Back to Journals » Journal of Blood Medicine » Volume 13
Low Rate of Clinically Evident Extravascular Hemolysis in Patients with Paroxysmal Nocturnal Hemoglobinuria Treated with a Complement C5 Inhibitor: Results from a Large, Multicenter, US Real-World Study
Authors Shammo J, Gajra A, Patel Y, Tomazos I ![]() , Kish J, Hill A, Sierra JR
, Kish J, Hill A, Sierra JR ![]() , Araten D
, Araten D
Received 22 February 2022
Accepted for publication 29 June 2022
Published 12 August 2022 Volume 2022:13 Pages 425—437
DOI https://doi.org/10.2147/JBM.S361863
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Jamile Shammo,1 Ajeet Gajra,2 Yogesh Patel,3 Ioannis Tomazos,3 Jonathan Kish,2 Anita Hill,4 J Rafael Sierra,3 David Araten5
1Rush Hematology, Oncology and Cell Therapy, Rush University Medical Center, Chicago, IL, USA; 2Cardinal Health Inc., Charlotte, NC, USA; 3Alexion, AstraZeneca Rare Disease, Boston, MA, USA; 4Alexion, AstraZeneca Rare Disease, Inc., Leeds, UK; 5Division of Hematology, New York University School of Medicine, New York, NY, USA
Correspondence: Jamile Shammo, Email [email protected]
Purpose: Most patients with paroxysmal nocturnal hemoglobinuria (PNH) treated with a complement protein 5 (C5) inhibitor achieve full control of terminal complement activity and intravascular hemolysis. The minority remains anemic and transfusion dependent despite this control. Etiology for ongoing anemia is multifactorial and includes bone marrow failure, breakthrough hemolysis, extravascular hemolysis (EVH) and nutritional deficiencies.
Patients and Methods: To evaluate the potential etiologies of hemoglobin levels < 10 g/dL despite receiving C5 inhibitor therapy, we performed a retrospective US chart review of adult patients with PNH and treated for at least 12 months with eculizumab (n=53), ravulizumab (n=32), or eculizumab followed by ravulizumab (n=15). Clinically evident EVH was defined as at least one transfusion, reticulocyte count ≥ 120× 109/L and hemoglobin level ≤ 9.5 g/dL. Safety data were not collected. Mean treatment duration was 26.5± 17.2 months.
Results: Treatment with C5 inhibitors significantly improved hemoglobin, lactate dehydrogenase, and number of transfusions versus baseline. Among the patients with hemoglobin < 10 g/dL during the last 6 months of treatment (n=38), one patient (eculizumab) had clinically evident EVH, and 10 patients had active concomitant bone marrow failure. Bone marrow failure was a major contributor to hemoglobin < 10 g/dL and transfusion dependence; clinically evident EVH was uncommon.
Conclusion: A range of hematologic causes need to be considered when evaluating anemia in the presence of treatment with a C5 inhibitor.
Keywords: real-world, anemia, intravascular hemolysis, bone marrow failure
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired, hematologic disorder caused by somatic mutations in the PIGA gene in bone marrow stem cells, which disrupt glycosylphosphatidylinositol (GPI) biosynthesis.1 In the absence of functional GPI, the terminal complement pathway becomes uncontrolled on the cell surface of erythrocytes, rendering them particularly susceptible to lysis by the membrane attack complex (C5b-9).1–5 In addition, activity of the membrane attack complex on PNH white blood cells and platelets, as well as the accompanying release of complement protein (C) 5a, promotes increases in inflammatory mediators and a prothrombotic state.2 In this context, uncontrolled terminal complement activation causes intravascular hemolysis (IVH), increased risk of thromboembolic events, organ damage, and eventually death.2,6,7
First approved in 2007, eculizumab, a C5 inhibitor, has been utilized for the treatment of PNH for more than 15 years;8 it inhibits terminal complement pathway activation thereby preventing the formation of C5a and the membrane attack complex. Ravulizumab-cwvz (ravulizumab), the first long-acting C5 inhibitor, was approved in the USA in 2018 and has become the standard of care in countries where it is available.9 In large, phase 3, randomized controlled trials and in real-world studies, treatment with either C5 inhibitor has been shown to reduce IVH, thromboembolism, organ damage, and mortality.10–22
However, some patients who receive ongoing treatment with a C5 inhibitor remain anemic and transfusion dependent. Etiologies for hemoglobin <10 g/dL include bone marrow failure, breakthrough hemolysis, extravascular hemolysis (EVH) and nutritional deficiencies in iron/folate. EVH is a mechanistic consequence of treatment with C5 inhibitors and is believed to be caused by ongoing C3 deposition on surviving yet defective red blood cells, which renders them susceptible to phagocytosis in the liver or spleen as they are no longer destroyed by IVH.23–28 As a result, lactate dehydrogenase (LDH) is not released in the blood and cannot be used to assess EVH. In eculizumab-treated patients, C3 deposition on red blood cells has been reported to occur in most patients to varying degrees.25,26 However, the clinical impact of C3 deposition remains unclear because the amount of C3 deposition does not correlate with levels of hemoglobin or transfusion dependence.25,29 The primary objective of this retrospective chart review was to assess the real-world incidence of clinically evident EVH or EVH requiring an intervention, in patients treated for PNH with eculizumab or ravulizumab.
