Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 9 » Issue 1
Lower airway colonization and inflammatory response in COPD: a focus on Haemophilus influenzae
Authors Finney L ![]() , Ritchie A, Pollard E, Johnston S
, Ritchie A, Pollard E, Johnston S ![]() , Mallia P
, Mallia P ![]()
Received 28 June 2014
Accepted for publication 18 August 2014
Published 13 October 2014 Volume 2014:9(1) Pages 1119—1132
DOI https://doi.org/10.2147/COPD.S54477
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Richard Russell
Lydia J Finney,1 Andrew Ritchie,1 Elizabeth Pollard,2 Sebastian L Johnston,1 Patrick Mallia1
1Airway Disease Infection Section, National Heart and Lung Institute, Imperial College, London, United Kingdom; 2King's College London, London, United Kingdom
Abstract: Bacterial infection of the lower respiratory tract in chronic obstructive pulmonary disease (COPD) patients is common both in stable patients and during acute exacerbations. The most frequent bacteria detected in COPD patients is Haemophilus influenzae, and it appears this organism is uniquely adapted to exploit immune deficiencies associated with COPD and to establish persistent infection in the lower respiratory tract. The presence of bacteria in the lower respiratory tract in stable COPD is termed colonization; however, there is increasing evidence that this is not an innocuous phenomenon but is associated with airway inflammation, increased symptoms, and increased risk for exacerbations. In this review, we discuss host immunity that offers protection against H. influenzae and how disturbance of these mechanisms, combined with pathogen mechanisms of immune evasion, promote persistence of H. influenzae in the lower airways in COPD. In addition, we examine the role of H. influenzae in COPD exacerbations, as well as interactions between H. influenzae and respiratory virus infections, and review the role of treatments and their effect on COPD outcomes. This review focuses predominantly on data derived from human studies but will refer to animal studies where they contribute to understanding the disease in humans.
Keywords: chronic obstructive pulmonary disease, Haemophilus influenzae, nontypeable Haemophilus influenzae, respiratory viruses, vaccination
COPD overview
Chronic obstructive pulmonary disease (COPD) is a major cause of morbidity and is the fourth leading cause of mortality worldwide.1 Conservative estimates suggest a 10% prevalence in the general population, but among heavy smokers, this figure increases to almost 50%.2 The clinical course of COPD is characterized by a progressive decline in lung function, accompanied by worsening exercise tolerance and health status. The pathological hallmark of COPD is an abnormal inflammatory response in the airways and alveoli. The principal abnormalities in airways are the presence of an inflammatory cellular infiltrate and remodeling that thickens the airway wall, thereby reducing the airway diameter and increasing resistance to flow.2 In the lung parenchyma, COPD is characterized by prominent inflammatory infiltrates in the alveolar walls, destruction of alveoli, and enlargement of air spaces. Cigarette smoking is a predominant etiological factor in the development of COPD, but other factors such as burning biomass fuels for cooking and heating are important causes, particularly in developing countries. Although exposure to cigarette smoke/biomass smoke is required for the development of COPD, disease progression and airway inflammation persist even after smoking cessation.3 Therefore, other factors contribute to persistence of inflammation and progression of the disease. One such factor is respiratory infection, with both bacterial and viral infections implicated in contributing to the pathogenesis of COPD.4,5 Bacteria are commonly detected in COPD, with the most common organism isolated being Haemophilus influenzae in most studies. The chronic clinical course of COPD is punctuated by periods of increased symptoms, termed acute exacerbations,6 which have both short- and long-term adverse effects in COPD patients. They have immediate effects resulting in considerable morbidity and mortality in COPD and are a major cause of excess health care costs, as they often result in unscheduled health care visits, treatment costs, and hospitalizations. Exacerbations also have long-term effects, as frequent exacerbations are associated with more rapid decline in lung function, airway and systemic inflammation, and impaired quality of life.7–9 Approximately half of all COPD exacerbations are associated with bacterial infections, and as is the case in stable COPD, the most common bacteria detected is H. influenzae.10 Therefore, H. influenzae may have a major pathogenic role both in stable COPD and in COPD exacerbations and has been the focus of much research interest.
H. influenzae: bacteriology
H. influenzae is a pleomorphic Gram-negative coccobacillus that is isolated exclusively from humans, predominantly from the respiratory tract. It is a member of the Pasteurellaceae family and is capable of growing either aerobically or anaerobically,11 and strains are divided into two groups on the basis of the presence of a polysaccharide capsule. Encapsulated strains are reactive with typing antisera (typeable), whereas unencapsulated strains are nonreactive (nontypeable H. influenzae [NTHi]). Six encapsulated serotypes (a–f) have been identified and account for the majority of invasive Haemophilus infections such as septicemia, pneumonia, and meningitis. NTHi, in contrast, rarely causes invasive disease but commonly colonizes the upper respiratory tract and can cause mucosal infections in both children and adults. The vast majority of Haemophilus strains isolated from the respiratory tract in COPD patients are NTHi. H. influenzae is a common commensal of the upper respiratory tract, with 20% of children colonized in the first year of life and up to 50% colonized by age 5 years.12 Disease caused by NTHi is predominantly by contiguous spread from the nasopharynx to adjacent structures such as sinuses, the middle ear, and trachea. In contrast to the frequent detection of H. influenzae in the upper respiratory tract, lower respiratory tract colonization appears rare in healthy individuals. In 70 healthy subjects from six different studies undergoing bronchoscopy, H. influenzae was detected in only 4%.13 Two subsequent studies that were not included in this analysis have been published more recently. In the first, H. influenzae was not detected in any of 26 healthy individuals undergoing bronchoscopy during anesthesia for elective surgery.14 In the second, H. influenzae was isolated in two (13.3%) of 15 healthy subjects who had never smoked, but in zero of 20 exsmokers.15 Therefore, from these results, the true prevalence of lower respiratory tract colonization with H. influenzae in healthy individuals is unclear, but it is undoubtedly lower than that in the upper respiratory tract. These studies were small, and therefore it is difficult to draw conclusions from them regarding the prevalence of H. influenzae colonization in the general population. Discrepancies in detection rates between studies are likely to be related to differences in characteristics of the populations studied, such as age, sex, smoking history, and so on, and further studies with greater numbers of participants are required.
Methods to detect H. influenzae
For many years, the standard method for detection of H. influenzae in respiratory samples was growth on culture plates and identification using morphological characteristics and growth requirements. However, culture has a number of drawbacks, including difficulty in distinguishing H. influenzae from other bacterial species, such as Haemophilus haemolyticus and Haemophilus parainfluenzae, as well as low sensitivity. H. influenzae possesses the ability to persist in biofilms and within host cells, and organisms in these niches may not be detected using culture of airway samples such as sputum, bronchial wash, and bronchoalveolar lavage.16 Culture-independent techniques based on detection and amplification of nucleic acid sequences using polymerase chain reaction (PCR) have been developed during the past 2 decades to detect pathogens such as H. influenzae. Protein D is a highly conserved surface lipoprotein present in all encapsulated and nonencapsulated H. influenzae strains,17 making its gene (hpd) an attractive target for the development of a PCR assay. An hpd real-time PCR assay can detect both encapsulated H. influenzae and NTHi strains with high sensitivity and specificity.18 Studies comparing bacterial detection rates using culture and PCR have consistently demonstrated greater sensitivity with PCR. Detection rates of H. influenzae in nasopharyngeal swabs collected from healthy individuals are 2.5–3 times greater with PCR compared with culture.19,20 As will be described in the following section, this has also been reported in COPD patients. However even with PCR, distinguishing H. influenzae from other species such as H. haemolyticus can be difficult,21 and more sophisticated techniques such as proteomic profiling may be required.22 More recently, newer techniques have been developed based on the sequencing of the 16S gene encoding bacterial ribosomal RNA. The 16S gene contains invariant regions used for priming the sequencing reaction and variable regions that permit genus- and species-level identification. High-throughput sequencing and sorting of the amplicons generated enables the identification of entire microbial communities. These techniques have revealed the presence of a respiratory “microbiome,” even in the respiratory tracts of healthy individuals, and overturned the previous dogma that the lower airways are sterile in health.23 Initial studies have revealed that the genus Proteobacteria that includes Haemophilus is overrepresented in the airways of subjects with asthma and COPD compared with in healthy individuals.24 Further studies of the respiratory microbiome are likely to radically change our understanding of the role of bacteria such as H. influenzae in both health and disease.
