:10.1016/j.chembiol.2014.12.009
20. He D, Su Y, Usatyuk PV, et al. Lysophosphatidic acid enhances pulmonary epithelial barrier integrity and protects endotoxin-induced epithelial barrier disruption and lung injury. J Biol Chem. 2009;284(36):24123–24132. doi:10.1074/jbc.M109.007393
21. Jiang J, Huang K, Xu S, Garcia JG, Wang C, Cai H. Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox Biol. 2020;36:101638. doi:10.1016/j.redox.2020.101638
22. Yeomyeong K, Sungwoon L, Haiying Z, et al. CLEC14A deficiency exacerbates neuronal loss by increasing blood-brain barrier permeability and inflammation. J Neuroinflammation. 2020;17(1):48. doi:10.1186/s12974-020-1727-6
23. Im K, Mareninov S, Diaz MF, Yong WH. An introduction to performing immunofluorescence staining. Methods Mol Biol. 2019;1897:299–311. doi:10.1007/978-1-4939-8935-5_26
24. Yang R, Yang H, Wei J, et al. Mechanisms underlying the effects of lianhua qingwen on sepsis-induced acute lung injury: a network pharmacology approach. Front Pharmacol. 2021;12:717652. eCollection 2021. doi:10.3389/fphar.2021.717652
25. Chrzanowska-Wodnicka M, Kraus AE, Gale D, et al. Defective angiogenesis, endothelial migration, proliferation, and MAPK signaling in Rap1b-deficient mice. Blood. 2008;111(5):2647–2656. doi:10.1182/blood-2007-08-109710
26. Kuck JL, Bastarache JA, Shaver CM, Fessel JP, Dikalov SI, May JM. Ascorbic acid attenuates endothelial permeability triggered by cell-free hemoglobin. Biochem Biophys Res Commun. 2018;495(1):433–437. doi:10.1016/j.bbrc.2017.11.058
27. Li Z, Yin M, Zhang H, et al. BMX represses thrombin-PAR1-mediated endothelial permeability and vascular leakage during early sepsis. Circ Res. 2020;126(4):471–485. doi:10.1161/CIRCRESAHA.119.315769
28. Chen X, Yang X, Wang ND, et al. Serum lysophosphatidic acid concentrations measured by dot immunogold filtration assay in patients with acute myocardial infarction. Scand J Clin Lab Invest. 2003;63(7–8):497–503. doi:10.1080/00365510310003265
29. Panchatcharam M, Miriyala S, Yang F, et al. Lysophosphatidic acid receptors 1 and 2 play roles in regulation of vascular injury responses but not blood pressure. Circ Res. 2008;103(6):662–670. doi:10.1161/CIRCRESAHA.108.180778
30. Dohi T, Miyauchi K, Ohkawa R, et al. Increased lysophosphatidic acid levels in culprit coronary arteries of patients with acute coronary syndrome. Atherosclerosis. 2013;229(1):192–197. doi:10.1016/j.atherosclerosis.2013.03.038
31. Kurano M, Kano K, Dohi T, et al. Different origins of lysophospholipid mediators between coronary and peripheral arteries in acute coronary syndrome. J Lipid Res. 2017;58(2):433–442. doi:10.1194/jlr.P071803
32. Sumitomo A, Siriwach R, Thumkeo D, et al. LPA induces keratinocyte differentiation and promotes skin barrier function through the LPAR1/LPAR5-RHO-ROCK-SRF Axis. J Invest Dermatol. 2019;139(5):1010–1022. doi:10.1016/j.jid.2018.10.034
33. Alexander JS, Patton WF, Christman BW, Cuiper LL, Haselton FR. Platelet-derived lysophosphatidic acid decreases endothelial permeability in vitro. Am J Physiol. 1998;274(1):H115–H122. doi:10.1152/ajpheart.1998.274.1
34. Van Nieuw Amerongen GP, Vermeer MA, van Hinsbergh VW. Role of RhoA and Rho kinase in lysophosphatidic acid-induced endothelial barrier dysfunction. Arterioscler Thromb Vasc Biol. 2000;20(12):E127–E133. doi:10.1161/01.atv.20.12.e127
35. Schulze C, Smales C, Rubin LL, Staddon JM. Lysophosphatidic acid increases tight junction permeability in cultured brain endothelial cells. J Neurochem. 1997;68(3):991–1000. doi:10.1046/j.1471-4159.1997.68030991.x
36. Cai J, Wei J, Li S, Suber T, Zhao J. AM966, an antagonist of lysophosphatidic acid receptor 1, increases lung microvascular endothelial permeability through activation of rho signaling pathway and phosphorylation of VE-Cadherin. Mediators Inflamm. 2017;2017:6893560. doi:10.1155/2017/6893560
37. Inoue A, Arima N, Ishiguro J, et al. LPA-producing enzyme PA-PLA 1 α regulates hair follicle development by modulating EGFR signalling. EMBO J. 2011;30(20):4248–4260. doi:10.1038/emboj.2011.296
38. Nitzsche A, Poittevin M, Benarab A, et al. Endothelial S1P (1) signaling counteracts infarct expansion in ischemic stroke. Circ Res. 2021;128(3):363–382. doi:10.1161/CIRCRESAHA.120.316711
39. Chun J, Kihara Y, Jonnalagadda D, Blaho VA. Fingolimod: lessons learned and new opportunities for treating multiple sclerosis and other disorders. Annu Rev Pharmacol Toxicol. 2019;59(1):149–170. doi:10.1146/annurev-pharmtox-010818-021358
40. Cohan SL, Benedict RH, Cree BA, Deluca J, Hua LH, Chun J. The two sides of siponimod: evidence for brain and immune mechanisms in multiple sclerosis. CNS Drugs. 2022;36(7):703–719. doi:10.1007/s40263-022-00927-z
41. Zhang N, Tang S, Zhang J, Pei B, Pang T, Sun G. The dipeptidyl peptidase-4 inhibitor linagliptin ameliorates LPS-induced acute lung injury by maintenance of pulmonary microvascular barrier via activating the Epac1/AKT pathway. Biomed Pharmacother. 2022;155:113704. doi:10.1016/j.biopha.2022.113704
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.