Materials and Methods
In this retrospective, non-interventional study of community-based medical practices in the USA, 796 hematologists and oncologists who were part of the Cardinal Health Specialty Solutions Oncology Provider Extended Network were invited electronically to participate in a chart review of adult patients with diagnosed PNH and treated with a C5 inhibitor (eculizumab or ravulizumab) for at least 12 months. To minimize selection bias, providers were asked to identify all eligible patients starting with the first chronological patient meeting eligibility criteria and then randomly selecting up to 10 patients from that point forward. Chart data were entered by providers into electronic case report forms for analysis. The study was conducted in accordance with International Society for Pharmacoepidemiology Guidelines for Good Pharmacoepidemiology Practices and applicable regulatory requirements. A waiver from the central institutional review board was received prior to the start of the study. Patient-protected health information was kept confidential at all times and in accordance with the Health Insurance Portability and Accountability Act of 1996; no data that could directly identify patients were extracted. As such, patient informed consent was not needed. Providers were compensated at fair market value for the time needed to complete data abstraction and quality control procedures.
Patient records could be included if the patient was an adult who was being managed and treated by the provider and if the patient had a PNH diagnosis with a known percentage of GPI-deficient granulocytes or monocytes confirmed via flow cytometry. Treatment of PNH needed to have been ongoing for at least 12 consecutive months (unless deceased) and needed to have been initiated during the index period: between January 1, 2014 and November 1, 2019 for eculizumab and between December 21, 2018 and November 1, 2019 for ravulizumab. Ongoing treatment was defined based on the prescribing information with intervals between infusions of not more than 16 days for eculizumab and 63 days for ravulizumab.8,9 For patients who were switched from eculizumab to ravulizumab (the “switch” subgroup), eculizumab treatment needed to have been initiated between January 1, 2014 and November 1, 2019 and have been switched to ravulizumab between December 21, 2018 and May 1, 2020. Switch patients needed to have been treated with ravulizumab for at least 6 consecutive months. At least one routine clinic visit needed to have taken place during the last 6 months of eculizumab or ravulizumab therapy. In addition, laboratory values from at least three separate clinic visits (routine or acute) during treatment needed to be available. Records were excluded if the patient had been enrolled in a clinical trial investigating PNH treatment or if C5 inhibitor treatment was initiated prior to regulatory drug approval.
Laboratory results from hematological evaluations were analyzed at baseline and at follow-up. Baseline laboratory data were collected from tests performed within 14 days prior to the initiation of treatment with the C5 inhibitor (eculizumab in the switch subgroup). The follow-up period was defined as the window of time directly preceding data entry, treatment discontinuation, or patient death (Figure 1). For all variables other than transfusion, the follow-up period was 6 months. Transfusion follow-up was assessed over 12 months in patients treated with eculizumab or ravulizumab only and over 6 months for patients in the switch subgroup.
 |
Figure 1 Study design. In this retrospective US chart review of adults diagnosed with PNH, patients needed to have been treated with a C5 inhibitor (eculizumab or ravulizumab) for at least 12 consecutive months. For patients who were switched from eculizumab to ravulizumab, patients needed to have been treated with ravulizumab for at least 6 consecutive months. The follow-up period was defined as the window of time directly preceding data entry, treatment discontinuation, or patient death. For all variables except transfusion, the follow-up period was 6 months. Transfusion follow-up was assessed over 12 months in patients treated with eculizumab or ravulizumab only and over 6 months for patients in the switch subgroup. C5, complement protein C5; PNH, paroxysmal nocturnal hemoglobinuria. |
Anemia was differentiated as hemoglobin levels <12 g/dL or <10 g/dL. Intravascular hemolysis was defined as LDH levels ≥1.5 times the upper limit of normal (ULN). Transfusion dependence was defined as at least one transfusion of packed red blood cells during follow-up. Breakthrough hemolysis (BTH) was defined as hemoglobin levels <10 g/dL and LDH elevation >2×ULN when the previous measurement was <1.5×ULN.19,20 According to the protocol, BTH was evaluated only in eculizumab-treated patients. Clinically evident EVH was defined as at least one transfusion, reticulocyte count ≥120×109/L, and hemoglobin level ≤9.5 g/dL during follow-up and was evaluated in patients who had data for all three variables. Because no consensus definition for EVH has been established in guidelines,30–33 this definition was based on the phase 3 ALPHA trial that is assessing danicopan (factor D inhibitor) treatment in patients with clinically evident EVH while being treated with eculizumab or ravulizumab.34 Concomitant active bone marrow failure was defined according to Camitta criteria as at least two of the following: hemoglobin level <10 g/dL, platelet count <50×109/L, neutrophil count <1.5×109/L.32 Reticulocytopenia was defined as a reticulocyte count <60×109. Safety data, including treatment-related adverse events and cause of death (all-cause), were not collected. Data describing other reasons for anemia, such as folate, erythropoietin, and iron deficiency, were not collected.
This is a descriptive analysis of retrospectively collected patient-level data. Data were analyzed for the whole cohort as well as for each subgroup (eculizumab only, ravulizumab only, and switch). Categorical variables are presented as number of patients and percentage. Continuous variables are presented as means ± standard deviation. Where applicable, medians, interquartile range (IQR), and minimum–maximum range are presented. Baseline and follow-up laboratory values and the number of transfusions were compared using paired t-tests with a statistical significance set at p≤0.05. If multiple visits of the same type (routine or non-routine) occurred within the follow-up period, the arithmetic mean for the data point was taken. Sensitivity analyses for EVH were performed by varying the cut-offs for hemoglobin level (<10 g/dL) and absolute reticulocyte count (≥100×109/L and ≥150×109/L) and by removing transfusion as a variable from the definition. Because no a priori hypotheses were specified for this study, a sample size power calculation was not performed.