The epidemiology of H. influenzae in COPD
It is well established that bacteria are often present in the lower airways in patients with stable COPD, although colonization rates vary between different studies and are affected by factors such as disease severity, treatments used, and sampling methods used. A pooled analysis of studies using bronchoscopic sampling reported that potentially pathogenic organisms were present in 29% of stable COPD patients,13 and studies using sputum have reported that potentially pathogenic organisms are present in up to half of COPD patients.25–27 The most common organisms detected in COPD patients are consistently H. influenzae, Streptococcus pneumoniae, and Moraxella catarrhalis, with H. influenzae accounting for around half of all isolates in the airways of COPD patients.13,25–27 However, the true prevalence of infection with H. influenzae in COPD may be underestimated by studies using culture for bacterial detection. In a longitudinal study over the course of 7 years, Murphy et al reported frequent episodes when sputum cultures were negative that were preceded and followed by positive cultures with the same strains of H. influenzae, and strain-specific H. influenzae DNA was detected in some of the sputum samples that had negative cultures.28 Other studies using PCR have confirmed higher detection rates compared with culture alone in COPD,29–31 with H. influenzae detected in 60% of sputum samples using PCR compared with only 30% using culture.31
Bacteria can be detected in sputum in 50%–60% of COPD patients during acute exacerbations, and H. influenzae is consistently among the most common bacteria reported.10,13,32 As discussed previously, even this may be an underestimate because of the limitations of culture. In a study of 15 chronic bronchitis patients with acute respiratory failure, NTHi was detected in only one bronchial wash specimen, whereas using in situ hybridization, intracellular NTHi was present in 13 (86.7%) of 15 bronchial biopsies.14 Sequencing studies have revealed that Proteobacteria are a key component of the respiratory microbiome in COPD exacerbations.33,34
Therefore, although it is clear that lower respiratory tract infection with H. influenzae is much more prevalent in COPD patients compared with healthy individuals, the exact prevalence remains to be determined and is highly dependent on the diagnostic method used.
Host immunity and immune evasion in H. influenzae infection
H. influenzae commonly colonizes the nasopharynx and causes upper respiratory tract disease in healthy individuals, but infection of the lower airways is rare because of host immune responses that prevent spread to the lower respiratory tract. In contrast, H. influenzae is commonly found in the lower airways of patients with COPD, implying that mucosal host immune mechanisms are impaired. H. influenzae possesses a number of mechanisms to avoid or neutralize host immune responses, and therefore it is likely that the combination of pathogen immune evasion and impaired host immunity combine to promote lower airway infection in COPD.
Host immunity to H. influenzae in healthy individuals
Structural components
The structural integrity of the respiratory tract is vital to preventing infection with inhaled microorganisms. Important mechanisms including an intact respiratory epithelium, mucus production, and mucociliary clearance via the action of cilia and cough act to prevent infection with respiratory pathogens such as H. influenzae.4,11 The importance of these mechanisms in host defense is highlighted by high rates of H. influenzae infection in conditions such as cystic fibrosis35 and immotile cilia syndromes36 that are characterized by abnormal mucociliary function.
Cellular immunity
Immune cells such as neutrophils and alveolar macrophages are present in large numbers in the airways and constitute a first line of defense against respiratory pathogens. Macrophages recognize microorganisms via the presence of surface receptors such as toll-like receptors leading to phagocytosis, microbicidal killing, and initiation of immune responses.11 A key component of the NTHi cell wall is lipooligosaccharide, which is recognized by toll-like receptor 4, leading to macrophage activation and phagocytosis,37 and the membrane protein P2, recognized by toll-like receptor 2.38,39 P6 is a specific trigger of macrophage activation, with secretion of interleukin 8 (IL-8) and tumor necrosis factor α being key effectors of P6-induced macrophage responses.40
Complement and immunoglobulin A
The complement system is an essential part of the innate immune system, and activation of complement results in protein activation and deposition on the surface of pathogens, resulting in opsonization and phagocytosis. Invading pathogens activate complement either through antibody binding (classical pathway) or spontaneously via the alternative pathway.41 Although complement is not present in the healthy human respiratory tract, after infection and inflammation, the permeability of the mucosa increases and plasma, including complement proteins, enters the airway lumen.42,43 Immunoglobulin A (IgA) is the main element of the humoral immune response that provides protection against microbial antigens at mucosal surfaces, including the lung. Mucosal IgA can bind to bacteria and prevent mucosal attachment and facilitate cytotoxicity.44 The importance of complement and IgA in host defense against pathogens is illustrated by the increased susceptibility to bacterial infections, including H. influenzae, seen in patients with complement deficiencies and hypogammaglobulinemia.45,46
Adaptive immunity
The adaptive immune response consists of humoral (B-cell-mediated) and cellular (T-cell-mediated) immunity.4,47 Hypogammaglobulinemia is a risk factor for systemic infection by NTHi, illustrating the importance of antibodies.48 T-helper cells and cytotoxic T lymphocytes contribute by producing interferon γ,4 which enhances the macrophage-induced killing of NTHi.49 Cytotoxic T cells and natural killer cells may be particularly important in the control of intracellular pathogens,50 and there is evidence that impaired T-helper responses to H. influenzae contribute to the failure to eradicate intracellular infection,49 resulting in chronic airway infection.47
Mechanisms of immune evasion by H. influenzae
Phase variation and antigenic change
H. influenzae has the ability to lose or gain cell structures, thereby allowing it to evade host immune responses, particularly antibody-mediated immunity. This property is called phase variation and has been reported for a number of different components of H. influenzae, including adhesins, pili, and lipooligosaccharide.51,52 Furthermore, some strains of H. influenzae undergo permanent changes in the amino acid sequences of important immunogenic surface proteins such as P2 and P5 in a manner reminiscent of antigenic drift in influenza viruses.53 There is evidence that phase variation may contribute to persistent infection in COPD, as it has been reported that serial isolates of H. influenzae from patients with COPD demonstrate decreased levels of HMW1 and HMW2 in association with persistence.54
Intracellular growth/biofilms
H. influenzae exhibits a number of different growth strategies that promote survival, including intracellular growth and the formation of biofilms.52 Biofilms are microbial communities that are encased within a matrix and can be found in a number of environments, including the human respiratory tract. Biofilms are believed to confer protection from a number of immune mechanisms such as antimicrobial peptides and antibodies, as well as from antibiotics, thereby promoting survival and persistence. There is evidence from both human studies and animal models that H. influenzae forms biofilms, which is likely to be an important mechanism in promoting persistent infection in the human respiratory tract.55–58 Although H. influenzae is predominantly an extracellular pathogen, there is evidence it is able to enter cells of the respiratory tract. H. influenzae has been detected in epithelial cells,59,60 adenoidal tissue,61,62 and monocytes/macrophages.63 As is the case for biofilm formation, intracellular growth may offer H. influenzae protection from antibody-mediated immune responses and antibiotics, and therefore may be another important mechanism of persistent infection.64 As discussed previously, both biofilms and intracellular growth may also contribute to underdetection of H. influenzae using sputum culture.
Complement/IgA
Complement and IgA are important mechanisms of host protection against H. influenzae. Some strains of H. influenzae demonstrate reduced susceptibility to complement, and although the exact mechanisms of this are unknown, a number of factors including lipooligosaccharide and surface proteins P2 and P5 are believed to contribute.41 In addition, it has been demonstrated that NTHi can acquire host complement inhibitors such as vitronectin,65,66 and factor H.67 NTHi can also secrete proteases that cleave the main IgA subclass IgA1,68 thus offering protection against IgA-mediated cytotoxicity.
Metabolic adaptations
In addition to changes to structural components, organisms in the human respiratory tract may undergo adaptations in metabolic pathways to survive in a hostile environment with limited nutrients, low pH, and high levels of reactive oxygen species. Such adaptations adopted by H. influenzae include production of urease,69 antioxidants,70 and proteins involved in iron and heme metabolism.71 There is evidence that upregulation of these metabolic pathways is related to the ability of NTHi strains to survive in the respiratory tract.72–74
Mechanisms of H. influenzae infection in COPD
There are a number of structural and functional changes in the airways of COPD patients that compromise host immunity to respiratory pathogens. H. influenzae has developed a number of mechanisms to evade host immune responses, many of which are already impaired in COPD. Therefore, among respiratory pathogens, H. influenzae appears to be uniquely adapted to exploit the already-impaired host immunity in COPD and establish persistent infection.
Bacterial adhesion
The first step in establishing infection in the respiratory tract by a pathogen is adhesion to the respiratory epithelium. H. influenzae possesses a number of cell surface proteins and structures that facilitate adhesion to respiratory mucosal surfaces. These include the presence of pili (rodlike projections present on a small subset of strains75) and a number of surface proteins that facilitate adhesion. The membrane proteins P2 and P5 facilitate binding of the bacteria to mucus,38 and the adhesins HMW1, HMW2, protein E, and protein F bind to epithelial cells and extracellular matrix proteins.76–78 Protein D is a highly conserved 42 kDa surface lipoprotein identified in all strains of H. influenzae. The activity of protein D as a virulence factor is as a glycerophosphodiesterase that promotes adherence of NTHi to airway epithelial cells and internalization by macrophages63 and impairs ciliary function.79–81 The airways in COPD patients are characterized by damaged epithelium, mucous hypersecretion, and reductions in cilia numbers and function. Therefore, this combination of exposed extracellular matrix, excess mucus, and impaired mucociliary clearance provides an ideal environment for the adhesion and subsequent growth of H. influenzae. Strains of H. influenzae isolated from COPD patients are more likely to express adhesion molecules such as HMW.82
Cellular and humoral immunity
Inflammatory cells in the airways such as neutrophils and macrophages are increased in COPD, which would be expected to result in more effective clearance of inhaled pathogens. However, the opposite is the case, as the function of these immune cells is impaired. Several studies have demonstrated that phagocytosis of H. influenzae and production of cytokines by macrophages is impaired in COPD.40,83–86 Impaired production of cytokines by macrophages in response to H. influenzae has been related to exacerbation frequency in COPD patients.83 As discussed previously, survival of H. influenzae has been documented in macrophages87 and epithelial cells,88 and intracellular H. influenzae is more common in patients with chronic bronchitis compared with in healthy subjects.14 Therefore, impaired macrophage responses to NTHi may promote intracellular survival, but further studies are required to prove this conclusively.
In addition to compromised macrophage function, T lymphocyte cell responses to H. influenzae in COPD are also impaired.89 Again, this may have clinical implications, as robust T-cell responses to the surface P6 lipoprotein are associated with a reduced risk for exacerbations.90 A recent paper reported that numbers of T regulatory cells are increased in COPD patients.91 Moreover T regulatory cells from COPD patients suppressed P6-specific T-cell proliferation to a greater extent than T regulatory cells from healthy controls; therefore, this may be an important mechanism of reduced effector T-cell responses to H. influenzae. Variations in antigenic components of the organism, including the membrane outer proteins P2 and P592,93 and the immunoglobulin A1 protease,94 may contribute to immune evasion and reduced host immunity.
There is evidence that IgA secretion is impaired in COPD95 and that reduced levels of Haemophilus-specific IgA may be related to chronic infection with H. influenzae.96,97
Inflammatory responses and H. influenzae in stable COPD
The presence of bacteria in the lower respiratory tract in stable COPD patients is termed colonization, but there is increasing evidence that this is not a benign phenomenon but is associated with adverse outcomes. Bacterial infection in stable COPD is associated with increased airways and systemic inflammation,98 more frequent exacerbations,27 and poorer health status.15,99 Therefore, chronic infection or persistent infection may be more accurate descriptions than colonization.100 Whether all bacterial species have adverse effects is unknown, as few studies have examined the role of specific bacteria such as H. influenzae. Data from animal models have suggested that NTHi can induce both inflammatory and structural changes in the lungs. Chronic instillation of NTHi in mice induced an inflammatory infiltrate similar to that seen in COPD, with increases in inflammatory cytokines and cells (neutrophils, macrophages, and CD8+ T cells) and airway collagen deposition.101 When exposed to both cigarette smoke and NTHi, mice developed structural changes typical of COPD, including emphysema, lung inflammation, and goblet cell metaplasia in both large and small airways.102
Human studies have reported that H. influenzae is associated with higher sputum levels of inflammatory mediators such as neutrophils, IL-1β, IL-12, IL-8, tumor necrosis factor α, and matrix metalloprotease 9.97,98,103,104 Moreover, COPD patients colonized with NTHi reported worse health status compared with noncolonized patients, and these effects were not seen in patients colonized with other bacteria.98 Therefore, the presence of H. influenzae in the lower airways of COPD patients is associated with increased inflammation and poorer health status. Whether the increases in inflammatory mediators contribute to long-term structural changes, as has been reported in animal studies, is unknown, but it is an important area for further research. Further studies also are needed to determine the effects of persistent infection with H. influenzae on other outcomes such as exacerbations, disease progression, and even mortality.
H. influenzae and COPD exacerbations
H. influenzae is frequently detected in airway samples collected from COPD patients during exacerbations,10 but this does not prove causation, as the organism may have been present when the patient was clinically stable. Studies comparing bacterial detection in stable and exacerbated patients have generally reported higher rates of bacterial infection in COPD exacerbations.10,29,105,106 Therefore, H. influenzae infection can result in either chronic infection or acute exacerbation. The mechanisms that determine the outcome of infection are poorly understood but are likely to involve both pathogen and host factors.
Exposure to new strains of NTHi to which the host has no preexisting immunity is one mechanism that may underlie the occurrence of acute exacerbations. Sethi et al carried out molecular typing on bacteria isolated from sputum samples from COPD patients collected when stable and during exacerbations over the course of 56 months. Isolation of H. influenzae using culture alone was not associated with exacerbations, but detection of a new strain was associated with increased risk for exacerbation.107 An exacerbation was diagnosed in 33% of visits at which a new bacterial strain was isolated compared with 15.4% of visits at which no new strain was present. Therefore, the majority of new strain acquisitions did not result in exacerbation, and other mechanisms must contribute. It was subsequently recognized that some strains of H. influenzae were actually H. haemolyticus species.108 When the results of the study were reanalyzed with the exclusion of H. haemolyticus, the association between acquisition of a new strain of H. influenzae and acute exacerbations was stronger.108 In another study from this group, an immune response to homologous H. influenzae occurred after 61% of exacerbations with newly acquired strains compared with 21.2% of exacerbations with preexisting strains.109
Factors relating to pathogen-related characteristics have been less well defined, but one study has reported a different genetic profile in strains associated with exacerbations compared with strains associated with asymptomatic chronic infection.110 Moreover, strains isolated from COPD patients during an exacerbation elicited greater inflammatory responses in mice and in vitro models compared with colonizing strains,111 although another study failed to replicate these findings.112 Therefore, it is likely there are complex interactions between host immunity and the pathogenicity of the infecting strain of H. influenzae that determine the outcomes of infection.
Respiratory viruses and Haemophilus influenzae
Other external factors may influence the interactions between host and H. influenzae; one such factor is infection with a respiratory virus. A relationship between influenza infection and H. influenzae pneumonia has long been recognized,113 and in fact, H. influenzae derives its name from the original mistaken belief that it was the causative organism of influenza. Synergism between the two organisms has been demonstrated in animal models.114 Respiratory virus infection can be detected in up to 40% of COPD exacerbations,115 and therefore virus infections in patients with chronic bacterial infection and concurrent virus/bacterial coinfections may be a relatively common occurrence. Data from in vitro studies and animal studies have suggested a number of potential mechanisms whereby virus and bacterial infections may interact. Mice infected with rhinovirus show impaired clearance of NTHi, which is related to impaired production of chemokines by both epithelial cells and macrophages in response to bacterial infection.116 Impaired chemokine responses to bacterial products in macrophages infected with rhinovirus have also been reported.117 Virus infection may also have effects on the airway epithelium that promote bacterial infection. Rhinovirus infection of epithelial cells in vitro disrupts the airway epithelial barrier, resulting in increased transmigration of NTHi across the epithelial barrier.118 In addition, rhinovirus infection increases expression of adhesion molecules on the surface of epithelial cells and enhances bacterial adhesion,119,120 and this effect has also been reported for other respiratory viruses.121 Whether these mechanisms are relevant in vivo is not known. Rhinovirus infection did not increase expression of the receptor platelet activating receptor in vivo in bronchial biopsies in COPD patients.122
Few studies have examined the role of concurrent virus-bacteria coinfection in COPD exacerbations. Those that are available have reported detection rates of between 12% and 25%.10,30 A study examining infection with respiratory viruses and H. influenzae found evidence of coinfection in only 9% of exacerbations.123 Our group has examined the relationship between viral and bacterial infections in COPD patients, using experimental rhinovirus infection as a model of COPD exacerbation.124 In COPD subjects infected with rhinovirus, secondary bacterial infections occurred in 60%, with H. influenzae the predominant organism (46% of bacterial isolates). The peak of virus infection occurred on day 5 postinoculation, whereas the peak of bacterial infection occurred later, on day 15. Therefore, studies in naturally occurring exacerbations in which a sample is collected at a single point are likely to underestimate the frequency of coinfections, as our data demonstrate that bacterial infection occurs up to 10 days after the initial viral infection. This was confirmed by two studies that carried out sequential sampling during COPD exacerbations. In one study, samples were collected at the onset of exacerbation and again 5–7 days later.125 In 19% of COPD exacerbations, a virus and bacteria were detected at exacerbation onset, but 36% of exacerbations in which a virus was detected on day 1 developed bacterial infection in the next 7 days. The commonest virus/bacteria combination was rhinovirus/H. influenzae; 78% of exacerbations in which H. influenzae was detected were preceded by symptoms of an upper respiratory tract infection. In the second study, COPD exacerbations associated with rhinovirus infection had bacteria detected in 73% of second samples collected 14 days later.126 Again, H. influenzae was the most frequent bacterial isolate. In both experimental124 and natural infections,106 virus infection is associated with higher bacterial loads.