Results
A total of 18 physicians responded to the invitation; 15 of them submitted at least one electronic case report. Provider practices (11 community practices, two academic medical centers, two affiliated teaching hospitals) were located throughout the USA and in a range of settings (six urban, seven suburban, two rural). The mean number of patients with PNH was 8.1±5.5 per physician in the year preceding this study (range 1–20).
Records for 100 patients were submitted and included 53 patients in the eculizumab subgroup, 32 patients in the ravulizumab subgroup, and 15 patients in the switch subgroup. The last day for data entry was February 22, 2021.
In the full cohort of 100 patients, 57.0% of patients were male (Table 1); mean ages at diagnosis and at C5 inhibitor treatment initiation were 54.3±10.5 and 54.6±10.6 years, respectively. The mean percentage of the GPI-deficient cells was 53.5±26.9% for granulocytes and 54.1±26.8% for monocytes. Fifteen percent of patients had a history of thromboembolism and 14.0% had a documented history of bone marrow disease (7.0% aplastic anemia; 7.0% myelodysplastic syndrome).
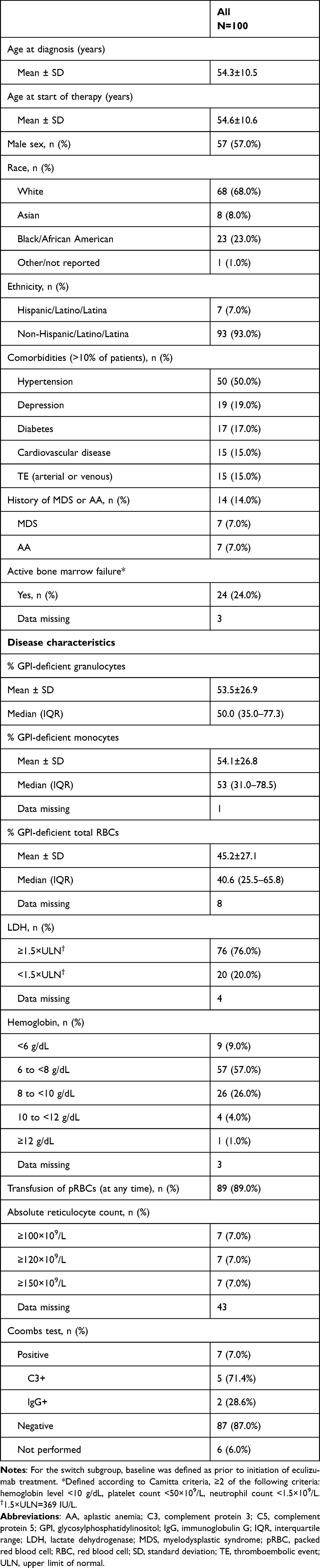 |
Table 1 Demographics and Disease Characteristics Prior to the Initiation of Any C5 Inhibitor |
Prior to initiating C5 inhibitor treatment, 60.0% of patients were taking medications to treat PNH or associated bone marrow failure. These included folic acid (41.0% of patients), steroids (26.0%), anticoagulants (17.0%), iron (15.0%), and immunosuppressive therapy (7.0%). Prior to initiating C5 inhibitor treatment, most patients were anemic (96.0%; hemoglobin level <12 g/dL; 92.0%; hemoglobin level <10 g/dL), had elevated LDH (76.0%; LDH level ≥1.5×ULN), were transfusion dependent (89.0%), or had active bone marrow failure (24.0%; Camitta criteria; data missing in three patients). Eighty-seven percent of patients had a negative Coombs test at baseline (87/100; data missing for six patients). Of the seven patients with a documented positive Coombs test, five were C3+ and two were immunoglobulin G+. Baseline characteristics of patients by treatment subgroup are presented in Supplemental Table 1.
In the full cohort, the most frequent reasons reported by the provider for initiating treatment with a C5 inhibitor (Table 2) were symptoms of hemolysis (86.0%), LDH level ≥1.5×ULN (83.0%), high percentage of GPI-deficient cells (75.0%), previous thromboembolic event (25.0%), and hemolytic crisis requiring hospitalization (19.0%). Mean duration of therapy (from initiation of C5 inhibitor to last encounter) was 26.5±17.2 months (range 5.4–84.0). Treatment characteristics by treatment subgroup are presented in Supplemental Table 2. In the switch subgroup (n=15), the most frequently reported reasons for switching from eculizumab to ravulizumab treatment were patient preference (n=11; 73.3%), reduced treatment frequency (n=10; 66.7%), and reduced time spent getting treatment (n=8; 53.3%).
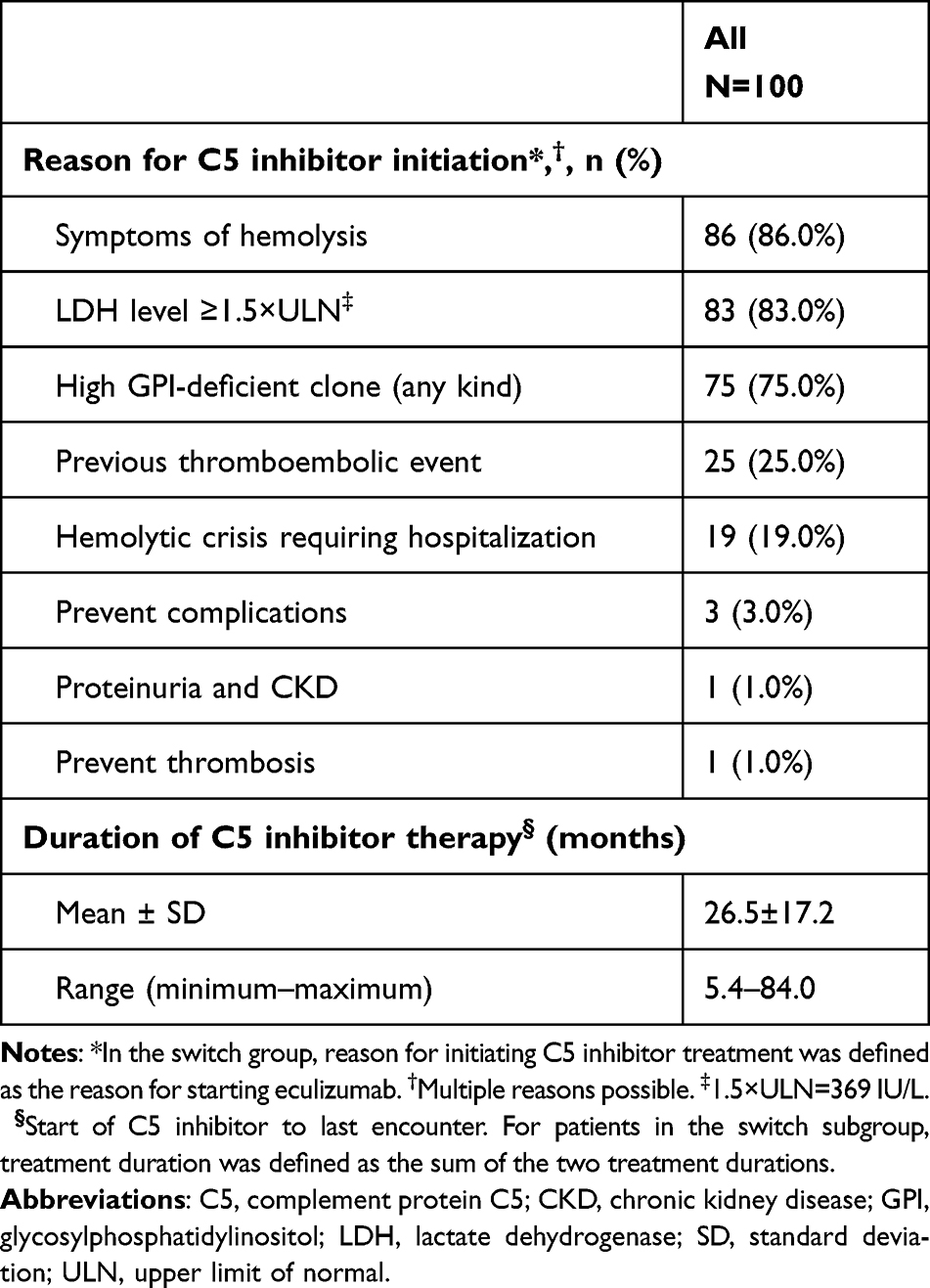 |
Table 2 Treatment Characteristics |
Of the 100 patients included, 74 were actively receiving C5 inhibitor at the time of data entry. Among the 26 patients who were not receiving active treatment, 11 patients had died. The mean time from initiation of therapy until death for these patients was 15.0 months. The mean time from discontinuation of therapy to death was 0.3 months. Five patients were no longer taking therapy owing to allo-transplantation, 4 patients were alive with disease but not receiving active therapy or palliative care, 1 patient was categorized as having normal blood count, 1 patient had changed practices, and 4 patients were lost to follow-up/unknown. The mean time from discontinuation of therapy to last seen encounter for these patients was 12.3 months.
Compared with baseline, routine laboratory measurements collected during follow-up indicated improvement in hemoglobin (p<0.001), LDH (p<0.001), aspartate transaminase (p<0.001), alanine transaminase (p<0.001), bilirubin (p<0.001), indirect bilirubin (p<0.001), ferritin (p=0.080), D-dimers (p=0.009), platelet count (p<0.001), and number of transfusions (p<0.001) (Table 3). Results by treatment subgroup are presented in Supplemental Table 3.
 |
Table 3 Change in Laboratory Values Between Baseline and Follow-Up (Routine Visits) |
During follow-up, 86 patients (86.0%; data missing for three patients) had an LDH level <1.5×ULN. Among the 11 patients (11.0%) who had an LDH level ≥1.5×ULN, 10 patients were in the eculizumab subgroup and 1 patient was in the ravulizumab subgroup. Four patients in the eculizumab group (7.5%; 4/53) met the criteria for BTH. A Coombs test was performed during the last 6 months of follow-up in 46 patients. Results were negative in all patients. One patient who had a positive Coombs test at baseline was negative at follow-up (switch subgroup). Follow-up Coombs tests were not available for the six other patients with positive baseline tests. Reticulocyte counts were available in 58 patients. Eight of these patients had an absolute reticulocyte count ≥120×109/L (four in the eculizumab subgroup, three in the ravulizumab subgroup, one in the switch subgroup).