Participants who developed secondary bacterial infection after experimental rhinovirus infection had lower airway levels of the antimicrobial peptides elafin and secretory leukoprotease inhibitor,124 suggesting another mechanism whereby virus infections may impair antibacterial immune responses.
In the experimental rhinovirus model, no subjects had bacteria detected by sputum culture at baseline before inoculation with rhinovirus; however, using sequencing techniques to analyze the bacterial microbiome, bacterial DNA could be detected. We further examined whether virus infection had an effect on the respiratory microbiome. After virus infection, there was a 16% increase in the number of proteobacterial sequences on day 15 postinoculation.33 Within the Proteobacteria phylum, there was a 21% increase in the average relative abundance of H. influenzae. At day 42 postinoculation, H. influenzae remained elevated compared with baseline, despite no organisms being detected by culture at this point. Therefore, this is the first in vivo evidence that a virus infection can induce a profound and prolonged change in the bacterial microbiome in the lungs and that there appears to be a specific interaction between rhinovirus and H. influenzae. The subjects included in the experimental infection studies had GOLD stage II COPD, had negative baseline sputum cultures, were not using inhaled corticosteroids, and had few previous exacerbations. In patients with more severe COPD with positive sputum cultures, the effect of virus infection on the bacterial microbiome may be even more pronounced. This holds out the prospect that treating or preventing viral infections may also reduce secondary bacterial infections and excessive antibiotic use.
Most research has focused on bacterial infections occurring after initial virus infection. However, chronic bacterial infection may also influence host responses to respiratory virus infections. H. influenzae infection of human epithelial cells potentiates cytokine responses and increases virus binding after subsequent rhinovirus infection.127 There are as yet no data investigating whether chronic bacterial infection influences responses to virus infection in vivo in COPD. The presence of bacteria in the lower airways is associated with increased exacerbations, and increased susceptibility to virus infection may be one mechanism mediating this effect, but further studies are needed to investigate this. Potential mechanisms of interactions between COPD, Haemophilus influenzae and respiratory virus infection are summarized in Figure 1.
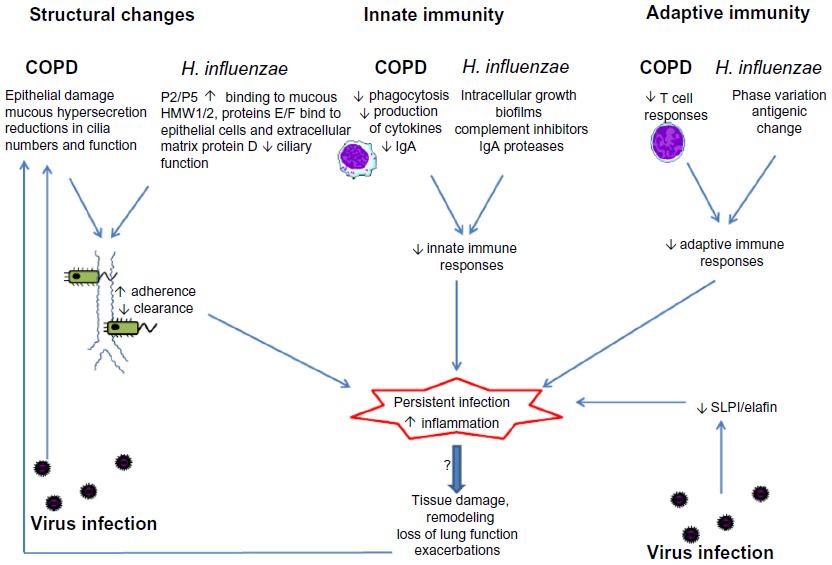 | Figure 1 Interactions between chronic obstructive pulmonary disease (COPD), virus infection, and persistent infection with Haemophilus influenzae. Structural changes in the airways and impaired innate and acquired immunity in COPD are exploited by H. influenzae, leading to failure to eradicate the organism and persistent infection. Virus infections may also cause epithelial damage and immune suppression and may favor persistent infection. Persistent infection with H. influenzae enhances airway inflammation and may contribute to disease progression in COPD. |
Therapies
In view of the evidence that persistent H. influenzae infection is associated with adverse outcomes in COPD, treating H. influenzae holds out the possibility of clinical benefit in COPD patients. Current anti-inflammatory therapies in COPD such as inhaled corticosteroids are not very effective. As H. influenzae infection is associated with greater airway inflammation in COPD, eradication of H. influenzae may have an anti-inflammatory effect. In this review, we focus on two interventions that are specifically relevant to Haemophilus influenzae infection: vaccination and long-term antibiotics.
Vaccination
Because of the significant mortality associated with invasive capsulated H. influenzae type b (Hib) disease in infants and children, conjugate Hib vaccines were developed that induce bactericidal antibodies to the capsular polysaccharide polyribitol ribose phosphate. Conjugation of polyribitol ribose phosphate to a carrier protein promotes antibody responses in infants younger than 2 years.128 After introduction of the conjugate Hib vaccine in the United States, the incidence of invasive H. influenzae disease has decreased by 99%, reaching less than one case per 100,000 children younger than 5 years.128 A vaccine for nontypeable H. influenzae is attractive because of its role in persistent infection and exacerbations in COPD. However, developing such a vaccine is likely to prove more challenging than was the case with Hib in view of the extensive antigenic variation among membrane proteins, lipopolysaccharides, and secreted virulence factors displayed by NTHi. Protein P2 is the major immunogenic outer membrane protein of NTHi and a target for human bactericidal immune responses. However, the extracellular portions of P2 demonstrate a high degree of sequence variability among strains, and antibodies that develop after a COPD exacerbation are highly strain-specific and do not prevent infection with different strains.109 Point mutations of genes coding for P2 or exchange of genes between strains colonizing the respiratory tract contribute to immune evasion and bacterial persistence in the respiratory tract.93,129 Therefore, although a P2 vaccine candidate has shown correlates with immune protection in animal models,130 it is likely to be a poor target in humans.
Protein D is a highly conserved surface lipoprotein identified in all strains of H. influenzae. Rats vaccinated with protein D have been shown to clear NTHi more effectively than controls in middle ear and pulmonary clearance models.131 In a chinchilla model of otitis media, protein D vaccination conferred a 34% protection against development of NTHI-induced otitis media.132 Children acquire serum antibodies to protein D within the first 2–3 years of life, and vaccination with a pneumococcal vaccine conjugated with H. influenzae protein D has been shown to induce high circulating levels of antibodies and a 35.5% reduction in otitis media caused by NTHi.133 Hawdon et al also demonstrated that serum immunoglobulin G antibodies to protein D were significantly reduced in adults with COPD when compared with healthy, age-matched controls.134 Therefore, on the basis of these observations, protein D-containing vaccines may be a possible candidate for providing defense against NTHi in COPD, but no clinical trials with candidate vaccines are yet available.
Oral vaccines
An alternative approach to vaccination has been the development of oral vaccines using whole killed NTHi. The mechanisms proposed for protection derived from oral vaccines are from rodent models, which demonstrated that they stimulated T-lymphocytes derived from gut-associated lymphoid tissue to translocate to bronchus-associated lymphoid tissue, thereby stimulating the respiratory epithelial host response.135 A number of studies using oral vaccinations of whole killed NTHi vaccine have been published, and analyzed in a recent Cochrane review. The reviewers concluded that oral NTHi vaccination in patients with recurrent exacerbations of COPD did not significantly reduce either the severity or frequency of exacerbations.136
A more recent placebo-controlled study using a new NTHi preparation (HI-1640V) in 38 COPD patients showed no effect on exacerbation frequency but did report reductions in mean duration of exacerbations, antibiotic use, and sputum cultures.137
The oral administration of bacterial lysates to stimulate respiratory immune responses has been used in Europe and China for many years. The OM-85 BV bacterial extract, a lysate of eight pulmonary pathogens including H. influenzae, is the most commonly studied.138,139 A systematic review of published trials of oral immunization with bacterial extracts in COPD concluded that the evidence for a reduction in exacerbations was inconclusive, but there is some evidence for reduction in exacerbation severity and duration.140 To date, oral vaccines have not been included in international guidelines for COPD.