The percentage of patients who had hemoglobin levels <12 g/dL decreased from 96.0% at baseline to 76.0% at follow-up; and the percentage of patients with hemoglobin levels <10 g/dL decreased from 92.0% at baseline to 38.0% (Figure 2A). Hemoglobin levels by treatment subgroup are presented in Figure 2B–D.
 |
Figure 2 Improvement in hemoglobin levels with C5 inhibitor treatment. Percentage of patients by hemoglobin category at baseline and during follow-up (last 6 months of treatment with eculizumab or ravulizumab). Patients needed to have been treated with a C5 inhibitor (eculizumab or ravulizumab) for at least 12 consecutive months. For patients who were switched from eculizumab to ravulizumab, patients needed to have been treated with ravulizumab for at least 6 consecutive months. (A) All patients (N=100); data missing for three patients at baseline and one patient at follow-up. (B) Eculizumab subgroup (n=53); data missing for two patients at baseline and one patient at follow-up. (C) Ravulizumab subgroup (n=35). (D) Switch subgroup (n=15); data missing for one patient at baseline. |
Among the 38 patients who had hemoglobin levels <10 g/dL during follow-up, 9 patients (23.7%) had an LDH level ≥1.5×ULN (all in the eculizumab subgroup) of whom 4 patients, as previously mentioned, met the BTH criteria. Ten out of 38 patients (26.3%) met the Camitta criteria; four patients had a hemoglobin level between 6 and <8 g/dL and six patients had a hemoglobin level between 8 g/dL and <10 g/dL. Only 3 of these 10 patients met the three criteria of having hemoglobin levels <10 g/dL, LDH level ≥1.5×ULN and the Camitta criteria; the remaining 7 patients had a hemoglobin level <10 g/dL and a neutrophil count <1.5×109/L.
The percentage of patients who were transfusion dependent decreased from 89.0% at baseline to 35.0% during follow-up (24 patients [45.3%] in the eculizumab subgroup and 11 patients [34.4%] in the ravulizumab subgroup). Only one patient, who was not transfusion dependent at baseline, became transfusion dependent; this patient had neither aplastic anemia nor myelodysplastic syndrome. Among the 35 patients who were transfusion dependent during follow-up, 10 patients (28.6%) had an LDH level ≥1.5×ULN (all in the eculizumab subgroup) and 8 patients (22.9%) fulfilled Camitta criteria for active bone marrow failure. Among the 35 patients who were transfusion dependent during follow-up, mean reticulocyte count was 25.6±30.4×109/L (median 13.9 [IQR: 8.5–33.9] ×109/L) and was <60×109/L in 21 patients (60.0%; 21/35; data missing for 8 patients). Only 1 patient of 35 had a reticulocyte count ≥120×109/L; this patient had an LDH level <1.5×ULN. Coombs tests were either negative (n=22) or not performed (n=13).
Fifty-eight patients were evaluable for the presence of EVH. One patient met the criteria for EVH (eculizumab subgroup); this patient had an LDH level <1.5×ULN. In the sensitivity analyses, the number of patients with EVH increased to two (one patient in the eculizumab subgroup and one patient in the ravulizumab subgroup) when the requirement for transfusion was removed from the definition but did not change when the hemoglobin level cut-off was changed to <10 g/dL or when the reticulocytosis cut-off was changed to ≥100×109/L or ≥150×109/L.
Discussion
We sought to evaluate the prevalence of anemia in a real-world, observational, medical chart review of 100 patients with PNH in the USA. Treatment with a C5 inhibitor improved anemia, transfusion dependence, and levels of LDH, platelets, and D-dimers. During follow-up (>6 months), LDH levels were <1.5×ULN in 86.0% of patients. The percentage of patients with hemoglobin levels <10 g/dL decreased from 92.0% at baseline to 38.0% at follow-up; and the percentage of patients who were transfusion dependent decreased from 89.0% to 35.0%. Among the 35 patients who were transfusion dependent, 21 patients had a reticulocyte count <60×109/L, 8 patients met Camitta criteria for bone marrow failure, and 1 patient met the criteria for clinically evident EVH. These results underscore the fact that in PNH, a range of hematologic causes needs to be considered when evaluating hemoglobin <10 g/dL and transfusion dependence in the setting of treatment with a C5 inhibitor.2,27
In prior publications, small sample sizes and inconsistent definitions have resulted in the reported incidence of clinically significant EVH ranging from 0% if defined as positive direct Coombs test plus at least one transfusion plus no aplastic anemia,26 to 12% if defined as C3+ red blood cells plus less than 50% reduction in transfusion dependence plus no aplastic anemia,25 to 76% if defined as a positive direct antiglobulin test plus at least one transfusion.24 Because there is no formal or agreed upon definition for EVH, we used the definition adopted in the danicopan (factor D inhibitor) phase 3 clinical trial that was specifically designed to evaluate treatment of the subpopulation of patients with PNH who develop EVH when treated with eculizumab or ravulizumab.34 In the present study, only one patient (1.7%; 1/58 evaluable patients) met the definition for clinically evident EVH. In the sensitivity analysis, removing transfusion dependence as a criterion increased the number of patients with EVH to two, whereas varying the reticulocytosis and the hemoglobin cut-offs had no impact on results. Thus, although more research is needed, our study, which takes a broad survey approach, suggests that the incidence of clinically evident EVH is likely to be lower than originally suspected.
Extravascular hemolysis is a secondary mechanistic consequence of treatment with C5 inhibitors. Because red blood cells are no longer rapidly destroyed by the membrane attack complex, C3 fragment deposition on the cell membrane can occur and, potentially, opsonization and extravascular clearance.24,25,35 The clinical impact of C3 deposition on red blood cells in eculizumab-treated patients remains unclear as the percentage of C3 deposition does not correlate clearly with levels of hemoglobin or transfusion dependence.25,29 Indeed, measuring C3 deposition and assessing patients using a Coombs test has not been included in recent recommendations on how to monitor patients with PNH on a complement inhibitor.27
Thrombosis is the leading cause of mortality in PNH.36 Long-term studies have shown that effective terminal complement inhibition significantly reduces and controls the terminal complement activity that leads to IVH, thrombotic risk, and mortality.18,21,22,37 Interventional studies are assessing proximal inhibitors to determine whether blocking of the complement pathway upwards of C5 has positive consequences for those patients who may be experiencing clinically evident EVH. Danicopan, a factor D inhibitor, is being evaluated in a phase 3 trial as an add-on therapy for patients with PNH treated with eculizumab or ravulizumab. A combination of factor D and C5 inhibitors will target the proximal and terminal complement pathways, and may offer a strategy to reduce EVH while maintaining established clinical benefits conferred by terminal complement inhibition.34 Recently, the findings of a 16-week phase 3 study (PEGASUS), which evaluated the efficacy of C3 inhibitor pegcetacoplan in a subset of eculizumab-treated patients who remained anemic and/or transfusion dependent, were published, demonstrating a significant improvement in hemoglobin level and resulting in US Food and Drug Administration approval.38 It should be noted that patients enrolled in the PEGASUS study do not represent a broad population of patients with PNH and, as such, the conclusions drawn should be interpreted accordingly and cannot be extended to the full PNH population.39–41 Additional studies will be needed to evaluate the long-term safety and efficacy of proximal complement inhibitors.38
Baseline characteristics prior to C5 inhibition suggest that the cohort of patients in this study was representative of patients encountered in real-world clinical practice. PNH disease burden was high: 96.0% of patients were anemic (hemoglobin level <12 g/dL), 76.0% of patients had an LDH level ≥1.5×ULN, 89.0% of patients were transfusion dependent, while most patients did not have severe bone marrow failure (no patients had a platelet count below 45×109/L at baseline). These data are consistent with previously published studies, which also reported high disease burden in untreated patients.42 Moreover, treatment response was consistent with that reported in published studies.15,19,37,43,44
This real-world study was a retrospective chart review; therefore, accuracy and completeness of collected data was limited by the quality of data in the medical charts. In addition, this study also informs that prevailing standards of care and work-up in real-world settings may not always be met (eg missing absolute reticulocyte count data) which implies a need for ongoing education in the management of this disease. Absolute reticulocyte count during follow-up was not reported in 42.0% of patients. Because this variable informed the number of patients with EVH, EVH was assessed in 58 patients. In addition, our analysis focused on hematological parameters; other reasons for anemia, such as folate and relative erythropoietin deficiency, were not evaluated. As the analysis focused on laboratory results, data on thromboembolic events, treatment emergent adverse events, and cause of death were not collected. Lastly, because physician participation was self-selected and the randomness of patient selection was not monitored, there is a possibility of physician-introduced bias.
Conclusion
In this first, large, real-world study using clinical data in which eculizumab or ravulizumab improved hematologic parameters by reducing anemia, IVH, and transfusion dependence in patients with PNH, clinically evident EVH was uncommon (1.7%; 1/58). Concomitant bone marrow failure was identified as a major contributor to patients with PNH having hemoglobin <10 g/dL and transfusion dependence, and concomitant treatment may be required for these patients.45 Altogether, these data underscore the fact that evaluating patients for all causes of anemia, including active bone marrow failure, is important for the comprehensive management of patients with PNH on a complement inhibitor therapy.
Abbreviations
BTH, breakthrough hemolysis; C, complement protein; EVH, extravascular hemolysis; GPI, glycosylphosphatidylinositol; IVH, intravascular hemolysis; IQR, interquartile range; LDH, lactate dehydrogenase; PNH, paroxysmal nocturnal hemoglobinuria; ULN, upper limit of normal.
Data Sharing Statement
Alexion will consider requests for disclosure of clinical study participant-level data provided that participant privacy is assured through methods like data de-identification, pseudonymization, or anonymization (as required by applicable law), and if such disclosure was included in the relevant study informed consent form or similar documentation. Qualified academic investigators may request participant-level clinical data and supporting documents (statistical analysis plan and protocol) pertaining to Alexion-sponsored studies. Further details regarding data availability and instructions for requesting information are available in the Alexion Clinical Trials Disclosure and Transparency Policy at https://alexionclinicaltrials.com/Disclosure-and-Transparency-Policy. Link to Data Request Form (https://alexion.com/contact-alexion/medical-information).
Ethics Approval and Consent to Participate
The study was conducted in accordance with International Society for Pharmacoepidemiology Guidelines for Good Pharmacoepidemiology Practices and applicable regulatory requirements. A waiver from the WCG Institutional Review Board under Privacy Rule: 45 CFR 164.512 was received prior to the start of the study. Patient-protected health information was kept confidential at all times and in accordance with the Health Insurance Portability and Accountability Act of 1996; no data that could directly identify patients were extracted. As such, patient informed consent was not needed. Providers were compensated at fair market value for the time needed to complete data abstraction and quality control procedures.
Acknowledgments
We thank Talia Miller (employee of Cardinal Health) for her contribution to the study, Hélène Dassule, PhD (employee of Alexion, AstraZeneca Rare Disease), for her medical writing support, and Oxford PharmaGenesis for their help with manuscript copy editing and formatting.