Antibiotics
A number of studies have investigated the effects of long-term or pulsed antibiotics in COPD and have reported that long-term use of macrolide antibiotics is associated with a reduction in exacerbations.141,142 The largest double-blinded, randomized, placebo-controlled trial of azithromycin in COPD patients demonstrated an increase in time to first exacerbation, reduced exacerbation frequency, and made greater improvement in health-related quality of life in the treatment group.143 Sputum cultures were not carried out, but nasopharyngeal swabs were collected in a subset of patients and showed reduced colonization in the treatment group, but increased incidence of macrolide-resistant organisms. A study of pulsed moxifloxacin also showed a reduction in exacerbations and a trend toward a reduction in subjects with bacteria cultured in sputum in the treatment group.144 Macrolide antibiotics have a number of effects apart from their antibacterial effects, including anti-inflammatory and antiviral effects.145 Therefore, the mechanisms whereby they reduce exacerbations in COPD are unclear. In fact, in those studies in which sputum microbiology was examined, antibiotic therapy appeared to have no effect on prevalence of positive sputum cultures.141,142 Therefore, the beneficial effects of macrolides may be mediated by mechanisms other than their antibacterial effects. Concerns remain regarding the development of antibiotic resistance with long-term use of antibiotics and may limit their use as a therapeutic strategy in COPD.146
Conclusion
Bacterial infection is common in COPD, both as persistent infection in stable patients and in exacerbated patients. The most common bacteria detected in COPD patients is H. influenzae, and this organism appears to be uniquely adapted to exploit structural and immunological abnormalities in the lungs in COPD to establish chronic infection. Persistent infection has been termed colonization, but there is now a wealth of evidence that outcomes are worse in COPD patients with chronic bacterial infection. Most studies to date have defined the presence or absence of H. influenzae, using sputum cultures, but studies using newer molecular diagnostic methods suggest that culture underestimates the true prevalence of H. influenzae infections. Moreover, with the advent of the ability to sequence the entire respiratory microbiome, defining infection as either the presence or absence of an organism is clearly overly simplistic. Increasingly, concepts such as the diversity, richness, evenness, and dominance of bacterial populations will be used to describe the respiratory microbiome. Therefore, current thinking regarding the relationships between H. influenzae and COPD will change radically on the basis of these new concepts. This, in turn, will lead to novel therapeutic strategies that may be based on particular microbial patterns in the airways in individual COPD patients. Eradication or prevention of H. influenzae infection may lead to improved clinical outcomes in COPD, and future therapies may include vaccination, antimicrobials, augmentation of immune responses, or even manipulation of the respiratory microbiome, as has already been trialed in the gastrointestinal tract. In addition, the recognition of the relationship between respiratory virus infection and H. influenzae, coupled with the development of antiviral agents, offers the possibility of reducing the burden of bacterial infection by treating or preventing respiratory virus infections. Current therapies for COPD (inhaled bronchodilators and corticosteroids) have only modest effects and do not prevent disease progression. New therapies targeting H. influenzae offer a novel therapeutic strategy that provides the possibility of improving clinical outcomes and modifying disease activity in COPD.
Disclosure
SLJ has received institutional funding for clinical trials and research grants and consultant compensation from Centocor, Sanofi, Pasteur, GlaxoSmithKline, Chiesi, Boehringer Ingelheim, Novartis, and Synairgen, as well as consultant compensation from Grünenthal, and has share options in Synairgen. PM has received speaker’s fees and travel grants from GlaxoSmithKline and Novartis. The other authors report no conflicts of interest in this work.
References
Halbert RJ, Natoli JL, Gano A, Badamgarav E, Buist AS, Mannino DM. Global burden of COPD: systematic review and meta-analysis. Eur Respir J. 2006;28(3):523–532. | |
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23):2445–2454. | |
Lapperre TS, Postma DS, Gosman MM, et al. Relation between duration of smoking cessation and bronchial inflammation in COPD. Thorax. 2006;61(2):115–121. | |
Alikhan MM, Lee FE. Understanding nontypeable Haemophilus influenzae and chronic obstructive pulmonary disease. Curr Opin Pulm Med. 2014;20(2):159–164. | |
Singanayagam A, Joshi PV, Mallia P, Johnston SL. Viruses exacerbating chronic pulmonary disease: the role of immune modulation. BMC Med. 2012;10(1):27. | |
Beasley V, Joshi PV, Singanayagam A, Molyneaux PL, Johnston SL, Mallia P. Lung microbiology and exacerbations in COPD. Int J Chron Obstruct Pulmon Dis. 2012;7:555–569. | |
Donaldson GC, Seemungal TA, Patel IS, et al. Airway and systemic inflammation and decline in lung function in patients with COPD. Chest. 2005;128(4):1995–2004. | |
Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57(10):847–852. | |
Seemungal TA, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(5 Pt 1):1418–1422. | |
Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. 2006;173(10):1114–1121. | |
King P. Haemophilus influenzae and the lung (Haemophilus and the lung). Clin Transl Med. 2012;1(1):10. | |
Fontanals D, Bou R, Pons I, et al. Prevalence of Haemophilus influenzae carriers in the Catalan preschool population. Working Group on Invasive Disease Caused by Haemophilus influenzae. Eur J Clin Microbiol Infect Dis. 2000;19(4):301–304. | |
Rosell A, Monsó E, Soler N, et al. Microbiologic determinants of exacerbation in chronic obstructive pulmonary disease. Arch Intern Med. 2005;165(8):891–897. | |
Bandi V, Apicella MA, Mason E, et al. Nontypeable Haemophilus influenzae in the lower respiratory tract of patients with chronic bronchitis. Am J Respir Crit Care Med. 2001;164(11):2114–2119. | |
Sethi S, Maloney J, Grove L, Wrona C, Berenson CS. Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173(9):991–998. | |
Clementi CF, Murphy TF. Non-typeable Haemophilus influenzae invasion and persistence in the human respiratory tract. Front Cell Infect Microbiol. 2011;1:1. | |
Janson H, Ruan M, Forsgren A. Limited diversity of the protein D gene (hpd) among encapsulated and nonencapsulated Haemophilus influenzae strains. Infect Immun. 1993;61(11):4546–4552. | |
Wang X, Mair R, Hatcher C, et al. Detection of bacterial pathogens in Mongolia meningitis surveillance with a new real-time PCR assay to detect Haemophilus influenzae. Int J Med Microbiol. 2011;301(4):303–309. | |
Ueyama T, Kurono Y, Shirabe K, Takeshita M, Mogi G. High incidence of Haemophilus influenzae in nasopharyngeal secretions and middle ear effusions as detected by PCR. J Clin Microbiol. 1995;33(7):1835–1838. | |
Tian GZ, Zhang LJ, Wang XL, et al. Rapid detection of Haemophilus influenzae and Haemophilus parainfluenzae in nasopharyngeal swabs by multiplex PCR. Biomed Environ Sci. 2012;25(3):367–371. | |
Binks MJ, Temple B, Kirkham LA, et al. Molecular surveillance of true nontypeable Haemophilus influenzae: an evaluation of PCR screening assays. PLoS ONE. 2012;7(3):e34083. | |
Zhu B, Xiao D, Zhang H, et al. MALDI-TOF MS distinctly differentiates nontypable Haemophilus influenzae from Haemophilus haemolyticus. PLoS ONE. 2013;8(2):e56139. | |
Dickson RP, Erb-Downward JR, Huffnagle GB. The role of the bacterial microbiome in lung disease. Expert Rev Respir Med. 2013;7(3):245–257. | |
Hilty M, Burke C, Pedro H, et al. Disordered microbial communities in asthmatic airways. PLoS ONE. 2010;5(1):e8578. | |