Author Contributions
All authors made a significant contribution to the work reported, have substantially revised and critically reviewed the article; have agreed on the journal to which the article has been submitted, have reviewed and agreed all versions of the article before submission and publication, agree to be accountable and take responsibility for the contents of the article.
Funding
This study was sponsored by Alexion Pharmaceuticals, Inc., Boston, MA (now Alexion, AstraZeneca Rare Disease).
Disclosure
JS has received research support from Alexion, AstraZeneca Rare Disease, BMS, CTI, Novartis, Kartos Pharma and Stemline Therapeutics; consulting fees from Novartis; has been a speaker for Incyte, Alexion, BMS and Sanofi. She also owns stocks from Abbvie, chairs the Data and Safety Monitoring Board (DSMB) for NS Pharma and is a member of the DSMB for Apellis. AG and JK are employees of Cardinal Health Inc. YP, IT, AH and JRS are employees and shareholders of Alexion, AstraZeneca Rare Disease. DA provides consulting to Alexion, AstraZeneca Rare Disease. The authors report no other conflicts of interest in this work.
References
1. Takeda J, Miyata T, Kawagoe K, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73(4):703–711. doi:10.1016/0092-8674(93)90250-t
2. Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014;124(18):2804–2811. doi:10.1182/blood-2014-02-522128
3. Hillmen P, Bessler M, Mason PJ, Watkins WM, Luzzatto L. Specific defect in N-acetylglucosamine incorporation in the biosynthesis of the glycosylphosphatidylinositol anchor in cloned cell lines from patients with paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci USA. 1993;90(11):5272–5276. doi:10.1073/pnas.90.11.5272
4. Hillmen P, Hows JM, Luzzatto L. Two distinct patterns of glycosylphosphatidylinositol (GPI) linked protein deficiency in the red cells of patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1992;80(3):399–405. doi:10.1111/j.1365-2141.1992.tb08151.x
5. Holguin MH, Fredrick LR, Bernshaw NJ, Wilcox LA, Parker CJ. Isolation and characterization of a membrane protein from normal human erythrocytes that inhibits reactive lysis of the erythrocytes of paroxysmal nocturnal hemoglobinuria. J Clin Invest. 1989;84(1):7–17. doi:10.1172/JCI114172
6. Moyo VM, Mukhina GL, Garrett ES, Brodsky RA. Natural history of paroxysmal nocturnal haemoglobinuria using modern diagnostic assays. Br J Haematol. 2004;126(1):133–138. doi:10.1111/j.1365-2141.2004.04992.x
7. Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. 2005;293(13):1653–1662. doi:10.1001/jama.293.13.1653
8. SOLIRIS® (Eculizumab) Injection, for Intravenous Use. Alexion Pharmaceuticals, Boston MA. 11/2020.
9. ULTOMIRIS® (Ravulizumab-cwvz) Injection, for Intravenous Use. Alexion Pharmaceuticals, Boston MA. 10/2019.
10. Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233–1243. doi:10.1056/NEJMoa061648
11. Hillmen P, Hall C, Marsh JC, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2004;350(6):552–559. doi:10.1056/NEJMoa031688
12. Hillmen P, Muus P, Duhrsen U, et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;110(12):4123–4128. doi:10.1182/blood-2007-06-095646
13. Hill A, Rother RP, Wang X, et al. Effect of eculizumab on haemolysis-associated nitric oxide depletion, dyspnoea, and measures of pulmonary hypertension in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2010;149(3):414–425. doi:10.1111/j.1365-2141.2010.08096.x
14. Hillmen P, Elebute M, Kelly R, et al. Long-term effect of the complement inhibitor eculizumab on kidney function in patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2010;85(8):553–559. doi:10.1002/ajh.21757
15. Hillmen P, Muus P, Roth A, et al. Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2013;162(1):62–73. doi:10.1111/bjh.12347
16. Brodsky RA, Young NS, Antonioli E, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111(4):1840–1847. doi:10.1182/blood-2007-06-094136
17. Ninomiya H, Obara N, Chiba S, et al. Interim analysis of post-marketing surveillance of eculizumab for paroxysmal nocturnal hemoglobinuria in Japan. Int J Hematol. 2016;104(5):548–558. doi:10.1007/s12185-016-2065-4
18. Loschi M, Porcher R, Barraco F, et al. Impact of eculizumab treatment on paroxysmal nocturnal hemoglobinuria: a treatment versus no-treatment study. Am J Hematol. 2016;91(4):366–370. doi:10.1002/ajh.24278
19. Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133(6):530–539. doi:10.1182/blood-2018-09-876136
20. Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study. Blood. 2019;133(6):540–549. doi:10.1182/blood-2018-09-876805
21. Schrezenmeier H, Kulasekararaj A, Mitchell L, et al. One-year efficacy and safety of ravulizumab in adults with paroxysmal nocturnal hemoglobinuria naive to complement inhibitor therapy: open-label extension of a randomized study. Ther Adv Hematol. 2020;11:2040620720966137. doi:10.1177/2040620720966137
22. Kulasekararaj AG, Hill A, Langemeijer S, et al. One-year outcomes from a phase 3 randomized trial of ravulizumab in adults with paroxysmal nocturnal hemoglobinuria who received prior eculizumab. Eur J Haematol. 2021;106(3):389–397. doi:10.1111/ejh.13564