Patel IS, Seemungal TA, Wilks M, Lloyd-Owen SJ, Donaldson GC, Wedzicha JA. Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax. 2002;57(9):759–764. | |
Wilkinson TM, Patel IS, Wilks M, Donaldson GC, Wedzicha JA. Airway bacterial load and FEV1 decline in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;167(8):1090–1095. | |
Hurst JR, Wilkinson TM, Perera WR, Donaldson GC, Wedzicha JA. Relationships among bacteria, upper airway, lower airway, and systemic inflammation in COPD. Chest. 2005;127(4):1219–1226. | |
Murphy TF, Brauer AL, Schiffmacher AT, Sethi S. Persistent colonization by Haemophilus influenzae in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;170(3):266–272. | |
Garcha DS, Thurston SJ, Patel AR, et al. Changes in prevalence and load of airway bacteria using quantitative PCR in stable and exacerbated COPD. Thorax. 2012;67(12):1075–1080. | |
Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med. 2011;184(6):662–671. | |
Curran T, Coyle PV, McManus TE, Kidney J, Coulter WA. Evaluation of real-time PCR for the detection and quantification of bacteria in chronic obstructive pulmonary disease. FEMS Immunol Med Microbiol. 2007;50(1):112–118. | |
Groenewegen KH, Wouters EF. Bacterial infections in patients requiring admission for an acute exacerbation of COPD; a 1-year prospective study. Respir Med. 2003;97(7):770–777. | |
Molyneaux PL, Mallia P, Cox MJ, et al. Outgrowth of the bacterial airway microbiome after rhinovirus exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(10):1224–1231. | |
Huang YJ, Sethi S, Murphy T, Nariya S, Boushey HA, Lynch SV. Airway microbiome dynamics in exacerbations of chronic obstructive pulmonary disease. J Clin Microbiol. 2014;52(8):2813–2823. | |
Rogers GB, van der Gast CJ, Serisier DJ. Predominant pathogen competition and core microbiota divergence in chronic airway infection. ISME J. 2014. Epub ahead of print http://dx.doi.org/10.1038/ismej.2014.124. | |
Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med. 2013;188(8):913–922. | |
Wieland CW, Florquin S, Maris NA, et al. The MyD88-dependent, but not the MyD88-independent, pathway of TLR4 signaling is important in clearing nontypeable haemophilus influenzae from the mouse lung. J Immunol. 2005;175(9):6042–6049. | |
Reddy MS, Murphy TF, Faden HS, Bernstein JM. Middle ear mucin glycoprotein: purification and interaction with nontypable Haemophilus influenzae and Moraxella catarrhalis. Otolaryngol Head Neck Surg. 1997;116(2):175–180. | |
Miyamoto N, Bakaletz LO. Selective adherence of non-typeable Haemophilus influenzae (NTHi) to mucus or epithelial cells in the chinchilla eustachian tube and middle ear. Microb Pathog. 1996;21(5):343–356. | |
Berenson CS, Garlipp MA, Grove LJ, Maloney J, Sethi S. Impaired phagocytosis of nontypeable Haemophilus influenzae by human alveolar macrophages in chronic obstructive pulmonary disease. J Infect Dis. 2006;194(10):1375–1384. | |
Hallström T, Riesbeck K. Haemophilus influenzae and the complement system. Trends Microbiol. 2010;18(6):258–265. | |
Marc MM, Korosec P, Kosnik M, et al. Complement factors c3a, c4a, and c5a in chronic obstructive pulmonary disease and asthma. Am J Respir Cell Mol Biol. 2004;31(2):216–219. | |
Marc MM, Kristan SS, Rozman A, et al. Complement factor C5a in acute exacerbation of Chronic Obstructive Pulmonary Disease. Scand J Immunol. 2010;71(5):386–391. | |
Woof JM, Kerr MA. The function of immunoglobulin A in immunity. J Pathol. 2006;208(2):270–282. | |
Ram S, Lewis LA, Rice PA. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clin Microbiol Rev. 2010;23(4):740–780. | |
Kainulainen L, Nikoskelainen J, Vuorinen T, Tevola K, Liippo K, Ruuskanen O. Viruses and bacteria in bronchial samples from patients with primary hypogammaglobulinemia. Am J Respir Crit Care Med. 1999;159(4 Pt 1):1199–1204. | |
King PT, Hutchinson PE, Johnson PD, Holmes PW, Freezer NJ, Holdsworth SR. Adaptive immunity to nontypeable Haemophilus influenzae. Am J Respir Crit Care Med. 2003;167(4):587–592. | |
King PT, Ngui J, Gunawardena D, Holmes PW, Farmer MW, Holdsworth SR. Systemic humoral immunity to non-typeable Haemophilus influenzae. Clin Exp Immunol. 2008;153(3):376–384. | |
King P, Ngui J, Oppedisano F, Robins-Browne R, Holmes P, Holdsworth S. Effect of interferon gamma and CD40 ligation on intracellular monocyte survival of nontypeable Haemophilus influenzae. APMIS. 2008;116(12):1043–1049. | |
King PT, Ngui J, Farmer MW, Hutchinson P, Holmes PW, Holdsworth SR. Cytotoxic T lymphocyte and natural killer cell responses to non-typeable Haemophilus influenzae. Clin Exp Immunol. 2008;152(3):542–551. | |
van Ham SM, van Alphen L, Mooi FR, van Putten JP. Phase variation of H. influenzae fimbriae: transcriptional control of two divergent genes through a variable combined promoter region. Cell. 1993;73(6):1187–1196. | |
Erwin AL, Smith AL. Nontypeable Haemophilus influenzae: understanding virulence and commensal behavior. Trends Microbiol. 2007;15(8):355–362. | |
Duim B, Vogel L, Puijk W, et al. Fine mapping of outer membrane protein P2 antigenic sites which vary during persistent infection by Haemophilus influenzae. Infect Immun. 1996;64(11):4673–4679. | |
Cholon DM, Cutter D, Richardson SK, et al. Serial isolates of persistent Haemophilus influenzae in patients with chronic obstructive pulmonary disease express diminishing quantities of the HMW1 and HMW2 adhesins. Infect Immun. 2008;76(10):4463–4468. | |
Swords WE. Nontypeable Haemophilus influenzae biofilms: role in chronic airway infections. Front Cell Infect Microbiol. 2012;2:97. | |
Murphy TF, Kirkham C. Biofilm formation by nontypeable Haemophilus influenzae: strain variability, outer membrane antigen expression and role of pili. BMC Microbiol. 2002;2(1):7. | |
Murphy TF, Kirkham C, Sethi S, Lesse AJ. Expression of a peroxiredoxin-glutaredoxin by Haemophilus influenzae in biofilms and during human respiratory tract infection. FEMS Immunol Med Microbiol. 2005;44(1):81–89. | |
Langereis JD, Hermans PW. Novel concepts in nontypeable Haemophilus influenzae biofilm formation. FEMS Microbiol Lett. 2013;346(2):81–89. | |
St Geme JW III, Falkow S. Loss of capsule expression by Haemophilus influenzae type b results in enhanced adherence to and invasion of human cells. Infect Immun. 1991;59(4):1325–1333. | |
St Geme JW III, Falkow S. Haemophilus influenzae adheres to and enters cultured human epithelial cells. Infect Immun. 1990;58(12):4036–4044. | |
Forsgren J, Samuelson A, Ahlin A, Jonasson J, Rynnel-Dagöö B, Lindberg A. Haemophilus influenzae resides and multiplies intracellularly in human adenoid tissue as demonstrated by in situ hybridization and bacterial viability assay. Infect Immun. 1994;62(2):673–679. | |
Forsgren J, Samuelson A, Lindberg A, Rynnel-Dagöö B. Quantitative bacterial culture from adenoid lymphatic tissue with special reference to Haemophilus [corrected]. [corrected]. Acta Otolaryngol. 1993;113(5):668–672. | |
Ahrén IL, Janson H, Forsgren A, Riesbeck K. Protein D expression promotes the adherence and internalization of non-typeable Haemophilus influenzae into human monocytic cells. Microb Pathog. 2001;31(3):151–158. | |
van Schilfgaarde M, Eijk P, Regelink A, et al. Haemophilus influenzae localized in epithelial cell layers is shielded from antibiotics and antibody-mediated bactericidal activity. Microb Pathog. 1999;26(5):249–262. | |
Su YC, Jalalvand F, Mörgelin M, Blom AM, Singh B, Riesbeck K. Haemophilus influenzae acquires vitronectin via the ubiquitous Protein F to subvert host innate immunity. Mol Microbiol. 2013;87(6):1245–1266. | |
Hallström T, Trajkovska E, Forsgren A, Riesbeck K. Haemophilus influenzae surface fibrils contribute to serum resistance by interacting with vitronectin. J Immunol. 2006;177(1):430–436. | |
Hallström T, Zipfel PF, Blom AM, Lauer N, Forsgren A, Riesbeck K. Haemophilus influenzae interacts with the human complement inhibitor factor H. J Immunol. 2008;181(1):537–545. | |