23. Brodsky RA, de Latour R Peffault, ST Rottinghaus, et al. Characterization of breakthrough hemolysis events observed in the phase 3 randomized studies of ravulizumab versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria. Haematologica. 2021;106(1):230–237. doi:10.3324/haematol.2019.236877
24. Hill A, Rother RP, Arnold L, et al. Eculizumab prevents intravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria and unmasks low-level extravascular hemolysis occurring through C3 opsonization. Haematologica. 2010;95(4):567–573. doi:10.3324/haematol.2009.007229
25. Risitano AM, Notaro R, Marando L, et al. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood. 2009;113(17):4094–4100. doi:10.1182/blood-2008-11-189944
26. Subias Hidalgo M, Martin Merinero H, Lopez A, et al. Extravascular hemolysis and complement consumption in paroxysmal nocturnal hemoglobinuria patients undergoing eculizumab treatment. Immunobiology. 2017;222(2):363–371. doi:10.1016/j.imbio.2016.09.002
27. Kulasekararaj AG, Brodsky RA, Hill A. Monitoring of patients with paroxysmal nocturnal hemoglobinuria on a complement inhibitor. Am J Hematol. 2021;96(7):E232–E235. doi:10.1002/ajh.26176
28. Debureaux PE, Kulasekararaj AG, Cacace F, et al. Categorizing hematological response to eculizumab in paroxysmal nocturnal hemoglobinuria: a multicenter real-life study. Bone Marrow Transplant. 2021;56(10):2600–2602. doi:10.1038/s41409-021-01372-0
29. Risitano AM, Notaro R, Luzzatto L, Hill A, Kelly R, Hillmen P. Paroxysmal nocturnal hemoglobinuria–hemolysis before and after eculizumab. N Engl J Med. 2010;363(23):2270–2272. doi:10.1056/NEJMc1010351
30. Patriquin CJ, Kiss T, Caplan S, et al. How we treat paroxysmal nocturnal hemoglobinuria: a consensus statement of the Canadian PNH Network and review of the national registry. Eur J Haematol. 2019;102(1):36–52. doi:10.1111/ejh.13176
31. Dezern AE, Borowitz MJ. ICCS/ESCCA consensus guidelines to detect GPI-deficient cells in paroxysmal nocturnal hemoglobinuria (PNH) and related disorders part 1 - clinical utility. Cytometry B Clin Cytom. 2018;94(1):16–22. doi:10.1002/cyto.b.21608
32. Killick SB, Bown N, Cavenagh J, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016;172(2):187–207. doi:10.1111/bjh.13853
33. Schubert J, Röth A, Bettelheim P, et al. Paroxysmale nächtliche Hämoglobinurie (PNH). Available from: https://www.onkopedia.com/de/onkopedia/guidelines/paroxysmale-naechtliche-haemoglobinurie-pnh/@@guideline/html/index.html.
34. ClinicalTrials.gov. Danicopan as add-on therapy to a C5 inhibitor in paroxysmal nocturnal hemoglobinuria (PNH) participants who have clinically evident extravascular hemolysis (EVH)(ALPHA). Available from: https://clinicaltrials.gov/ct2/show/NCT04469465?term=danicopan&draw=2&rank=5.
35. Sica M, Rondelli T, Ricci P, De Angioletti M, Risitano AM, Notaro R. Eculizumab treatment: stochastic occurrence of C3 binding to individual PNH erythrocytes. J Hematol Oncol. 2017;10(1):126. doi:10.1186/s13045-017-0496-x
36. Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333(19):1253–1258. doi:10.1056/NEJM199511093331904
37. Kelly RJ, Hill A, Arnold LM, et al. Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood. 2011;117(25):6786–6792. doi:10.1182/blood-2011-02-333997
38. Hillmen P, Szer J, Weitz I, et al. Pegcetacoplan versus eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2021;384(11):1028–1037. doi:10.1056/NEJMoa2029073
39. Gurnari C, Pagliuca S, Maciejewski JP. Pegcetacoplan versus eculizumab in PNH. N Engl J Med. 2021;385(18):1725. doi:10.1056/NEJMc2106424
40. Kulasekararaj AG, Gandhi S, Brodsky RA. Pegcetacoplan versus eculizumab in PNH. N Engl J Med. 2021;385(18):1724–1725. doi:10.1056/NEJMc2106424
41. Ueda Y, Takamori H, Nishimura JI. Pegcetacoplan versus eculizumab in PNH. N Engl J Med. 2021;385(18):1723–1724. doi:10.1056/NEJMc2106424
42. Schrezenmeier H, Roth A, Araten DJ, et al. Baseline clinical characteristics and disease burden in patients with paroxysmal nocturnal hemoglobinuria (PNH): updated analysis from the international PNH registry. Ann Hematol. 2020;99(7):1505–1514. doi:10.1007/s00277-020-04052-z
43. Nakayama H, Usuki K, Echizen H, Ogawa R, Orii T. Eculizumab dosing intervals longer than 17 days may be associated with greater risk of breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria. Biol Pharm Bull. 2016;39(2):285–288. doi:10.1248/bpb.b15-00703
44. Peffault de Latour R, Fremeaux-Bacchi V, Porcher R, et al. Assessing complement blockade in patients with paroxysmal nocturnal hemoglobinuria receiving eculizumab. Blood. 2015;125(5):775–783. doi:10.1182/blood-2014-03-560540
45. Griffin M, Kulasekararaj A, Gandhi S, et al. Concurrent treatment of aplastic anemia/paroxysmal nocturnal hemoglobinuria syndrome with immunosuppressive therapy and eculizumab: a UK experience. Haematologica. 2018;103(8):e345–e347. doi:10.3324/haematol.2017.183046
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.