Kilian M, Mestecky J, Russell MW. Defense mechanisms involving Fc-dependent functions of immunoglobulin A and their subversion by bacterial immunoglobulin A proteases. Microbiol Rev. 1988;52(2):296–303. | |
Murphy TF, Brauer AL. Expression of urease by Haemophilus influenzae during human respiratory tract infection and role in survival in an acid environment. BMC Microbiol. 2011;11(1):183. | |
Whitby PW, Morton DJ, Vanwagoner TM, et al. Haemophilus influenzae OxyR: characterization of its regulation, regulon and role in fitness. PLoS ONE. 2012;7(11):e50588. | |
Whitby PW, VanWagoner TM, Seale TW, Morton DJ, Stull TL. Comparison of transcription of the Haemophilus influenzae iron/heme modulon genes in vitro and in vivo in the chinchilla middle ear. BMC Genomics. 2013;14(1):925. | |
Qu J, Lesse AJ, Brauer AL, Cao J, Gill SR, Murphy TF. Proteomic expression profiling of Haemophilus influenzae grown in pooled human sputum from adults with chronic obstructive pulmonary disease reveal antioxidant and stress responses. BMC Microbiol. 2010;10(1):162. | |
Zhang L, Patel M, Xie J, Davis GS, Marrs CF, Gilsdorf JR. Urease operon and urease activity in commensal and disease-causing nontypeable Haemophilus influenzae. J Clin Microbiol. 2013;51(2):653–655. | |
Zhang L, Xie J, Patel M, et al. Nontypeable Haemophilus influenzae genetic islands associated with chronic pulmonary infection. PLoS ONE. 2012;7(9):e44730. | |
St Geme JW, Pinkner JS III, Krasan GP, et al. Haemophilus influenzae pili are composite structures assembled via the HifB chaperone. Proc Natl Acad Sci USA. 1996;93(21):11913–11918. | |
Hallström T, Singh B, Resman F, Blom AM, Mörgelin M, Riesbeck K. Haemophilus influenzae protein E binds to the extracellular matrix by concurrently interacting with laminin and vitronectin. J Infect Dis. 2011;204(7):1065–1074. | |
Jalalvand F, Su YC, Mörgelin M, et al. Haemophilus influenzae protein F mediates binding to laminin and human pulmonary epithelial cells. J Infect Dis. 2013;207(5):803–813. | |
St Geme JW III, Falkow S, Barenkamp SJ. High-molecular-weight proteins of nontypable Haemophilus influenzae mediate attachment to human epithelial cells. Proc Natl Acad Sci U S A. 1993;90(7):2875–2879. | |
Johnson RW, McGillivary G, Denoël P, Poolman J, Bakaletz LO. Abrogation of nontypeable Haemophilus influenzae protein D function reduces phosphorylcholine decoration, adherence to airway epithelial cells, and fitness in a chinchilla model of otitis media. Vaccine. 2011;29(6):1211–1221. | |
Denny FW. Effect of a toxin produced by Haemophilus influenzae on ciliated respiratory epithelium. J Infect Dis. 1974;129(2):93–100. | |
Janson H, Carln B, Cervin A, et al. Effects on the ciliated epithelium of protein D-producing and -nonproducing nontypeable Haemophilus influenzae in nasopharyngeal tissue cultures. J Infect Dis. 1999;180(3):737–746. | |
van Schilfgaarde M, van Ulsen P, Eijk P, et al. Characterization of adherence of nontypeable Haemophilus influenzae to human epithelial cells. Infect Immun. 2000;68(8):4658–4665. | |
Berenson CS, Kruzel RL, Eberhardt E, et al. Impaired innate immune alveolar macrophage response and the predilection for COPD exacerbations. Thorax. 2014;69(9):811–818. | |
Berenson CS, Kruzel RL, Eberhardt E, Sethi S. Phagocytic dysfunction of human alveolar macrophages and severity of chronic obstructive pulmonary disease. J Infect Dis. 2013;208(12):2036–2045. | |
Taylor AE, Finney-Hayward TK, Quint JK, et al. Defective macrophage phagocytosis of bacteria in COPD. Eur Respir J. 2010;35(5):1039–1047. | |
Berenson CS, Wrona CT, Grove LJ, et al. Impaired alveolar macrophage response to Haemophilus antigens in chronic obstructive lung disease. Am J Respir Crit Care Med. 2006;174(1):31–40. | |
Craig JE, Cliffe A, Garnett K, High NJ. Survival of nontypeable Haemophilus influenzae in macrophages. FEMS Microbiol Lett. 2001;203(1):55–61. | |
Clementi CF, Håkansson AP, Murphy TF. Internalization and trafficking of nontypeable Haemophilus influenzae in human respiratory epithelial cells and roles of IgA1 proteases for optimal invasion and persistence. Infect Immun. 2014;82(1):433–444. | |
Knobloch J, Schild K, Jungck D, et al. The T-helper cell type 1 immune response to gram-negative bacterial infections is impaired in COPD. Am J Respir Crit Care Med. 2011;183(2):204–214. | |
Abe Y, Murphy TF, Sethi S, et al. Lymphocyte proliferative response to P6 of Haemophilus influenzae is associated with relative protection from exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;165(7):967–971. | |
Kalathil SG, Lugade AA, Pradhan V, et al. T-regulatory cells and programmed death 1+ T cells contribute to effector T-cell dysfunction in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;190(1):40–50. | |
Duim B, Bowler LD, Eijk PP, Jansen HM, Dankert J, van Alphen L. Molecular variation in the major outer membrane protein P5 gene of nonencapsulated Haemophilus influenzae during chronic infections. Infect Immun. 1997;65(4):1351–1356. | |
Duim B, van Alphen L, Eijk P, Jansen HM, Dankert J. Antigenic drift of non-encapsulated Haemophilus influenzae major outer membrane protein P2 in patients with chronic bronchitis is caused by point mutations. Mol Microbiol. 1994;11(6):1181–1189. | |
Lomholt H, van Alphen L, Kilian M. Antigenic variation of immunoglobulin A1 proteases among sequential isolates of Haemophilus influenzae from healthy children and patients with chronic obstructive pulmonary disease. Infect Immun. 1993;61(11):4575–4581. | |
Polosukhin VV, Cates JM, Lawson WE, et al. Bronchial secretory immunoglobulin a deficiency correlates with airway inflammation and progression of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;184(3):317–327. | |
Murphy TF, Lesse AJ, Kirkham C, Zhong H, Sethi S, Munson RS Jr. A clonal group of nontypeable Haemophilus influenzae with two IgA proteases is adapted to infection in chronic obstructive pulmonary disease. PLoS ONE. 2011;6(10):e25923. | |
Millares L, Marin A, Garcia-Aymerich J, Sauleda J, Belda J, Monsó E; PAC-COPD Study Group. Specific IgA and metalloproteinase activity in bronchial secretions from stable chronic obstructive pulmonary disease patients colonized by Haemophilus influenzae. Respir Res. 2012;13(1):113. | |
Marin A, Garcia-Aymerich J, Sauleda J, et al; PAC-COPD Study Group. Effect of bronchial colonisation on airway and systemic inflammation in stable COPD. COPD. 2012;9(2):121–130. | |
Desai H, Eschberger K, Wrona C, et al. Bacterial colonization increases daily symptoms in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2014;11(3):303–309. | |
Beasley V, Joshi PV, Singanayagam A, Molyneaux PL, Johnston SL, Mallia P. Lung microbiology and exacerbations in COPD. Int J Chron Obstruct Pulmon Dis. 2012;7:555–569. | |
Moghaddam SJ, Clement CG, De la Garza MM, et al. Haemophilus influenzae lysate induces aspects of the chronic obstructive pulmonary disease phenotype. Am J Respir Cell Mol Biol. 2008;38(6):629–638. | |
Ganesan S, Comstock AT, Kinker B, Mancuso P, Beck JM, Sajjan US. Combined exposure to cigarette smoke and nontypeable Haemophilus influenzae drives development of a COPD phenotype in mice. Respir Res. 2014;15(1):11. | |
Bresser P, Out TA, van Alphen L, Jansen HM, Lutter R. Airway inflammation in nonobstructive and obstructive chronic bronchitis with chronic haemophilus influenzae airway infection. Comparison with noninfected patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;162(3 Pt 1):947–952. | |
Marin A, Monsó E, Garcia-Nuñez M, et al. Variability and effects of bronchial colonisation in patients with moderate COPD. Eur Respir J. 2010;35(2):295–302. | |
Hurst JR, Perera WR, Wilkinson TM, Donaldson GC, Wedzicha JA. Systemic and upper and lower airway inflammation at exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173(1):71–78. | |
Wilkinson TM, Hurst JR, Perera WR, Wilks M, Donaldson GC, Wedzicha JA. Effect of interactions between lower airway bacterial and rhinoviral infection in exacerbations of COPD. Chest. 2006; 129(2):317–324. | |
Sethi S, Evans N, Grant BJ, Murphy TF. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 2002;347(7):465–471. | |
Murphy TF, Brauer AL, Sethi S, Kilian M, Cai X, Lesse AJ. Haemophilus haemolyticus: a human respiratory tract commensal to be distinguished from Haemophilus influenzae. J Infect Dis. 2007;195(1):81–89. | |
Sethi S, Wrona C, Grant BJ, Murphy TF. Strain-specific immune response to Haemophilus influenzae in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;169(4):448–453. | |
Fernaays MM, Lesse AJ, Sethi S, Cai X, Murphy TF. Differential genome contents of nontypeable Haemophilus influenzae strains from adults with chronic obstructive pulmonary disease. Infect Immun. 2006;74(6):3366–3374. | |
Chin CL, Manzel LJ, Lehman EE, et al. Haemophilus influenzae from patients with chronic obstructive pulmonary disease exacerbation induce more inflammation than colonizers. Am J Respir Crit Care Med. 2005;172(1):85–91. | |
Geelen TH, Gaajetaan GR, Wouters EF, et al. The host immune response contributes to Haemophilus influenzae virulence. Respir Med. 2014;108(1):144–152. | |
Wang XY, Kilgore PE, Lim KA, et al. Influenza and bacterial pathogen coinfections in the 20th century. Interdiscip Perspect Infect Dis. 2011;2011:146376. | |
Lee LN, Dias P, Han D, et al. A mouse model of lethal synergism between influenza virus and Haemophilus influenzae. Am J Pathol. 2010;176(2):800–811. | |
Zwaans WA, Mallia P, van Winden ME, Rohde GG. The relevance of respiratory viral infections in the exacerbations of chronic obstructive pulmonary disease-A systematic review. J Clin Virol. 2014. Epub ahead of print http://dx.doi.org/10.1016/j.jcv.2014.06.025. | |
Unger BL, Faris AN, Ganesan S, Comstock AT, Hershenson MB, Sajjan US. Rhinovirus attenuates non-typeable Hemophilus influenzae-stimulated IL-8 responses via TLR2-dependent degradation of IRAK-1. PLoS Pathog. 2012;8(10):e1002969. | |
Oliver BG, Lim S, Wark P, et al. Rhinovirus exposure impairs immune responses to bacterial products in human alveolar macrophages. Thorax. 2008;63(6):519–525. | |
Comstock AT, Ganesan S, Chattoraj A, et al. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J Virol. 2011;85(13):6795–6808. | |
Wang JH, Kwon HJ, Jang YJ. Rhinovirus enhances various bacterial adhesions to nasal epithelial cells simultaneously. Laryngoscope. 2009;119(7):1406–1411. | |
Wang JH, Lee SH, Kwon HJ, Jang YJ. Clarithromycin inhibits rhinovirus-induced bacterial adhesions to nasal epithelial cells. Laryngoscope. 2010;120(1):193–199. | |
Avadhanula V, Rodriguez CA, Devincenzo JP, et al. Respiratory viruses augment the adhesion of bacterial pathogens to respiratory epithelium in a viral species- and cell type-dependent manner. J Virol. 2006;80(4):1629–1636. | |
Suri R, Mallia P, Martin JE, et al. Bronchial platelet-activating factor receptor in chronic obstructive pulmonary disease. Respir Med. 2014;108(6):898–904. | |
Bandi V, Jakubowycz M, Kinyon C, et al. Infectious exacerbations of chronic obstructive pulmonary disease associated with respiratory viruses and non-typeable Haemophilus influenzae. FEMS Immunol Med Microbiol. 2003;37(1):69–75. | |
Mallia P, Footitt J, Sotero R, et al. Rhinovirus infection induces degradation of antimicrobial peptides and secondary bacterial infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186(11):1117–1124. | |
Hutchinson AF, Ghimire AK, Thompson MA, et al. A community-based, time-matched, case-control study of respiratory viruses and exacerbations of COPD. Respir Med. 2007;101(12):2472–2481. | |
George SN, Garcha DS, Mackay AJ, et al. Human rhinovirus infection during naturally occurring COPD exacerbations. Eur Respir J. 2014;44(1):87–96. | |
Sajjan US, Jia Y, Newcomb DC, et al. H. influenzae potentiates airway epithelial cell responses to rhinovirus by increasing ICAM-1 and TLR3 expression. FASEB J. 2006;20(12):2121–2123. | |
Agrawal A, Murphy TF. Haemophilus influenzae infections in the H. influenzae type b conjugate vaccine era. J Clin Microbiol. 2011;49(11):3728–3732. | |
Hiltke TJ, Schiffmacher AT, Dagonese AJ, Sethi S, Murphy TF. Horizontal transfer of the gene encoding outer membrane protein P2 of nontypeable Haemophilus influenzae, in a patient with chronic obstructive pulmonary disease. J Infect Dis. 2003;188(1):114–117. | |
Ostberg KL, Russell MW, Murphy TF. Mucosal immunization of mice with recombinant OMP P2 induces antibodies that bind to surface epitopes of multiple strains of nontypeable Haemophilus influenzae. Mucosal Immunol. 2009;2(1):63–73. | |
Poolman JT, Bakaletz L, Cripps A, et al. Developing a nontypeable Haemophilus influenzae (NTHi) vaccine. Vaccine. 2000; 19(Suppl 1):S108–S115. | |
Novotny LA, Jurcisek JA, Godfroid F, Poolman JT, Denoël PA, Bakaletz LO. Passive immunization with human anti-protein D antibodies induced by polysaccharide protein D conjugates protects chinchillas against otitis media after intranasal challenge with Haemophilus influenzae. Vaccine. 2006;24(22):4804–4811. | |
Prymula R, Peeters P, Chrobok V, et al. Pneumococcal capsular polysaccharides conjugated to protein D for prevention of acute otitis media caused by both Streptococcus pneumoniae and non-typable Haemophilus influenzae: a randomised double-blind efficacy study. Lancet. 2006;367(9512):740–748. | |
Hawdon N, Biman B, McCready W, et al. Antibody against Haemophilus influenzae protein D in patients with chronic conditions causing secondary immunodeficiency. Vaccine. 2012;30(7):1235–1238. | |
Clancy RL, Dunkley M. A vaccine to prevent exacerbations in COPD. Med J Aust. 2011;195(2):99–100. | |
Teo E, House H, Lockhart K, Purchuri SN, Pushparajah J, Cripps AW, van Driel ML. Haemophilus influenzae oral vaccination for preventing acute exacerbations of chronic bronchitis and chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2014;9:CD010010. | |
Tandon MK, Phillips M, Waterer G, Dunkley M, Comans P, Clancy R. Oral immunotherapy with inactivated nontypeable Haemophilus influenzae reduces severity of acute exacerbations in severe COPD. Chest. 2010;137(4):805–811. | |
Collet JP, Shapiro P, Ernst P, Renzi T, Ducruet T, Robinson A. Effects of an immunostimulating agent on acute exacerbations and hospitalizations in patients with chronic obstructive pulmonary disease. The PARI-IS Study Steering Committee and Research Group. Prevention of Acute Respiratory Infection by an Immunostimulant. Am J Respir Crit Care Med. 1997;156(6):1719–1724. | |
Cvoriscec B, Ustar M, Pardon R, Palecek I, Stipic-Markovic A, Zimic B. Oral immunotherapy of chronic bronchitis: a double-blind placebo-controlled multicentre study. Respiration. 1989;55(3):129–135. | |
Arandjus C, Black PN, Poole PJ, Wood Baker R, Steurer-Stey C. Oral bacterial vaccines for the prevention of acute exacerbations in chronic obstructive pulmonary disease and chronic bronchitis. Respir Med. 2006;100(10):1671–1681. | |
He ZY, Ou LM, Zhang JQ, et al. Effect of 6 months of erythromycin treatment on inflammatory cells in induced sputum and exacerbations in chronic obstructive pulmonary disease. Respiration. 2010;80(6):445–452. | |
Seemungal TA, Wilkinson TM, Hurst JR, Perera WR, Sapsford RJ, Wedzicha JA. Long-term erythromycin therapy is associated with decreased chronic obstructive pulmonary disease exacerbations. Am J Respir Crit Care Med. 2008;178(11):1139–1147. | |
Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med. 2011;365(8):689–698. | |
Sethi S, Jones PW, Theron MS, et al; PULSE Study group. Pulsed moxifloxacin for the prevention of exacerbations of chronic obstructive pulmonary disease: a randomized controlled trial. Respir Res. 2010;11(1):10. | |
Gielen V, Johnston SL, Edwards MR. Azithromycin induces anti-viral responses in bronchial epithelial cells. Eur Respir J. 2010;36(3):646–654. | |
Serisier DJ. Risks of population antimicrobial resistance associated with chronic macrolide use for inflammatory airway diseases. Lancet Respir Med. 2013;1(3):262–274. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.